

See Sastre et al. (doi:10.1093/brain/awy225) for a scientific commentary on this article.
Traumatic brain injury (TBI) is a risk factor for subsequent neurodegenerative disease. Zanier et al. report that single severe TBI induces tau pathology in humans and mice. Studies in the latter provide evidence that this pathology is self-propagating and can be transmitted between animals, causing brain dysfunction like a prion.
Keywords: traumatic brain injury, tau pathology, prion, neurological dysfunction, neurodegeneration
Abstract
Traumatic brain injury is a risk factor for subsequent neurodegenerative disease, including chronic traumatic encephalopathy, a tauopathy mostly associated with repetitive concussion and blast, but not well recognized as a consequence of severe traumatic brain injury. Here we show that a single severe brain trauma is associated with the emergence of widespread hyperphosphorylated tau pathology in a proportion of humans surviving late after injury. In parallel experimental studies, in a model of severe traumatic brain injury in wild-type mice, we found progressive and widespread tau pathology, replicating the findings in humans. Brain homogenates from these mice, when inoculated into the hippocampus and overlying cerebral cortex of naïve mice, induced widespread tau pathology, synaptic loss, and persistent memory deficits. These data provide evidence that experimental brain trauma induces a self-propagating tau pathology, which can be transmitted between mice, and call for future studies aimed at investigating the potential transmissibility of trauma associated tau pathology in humans.
See Sastre et al. (doi:10.1093/brain/awy225) for a scientific commentary on this article.
Introduction
Traumatic brain injury (TBI) is a recognized risk factor for delayed neurodegeneration and dementia, particularly chronic traumatic encephalopathy (CTE). The neuropathological picture that is emerging with late survival from TBI is of a complex of pathologies featuring abnormalities in tau, amyloid-beta, neuronal loss, axonal degeneration, neuroinflammation and blood–brain barrier disruption (Johnson et al., 2012, 2013; Hay et al., 2016). Of particular note is the emergence of hyperphosphorylated forms of the microtubule-associated protein tau in a stereotypical pattern and distribution regarded as pathognomonic of CTE (McKee et al., 2016). Specifically, in its early stage, CTE is recognized through the demonstration of clusters of perivascular neurons and glia containing hyperphosphorylated tau (P-tau), typically with patchy distribution and concentrated towards the depths of cortical sulci (Geddes et al., 1999; McKee et al., 2014, 2016; Mez et al., 2017). There is a suggestion of hierarchical progression of pathology from these initially focal and patchy cortical P-tau deposits to more widespread cortical, medial temporal and subcortical grey matter in late-stage disease (McKee et al., 2014). However, the mechanisms by which this initially localized cortical pathology progresses to more widespread and generalized disease are still not known.
In Alzheimer’s disease, tau pathology appears to progress along neural networks, suggesting transcellular spread of tau through neural connections (Goedert et al., 2017). In vivo studies have described the spreading of Alzheimer’s disease-related tau pathology from local sites to distant regions of the brain (Clavaguera et al., 2009, 2013; de Calignon et al., 2012; Ahmed et al., 2014; Guo et al., 2016), and it has been suggested that tau aggregates—or seeds—serve as the agent of this spread, transmitting the aggregated/hyperphosphorylated state from cell to cell in a prion-like fashion (Prusiner, 2012; Clavaguera et al., 2013; Iba et al., 2013, 2015; Woerman et al., 2016). Prions are proteinaceous infectious agents consisting of misfolded prion protein (PrPSc) that propagate by imposing their abnormal structures on host-encoded cellular prion protein (PrPC) (Prusiner, 1982). As PrPSc prions colonize the affected patient, they perturb the function of the nervous system, leading to motor and neurophysiological deficits, cognitive decline, and ultimately death (Chiesa, 2015). Whether TBI generates misfolded or aggregated forms of tau protein that propagate by inducing misfolding of native tau in injured tissue and can be transmitted from one individual to another (tau prions), is not clear (Prusiner, 2012; Mudher et al., 2017). We hypothesized that late P-tau deposition after severe TBI would show prion behaviour, and addressed this hypothesis in sequential steps. First, we set out to demonstrate and characterize the deposition of P-tau late after severe TBI as distinct from P-tau deposition in ‘normal’ aged human controls. Second, we sought the emergence of self-propagating P-tau pathology in wild-type mice subjected to severe TBI that replicated P-tau pathology observed in humans long after a single severe TBI. Finally, we went on to establish whether this TBI-induced tau pathology could be transmitted between mice and cause neurological deficits, satisfying the requirements for a bona fide tau prion.
Materials and methods
Study design
The aim of the study was to test whether a single severe TBI was able to induce a self-propagating tau pathology. First, we analysed the deposition of P-tau late after single TBI in humans. Next, we characterized the temporal evolution of tau pathology in wild-type mice subjected to severe controlled cortical impact. Finally, we tested whether authentic tau prions are generated after trauma by attempting the transmission of tauopathy to naïve mice by intracerebral inoculation of brain homogenates from TBI mice.
Male C57BL/6J mice (8 to 9 weeks of age) from Envigo were used in this study. The animals were housed four per cage at controlled temperature (22 ± 2°C) and relative humidity (60 ± 5%) with a 12/12-h light/dark cycle and free access to pelleted food and water.
All animal experiments were designed in accordance with the ARRIVE guidelines (Kilkenny et al., 2010), with a commitment to refinement, reduction, and replacement, minimizing the numbers of mice, while using biostatistics to optimize mouse numbers [as in our previously published work using the mouse TBI model (Zanier et al., 2016) and prion inoculation in mice (Bouybayoune et al., 2015)]. Thus, for statistical validity, we used 8 to 12 mice for the behavioural tests, and three to eight mice for biochemical analysis and histology. The mice were assigned to the different experimental groups using randomization lists (www.randomizer.org). Inoculations, behavioural and histological assessments were done by researchers blinded to the experimental groups.
Ethics statement
Human tissue was obtained from the Glasgow TBI Archive of the Department of Neuropathology, Queen Elizabeth University Hospital, Glasgow, UK. Tissue samples were acquired at routine diagnostic autopsy, with approval from the Greater Glasgow and Clyde Biorepository Governance Committee for their use in research. Procedures involving animals and their care were conducted in conformity with the institutional guidelines at the IRCCS – Mario Negri Institute for Pharmacological Research in compliance with national (D.lgs 26/2014; Authorization n. 19/2008-A issued March 6, 2008 by Ministry of Health) and international laws and policies (EEC Council Directive 2010/63/UE; the NIH Guide for the Care and Use of Laboratory Animals, 2011 edition). They were reviewed and approved by the Mario Negri Institute Animal Care and Use Committee that includes ad hoc members for ethical issues, and by the Italian Ministry of Health (Decreto no. D/07/2013-B and 301/2017-PR). Animal facilities meet international standards and are regularly checked by a certified veterinarian who is responsible for health monitoring, animal welfare supervision, experimental protocols and review of procedures.
Human studies
At the time of the original diagnostic autopsy, whole brains were immersion-fixed in 10% formal saline for a minimum of 3 weeks, then the specimens were examined, sampled and processed to paraffin tissue blocks. For this study, 15 patients surviving a year or more from single moderate or severe, closed TBI (defined as Glasgow Coma Scale at presentation <13) and 15 age-matched controls with no history of TBI or of known neurological disease were randomly selected from the comprehensive databases of the Glasgow TBI Archive by a laboratory research assistant with no knowledge of study purpose or design or of the pathologies in each of the TBI cases and controls selected. Full demographic information for late TBI cases and controls is provided in Supplementary Table 1. Information on TBI and controls’ history was obtained from review of diagnostic post-mortem and/or forensic reports. The TBI survival cases and the age-matched controls had no recorded history of exposure to contact sports or military service. The mechanism of injury is summarized in Supplementary Table 1. None were penetrating. From review of the original hospital records, at presentation, intracerebral haemorrhage was present in five cases, subdural a further seven, with three cases having no evidence of intracranial haemorrhage. Following the original TBI, all cases were discharged from hospitalization following recovery and, ultimately, died from causes of death unrelated to TBI. None were in a persistent vegetative state prior to death. Each case was sampled and assessed for this study as standardized regions, no matter the injury severity or location. As this study accesses archival tissue blocks, there were necessary restrictions on availability of sampled regions. Nevertheless, the standardized sampling protocols used at the original diagnostic autopsy included a coronal slice of the cerebral hemispheres at mid-thalamic level from which tissue blocks from multiple brain regions were selected for the purposes of this study to include: the corpus callosum with adjacent cingulate gyrus; the medial temporal lobe to include the hippocampus and entorhinal cortex; the insular cortex and the thalamus. From these tissue blocks 8 μm sections were prepared for immunohistochemistry procedures. After deparaffinization and rehydration, endogenous peroxidase was quenched by immersing sections in 3% aqueous H2O2 for 15 min. Thereafter, heat-induced antigen retrieval was performed using a microwave pressure cooker for 8 min in preheated 0.1 M Tris EDTA buffer (pH 8), followed by blocking in 50 ml of normal horse serum (Vector Labs) per 5 ml of OptiMax™ buffer (BioGenex) for 30 min. The sections were then incubated in antibody to phosphorylated tau (PHF1; 1:1000; gift from P. Davies, Feinstein Institute for Medical Research, Manhasset, NY, USA) at 4°C for 20 h prior to washing and application of biotinylated secondary antibody for 30 min, followed by an avidin biotin complex, as per the manufacturer’s instructions (Vectastain Universal Elite Kit, Vector Labs). The target antigen was visualized using the DAB peroxidase substrate kit (Vector Labs), with the sections then counterstained with haematoxylin. Control sections (positive and negative) for each antibody were run in parallel with the test sections to confirm antibody specificity. Sections were viewed, blind to whether they were TBI or control, using a Leica DMRB light microscope (Leica Microsystems) and the extent and distribution of tau pathology was assessed and semiquantitatively scored as: score 0, where no tau pathology was present; score 1, PHF1 pathology comprising immunoreactive neurofibrillary tangles and neurites of sparse to moderate density in the transentorhinal cortex with or without small numbers in the CA1 sector of the hippocampus; score 2, PHF1-immunoreactive neurofibrillary tangles and neurites as described for score 1 together with neurofibrillary tangles extending into the fusiform gyrus (lateral to entorhinal cortex) and sparse tangles in the cortex (cingulate/insular blocks); and score 3, contained PHF1-immunoreactive neurofibrillary tangles and neurites in more widespread sectors of the hippocampus and subiculum, with extensive medial temporal, cingulate and insular cortical involvement.
Mouse model of traumatic brain injury
The controlled cortical impact brain injury mouse model used in this study replicates both the mechanical forces and the main secondary injury processes observed in severe TBI patients with brain contusion and is associated with clinically-relevant behavioural and histopathological outcomes (Smith et al., 1995; Pischiutta et al., 2018). Mice were anaesthetized by isoflurane inhalation (induction 3%; maintenance 1.5%) in an N2O/O2 (70%/30%) mixture and placed in a stereotaxic frame. Rectal temperature was maintained at 37°C. Mice were then subjected to craniectomy followed by induction of controlled cortical impact brain injury as previously described (Zanier et al., 2016). Briefly, the injury was induced using a 3-mm rigid impactor driven by a pneumatic piston rigidly mounted at an angle of 20° from the vertical plane and applied to the exposed dura mater, between bregma and lambda, over the left parieto-temporal cortex (antero-posteriority: −2.5 mm, laterality: −2.5 mm), at an impactor velocity of 5 m/s and deformation depth 1 mm, resulting in a severe level of injury (Smith et al., 1995). The craniotomy was then covered with a cranioplasty and the scalp sutured. Sham mice received identical anaesthesia and surgery without brain injury.
Transmission studies
Brain homogenates (10% w/v in PBS) from the ipsi- and contralateral cerebral cortex (including all the cortical tissue above the rhinal fissure) of mice 12 months from TBI or sham injury were inoculated in the hippocampus (A/P, −2.5 mm from bregma; L, ±2.0 mm; D/V, −1.8 mm) and overlying cerebral cortex (A/P, −2.5 mm from bregma; L, ±2.0 mm; D/V, −0.8 mm). Inocula were from single brains. The TBI brain homogenates showing the highest amounts of P-tau by western blot were chosen for inoculation. Mice were injected bilaterally (2.5 µl each, injection speed of 1.25 μl/min).
Novel object recognition
Mice were tested in an open-square grey arena (40 × 40 cm), 30 cm high, with the floor divided into 25 squares by black lines. The following objects were used: a black plastic cylinder (4 × 5 cm), a glass vial with a white cap (3 × 6 cm) and a metal cube (3 × 5 cm). The task started with a habituation trial during which mice were placed in the empty arena for 5 min and their movements were recorded as the number of line-crossings. The next day, mice were placed in the same arena containing two identical objects (familiarization phase). Exploration was recorded in a 10-min trial by an investigator blinded to the experimental group. Sniffing, touching and stretching the head toward the object at a distance of no more than 2 cm were scored as object investigation. Twenty-four hours later (test phase) mice were placed in the arena containing two objects: one identical to one of the objects presented during the familiarization phase (familiar object), and a new, different one (novel object), and the time spent exploring the two objects was recorded for 10 min. Memory was expressed as a discrimination index, i.e. the time spent exploring the novel object minus the time spent exploring the familiar one, divided by the total time spent exploring both objects; the higher the discrimination index, the better the performance.
Immunohistochemistry and immunofluorescence
Mice were euthanized by cervical dislocation, brains were removed and fixed in 10% formalin for 24 h, dehydrated in graded ethanol solutions, cleared in xylene, and embedded in paraffin. Serial sections (5 μm thick) were cut onto glass slides and boiled in citrate buffer (10 mM pH 6.0, Dako) for antigen retrieval, followed by incubation with the following anti-tau monoclonal antibodies: AT8 (phospho-tau Ser202/Thr205; Thermo Fisher; 1:1000), AT180 (phospho-tau Thr231; Thermo Fisher; 1:1000), PHF1 (Ser396/Ser404; from P. Davies; 1:4000) and MC1 (aa 312–322; from P. Davies; 1:100). AT8 and AT180 immunostaining was visualized using the ARK kit (Dako). For secondary staining of MC1 and PHF1, slices were incubated with biotinylated goat anti-mouse IgG1 in Superblock Solution (Pierce), followed by visualization with the TSA Biotin System (Perkin Elmer) and DAB (diaminobenzidine) Substrate Chromogen System (Dako).
Slices were examined by an Olympus BX-61 Virtual Stage microscope, interfaced with VS-ASW-FL software (Olympus). For quantification of the AT8-positive staining in TBI mice regions of interest were selected according to the Paxinos atlas (Paxinos and Franklin, 2004) in coronal (bregma −1.6 mm) and sagittal (lateral 1.5 mm) sections. In TBI mice the ipsilateral cortex was analysed over an area 1 mm deep from the edge of the contusion (Pischiutta et al., 2018) and the contralateral from midline to the entorhinal cortex. In the inoculated mice, regions of interest included whole cortex, hippocampus, thalamus and cerebellum. The same regions of interest were selected in sham mice. Images were acquired using a 60× oil objective with pixel size 0.115 μm. Acquisition was done over 5-μm thick stacks, with step size 1 μm. The different focal planes were merged into a single stack by mean intensity projection to ensure consistent focus throughout the sample. Images were analysed using Fiji software (http://fiji.sc/Fiji).
The digital separation of DAB staining from haematoxylin signal was done by colour deconvolution (http://www.mecourse.com/landinig/software/cdeconv/cdeconv.html) and the DAB channel was used for quantification. To ensure consistency of the segmentation in the image processing, we subtracted the intensity background from the image calculated by averaging non-specific signal in identified region of interest. Specific signal after the image processing was computed as positive pixels/total pixels, and reported as the percentage stained area for statistical analysis.
For immunofluorescence, brain sections on glass slides were boiled in citrate buffer and incubated in blocking solution with the following primary antibodies: anti-drebrin (Fitzgerald, 1:200) an actin-binding protein enriched in dendritic spines, (Sekino et al., 2017) and anti-vesicular glutamate transporter-1 (VGLUT-1, Synaptic System, 1:6000), for analysis of the presynaptic compartment (Senatore et al., 2012). Slices were then incubated with biotinylated secondary antibody (Vecstain, 1:500) and streptavidin 647 (Molecular Probes, 1:500), and examined by confocal microscopy (FV-500 Olympus) acquiring images in two different areas of CA1 field of the hippocampus using a 60× oil objective. Z-stack images were acquired: 0.2 μm Z-distance, 3-μm thick confocal z-stacks (optical section depth 0.2 μm, 21 sections/z-stack) and 1024 × 1024 pixel resolution. Confocal images were deconvoluted (CellP) and puncta were identified with Imaris software using the ‘quality’ parameter (Heise et al., 2016). Only puncta stringently conforming with the quality parameter entered analysis.
Biochemical analysis
Frozen brain tissue was homogenized in 10 volumes of phosphate-buffered saline (PBS). Homogenates were aliquoted and stored at −80°C until use. Western blot analysis of total brain homogenates with monoclonal antibody AT8 was done on heat-stable protein fractions prepared according to Petry et al. (2014) to avoid the interfering signal due to endogenous immunoglobulins. Proteins (50 µg) were separated on NuPAGE 4–12% gels (Thermo Scientific) and blotted onto nitrocellulose membranes (Hybond, GE Healthcare). After blocking with 5% dry milk for 1 h at room temperature, membranes were incubated overnight at 4°C with anti-total tau (1:10 000, Dako) or AT8 (1:500, Thermo Scientific), then with HRP-conjugated secondary antibodies (Santa Cruz). Peroxidase activity was detected using an ECL (Iluminata Forte) kit (Millipore) and visualized with a ChemiDoc imaging system (Bio-Rad). For RIPA insoluble preparation 100 µl of brain homogenate were incubated for 20 min on an orbital shaker with 1% RIPA buffer (50 mM Tris-HCl, pH 7.4, 1% NP-40, 150 mM NaCl, 0.25% Na-deoxycholate, 1 mM EDTA, 1 mM Na3VO4, 1 mM NaF, 1 mM PMSF and protease inhibitors). Brain extract was ultracentrifuged for 60 min at 100 000g at room temperature. The pellet was resuspended in 450 µl of RIPA buffer and ultracentrifuged at 100 000g for 30 min at room temperature. The supernatant was carefully removed and the pellet was resuspended in 100 µl of 1× Laemmli sample buffer and proteins analysed by western blot.
For assay of protease resistance brain lysates prepared in 0.05% Triton™ X-100 in PBS were centrifuged at 300g for 5 min. Proteins (20 µg) in the supernatants were digested with pronase (0–10 µg/ml; Sigma) in a final volume of 20 µl for 1 h at 37°C, then resolved by NuPAGE 4–12% gels and analysed by western blot with primary anti-tau antibody BR135 (1:1000, gift from M. Goedert, MRC Laboratory of Molecular Biology, Cambridge, UK; Goedert et al., 1989).
Electron microscopy
Three controls and three TBI inoculated mice were deeply anaesthetized and perfused through the ascending aorta with PBS (0.1 M; pH 7.4) followed by a 5-min perfusion with 4% paraformaldehyde. Hippocampus was excised and cut in sagittal plane with a razor blade and postfixed in 4% paraformaldehyde and 2% glutaraldehyde in phosphate buffer 0.12 M, then for 2 h in OsO4. After dehydration in graded series of ethanol, tissue samples were cleared in propylene oxide, embedded in epoxy medium (Epoxy Embedding Medium kit; Sigma-Aldrich) and polymerized at 60°C for 72 h. From each sample, one semi-thin (1 μm) section was cut with a Leica EM UC6 ultramicrotome and mounted on glass slides for light microscopic inspection to identify the stratum oriens of the hippocampus. Ultrathin (70-nm thick) sections of the areas of interest were obtained, counterstained with uranyl acetate and lead citrate, and examined with an Energy Filter Transmission Electron Microscope (Libra120, Carl Zeiss NTS) equipped with an yttrium aluminium garnet scintillator slow-scan charge-coupled device camera (Sharp eye, TRS). A region of interest including the postsynaptic density was manually traced and its area and thickness measured by iTem software (Olympus Soft Imaging Solutions, Germany) of 263 synapses in control inoculated mice and 262 in TBI inoculated mice fitting the following two criteria: (i) presence of a cluster of presynaptic vesicles (at least three of them); and (ii) presence of a defined synaptic cleft.
Statistical analysis
All graphs represent the mean ± standard error of the mean (SEM). GraphPad Prism 7 was used for statistical analyses. A categorical assessment score was used to assess human tau deposition. Results were dichotomized in either ‘low’ score (0–1) or ‘high’ score (2–3) and the chi-square test was used to assess differences between groups. Data in mice were analysed by one-way ANOVA, followed by Tukey’s test to assess differences in case of three or more groups (behavioural tests and temporal changes of tau deposition) or by unpaired t-test. P-values < 0.05 were considered to be statistically significant.
Data availability
The authors confirm that the data supporting the findings of this study are available within the paper and its Supplementary material.
Results
Phosphorylated tau deposition is widespread in patients surviving a year or more from single TBI
Representative tissue sections from the brains of patients surviving a median of 5 years (range: 1–18 years) following a single moderate or severe, closed TBI [n = 15; age 19–89 (median 60) years], or control subjects with no history of TBI or known neurological disease [n = 15; age 20–92 (median 60) years] (Supplementary Table 1) were stained for P-tau using antibody PHF1. In TBI patients surviving a year or more, PHF1-immunreactive profiles appeared as neurofibrillary tangles and immunoreactive neurites, astrocytes and plaques within all cortical layers, although often with higher density in the superficial cortical layers (Fig. 1A). Elsewhere, where hippocampal profiles were present, all sectors appeared involved, CA2 less frequent than other sectors in this series. Six cases showed involvement of structures in the region of the thalamus as sparse immunoreactive thalamic or nigral neurons, occasional neurites and immunoreactive astrocytes. Although the proportions of patients with PHF1-immunoractive tau pathology were similar in controls and TBI (12 cases with pathology in each group), the extent and distribution of the pathologies were considerably greater in survivors of TBI than in non-injured controls (Fig. 1B). Specifically, in controls, PHF1-immunoreactive neurofibrillary tangles and neurites were largely restricted to sparse profiles in the entorhinal cortex, with just four cases showing more extensive pathologies extending to the hippocampus and cortex. In contrast, in late survivors of TBI, 9 of 12 PHF1-immunoreactive cases displayed higher stage tau pathology, with PHF1-immunoreactive profiles in moderate to high density across the medial temporal lobe, including the hippocampus, and extending to the cortex. Neuritic P-tau-positive profiles were found in the neuropil, many with a truncated, granular appearance as illustrated in Fig 1A. In 3 of 12 TBI cases with P-tau pathology, immunoreactive neuronal and glial profiles showed focal clustering around cortical vessels, in two of these at the depths of sulci (Fig. 1A); appearances consistent with the pathognomonic pathology of CTE described in the preliminary NIH diagnostic criteria (McKee et al., 2016). One further TBI case, an 87-year-old male with a 5-year survival following a road traffic accident, showed clusters of P-tau immunoreactive subpial astrocytes in the medial temporal lobe, consistent with ageing-related tau astrogliopathy (Kovacs et al., 2016). Both in late TBI cases and controls the extent of P-tau pathology appeared to increase with increasing age; the higher score pathology encountered more often with increasing age. Specifically, the four controls with higher tau scores (2 or 3) were among the older aged (74 years or above) controls. Of note, while again typically older within the cohort, the nine late TBI cases with higher tau scores ranged from a younger age (age 59 years upwards) than controls. No clear association between survival duration and extent of tau pathology could be ascertained in this small series (Supplementary Fig. 1). In keeping with injury severity, evidence of healed contusion was near ubiquitous; present in all but one case. There was no association between presence or nature of intracranial haemorrhage, including contusions, and tau pathology.
Figure 1.

Tau pathology in survivors of a single moderate or severe TBI. (A) Typically, controls showed sparse P-tau immunoreactive neurofibrillary tangles and neurites localized to the entorhinal cortex (i; 59-year-old male, no history of TBI). In contrast, patients surviving a year or more from single moderate or severe TBI showed more extensive neurofibrillary, neuritic and glial P-tau immunoreactive profiles, with occasional clusters around cortical vessels in a ‘CTE-like’ manner (ii; 59-year-old male 17-year survival from single severe TBI), and extending beyond the entorhinal cortex to involve wider grey matter regions including the hippocampus (iii; 87-year-old male 5-year survival from single severe TBI). (B) While the proportion of patients in TBI and controls with tau pathologies was similar, following TBI the extent and distribution of pathology were considerably greater. Thus, using a semiquantitative scheme to score tau distribution, while all but four controls showed no or only localized tau pathology (score 0 or 1), in a majority of TBI cases (9 of 15) P-tau pathology was widespread (score 2 or 3; P = 0.0035; χ2). All sections stained for PHF-1. Scale bar = 100 µm.
Single severe TBI induces a progressive tau pathology that spreads from the site of injury in wild-type mice
We tested whether single focal brain trauma triggered propagating tau pathology in wild-type mice. Two-month-old C57BL/6J mice were subjected to severe TBI (16 mice) or to sham injury (14 mice). Eight TBI and seven sham mice were sacrificed at 3 months and used for immunohistochemistry (four TBI and four sham) or biochemical analysis (four TBI and three sham). One TBI mouse was found dead at ∼4 months, and the brain was not collected due to prolonged post-mortem delay. The remaining mice were culled at 12 months and used for immunohistochemistry (four TBI and five sham) and for biochemistry (three TBI and two sham).
Phospho-tau immunopositivity, assessed by AT8, AT180 and PHF1 antibodies, was evident in two of four mice at 3 months post-TBI and restricted to the hemisphere ipsilateral to injury, with concentration to the regions adjacent to injury [Fig. 2A (cf. i and ii), B, and Supplementary Fig. 2]. In contrast, at 12 months, both ipsi- and contralateral P-tau pathologies were present, supporting progressive spreading of tau pathology from the injured hemisphere [Fig. 2A–D (cf. Ai and Aiii; cf. Civ and Cvi) and Supplementary Fig. 2]. Western blot analysis found increased levels of total and AT8-reactive tau in one frozen TBI brain compared to sham-injured controls at 3 months and in two of three TBI brains at 12 months (an example is shown in Supplementary Fig. 3). Thus, three of seven TBI mice (∼40%) developed pathological tau at 3 months, and six of seven (∼85%) at 12 months.
Figure 2.

Evidence of tau pathology in mouse TBI. AT8 immunostaining and quantification of the AT8-positive area in the ipsilateral and contralateral cortex of sham and TBI mice at 3 and 12 months post-injury. (A and B) AT8 immunoreactivity is detected in the ipsilateral (ipsi) pericontusional cortical area in two of four mice at 3 months post-TBI, and in all mice at 12 months. (C and D) In the contralateral cortex AT8 immunoreactivity is detected only after 12 months post-TBI. Data are mean ± SEM; n = 4–5; *P < 0.05; **P < 0.01 by one-way ANOVA, Tukey post hoc test. Scale bar = 100 µm (B and D).
P-tau was observed as neuronal cytoplasmic, neuritic and glial profiles throughout the cerebral cortex, hippocampus (CA1 and CA3 fields) and thalamus, with pathology also in the striatum and zona incerta (Fig. 3). In addition, a notable observation was numerous small to medium sized, rounded, ‘grain-like’ profiles, particularly within the neuropil (Fig. 3C). Occasionally there was faint clustering of these granular immunoreactive profiles (Fig. 3G), with still other examples showing apparent localization along cortical vessels (Fig. 3H). Elsewhere, glial cytoplasmic P-tau immunoreactivity was present within white matter of the corpus callosum.
Figure 3.

Widespread tau pathology in mice 12 months post-TBI. Examples of tau immunostaining with the AT180 (A and F), PHF1 (B), and AT8 (C–E, G and H) antibodies in the cerebral cortex (A and B), CA1 (C) and CA3 (D) fields of the hippocampus, and in the zona incerta (E and F). Examples of clusters of small to medium sized, rounded, ‘grain-like’ profiles, in the zona incerta (G), and granular profiles along a cortical vessel (H; arrows). Scale bars: 20 µm (A–D and G); 10 µm (E and F); 50 µm (H).
Tau prions are generated in TBI mice, causing transmission of tau pathology, synaptic loss and behavioural deficits
The evidence that TBI induced a P-tau pathology that progressively spread throughout the brain suggested formation of a self-templating tau conformation. To test whether this could be horizontally transmitted like a prion, causing brain dysfunction, we intracerebrally inoculated 10% brain homogenates from TBI and sham-injured mice into naïve C57BL/6J mice. Brain homogenates of contused and contralateral hemispheres (TBIipsi and TBIcontra, respectively) showing increased levels of total and AT8-positive tau by western blot at 12 months post-TBI (Supplementary Fig. 3) were inoculated bilaterally into the hippocampus and overlaying cerebral cortex of the recipient mice (Fig. 4A). The mice were examined in the novel object recognition test 8 and 12 months post-inoculation (m.p.i.). There was an overt memory deficit in the TBIipsi- but not in the TBIcontra-inoculated mice at 8 m.p.i. (Fig. 4B). At 12 m.p.i the memory deficit persisted in the TBIipsi- and was also detected in the TBIcontra-inoculated mice (Fig. 4C). No memory deficit was seen in mice inoculated with sham-injured brain tissue.
Figure 4.

Inoculation of contused tissues induces memory deficits in wild-type mice. (A) Groups of mice used in the inoculation studies. C57BL/6J male mice were either left untreated (open bar) or inoculated bilaterally into the hippocampus and overlaying cerebral cortex with 10% brain homogenates from sham mice (grey) or from the contused (TBIipsi, indigo) or contralateral (TBIcontra, magenta) hemisphere at 12 months post-TBI. (B and C) Recognition memory was investigated by the novel object recognition (NOR) test at 8 (B) and 12 (C) months after inoculation. Performance in the novel object recognition task was expressed as discrimination index (the higher the discrimination index the better the performance). (D and E) Locomotor activity was similar in naïve, sham and TBI inoculated mice at 8 (D) and 12 (E) months after inoculation. Data are mean ± SEM; *P < 0.05, versus naïve; #P < 0.05 versus sham by one-way ANOVA, Tukey post hoc test.
This was confirmed in an independent experiment in which we tested the effect of the inoculation of TBI brain homogenate, obtained from the injured hemisphere 12 months post-injury, on recognition memory in the inoculated animals at an earlier time (4 m.p.i). Memory was already significantly impaired at 4 months, and this was confirmed at 8 m.p.i. (Fig. 5B and C). Locomotor activity was similar in sham and TBI inoculated mice, indicating that the deficit in the novel object recognition test was not due to motor abnormalities (Figs 4D, E, 5D and E).
Figure 5.
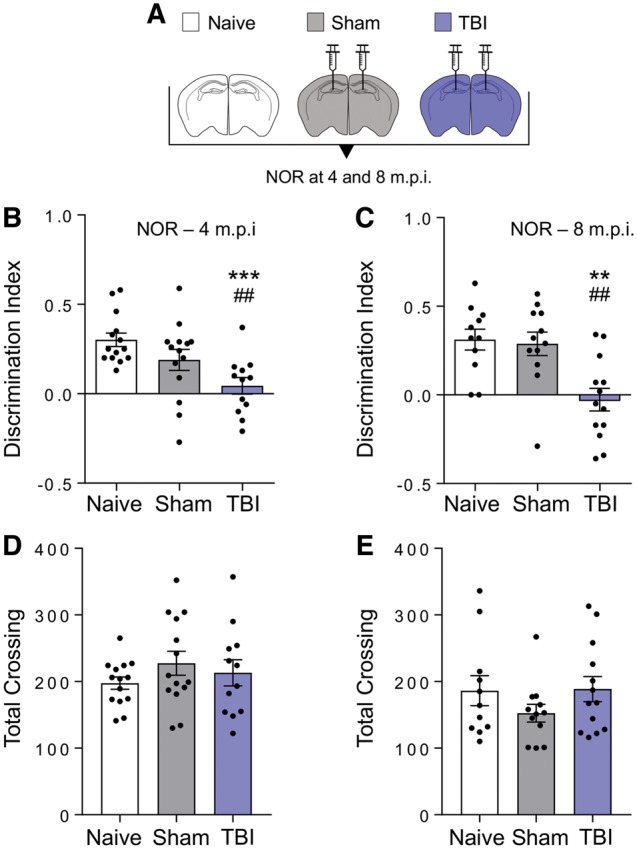
Memory deficits are already detected at 4 months post-inoculation. (A) Scheme of the groups of mice used in the inoculation studies. C57BL/6J male mice were either left untreated (open bar) or inoculated bilaterally into the hippocampus and overlaying cerebral cortex with 10% brain homogenates from sham (grey) or TBI (contused hemisphere, indigo) mice 12 months post-injury. (B and C) Recognition memory was investigated by novel object recognition (NOR) test at 4 (B) and 8 (C) months after inoculation. Histograms indicate the discrimination index (the higher the discrimination index the better the performance). (D and E) Locomotor activity was similar in sham and TBI inoculated mice at 4 (D) and 8 (E) months after inoculation. Data are mean ± SEM, n = 11–14. ***P < 0.001, **P < 0.01 versus naïve; ##P < 0.01 versus sham; by one-way ANOVA, Tukey post hoc test.
The memory deficit was associated with hippocampal synaptic pathology. Analysis of pre- and postsynaptic compartments by quantitative VGLUT-1 and drebrin immunofluorescence showed marked synaptic loss in the stratum oriens of the CA1 region at 12 m.p.i. (Fig. 6A and B). Electron microscopy examination showed a significant decrease in postsynaptic density thickness and area in hippocampal synapses in TBI inoculated mice (Fig. 6C) consistent with impaired synaptic efficacy. Analysis of P-tau deposition by antibody AT8 showed a significant difference between sham and TBI inoculated mice not only at the site of injection, but also widespread throughout the brain (Fig. 7).
Figure 6.

Synaptic alterations in the hippocampus of TBI inoculated mice. (A and B) Representative images of immunofluorescence staining and quantitative analysis of presynaptic (VGLUT-1; A) and postsynaptic (drebrin; B) markers in the hippocampus of the inoculated mice at 12 months post-inoculation. Data are mean ± SEM; n = 5–6; *P < 0.05; **P < 0.01 by unpaired t-test. Scale bar = 10 µm. (C) Representative electron micrographs of stratum oriens synapses, and quantification of the thickness and area of the postsynaptic density in sham and TBI inoculated mice. Data are mean ± SEM; n = 3. *P < 0.05 by unpaired t-test. Scale bar = 100 nm.
Figure 7.

Evidence of tau pathology in mice inoculated with TBI homogenates. Quantification of the per cent area covered by AT8 staining in the cortex (A–C), hippocampus (D–F), thalamus (G–I), and cerebellum (J–L). Data are mean ± SEM; n = 3–6; *P < 0.05; **P < 0.01 by unpaired t-test. Scale bars = 100 µm (B–I) and 200 µm (K and L).
Specifically, P-tau neurons, neurites and glia were identified in the cerebral cortex, hippocampus (CA1, CA3, and hilus of the dentate gyrus) and thalamus, with morphologies similar to those observed in mice exposed to TBI, including widespread ‘grain-like’ profiles (Fig. 8B–I). In the cerebellum, P-tau immunoreactive cytoplasmic and neuritic inclusions were noted in association with Purkinje cells and within the granule cell layer (Fig. 8J–K). Consistent with widespread tau pathology, there was an increase in the amount of aggregated tau in the thalamus of TBI-inoculated mice (Supplementary Fig. 4A and B). The tau conformer that accumulated in the brains of TBI-inoculated mice was partially resistant to protease digestion, yielding a pronase-resistant fragment of ∼10 kDa (Supplementary Fig. 4C), consistent with tau prion formation (Sanders et al., 2014; Kaufman et al., 2016).
Figure 8.

Widespread tau pathology in mice inoculated with TBI homogenates. Representative micrographs of tau immunostaining in the brains of mice inoculated with sham (A) and TBI (B–K) homogenates. Examples of immunostaining with AT8 (A–C, F–H and K), PHF1 (D, I and J) and conformation-dependent antibody MC1 (E) in the hippocampus (A, B, E and F), cerebral cortex (C and D), thalamus (G–I) and cerebellum (J and K). CA1 and CA3 = fields of the hippocampus; LMol = stratum lacunosum molecolare; M = molecular layer; PC = Purkinje cell layer; G = granule cell layer. Scale bars = 200 µm (A and B); 50 µm (C); 20 µm (E–G, J and K); 10 µm (D and H); 5 µm (I).
Discussion
The present study found that a single moderate or severe TBI in humans resulted in widespread tau pathology with a notable pattern and distribution. Further, a single severe TBI induced persistent and evolving tau pathology in wild-type mice, with characteristics similar to late post-TBI tau pathology in humans, which progressively spread from the site of injury to ipsi- and contralateral brain regions. This TBI generated tau pathology could be transmitted to wild-type recipient mice by intracerebral inoculation, inducing tau aggregation, memory deficits and synaptic alterations. These results indicate that tau prions are generated in TBI, providing a mechanistic explanation for how a biomechanical insult might trigger self-sustained neurodegeneration.
In the past decade, there has been growing recognition that repetitive mild TBI might set in motion brain pathological processes that place exposed individuals at risk for neurodegenerative disorders in mid-to-late life, particularly CTE. While there is also evidence that tau pathology may be seen many years after a single severe TBI, most attention has focused on the association between repeated mild TBI and CTE, with the common impression tau pathology is uniquely associated with repetitive mild TBI, which dictates a distinct neurodegenerative disease.
Here we confirm that widespread CTE-like tau pathology is present in survivors of a year or more after just a single moderate to severe TBI in humans, and in mice exposed to severe TBI surviving up to 1 year from injury. In our series of 15 patients who survived a year or more post-TBI, 12 showed tau pathology, the extent and distribution of which was considerably greater than that seen in age-matched, non-TBI controls. Among this small series of late post-TBI patients this tau pathology appeared to follow a stereotypic pattern of deposition, reminiscent of hierarchical P-tau deposition in other neurodegenerative diseases (Braak and Braak, 1995; Kovacs, 2015). These data provide one possible neuropathological substrate for the increased incidence of neurodegenerative disease in patients who survive a single moderate or severe TBI. Although the extent and distribution of P-tau pathology was greater in late TBI survivors, it was not possible to establish a clear association between survival interval and extent of tau pathology. Further, our data in this limited human study show similar percentages of cases with tau pathology in TBI and control groups, making it hard to establish whether TBI accelerates a pre-existing age-related tau pathology, or triggers de novo tau deposition.
A better understanding of the biology that underlies this process calls for tractable experimental models. Previous investigations have shown that TBI accelerates genetically determined tau deposition in transgenic mice overexpressing mutant or wild-type human tau (Yoshiyama et al., 2005; Tran et al., 2011a, b; Ojo et al., 2016), and can lead to tau pathology in wild-type mice (Goldstein et al., 2012; Kondo et al., 2015; Meabon et al., 2016). A few studies reported formation of tau aggregates shortly after severe TBI in rats (Hawkins et al., 2013; Gerson et al., 2016), with only one study showing persistent tau pathology in wild-type mice (Kondo et al., 2015). Our data not only confirm de novo tau deposition in wild-type animals after single TBI, but also document widespread tau immunopositivity at chronic stages, with a pattern echoing our observations in human TBI cases, including grain-like profiles in the neuropil and perivascular deposits. Interestingly we observed an increase in total tau after TBI. This could be due to increased tau expression or impaired tau clearance, perhaps because of TBI-induced alterations in proteostatic mechanisms, such as the glymphatic system (Iliff et al., 2014), and/or formation of an intrinsically protease-resistant tau conformer.
To establish a direct contribution of TBI-induced tau pathology to late symptomatology in a clean setting, we performed inoculation studies in which there are no confounding factors inherent to the biomechanical injury. We found that inoculation of mouse TBI homogenates induced tau pathology with formation of partially protease-resistant tau, memory deficits and synaptic alterations in wild-type recipient mice, providing evidence for generation of authentic tau prions. Previous studies showed that fibrillar forms of recombinant tau or tau aggregates from Alzheimer’s disease patients or other tauopathies can accelerate the genetically determined tau pathology in mutant tau cells and mice (Clavaguera et al., 2009; Sanders et al., 2014; Kaufman et al., 2016; Woerman et al., 2016). More recently, aggregated tau isolated from Alzheimer’s disease brains and other tauopathies was shown to induce tau pathology when inoculated intracerebrally in wild-type mice (Lasagna-Reeves et al., 2012; Guo et al., 2016; Narasimhan et al., 2017), supporting the generation of self-propagating tau in Alzheimer’s disease. However, to the best of our knowledge no long-lasting memory deficits or other signs of brain dysfunction were documented in these animals, leaving open the question of whether toxic tau prions are formed in these models. The observation that TBI-induced tau pathology can be transmitted between wild-type mice and cause persistent memory deficits satisfies the main requirement for a toxic, bona fide prion.
Oligomeric or fibrillar forms of tau have been identified in different tauopathies that may have distinct toxic potency and seeding properties (Goedert and Spillantini, 2017). Future studies should aim at identifying the abnormal tau species generated by TBI responsible for propagation of tau pathology and synaptic toxicity. It will also be important to immunodeplete tau from the inocula to rule out that persistent inflammatory processes and/or other TBI-related signalling pathways (Makinde et al., 2017; Pischiutta et al., 2018) may contribute to triggering tau pathology in the inoculated mice.
Host PrPC is required for PrPSc replication and pathogenic effect (Chiesa, 2015). It will be interesting to assess if the same holds true for tau by testing the amnestic effect of TBI-induced tau prions in tau knockout mice. Finally, the development of bioassays and/or biochemical methods for measuring tau prion titres as for PrPSc prions, will enable us to define the time lag between the biomechanical insult and the formation of the tau conformers responsible for seeding the spread of pathology. This will not only be important for therapeutic approaches targeting tau propagation (Holmes and Diamond, 2014), but also to assess the possible formation of infectious tau in humans post-TBI. The demonstration of tau prions in human TBI will urgently require studies to determine whether neurosurgical intervention in such patients should conform to well established precautions for transmission of prion disease, as suggested by the Centers for Disease Control (https://www.cdc.gov/prions/cjd/infection-control.html) and the WHO (http://www.who.int/csr/resources/publications/bse/WHO_CDS_CSR_APH_2000_3/en/).
Supplementary Material
Acknowledgements
We are grateful to Peter Davies for providing the PHF1 and MC1 antibodies and to Michael Goedert for the BR135 antibody. We also acknowledge Stefano Fumagalli and Pietro Veglianese for advice on quantification of immunofluorescent staining.
Glossary
Abbreviations
- CTE
chronic traumatic encephalopathy
- P-tau
hyperphosphorylated tau
- TBI
traumatic brain injury
Funding
This work was supported by the National Institutes of Health (NIH NS094003 and NS038104) (D.H.S., W.S.); by NHS Research Scotland Career- Researcher Fellowship (W.S.); and the Alzheimer’s Association (AARG-18-532633) (E.R.Z., R.C.); D.K.M. was supported by a Senior Investigator award from the National Institute for Health Research (UK).
References
- Ahmed Z, Cooper J, Murray TK, Garn K, McNaughton E, Clarke H et al. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol 2014; 127: 667–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouybayoune I, Mantovani S, Del Gallo F, Bertani I, Restelli E, Comerio L et al. Transgenic fatal familial insomnia mice indicate prion infectivity-independent mechanisms of pathogenesis and phenotypic expression of disease. PLoS Pathog 2015; 11: e1004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging 1995; 16: 271–8. [DOI] [PubMed] [Google Scholar]
- Chiesa R. The elusive role of the prion protein and the mechanism of toxicity in prion disease. PLoS Pathog 2015; 11: e1004745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci USA 2013; 110: 9535–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 2009; 11: 909–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012; 73: 685–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes JF, Vowles GH, Nicoll JA, Révész T. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol 1999; 98: 171–8. [DOI] [PubMed] [Google Scholar]
- Gerson J, Castillo-Carranza DL, Sengupta U, Bodani R, Prough DS, DeWitt DS et al. Tau oligomers derived from traumatic brain injury cause cognitive impairment and accelerate onset of pathology in Htau mice. J Neurotrauma 2016; 33: 2034–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Masuda-Suzukake M, Falcon B. Like prions: the propagation of aggregated tau and alpha-synuclein in neurodegeneration. Brain 2017; 140: 266–78. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG. Propagation of Tau aggregates. Mol Brain 2017; 10: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989; 3: 519–26. [DOI] [PubMed] [Google Scholar]
- Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med 2012; 4: 134ra60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, Narasimhan S, Changolkar L, He Z, Stieber A, Zhang B et al. Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J Exp Med 2016; 213: 2635–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins BE, Krishnamurthy S, Castillo-Carranza DL, Sengupta U, Prough DS, Jackson GR et al. Rapid accumulation of endogenous tau oligomers in a rat model of traumatic brain injury: possible link between traumatic brain injury and sporadic tauopathies. J Biol Chem 2013; 288: 17042–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay J, Johnson VE, Smith DH, Stewart W. Chronic traumatic encephalopathy: the neuropathological legacy of traumatic brain injury. Annu Rev Pathol 2016; 11: 21–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise C, Schroeder JC, Schoen M, Halbedl S, Reim D, Woelfle S et al. Selective localization of shanks to VGLUT1-positive excitatory synapses in the mouse hippocampus. Front Cell Neurosci 2016; 10: 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes BB, Diamond MI. Prion-like properties of Tau protein: the importance of extracellular Tau as a therapeutic target. J Biol Chem 2014; 289: 19855–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM. Synthetic Tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci 2013; 33: 1024–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iba M, McBride JD, Guo JL, Zhang B, Trojanowski JQ, Lee VM. Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC’s afferent and efferent connections. Acta Neuropathol 2015; 130: 349–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L et al. Impairment of glymphatic pathway function promotes Tau pathology after traumatic brain injury. J Neurosci 2014; 34: 16180–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 2013; 136: 28–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. Widespread τ and amyloid-β pathology many years after a single traumatic brain injury in humans. Brain Pathol 2012; 22: 142–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman SK, Sanders DW, Thomas TL, Ruchinskas AJ, Vaquer-Alicea J, Sharma AM et al. Tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo. Neuron 2016; 92: 796–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 2010; 8: e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, Chen CH et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 2015; 523: 431–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG. Invited review: neuropathology of tauopathies: principles and practice. Neuropathol Appl Neurobiol 2015; 41: 3–23. [DOI] [PubMed] [Google Scholar]
- Kovacs GG, Ferrer I, Grinberg LT, Alafuzoff I, Attems J, Budka H et al. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol 2016; 131: 87–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Guerrero-Munoz MJ, Kiritoshi T, Neugebauer V et al. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep 2012; 2: 700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinde HM, Just TB, Cuda CM, Perlman H, Schwulst SJ. The role of microglia in the etiology and evolution of chronic traumatic encephalopathy. Shock 2017; 48: 276–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 2016; 131: 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Daneshvar DH, Alvarez VE, Stein TD. The neuropathology of sport. Acta Neuropathol 2014; 127: 29–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meabon JS, Huber BR, Cross DJ, Richards TL, Minoshima S, Pagulayan KF et al. Repetitive blast exposure in mice and combat veterans causes persistent cerebellar dysfunction. Sci Transl Med 2016; 8: 321ra6. [DOI] [PubMed] [Google Scholar]
- Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR et al. Clinicopathological evaluation of chronic traumatic encephalopathy in players of American football. JAMA 2017; 318: 360–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudher A, Colin M, Dujardin S, Medina M, Dewachter I, Alavi Naini SM et al. What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol Commun 2017; 5: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan S, Guo JL, Changolkar L, Stieber A, McBride JD, Silva LV et al. Pathological tau strains from human brains recapitulate the diversity of tauopathies in non-transgenic mouse brain. J Neurosci 2017; 37: 11406–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo JO, Mouzon B, Algamal M, Leary P, Lynch C, Abdullah L et al. Chronic repetitive mild traumatic brain injury results in reduced cerebral blood flow, axonal injury, gliosis, and increased T-Tau and Tau oligomers. J Neuropathol Exp Neurol 2016; 75: 636–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. San Diego, CA: Academic Press; 2004. [Google Scholar]
- Petry FR, Pelletier J, Bretteville A, Morin F, Calon F, Hébert SS et al. Specificity of anti-tau antibodies when analyzing mice models of Alzheimer’s disease: problems and solutions. PLoS One 2014; 9: e94251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pischiutta F, Micotti E, Hay JR, Marongiu I, Sammali E, Tolomeo D et al. Single severe traumatic brain injury produces progressive pathology with ongoing contralateral white matter damage one year after injury. Exp Neurol 2018; 300: 167–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216: 136–44. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Cell biology. A unifying role for prions in neurodegenerative diseases. Science 2012; 336: 1511–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014; 82: 1271–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekino Y, Koganezawa N, Mizui T, Shirao T. Role of Drebrin in synaptic plasticity. In: Drebrin. Tokyo: Springer; 2017. p. 183–201. [DOI] [PubMed] [Google Scholar]
- Senatore A, Colleoni S, Verderio C, Restelli E, Morini R, Condliffe SB et al. Mutant PrP suppresses glutamatergic neurotransmission in cerebellar granule neurons by impairing membrane delivery of VGCC alpha(2)delta-1 subunit. Neuron 2012; 74: 300–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF et al. A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma 1995; 12: 169–78. [DOI] [PubMed] [Google Scholar]
- Tran HT, LaFerla FM, Holtzman DM, Brody DL. Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-beta accumulation and independently accelerates the development of tau abnormalities. J Neurosci 2011a; 31: 9513–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HT, Sanchez L, Esparza TJ, Brody DL. Distinct temporal and anatomical distributions of amyloid-β and tau abnormalities following controlled cortical impact in transgenic mice. PLoS One 2011b; 6: e25475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woerman AL, Aoyagi A, Patel S, Kazmi SA, Lobach I, Grinberg LT et al. Tau prions from Alzheimer’s disease and chronic traumatic encephalopathy patients propagate in cultured cells. Proc Natl Acad Sci USA 2016; 113: E8187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Uryu K, Higuchi M, Longhi L, Hoover R, Fujimoto S et al. Enhanced neurofibrillary tangle formation, cerebral atrophy, and cognitive deficits induced by repetitive mild brain injury in a transgenic tauopathy mouse model. J Neurotrauma 2005; 22: 1134–41. [DOI] [PubMed] [Google Scholar]
- Zanier ER, Marchesi F, Ortolano F, Perego C, Arabian M, Zoerle T et al. Fractalkine receptor deficiency is associated with early protection but late worsening of outcome following brain trauma in mice. J Neurotrauma 2016; 33: 1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the paper and its Supplementary material.