

Abstract
The vLAMPS is a human, biomimetic liver MPS, in which the ECM and cell seeding of the intermediate layer prior to assembly, simplifies construction of the model and makes the platform user-friendly. This primarily glass microfluidic device is optimal for real-time imaging, while minimizing the binding of hydrophobic drugs/biologics to the materials that constitute the device. The assembly of the three layered device with primary human hepatocytes and liver sinusoidal endothelial cells (LSECs), and human cell lines for stellate and Kupffer cells, creates a vascular channel separated from the hepatic channel (chamber) by a porous membrane that allows communication between channels, recapitulating the 3D structure of the liver acinus. The vascular channel can be used to deliver drugs, immune cells, as well as various circulating cells and other factors to a stand-alone liver MPS and/or to couple the liver MPS to other organ MPS. We have successfully created continuous oxygen zonation by controlling the flow rates of media in the distinct vascular and hepatic channels and validated the computational modeling of zonation with oxygen sensitive and insensitive beads. This allows the direct investigation of the role of zonation in physiology, toxicology and disease progression. The vascular channel is lined with human LSECs, recapitulating partial immunologic functions within the liver sinusoid, including the activation of LSECs, promoting the binding of polymorphonuclear leukocytes (PMNs) followed by transmigration into the hepatic chamber. The vLAMPS is a valuable platform to investigate the functions of the healthy and diseased human liver using all primary human cell types and/or iPSC-derived cells.
We developed a glass based, vascularized human biomimetic liver MPS recreating oxygen zonation present in the liver acinus.
Introduction
Organ-on-a-chip microphysiological systems (MPS) have emerged as a powerful platform to mimic a particular human tissue, organ and/or multiple organs for in vitro preclinical studies, drug discovery and drug development, as well as environmental toxicology.1–7 The liver gained particular interest for MPS development due to its central role in metabolism, liver-specific diseases and drug toxicity.6, 8, 9
The liver acinus is the basic structural and functional unit of the liver, where the blood flows from the portal triads through the network of sinusoidal capillaries (sinusoids) to the central vein.8, 10 The sinusoids are lined with liver sinusoidal endothelial cells (LSECs), a special type of endothelial cell that does not form tight junctions and is perforated by fenestrations, allowing rapid, “leaky” physical transfer of macromolecules and small molecules through the space of Disse, the thin protein matrix that together with the LSECs protects hepatocytes from the shear-stress of direct blood flow. Stellate cells in the inactive state reside in the hepatic layer with the hepatocytes and store vitamin A and fat, while the resident Kupffer cell macrophages reside in the endothelial wall of the sinusoid to aid digestion, detoxifying the portal blood and are a key contributor in liver damage or repair as a response to challenges.11–13 Upon activation by a variety of drugs and disease “molecular drivers”; such as lipopolysaccharides (LPS) and transforming growth factor beta (TGF-β) from the intestine, free fatty acids (FFA) from adipose tissue, and epidermal growth factor (EGF) from kidney14; the resident Kupffer cells, along with circulating immune x`cells, transmigrate into the hepatic layer where they either cause damage from inflammation or aid in damage repair, while the stellate cells undergo a transformation into myofibroblasts that dramatically change shape, proliferate and produce collagen. These changes are part of multiple liver disease phenotypes including non-alcoholic fatty liver disease (NAFLD) and type 2 diabetes.15–17
The unique organization of the liver sinusoid mixes blood from the oxygen poor, nutrient rich portal vein and the oxygen rich, nutrient poor hepatic artery, and creates an oxygen and metabolic gradient along the portal triad to the central vein axis, as hepatocytes consume oxygen and nutrients, remove wastes, regulate blood carbohydrates and secrete the needed proteins, lipids and bile acids to sustain body functions.12, 18, 19 The liver sinusoid is typically divided into three domains: zone 1 (oxygen rich), zone 2 (intermediate oxygen) and zone 3 (oxygen poor).10 The oxygen gradient is key to establishing a metabolic gradient, which in turn allows the liver to carry out many metabolic/secretory functions with maximum efficiency.12, 18, 19 For example, many liver functions are enhanced in specific zones: biochemical and physiological functions such as albumin synthesis, urea synthesis, oxidative phosphorylation and gluconeogenesis are elevated in zone 1; while glycolysis, lipogenesis and xenobiotic metabolism are higher in zone 3. Disease initiation and progression also display some zone specificity.12, 19
Conventional liver models such as single cell layers, liver slices and animal models mimic the human liver to only a limited extent, lacking either the tissue-level complexity, the longevity or the human-specificity to recapitulate clinically relevant tissue responses for predictive toxicology or disease modeling.8, 11, 20, 21 By utilizing microfabrication22–24, bio-printing or the combination of both25, 26, as well as sequential layering of cells 27–30, human liver MPS have been developed as promising alternative or complementary models.7, 31–33
There have been many engineering approaches to better approximate specific aspects of the highly sophisticated microenvironment of the liver.8 For example, co-culture of multiple cell types in the form of spheroids/organoids34, 35, heterogeneous seeding of hepatocytes and endothelial cells within a microfluidic microchannel36, 37, spatially arranging hepatocytes (with or without non-parenchymal cells (NPCs) in scaffolds38–41, porous membranes42, 43 or fabricated microstructures38, 44–46 all reconstitute, to a certain degree, the liver sinusoid-on-a-chip.
We have focused on creating a human, biomimetic liver MPS that includes multiple human liver cell types: hepatocytes, LSECs, Kupffer cells and human stellate cells (HSC) at physiological ratios and structured in a 3D organization that mimics the liver acinus.28, 47 Our approach has been to: evolve the complexity to improve on the model design; diminish the use of synthetic biomaterials, such as polydimethylsiloxane (PDMS), that bind lipophilic compounds in the microfluidic device; improve the patient specificity of the human cell types, optimize the contents of the media to reflect normal or disease conditions, reproduce the oxygen zonation; and expand the use of fluorescent biosensors for specific toxicological or disease specific cell functions.27–29 The first generation liver MPS was based on a single chamber, PDMS microfluidic device (PAR-V1, Nortis, Woodinville, WA) that allowed direct optical imaging for real-time monitoring of the cells. The sequentially layered, self-assembled liver (SQL-SAL) model was based on human primary hepatocytes, and human cell lines for the endothelial, stellate and Kupffer cells, incorporated fluorescent biosensors, and maintained functionality for at least one month, as indicated by real-time functional read-outs including ROS production, apoptosis and HSC activation, as well as biochemical and mass spectrometry measurement of secretions and metabolites in the perfused efflux media.28, 29 The next generation liver MPS, the liver acinus MPS (LAMPS), used the same PDMS microfluidic device but replaced the endothelial cell line with primary human microvascular endothelial cells (HMVEC) and added an extract of porcine liver extracellular matrix (LECM) between the endothelial cells and the hepatocytes to form a thin layer that mimics the space of Disse. In addition, separate zone 1 and zone 3 models were produced by controlling the flow rate of the media in the models, well within the shear-stress limits for hepatocytes.30 The oxygen tension in the models was computationally defined and confirmed by direct temporal and spatial measurements with oxygen sensitive and insensitive dyes.30 A range of biological parameters were compared in zone 1 and zone 3 devices and the expected zonation dependent liver sinusoidal acinus functions were identified.30
The vascularized liver acinus MPS (vLAMPS) described here continues the evolution of our previous models with five major improvements. First, the microfluidic device is primarily made of glass, minimizing the absorbance of hydrophobic molecules to polymers such as PDMS, while maintaining the ability to perform real-time imaging measurements using fluorescent biosensors. Second, separate flow channels for the sinusoid (vascular channel) and the hepatic compartment (hepatic chamber), which are separated by a porous membrane, recapitulates a major structural element of the liver sinusoid, allowing the vascular channel to be used to deliver drugs, immune cells and other factors to the liver, as well as to couple to other organ MPS. Third, we have engineered continuous zonation or zone specific oxygen tensions by controlling the flow rates of media in the vascular channel and hepatic chambers and have both computationally modeled and directly measured the oxygen tension with oxygen sensitive and insensitive fluorescent beads to define the temporal and spatial signals. Fourth, we introduced an endothelial layer with primary human LSECs to better mimic the permeability properties of the liver specific endothelium, the potential scavenger properties48 and immunological functions49 within the liver sinusoid. Fifth, we demonstrated the activation of LSECs as an initial step in the inflammatory response, followed by the binding of human PMNs to the activated LSECS as they were perfused into the vascular channel and then the transmigration of the PMNs into the hepatic chamber, induced by pro-inflammatory molecular drivers including LPS, TGF-β, and EGF.
The vLAMPS provides a biomimetic platform that optimizes the investigation of experimental models of disease and ADME/TOX. The vLAMPS allows the mechanistic study of the characteristics and functionality of the healthy or diseased liver, the interactions between hepatocytes and the NPCs, their individual contributions to the metabolism of nutrients and drugs, the inflammatory response, as well as hepatotoxicity and liver damage. The vLAMPS also allows the efficient physical coupling to other organ MPS through the vascular channel.
Material and methods
Microfluidic device preparation
The 3-layer borosilicate glass chips and the chip-holder were purchased from Micronit Microtechnologies (OOC 00739, OOC Platform 4 slots, Enschede, The Netherlands) (Fig.1A). The layers were sterilized in 70% ethanol (EtOH) overnight and air-dried. The intermediate layer (OOC 01206) contains 3 μm diameter pores in a polyethylene terephthalate (PET) membrane that was oriented with the membrane side down. A solution of 100 μg/mL fibronectin and 100 μg/mL collagen in phosphate buffered saline (PBS) was used to pre-coat the PET membrane at 4 °C overnight to provide a protein substrate that would promote cell adhesion. The glass syringes, syringe needles, polyetheretherketone (PEEK) tubing and FFKM connector ferrules (FC_PRO_FFKM_KIT.05) were all sterilized by autoclaving. These components were selected by virtue of low drug absorptive properties.29
Figure 1.
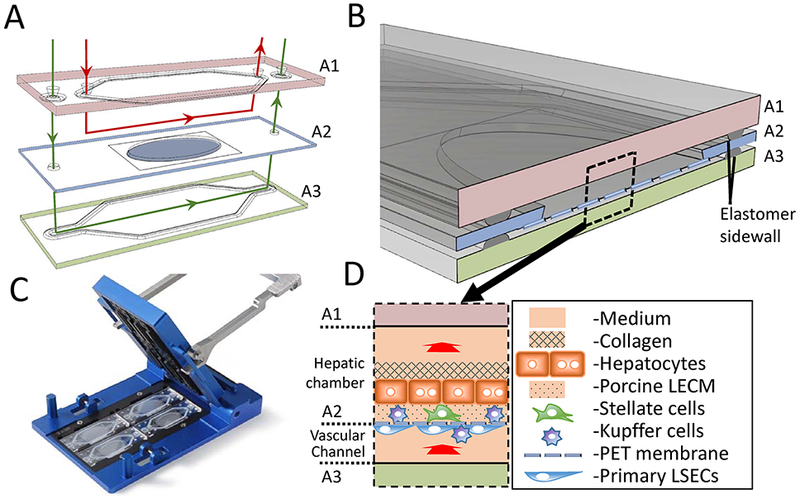
The Vascularized Liver Acinus Microphysiology System (vLAMPS). (A) The vLAMPS is constructed from 3 glass layers A1–A3 (15mm × 45mm). The intermediate layer (A2) contains an elliptical opening (8mm × 16mm) with a PET membrane with 3 μm pores that spans the opening and is attached to the bottom. Four human liver cell types (primary hepatocytes, primary liver sinusoidal endothelial cells (LSECS), LX-2 human stellate cell line and Kupffer cells derived from a THP-1 cell line are sequentially layered onto the two sides of the PET membrane. The top (A1) and bottom (A3) layers have polymer gaskets that seal against the intermediate layer to create two fluidic channels: the lower vascular channel lined with LSECS and Kupffer cells and the upper hepatic chamber containing the Hepatocytes and stellate cells, and optionally Kupffer cells, as well as a space of Disse matrix (see Table 1). (B) The assembled vLAMPS showing the intermediate layer (A2) sandwiched between the two resealable glass layers. The shape of the flow channels is defined by the gaskets on the top and bottom layers, with the height determined by the thickness of the gaskets (C) after all the three layers are compressed in the chip holder (85mm × 127mm). (D) Diagram of the vLAMPS showing the cells, the two distinct compartments (~100 μL hepatic and ~45 μL vascular), with the direction of flow into the page as indicated. The flow rate in the hepatic chamber is 80 μL/hr (shear-stress on hepatocytes = 2.5×10−7 dyne/cm2) and in the vascular channel is 100 μL/hr (shear-stress on LSECs = 4.0 × 10−3 dyne/cm2) under the flow rates reported here.
Cell sources and culture
A selected lot of cryopreserved “normal” primary human hepatocytes found to have >90% viability and re-plating efficiency post thaw, were obtained from ThermoFisher (HU1838, Waltham, MA) and used for the majority of the experiments. A second lot of cryopreserved “normal” primary human hepatocytes was obtained from ThermoFisher (HU8241) for evaluation of reproducibility between lots. Cryopreserved “normal” liver derived endothelial cells containing up to 80% LSECs50 were purchased from Samsara Science (HL160019, San Diego, CA). The human monoblast cell line THP-1, used to generate Kupffer cells, was purchased from ATCC (Rockville, MD). LX-2 human stellate cells were acquired from EMD Millipore (Billerica, MA, USA). Human dermal microvascular endothelial cells (HMVEC) were purchased from Lonza (CC-2505, Alpharetta, GA). Primary human PMNs were isolated from a heparin whole blood draw with informed consent from healthy donors in the Thomas E. Starzl Transplantation Institute, University of Pittsburgh, in accordance with protocol IRB0608014, approved by the University of Pittsburgh Institutional Review Board (IRB) as meeting Federal regulations 45 CFR 46.111 and 21 CFR 56.111.The isolation was performed with a gradient centrifugation kit (10771 and 11191, Sigma, St. Louis, MO) according to the procedure provided by the manufacturer.
LSECs were thawed and cultured in EGM-2 media, 5% FBS, 50 μg/mL gentamycin (Lonza, Alpharetta, GA). THP-1 monoblast cells were differentiated into macrophages by adding 200 ng/mL phorbol myristate acetate (PMA) in RPMI growth medium (10% FBS, 10 mg/mL penicillin and 10 mg/mL streptomycin [pen/strep] and 2 mM L-glutamine) for 48 hours (hr) followed an additional 6 hr in PMA-free RPMI growth medium before harvesting and seeding into the devices. LX-2 cells were cultured with DMEM medium supplemented with 2% FBS and 10 mg/mL pen/strep. All cells were maintained in a tissue culture incubator at 37°C, 5% CO2 and 18% O2. Isolated PMNs were maintained in RPMI with 5% FBS and used within 2 hr of isolation.
Fibronectin, LPS and EGF were purchased from Sigma Aldrich (St. Louis, Mo). Human recombinant TGF-β was purchased from R&D Systems (Minneapolis, MN). Rat tail collagen type 1 was purchased from Becton Dickinson (Franklin Lakes, NJ).
Flow set-up and computational fluid dynamics (CFD)
The oxygen consumption rate (OCR) of primary hepatocytes was a key factor in the flow design, as hepatocytes typically have much higher OCR than other cell types.51, 52 In hard plastic or glass sealed microfluidic devices, the sole source of oxygen for cells is the influx of cell culture media (~170–200 μM52, 53). In the vLAMPS, the hepatocytes and NPCs are fed by two independent flow channels, the vascular channel and the hepatic chamber. A 3D COMSOL model (COMSOL Multiphysics, version 5.2a) was used to estimate the flow fields in both flow channels as well as the distribution of the oxygen tension across the hepatocyte chamber. The Navier-Stokes equation (incompressible flows) was used to model the free flow in both channels, and the Brinkman equations were applied for the porous region (the hepatocyte layer, the endothelial cell layer, the PET membrane and all ECM layers). To simulate oxygen consumption, a transport of diluted species model (with a chemical reaction) was coupled with the flow model. We used an OCR for primary hepatocytes of 0.425 nmol/s/106 cells (Table S1), at the high end of previously published reports54, 55, to estimate that a flow rate of 180 μL/hr was required in the vLAMPS to support ~100,000 hepatocytes. We then empirically determined the optimal flow rates of 80 μL/hr in the hepatic channel, and 100 μL/hr in the vascular channel.
vLAMPS construction and cell seeding
The four cell types were sequentially seeded onto the PET membrane of the intermediate layer, which was maintained in media in a sterile Petri dish at 37°C, 5% CO2, in the following manner: On assembly day 1, the intermediate layer was inverted so that the membrane was on top, and 100,000 LSECs in EGM-2 media, (EGM-2, 2% FBS, 50 μg/mL gentamycin) were seeded onto the membrane and allowed to attach and spread in EGM-2 media for 24 hr. Optionally, some or all of the 36,000 differentiated THP-1-derived Kupffer cells can be seeded in the vascular channel on top of the LSECs, 12–15 hr after seeding the LSECs; On assembly day 2, the intermediate layer was inverted so that the LSECs and in some experiments Kupffer cells were on the bottom, and the top of the membrane was seeded with 15,000 LX-2 stellate cells and in other experiments 36,000 differentiated THP-1 Kupffer cells in EGM-2 media for 24 hr (see Table 1) . On assembly day 3, 70 μL of a 400 μg/mL (total protein) solution of porcine LECM (provided as a 10 mg/mL stock by Dr. Stephen Badylak, University of Pittsburgh) was added on top of the stellate and Kupffer cells (when introduced in the hepatic chamber) and incubated for 30 min at 37°C to create a thin ECM layer mimicking the space of Disse (perisinusoidal space).30 The cryopreserved primary human hepatocytes were thawed according to the supplier’s instructions and 100,000 were seeded onto the intermediate layer membrane as a 2.5 X106 hepatocytes/mL suspension in hepatocyte plating media (HPM, William’s E medium (Thermo Fisher, Waltham, MA ) supplemented with 5% fetal bovine serum (FBS), 10 mg/mL pen/strep and 2 mM L-glutamine. The hepatocytes were allowed to attach and spread in a tissue culture incubator (37o C, 5% CO2) for 16 – 18 hr without flow. The hepatocyte plating media was replaced by hepatocyte maintenance medium (HMM, Williams E supplemented with mL 1% FBS, 6.25 μg/mL human insulin, 100 nM dexamethasone, 6.25 μg/mL human transferrin, 6.25 ng/mL selenous acid, 2 mM L-glutamine, 15 mM HEPES, and 10 mg/mL pen/strep) (see Table 4). On assembly day 4, 40 μL of a 2 mg/mL rat tail collagen type 1 (Becton Dickinson, Franklin Lakes, NJ) solution in HMM was deposited on top of the hepatocytes and allowed to polymerize. A sufficient volume of collagen was used to form a ~50 μm thick overlay (measured by confocal imaging) to preserve the morphology and functions of the human primary hepatocytes for long-term culture.56, 57 A schematic of the fully assembled model is shown in Fig. 1B & 1D. The cell types and seeding numbers were guided by allometric scaling,58 and are summarized in Table 1. By allometric scaling the proportions of the cell types in the liver are ca. 60:20:15:5, hepatocytes:endothelial:Kupffer:stellate. However, the liver acinus is essentially a tube with the lumen lined with endothelial cells, surrounded by a Space of Disse and then surrounded by the hepatocytes. The larger diameter of the hepatocyte layer requires more cells than the endothelial layer. In the vLAMPS, the hepatic chamber communicates with the vascular channel through the PET filter in middle plate (A2 in Fig. 1A). The vascular channel requires a larger fraction of endothelial cells relative to hepatocytes as they occupy the same area. This was confirmed when constructing the model by plating varying numbers of hepatocytes and endothelial cells to determine the number required to create a confluent layer (data not shown). The fractions of Kupffer and stellate cells are consistent with allometric scaling and our previously published models.28, 30 The whole cell seeding process is also illustrated in the supplement (Fig.S1).
Table 1.
The cell seeding number in the vLAMPs
| Cell type | Seeding number | % of Total Cells | |
|---|---|---|---|
| Hepatic chamber | Hepatocytes | 100,000 | 40% |
| Kupffer cells | 36,000 | 14% | |
| Stellate cells | 15,000 | 6% | |
| Vascular channel | LSEC | 100,000 | 40% |
Table 4.
| Model Component | Further Development |
|---|---|
| Cells | Use patient derived primary cells or patient-derived iPSCs for all cells |
| Bile Ducts | Integration of Cholangiocytes and other cells required to create bile ducts |
| Media | Optimize media for normal vs disease states (e.g. modulate glucose, hormones, growth factors, free fatty acids, lipopolysaccharides, etc.) |
| Carrier Proteins | Evaluate the need for additional carrier proteins for small molecule drugs and biologics to minimize retention in the microfluidic devices |
| Flow Rate in Vascular Channel | Balance the optimal shear-stress in vascular chamber to stimulate LSECs with the use of flow rate to optimize zonation |
| Design of the Geometry and Volumes in the Vascular and Hepatic channels | Optimize the total volumes of the hepatic and vascular chambers and the geometry of the opening in the middle layer (A2) |
| Functional Scaling for Multiple Organs | Evaluate the maximal output of each coupled organ for factors required for the optimal function of the next organ in the series. Adjust the scaling of each organ to optimize the functional coupling |
Device assembly and perfusion
After the 4-cell liver model was constructed and stabilized on the intermediate membrane layer, the device was assembled into a two channel, microfluidic device by sandwiching the 0.4 mm thick intermediate layer between the 1.1 mm thick top layer and the 0.7 mm thick bottom layer, with spacing provided by the thin elastomer gasket (Fig. 1B). The three layers were “clamped” in the Micronit chip holder (Fig. 1B) sealing the flow channels in the proper alignment for receiving the tubing connection inserts. Flow connections to the 3-layer unit were made by compressing the FFKM ferrules into the tubing inserts. The widths of the two flow channels (11 mm at the widest) were established by the thin elastomer gaskets bonded on the top and bottom layer, as shown in Fig. 1A. The height of the flow channels was established by the thickness of the elastomer after compression (Fig. 1B) and was ~200 μm. However, due to the bonding of the PET membrane (10 μm thick) to the bottom of the 400 μm thick intermediate layer, the total depth of the hepatic chamber within the hepatic channel, bounded by the intermediate layer and the PET membrane, was ~500 μm, the bottom ~50 μm of which contained the hepatocytes, stellate and some Kupffer cells in the protein matrices, leaving ~100 μm for the hepatic flow channel. Also because of the membrane and adhesive, the depth of the vascular flow channel just below the membrane is ~100 μm. The volume of the hepatic chamber is ~100 μL, while the volume of the vascular channel is ~45 μL.
Once assembled, PEEK tubing was inserted into the inlets/outlets of the chip holder with compressed FFKM ferrules ensuring tight tubing connections. The media (Table 2) was pre-equilibrated for 16–18 hr at 37 °C, 5% CO2, and ambient (18%) O2. Flow was initiated with perfusion of HMM into the hepatic chamber and HMM supplemented with 1% FBS into the vascular channel using sterile glass syringes and syringe pumps. The initiation of flow in the vLAMPS starts Day 1 of experimental treatment.
Table 2.
The medium components for the hepatic and vascular channels
| Standard medium | Medium with molecular drivers |
|
|---|---|---|
| Hepatic channel | HMM | +LPS (1 μg/ml) +EGF (15 nM) +TGF-β (10 ng/ml) |
| Vascular channel | HMM +1% FBS | |
Oxygen tension measurement
Oxygen tension was measured by ratiometric imaging of oxygen sensitive and oxygen insensitive fluorescent beads. The oxygen sensitive beads were made in house by loading palladium(II)-5,10,15,20-tetrakis-(2,3,4,5,6-pentafluorphenyl)-porphyrin (PdTFPP, ex/em 405/706 nm, Sigma Aldrich, St Louis, MO) into 4.6 μm diameter polystyrene beads (Bangs Laboratories, Fishers, IN) as follows: A 4 mM stock solution of PdTFPP was freshly prepared in tetrahydrofuran (Sigma Aldrich, St Louis, MO). The loading solution was made by diluting 10 μL of bead emulsion (containing 10 μg of the polymer beads) with a mixture of 225 μL PdTFPP stock, 150 μL 2% Pluronic and 515 μL of sterile filtered water. The loading solution was protected from light and placed on a plate shaker for 20 min. The PdTFPP loaded beads were then rinsed 4 times in 0.1% Pluronic solution by centrifugation and resuspension. The beads were stored at 4°C in 0.1% Pluronic solution. The 5 μm diameter oxygen insensitive beads (ex/em 405/455 nm) were purchased directly from Bangs Laboratories.
In order to calibrate the fluorescence intensity (FI) ratio against oxygen concentration, three media standards were prepared: oxygen-depleted media (0%); ambient oxygen media (18%); and mid-range oxygen media (9%). The oxygen depleted media (0%) was generated by adding 4 U/mL glucose oxidase (Sigma, St Louis, MO). The mid-range media was prepared by adding 0.14 U/mL of glucose oxidase. This enzyme-reaction based approach to control of oxygen tension can efficiently maintain an oxygen deficient condition (e.g. 0% or 9%) in a sealed system over a period of hours,53 which served well for calibration purposes.
Real-time imaging of fluorescent biosensors, imaging immunofluorescence (IF) and scanning electron microscopy
A mitochondrial targeted Reactive Oxygen Species (ROS) sensitive GFP- fluorescent protein biosensor was transduced into primary hepatocytes by previously reported methods.28 The functionality of the biosensor was confirmed by treating the vLAMPS for 24 hr with 100 μM Menadione, a known ROS producing compound. Images were collected hourly and fluorescence intensity analysis was performed using Fiji (ImageJ version 1.151t).59
At the end of each experiment, the devices were fixed with 2% paraformaldehyde60 at 4°C for 30 min either by dissembling the system and fixing the intermediate layer or by perfusion of the fixative into the device. E-cadherin, principally in hepatocytes, or α-smooth muscle actin (α-SMA) in Stellate cells were labeled in separate devices by indirect-immunofluorescence of the fixed cells as follows. Cells were permeabilized with 0.1% Triton X-100 in PBS for 7 min, blocked 30 min. in 1% BSA/PBS and then reacted overnight at 4o C with 2.5 μg/mL mouse anti-E-cadherin (Becton Dickinson, Franklin Lakes, NJ) or 2.5 μg/mL anti-α-SMA (Sigma Aldrich, St Louis, MO). The primary antibody labeled cells were rinsed in PBS and incubated for 1 hr at room temperature in 6.7 μg/mL Alexa 488 conjugated goat anti-mouse secondary antibody (ThermoFisher, Waltham, MA) in PBS. The F-actin in the endothelial cells was labeled with 4 μg/mL TRITC-phalloidin (Sigma, St. Louis, MO) for 10 min. The mitochondrial membrane potential was monitored by perfusing media containing 200 nM Mitotracker Red (ThermoFisher, Waltham, MA) into the device for 30 minutes just prior to fixation. Steatosis was determined by staining cells with the neutral lipid dye LipidTox (ThermoFisher, Waltham, MA) prepared according to the supplier’s instructions. For VE-cadherin and ICAM-1 staining of the endothelial cells, the devices were fixed, permeabilized, and blocked with the procedure described above. The endothelial cells were then incubated with rabbit anti-VE-cadherin or mouse anti-ICAM-1 (PA5–17401 and MA5407, Invitrogen, Carlsbad, CA) for 24 hr. The primary antibody labeled endothelial cells were rinsed in PBS and incubated for 1 hr at room temperature in 6.7 μg/mL Alexa 488 conjugated donkey anti-rabbit or goat anti-mouse secondary antibody (Thermo Fisher, Waltham, MA) in PBS. PMNs were labeled for 30 min with 5 μM CellTracker Red CMPTX (ThermoFisher, Waltham, MA). For scanning PMN distributions within the fixed devices the PMNs were labeled with a mouse anti-CD24 (BD Biosciences, Franklin Lakes, NJ) followed by a secondary antibody as above. All images were collected using either a 20X (0.45NA) or a 40X (0.9 NA) objective on the IN Cell 6000 (GE Healthcare, Chicago, IL) in confocal mode with the aperture set at ≈ 1 airy unit. Fluorescence intensity analysis was performed using Fiji (ImageJ). The images were segmented with an intensity threshold of 50 to exclude background fluorescence and by object size (10 μm2) to exclude small objects, in order to evaluate the fluorescence intensity of the mitochondria and steatotic vacuoles.
The protocol for the scanning electron microscopy (SEM) was provided by the Center for Biologic Imaging (CBI, University of Pittsburgh). Briefly, the samples were fixed with 2.5% Glutaraldehyde for 1 hr, followed with 1% OsO4 treatment for 1 hr. Samples were dehydrated using a graded series of ethanol solutions and then air-dried overnight. After removing the PET membranes from the intermediate layer and mounting them on specimen stubs, the samples were sputter-coated with 3.5 nm platinum (Cressington Scientific, Watford, UK), then imaged on a JSM-6330F electron microscope (JEOL, Peabody, MA).
Bile Efflux Assay
The viability dye CMFDA (5-chloromethylfluorescein diacetate, Thermo Fisher, Waltham, MA) was used to demonstrate polarized bile efflux in the hepatocyte layer. Briefly, on Day 6 of the vLAMPS experiment, 2 μM CMFDA in HMM media was manually injected into the vLAMPS. After 20 min of static incubation allowing the dye to diffuse into the collagen layers, perfusion flow of non-CMFDA media was restarted and images were collected with a 20X (0.45NA) objective on the IN Cell 6000 as outlined above.
Permeability assay
Permeability of the endothelial cell layers was assessed by measuring the rate of FITC-albumin transport across a confluent monolayer cultured for 48 hr on the bottom surface of a 24 well, 3 μm pore size, PET Transwell® plate (Corning Costar, Tewksbury, MA).61 A solution of 10 μg/mL FITC-albumin was added to the lower chamber. After 2 hr incubation, 10 μL aliquots of medium were withdrawn from the upper chamber and diluted into 100 μL of PBS in a 96 well plate. The fluorescence intensity was measured with the IN Cell 6000 using a 20× (0.45 NA) objective, focused 50 μm above the plate bottom. After subtracting the background fluorescence, the permeability coefficient (P, cm/sec) was calculated by the equation62
where V is the total volume of the upper chamber (100 μL), S is the total surface area of the insert (100 mm2), dFI is the total fluorescence intensity change per unit time, and FI0 is the initial fluorescence intensity of the lower chamber.
Determination of Drug Binding/Recovery in the PDMS Device (LAMPS) vs the Micronit Glass Device (vLAMPS)
For comparison of drug binding between the vLAMPS and a PDMS device, we selected the single chamber commercial microfluidic device (HAR-V1 single chamber device, SCC-001, Nortis, Inc. Seattle, WA) used in the LAMPS model.30 The devices were stored in PBS until use. The interior of the device was dried under vacuum, followed by treatment with the standard LAMPS coating solution of 100 μg/mL fibronectin and collagen for 1 hr at room temperature. For all steps involving injection of fluid into the devices, 100–150 μL per device was used to ensure complete filling of fluidic pathways, chamber and bubble traps.
The preparation of the vLAMPS devices is described above. Cell-free devices were assembled, the membrane was coated with the standard ECM layers (LECM and collagen), and three different compounds with different values of LogP were perfused for up to seven days (Table 3). Compounds were run with and without low density lipoprotein (LDL) as a ‘carrier’ lipoprotein, to determine whether the use of a carrier would attenuate the loss due to absorption.29
Table 3.
The drug recovery rates (%) in MPS devices with or without LDL carrier
| Acetaminophen | Nefazodone | Terfenadine | |
|---|---|---|---|
| LogP | 0.46 | 4.7 | 7.1 |
| PDMS-based MPS | 100 ± 1 % | 0.50 ± 0.01 % | 5.4 ± 3.0 % |
| PDMS-based MPS with LDL Carrier |
100 ± 5 % | 2.7 ± 0.3 % | 8.7 ± 5.0 % |
| Glass-based vLAMPs | 95 ± 7 % | 63 ± 1 % | 27 ± 1.8 % |
| Glass-based vLAMPs with LDL Carrier |
95 ± 0 % | 100 ± 8 % | 100 ± 7 % |
The drugs Nefazodone, Terfenadine and Acetaminophen were solubilized in DMSO at 1, 10 and 100 mM, respectively. The stock drug solutions were added at 1:1000 dilution into HMM medium (plus 1% FBS) for perfusion into the vascular channel of a fully assembled cell free device. A portion of each drug cocktail was stored at −20°C for later analysis as the influx concentration. The efflux media was collected daily for 3 days and stored at −20°C for mass spectroscopy analysis. The efflux samples were extracted at room temperature by adding 100 μL of each sample to 200 μL of acetonitrile containing 10 μM Diclofenac as an internal standard (IS) in a 1.5 mL Eppendorf tube. The samples were vortexed for 30 sec, and then centrifuged at 15,000 rpm for 2.5 minutes in an Eppendorf minifuge to pellet the acetonitrile insoluble proteins. Then 20 μL of the protein free supernatant was transferred to a 96-well Acquity Collection plate (Waters, Cheshire, UK) to which 180 μL of a 20/80 % acetonitrile/H2O solution was added. The solutions were analyzed as previously described.29 The recovery of each drug was calculated as the ratio of the area under the curve (AUC) for the drug in the efflux media to the AUC for the drug in the influx media, both normalized to the IS.
Statistical analysis of the data
The data are presented as mean ± standard error (SEM) for n = 4 – 6 devices per test. For the oxygen tension measurements, beads were imaged with the 20x objective in 4 regions of interest (ROIs) per zone, and mitochondrial membrane potential and steatosis were imaged at 20x in 6 – 9 ROIs per zone. A second lot of cryopreserved human hepatocytes was used to evaluate lot-to-lot reproducibility of the oxygen and functional gradients in the vLAMPS under the described conditions. Differences between lots were evaluated using the T.Test function in Excel (Microsoft, Redmond, WA). For the mitochondrial targeted ROS biosensor experiment, images were collected in 4 ROIs per zone in 2 devices.
Results
The Design and Assembly of the vLAMPS
The vLAMPS has been developed around the Micronit three-layer chip (Fig.1 A). The three layers A1 (top), A2 (middle) and A3 (bottom) are borosilicate glass. A porous PET membrane (3 μm pores) is bonded over a ca. 8mm × 16 mm oval hole in the middle layer to allow diffusion of molecules and the transmigration of cells from the lower vascular channel (between A2 and A3), while serving as a support for the endothelial cells in the vascular channel and the stellate cells, the space of Disse matrix and the hepatocytes in the hepatic chamber (materials and methods). The hepatic chamber is enclosed by the A1 and A2 layers. A small ring of elastomer is used to seal the layers when assembled into the chip holder (Fig.1 B and C). Fig. 1 D is a diagram of the assembled device showing the hepatic chamber and vascular channel with the direction of flow. Table 1 lists the cell types: primary human hepatocytes, LSECs, stellate and Kupffer cells (see material and methods) and the number of each cell type plated in the device. Fig. S1 outlines the steps in seeding the cells on the A2 layer.
Reduced Drug Binding in the vLAMPS Compared to the LAMPS
PDMS is still the most widely used material for the fabrication of organ-on-chip devices as it is an oxygen-permeable, non-cytotoxic, and transparent elastomer that can be easily molded into complex structures.63, 64 However, the strong absorption of hydrophobic molecules/biologics to PDMS makes it quite challenging for use with many drugs, chemicals and biologics such as hormones. Based on a previous report, molecules with LogP less than 2.47 usually exhibit minimal absorption (<10%), whereas molecules with LogP greater than 2.62 may exhibit extensive absorption (>90%) into PDMS channels.65 However, it is critical to test all drugs and biologics for retention in microfluidic devices, typically by performing mass spectrometry analyses of the efflux to determine the effective concentration. As a result, it is difficult to reliably evaluate the dose-response and the sensitivity to drugs, and especially test compounds, for which the LogP may be unknown, in MPS that utilize high levels of elastomers.66, 67 To address this issue, surface treatments or alternative materials have been introduced, but the trade-offs in binding and oxygen diffusion are still being evaluated.68
In perfusion flow tests performed in the PDMS based LAMPS30 and the primarily glass vLAMPS, all-glass syringes and PEEK tubing were used to minimize binding in the influx and efflux (see materials and methods).29, 69 Cell-free devices were assembled and coated with the ECM layers (LECM and collagen), and three different compounds with different values of LogP were perfused for up to seven days. Three drugs, Nefazodone (LogP=4.7), Terfenadine (LogP=7.1), and Acetaminophen (LogP=0.46) were perfused into each type of device with and without an additional carrier protein (0.1 mg/mL LDL).70 Table 3 summarizes the results. Acetaminophen, a hydrophilic drug, exhibited complete recovery in both the LAMPS and vLAMPS. The recovery rates for Nefazodone (63%) and Terfenadine (27%) in vLAMPS were much higher than in LAMPS (0.5% and 5.38% respectively). When low density lipoprotein (LDL, 0.1 mg/mL) was added as a carrier molecule, the drug recovery rates for both drugs in vLAMPS were ca. 100%. For the PDMS based LAMPS however, there was only a slight improvement. This may be due to the absorbance of LDL (recover rate of LDL: 39.05 ± 2.5 %) into the bulk PDMS material.
3D structural organization of cells in the vLAMPS mimics the structure of the liver sinusoid
The sequential plating of the four cell types on the two sides of the intermediate layer (Fig. 1A, A2 layer) creates the polarity of the hepatic chamber and vascular channels when the whole chip is assembled (Fig. 1 B, C and D). Fig. 2 illustrates several key elements of the organization, structure and function of the vLAMPS using different imaging modalities. Fig. 2A shows the hepatocytes in the hepatic chamber labeled with E-cadherin (green), which is involved in the formation of junctions between the cells.71 Fig. 2B shows the distribution of the stellate cells (red) and Kupffer cells (yellow). The stellate and Kupffer cells are located on the lower surface of the hepatic chamber in the space of Disse between the hepatocytes and the LSECs. The Kupffer cells can initially be placed in either or both of the vascular channel and hepatic chamber since they can migrate between these compartments. Fig. 2C shows the F-actin stress fibers in the LSECs (red) labeled with rhodamine phalloidin and nuclei labeled with Hoechst (blue), illustrating the confluence of the LSEC layer and the orientation of the stress fibers. The confocal INCell 6000 permits focusing on the LSECs within the vascular channel. Although the LSECs form a confluent layer, they lack the tight junctions formed by other vascular endothelial cells such as HMVECs, as determined by labeling with VE-Cadherin (green, Fig. 2D). Further analysis of the LSEC layer by scanning electron microscopy showed the presence of fenestrations (arrows, average pore diameter 170 nm), characteristic of LSECs (Fig. 2E). Fenestrations were maintained for at least two weeks, unlike simpler 2D monocultures of LSECs in the absence of flow stimulation, in which fenestrations are rapidly lost.72–74 As a result, the permeability of the LSEC layer, as measured by diffusion of fluorescently labeled albumin, is much higher than the permeability of a HMVEC layer (Fig.2F), consistent with the physiology of LSECs in the liver. We plan to investigate other unique characteristics of LSECs including the relatively high level of receptor-mediated endocytosis, a role in portal hypertension71, 72, 75 and immune functions such as the production of cytokines and eicosanoids.76, 77
Figure 2.
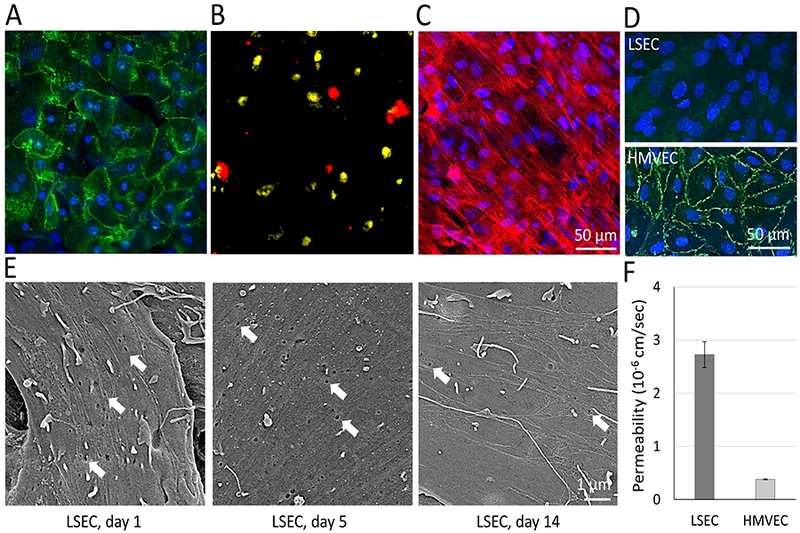
Characteristics of the 4 human cell types in the vLAMPS under flow. (A) Immunofluorescence of human primary hepatocytes labeled with an E-cadherin antibody (green) and nuclei with Hoechst (blue) in the hepatic chamber. (B) Stellate cells (red) and Kupffer cells (yellow) were pre-labeled with CellTracker deep red and CellTracker red, respectively, before assembly into the device in the hepatic chamber. (C) F-actin in Human LSECs in the vascular channel was labeled with rhodamine-phalloidin (red) and nuclei with Hoechst (blue). (D) Images comparing VE-cadherin immunolabeling (green) in LSEC and HMVEC cells demonstrates lack of tight junctions in LSECs. Nuclei were labeled with Hoechst (blue). (E) Scanning electron microscopy of LSECs in the vascular channel after 1 day, 5 days and 2 weeks. Arrows show fenestrations (pore size: 118.7±7.1 nm) that decreased in density, but were still present after 2 weeks. (F) The permeability coefficient, measured by the rate of fluorescent albumin permeation through the endothelial cell layer was much greater for LSECs compared to HMVECs, as expected, due to the absence of tight junctions and presence of fenestrations in the LSECs (P<0.01 between LSEC and HMVEC, one tailed t-test).
Creating Continuous Oxygen Zonation in the vLAMPS
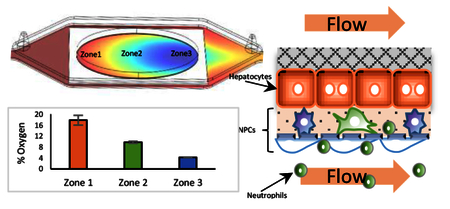
The spatial variation in oxygen tension is an important characteristic of the liver, and therefore important for recapitulating liver physiology in vitro, as metabolism, energy production, and gene expression in hepatocytes are highly oxygen dependent processes.21, 78, 79 In vivo, an oxygen gradient is generated by oxygen consumption, primarily by the hepatocytes, that depletes the oxygen transported by the blood, through diffusion and exchange as it flows through the liver sinusoids. It is technically challenging to reproduce a gradient in conventional cell culture dishes and even in microfluidic devices and MPS. Due to the excellent oxygen permeability of PDMS, oxygen rapidly reaches steady state by diffusion through the PDMS, precluding gradient formation without external environmental controls.80, 81 Modeling oxygen tension in media flowing through a device constructed of oxygen impermeable materials, such as glass, suggested that it was possible to generate physiologic oxygen gradients by controlling the flow rates in the vascular and hepatic channels.82, 83 Modeling oxygen tension, based on the maximal rate of oxygen consumption by the hepatocytes, Michaelis–Menten kinetics30 with 18% oxygen content in the influx media, and using 80 and 100 μL/hr flow rates, predicted that an oxygen tension gradient (Fig. 3) would be established and stabilized within 2 hr. Furthermore, establishing the physiologically accurate zone-specific oxygen environment in the device only required flow rates at which shear stress levels were orders of magnitude below those shown to negatively affect the health and functions of the hepatocytes and endothelial cells.30
Figure 3.
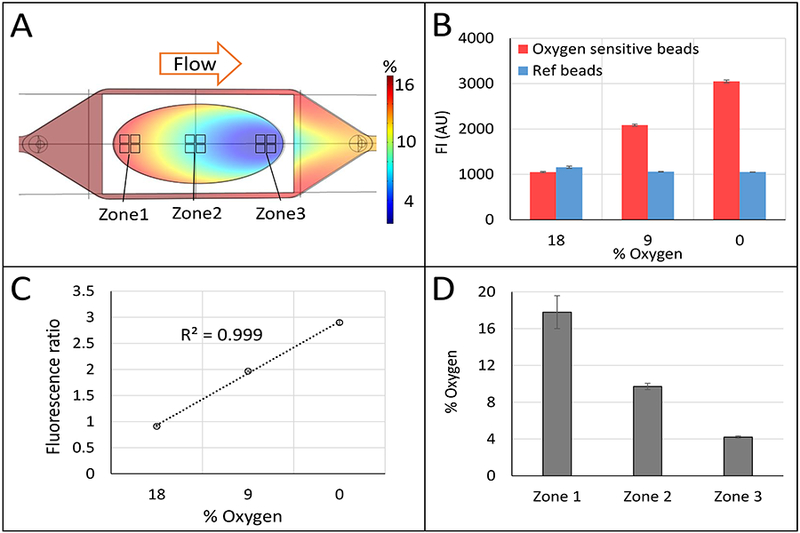
Computational and experimental demonstration of continuous zonation in the vLAMPS. (A) Comsol Multiphysics-based model of oxygen tension computed based on physical and biological parameters (see Table S1). The calculated oxygen tension ca. 4% to 18% is shown in a color scale bar. Regions corresponding to zone 1 (near the inlet to the hepatic chamber), zone 2 (the center of the chamber) and zone 3 (near the outlet of the chamber) are indicated. (B) Direct measurement of oxygen tension using a ratiometric method with oxygen sensitive and insensitive beads placed in the hepatic chamber of the vLAMPS (see also Fig. S2), (P<0.001 between three oxygen % conditions of oxygen sensitive beads, one-way ANOVA). (C) Ratiometric standard curve from 18% to 0% oxygen. (D) % oxygen tension measured in zones 1, 2 and 3 using the ratios of bead fluorescence in the regions depicted in (A) and the standard curve (C). The continuous zonation was created with a media flow rate of 100 μL/hr in the vascular channel and 80 μL/hr in the hepatic channel (P<0.001 between three zones, one-way ANOVA). See also Fig. S3.
We were able to generate continuous oxygen zonation within the vLAMPS, and validated the establishment of physiological zonation (Fig. 4) within 1 – 3 days after the initiation of flow. Fig. 3A depicts the spatial pattern of zonation based on computational modeling using the COMSOL simulation software (see materials and methods). Direct experimental validation of oxygen zonation was performed using fluorescence ratio imaging30 of oxygen sensitive and oxygen insensitive beads. After seeding the beads in the hepatic chamber, ~950 beads/mm2 for the oxygen sensitive beads and ~475 beads/mm2 for the insensitive beads, both bead types were parfocal and ca. 2 μm below the hepatocytes in the thin LECM layer. Fig. 3B shows that the fluorescence intensities of the beads that are quenched by oxygen (red bars), increase with decreasing oxygen tension, while the oxygen insensitive beads (blue bars) maintain a constant intensity. The ratio of fluorescence intensities of oxygen sensitive to oxygen insensitive beads is linear, providing a means to calibrate the % oxygen based on the fluorescence ratio (Fig. 3C). Using the standard curve, the measured % oxygen in three ROIs (Fig. 3A, near the input, the middle, and near the output) were found to correspond to physiological zones 1, 2 and 3 (Fig. 3D) with oxygen tensions of ca. 18%, 10% and 4%, respectively. Fig. S2 shows an image of the oxygen sensitive and insensitive beads with a line profile across a single oxygen sensitive bead. Fig. S3 demonstrates that the control of the flow of media in the vascular and hepatic channels can create distinct patterns of oxygen tension, including the creation of essentially a single zone within the device. By controlling the flow rates, we are able to generate a primarily zone 1 (Fig. S3C) or zone 3 (Fig. S3A) model. Due to the oval shape of the hepatocyte seeding area (8 × 16 mm), the oxygen consumption on the edge was lower due to fewer cells in the flow path, resulting in the increased oxygen tension along the edges of the chamber (Fig. 3A). We are working on an alternative flow channel design to mitigate such edge effects for the next iteration of the vLAMPS. Ideally, a single straight channel, or multiple straight channels, with less open space on the edge will be used to construct the cell layer in the next stage of the vLAMPS evolution that will include other advances (Table 4).
Figure 4.
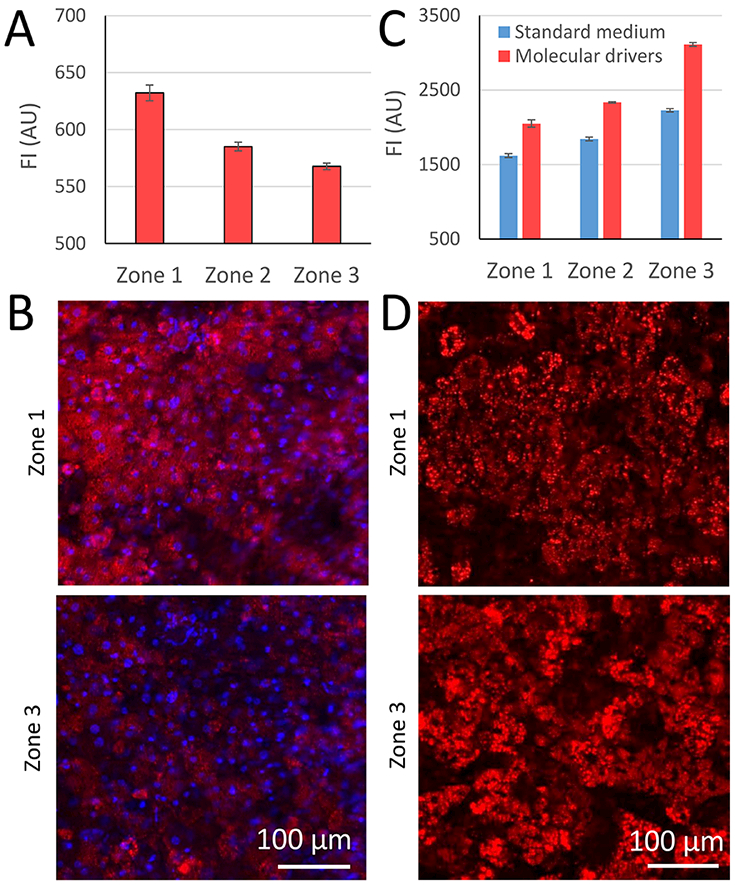
Biological demonstration of zonation in the vLAMPS. (A) Mitochondrial membrane potential quantified on Day 5 with MitoTracker Red, as a fluorescent indicator of the level of oxidative phosphorylation in the three zones, the locations of which are defined in Fig. 3A. Mitochondrial membrane potential is maximal in zone 1 (P<0.001 between Zone 1 and Zone 2&3; P<0.05 between Zone 2 and Zone 3, one-way ANOVA). (B) Representative images from zones 1 and 3 regions as defined in Figure 3A. (C) Steatosis quantified on Day 5 by imaging the fluorescent indicator LipidTox Deep Red. Steatosis is maximal in zone 3, both with standard media and media supplemented with molecular drivers associated with NAFLD including LPS, EGF and TGF-β (see Table 2) (P<0.001 between the three zones within each media condition; P<0.001 between two media conditions in each zone, one way ANOVA). (D) Representative images of LipidTox Deep Red staining in zone 1 and zone 3 as defined in Fig. 3A.
Biological function of hepatocytes in Zone 1 and 3 and the impact of molecular drivers
To investigate the oxygen zone-specific biology, mitochondrial membrane potential was measured with Mitotracker Red as an indicator that reflects the level of oxidative phosphorylation, and steatosis was quantified by labeling with LipidTox. The mean fluorescence intensity of the mitochondrial membrane potential in the vLAMPS was highest in Zone 1 and lowest in Zone 3, consistent with higher levels of oxidative phosphorylation in Zone 1 compared with Zone 3 (Fig. 4A). Example images of the mitochondrial membrane potential acquired in zone 1 and zone 3 are shown in Fig. 4B. This result is consistent with the physiological results from our published single zone LAMPS studies.30
The accumulation of lipid droplets in the hepatocytes was another important indicator of zone-dependent metabolism. Using LipidTox to label the fat droplets, we were able to show that on Day 5, lipogenesis in Zone 3 was higher than in Zone 1 (Fig. 4C). When molecular drivers of NAFLD (Table 2) were perfused into the vLAMPS over the same period, there was a proportional increase in fluorescence intensity in all 3 zones, and again, the highest intensity was in zone 3 compared to zone 1 (Fig. 4D). These results are consistent to the results from our published single zone LAMPS studies.30 Increased albumin and urea synthesis were demonstrated under flow compared to static culture of the fully assembled intermediate layer (Fig. S4A & B).
Testing Reproducibility
We used a single lot of cryopreserved human hepatocytes for consistency from experiment–to-experiment, to avoid the lot-to-lot variation in primary human cells. However, it is also important to understand the potential impact of lot-to-lot variability on the performance of the model in creating zonation. A second lot of cryopreserved human hepatocytes from ThermoFisher was used to evaluate the reproducibility of the oxygen and the functional impact of zonation on steatosis in the vLAMPS (Fig. S5). There was no significant difference in the fluorescence intensity ratio of the oxygen sensitive beads to insensitive beads between lots of cells in the vLAMPS (Fig. S5A). There was also no significant difference in the measured steatosis values between lots of hepatocytes, both of which exhibited a 25–28% elevated level of steatosis in zone 3 compared with zone 1,. (Fig S5B). Although testing with only two lots is admittedly limited, the minimal differences between lots is at least an indication that the model performance is not wholly dependent on the chosen lot of cells.
We also performed an initial analysis on the reproducibility of a selected physiological parameter between devices under the same conditions. This is made simple by employing the Microphysiology Systems Database (MPS-Db).84 For the steatosis measurements in Fig. S5B we analyzed data from 4–5 devices for each cell type that were investigated in two different experiments under exactly the same conditions. The coefficient of variation (CV) among replicate sets vLAMPS models with the same cells was 7–16%, with the exception of one set in which one of four devices had a steatosis value that was 33% lower than the other three, resulting in a CV of 25% (data not shown). There was no significant difference in the measured steatosis between experiments.
We also compared the time course of the secretion of albumin over 9 days in two different experiments (see vLAMPS data in Fig S4A). to compare the time courses, we used the intraclass correlation coefficient (ICC), which compares both the magnitude and the trend in the data, and has a value of 1 for perfectly correlated data.85 The albumin measurements for two sets of 4 vLAMPS models run in different experiments have an ICC =0.42 (good), increasing to 0.82 (excellent) if day 1 is not included. The vLAMPS models sometimes show more variation in the first day following assembly, becoming more consistent over the next couple of days.
All future studies using MPS should be captured in an analytical database like the MPS-Db in order to demonstrate the reproducibility of any measured parameter. This is critical since we expect to see biologically relevant heterogeneity between patient cells and treatments (primary or iPSC-derived) due to disease and/or genetic variation.
Bile Efflux in the vLAMPS
An additional characteristic of normally functioning hepatocytes is the establishment of polarized bile efflux transporter proteins in the canalicular spaces formed between adjacent hepatocytes. The accumulation of CMFDA in the canalicular spaces on Day 6 demonstrates the establishment of polarized transport in the vLAMPS model (Fig. S4C). Accumulations of CMFDA in canalicular spaces on days 10 and 14 were essentially the same as day 6 (data not shown). Future studies will evaluate inhibition of the bile salt exporter protein function in the vLAMPS, as it is an underlying cause of liver damage from cholestatic liver diseases or drugs.86
The activation of stellate and Kupffer cells by molecular drivers
The transactivation of the fat and vitamin A storing stellate cell into a proliferating, collagen depositing myofibroblast can be monitored by the increase in α-SMA expression and proliferation of the cells.87, 88 In the vLAMPS we measured an increase in α-SMA in stellate cells, higher in zone 3 than in zone 1, in response to exposure to molecular drivers (Fig. 5). TGF-β, a component of the molecular drivers, is one of the key profibrogenic cytokines that induces collagen deposition following liver injury.89, 90 The activation of stellate cells was also evaluated with F-actin staining (data not shown), which showed an increase in stress fibers and a more elongated spindle shape after 5 days of perfusion culture with molecular drivers, compared with standard media. We have also previously published LPS induced release of TNF-α from the Kupffer-like cells in the LAMPS model.28, 30
Figure 5.
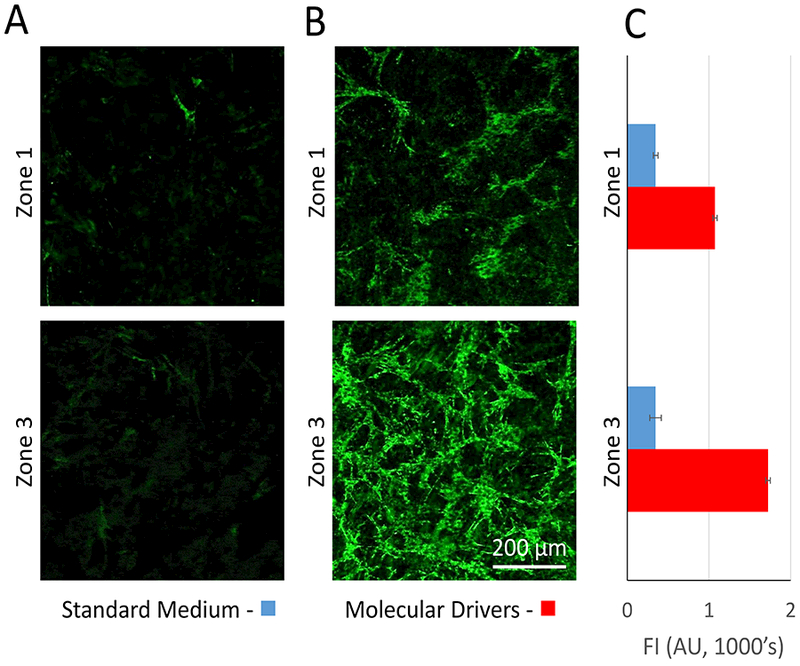
Demonstration of zonation specific effect on stellate cell activation in the vLAMPS. (A) Representative images showing minimal expression of α-SMA and proliferation of stellate cells in Zone 1 and zone 3 under control conditions with standard medium. (B) Representative images showing increased α-SMA expression and proliferation of stellate cells in zone 1 and extensive expression of α-SMA and proliferation of the activated stellate cells in zone 3 in the presence of molecular drivers after five days (see table 2). (C) Zone 3 stellate cells express higher levels of α-SMA in the presence of molecular drivers compared to zone 1 stellate cells.
The vascular inflammatory response to a molecular driver challenge was consistent with Non-Alcoholic Steatohepatitis (NASH)
We investigated the response of the LSECs in the vascular channel to the challenge of molecular drivers that are characteristic of conditions observed during disease progression of NAFLD. First, we established that the primary LSECs could be activated by molecular drivers (Table 2) in a microplate co-culture set-up, by imaging and quantitating ICAM-1 expression (Fig. S6). Following stimulation with molecular drivers for 6 hr, there was a 12–19% increase in ICAM-1 expression, with the largest increase involving all cell types (Fig. S6).
Fluorescently labeled, freshly isolated human PMNs were perfused into the vascular channel at the standard flow rate of 100 μL/hr in conditions of control media (no LSEC activation) and after the activation of LSECs with molecular drivers (Table 2). Fig. 6A demonstrates that very few PMNs bound to the LSECs in the absence of activation. However, perfusion of the vascular channel with the molecular drivers for 24 hr prior to perfusion with the PMNs induced a significant increase in the number of PMNs bound to the LSECs (Fig. 6B and 6C).
Figure 6.
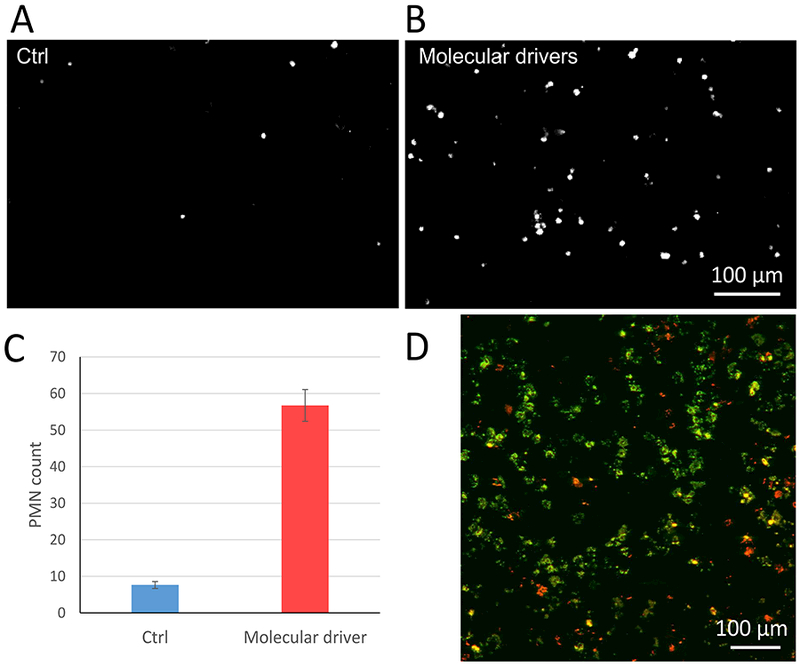
Binding of freshly isolated, fluorescently labeled polymorphonuclear leukocytes (PMNs) to activated LSECs and migration of PMNs from the vascular channel into the hepatic chamber in the vLAMPS. (A-B) Representative image of PMN adhesion in the vascular channel under the perfusion of standard media (A) or molecular drivers (B). (C) Comparison of the number of PMNs adhering to un-activated LSECs (standard media) and activated LSECs (molecular drivers) (P<0.001, one tailed t-test). (D) To evaluate transmigration of PMNs, the vLAMPS model was fixed, disassembled and labeled for immunofluorescence. A representative image field consisting of two 20x confocal image planes, one from the bottom of the membrane (green, the vascular channel) and the other 20 μm above (red, in the hepatic chamber) were overlaid to visualize the PMNs in each plane. No obvious zone-specific pattern of transmigration was detected in these studies. Cell tracker labeled PMNs can also be used to track the real-time dynamics of transmigration.
Following binding to the activated LSECs, the PMNs migrated into the hepatic chamber, also treated with molecular drivers, over a 1 ½ hr period. Fig. 6D depicts a representative image field consisting of two 20x confocal image planes, one from the bottom of the membrane (green) in the vascular channel and the other 20 μm above the membrane (red) in the hepatic chamber. The images from the two planes were overlaid in color to visualize the PMNs in each plane. These results demonstrate that the PMNs transmigrated from the vascular channel through the pores in the PET membrane and the space of Disse matrix into the hepatic chamber in response to the pro-inflammatory molecular drivers (Table 2). No zone-specific pattern of transmigration was detected in these experiments. The dynamics of the PMNs transmigration, like the binding of the PMNs to the activated LSECs (Fig. 6B and 6C), can also be tracked in real-time by time-lapse imaging of the fluorescently labeled PMNs.
Measuring Reactive Oxygen Species Generation in vLAMPS
The mitochondrial targeted ROS biosensor was used in live cell experiments to monitor generation of hydrogen peroxide in the hepatocytes.27, 91 Menadione, a redox cycling compound that directly produces H2O2 in mitochondria91 was used to induce ROS, as measured by the time dependent increase in the biosensor intensity (Fig. S7 A, B). The ROS biosensor response was greater in zone 1 compared to zone 3. This is consistent with the reports of higher oxidative phosphorylation and higher ROS level generated in mitochondria with higher transmembrane potential92 and consistent with our results showing higher oxidative phosphorylation in zone 1 hepatocytes [Fig. 4A].
Discussion
The evolution to the vLAMPS addresses several key challenges to creating an optimal, biomimetic liver MPS:
-
(1)
The optimal liver model will maximally recapitulate human liver functions. Zonation in the liver, established by the consumption of oxygen by hepatocytes, plays a key role in regulating hepatocyte functions. We have previously created zone 1 or zone 3 oxygen tension in separate LAMPS models to demonstrate that oxidative phosphorylation, albumin and urea production are highest in zone 1, whereas glycolysis, α−1 anti-trypsin, Cyp 2E1 expression and steatosis are elevated in zone 3.30 In addition, acetaminophen toxicity initially caused an early release of LDH from zone 3 compared to zone 1 models, a finding consistent with the known clinical effect of the drug.93, 94 In the vLAMPS we have created a liver MPS with continuous physiological zonation, and demonstrated that it induces the expected, zone dependent liver functions in the metabolic functions evaluated. Current animal models typically fail to reproduce the spectrum of human liver injury and it has been very difficult to study the effect of zonation in the in vivo human liver. The creation of a continuous oxygen gradient within one microfluidic platform built as a multi-cellular biomimetic of the human liver acinar structure, will ultimately allow the study of molecular level interactions between resident liver cells, the adaptive immune response and soluble molecular drivers in the initiation and progression of liver injury from drugs, toxins or disease, especially with the use of iPSC-derived cells from patients.
A second key characteristic of the human liver is the leaky ‘barrier’ created by liver sinusoidal endothelial cells, through a combination of reduced expression of cell junction proteins like VE-Cadherin, and fenestration of the cells. Liver derived endothelial cells are known to be heterogenous. Even within the sinusoid, two subpopulations of LSECs have been identified in liver sections by unique expression profiles.95 The variation in phenotypes among liver derived endothelial cells, and even among LSECs, makes it difficult to obtain a pure population of LSECs. To establish that the liver derived endothelial cells used in the vLAMPS contain a significant population of LSECs, we demonstrated that the LSECs used in the vLAMPS did not exhibit tight junctions compared to HMVEC cells (Fig 2D), and exhibited large pore fenestrations consistent with LSECs96 that persisted for at least 2 weeks (Fig 2E). The maintenance of fenestrations is likely due, in part, to the combination of flow stimulation and the communication between cells between the hepatic chamber and vascular channel.
A third key characteristic of the liver is the binding of migratory immune cells followed by transmigration into the hepatic chamber as a result of activation of LSECs by molecular drivers and a gradient of migration stimuli from the hepatic chamber. We have demonstrated in the vLAMPS that we can use molecular drivers to activate the LSECs and promote binding and migration of PMNs. These three major advancements in the design of a liver MPS will greatly enhance its utility for discovery of therapeutics, modeling diseases, and testing for toxicity of drugs and environmental agents.
-
(2)
The future of human MPS will involve their use in drug discovery and development, including clinical trials. Many drugs/drug candidates, including biologics, are hydrophobic which increases the potential for binding to polymers such as PDMS. Various approaches to minimizing binding to PDMS or related polymers have been employed, but the results are still being debated.68, 97, 98 The primarily glass device used for the vLAMPS model, with only a minimal amount of polymer to seal the layers, bound (retained) much less of the test drugs investigated here, than the PDMS-based device used for the LAMPS model (Table 3). Further tests of a wider range of small molecule drugs and biologics are required, but it is clear that the vLAMPS shows great potential for integrating drug testing in liver disease and ADME/TOX, as well as environmental chemicals. The use of PDMS devices can still be valuable, however, the drug/biologic retention must be evaluated in advance, with and without general and/or specific carrier molecules.29
-
(3)
Biomimetic organs on chips, where multiple cell types are organized in three dimensions with a separate but integrated vascular channel, are critical for recapitulating organ functions, including at least partial immune responses. The vLAMPS, with a vascular channel lined with LSECs and a porous membrane, allows the physiologically relevant liver sinusoid model to deliver cells and molecules from the vascular channel to the hepatic chamber, including immune cells and “molecular drivers” of disease. The activation of LSECs allows signaling between the LSECs, hepatocytes and other NPCs, and promotes binding of migratory immune cells such as PMNs, that is the first step in transmigration into the hepatic chamber, where immune cells can be involved in repair or induction of damaging inflammation. The vascular channel in the vLAMPS also provides a means for physical coupling to other organ MPS for ADME/TOX and disease modeling. For example, a more complete model of NAFLD will involve the integration of the vLAMPS with an intestine MPS, as well as a white adipose tissue MPS.
The evolution of the vLAMPS will continue in parallel to its application to the investigation of a variety of liver diseases including NAFLD, type 2 diabetes and the liver as a metastatic niche for a variety of cancers. Further improvements (Table 4) will include the integration of either isogenic patient-derived primary cells and/or iPSC-derived liver cells as they are validated as adult cells, cholangiocytes to guide the formation of bile ducts, the development of specific medias for “normal” and specific disease conditions (e.g. glucose, hormone free fatty acid levels), optimization of geometry and volumes in the hepatic and vascular channels and control of flow rates and scaling between organ MPS.29, 99 The progress in creating valuable human MPS over the last seven years has been impressive, but more effort is required to optimize the “normal” physiological and pathophysiological characteristics. In addition, for the organ MPS to be widely accepted, strict protocols must be adopted and validation/reproducibility must be quantified within and between laboratories.7
Conclusions
The vLAMPS is the next evolution of the human, biomimetic liver MPS. Compared to sealed microfluidic devices, the coating and cell seeding of the intermediate layer prior to assembly, simplifies construction of the model making the platform user-friendly to biomedical researchers. We have demonstrated that this primarily glass microfluidic device is optimal for real-time imaging to directly monitor cell/tissue functions with a range of fluorescent biosensors, as well as an optimal platform for minimizing the binding of hydrophobic drugs/biologics to the materials that constitute the device. The assembly of the three layered device with primary human hepatocytes, human cell lines for stellate and Kupffer cells and primary human LSECs creates a vascular channel separated from the hepatic channel by a porous membrane that allows communication between the flow channels, recapitulating the 3D structure of the liver acinus. The vascular channel can be used to deliver drugs, immune cells, other cells and other factors to a stand-alone liver MPS and/or to couple the liver MPS to other organ MPS. The assembly of the three-layer devices permits a high degree of control in adding matrix materials and cells on both sides of the porous membrane (hepatic and vascular). This enables a combination of bioprinting and manual or automated seeding. In addition, the three-layered devices can be disassembled at the end of a study to access specific cell types or layers for single cell studies such as genomics. We have successfully created continuous oxygen zonation by controlling the flow rates of media in the distinct vascular and hepatic flow channels and validated the computational modeling of zonation with oxygen sensitive and insensitive beads. This allows the direct investigation of the role of zonation in physiology, toxicology and disease progression. The vascular channel is lined with human LSECs that recapitulates partial immunologic functions within the liver sinusoid, including the activation of LSECs with “driver molecules”, promoting the binding of fluorescently labeled PMNs followed by transmigration into the hepatic chamber. The driver molecules also activate the stellate cells that express α-SMA, become fibroblastic in shape and proliferate. The infiltration of PMNs, as well as other migratory immune cells into the hepatic chamber is critical for the induction of inflammation in a variety of liver diseases and drug induced liver injury (DILI). The vLAMPS is a valuable platform to investigate the functions of the healthy and diseased human liver using all primary human cell types and/or iPSC-derived cells.
Supplementary Material
Acknowledgements
Albert Gough and D. Lansing Taylor are co-corresponding authors. The authors would like to acknowledge valuable discussions with Drs. Paul Monga, Ramon Bataller, Josepmaria Argemi, Andrew Stern and Alex Soto-Gutierrez, as well as with members of the Drug Discovery Institute and the Pittsburgh Liver Research Center (PLRC). Dr Hamza Yazdani and Dr Allan Tsung from the Pittsburgh Liver Research Center (PLRC) advised us on the isolation and handling of the human PMNs. In addition, we would like to acknowledge the invaluable contributions of Celeste Reese and Richard DeBiasio in carrying out the experiments presented here. This work was supported by NIH-NCATS grant #5UH3 TR000503–05, USEPA Star Center grant #83573601, the University of Pittsburgh Cancer Center Facility grant #2P30 CA047904, and an NIH S10 Instrumentation grant # S10OD12269.
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Godoy P, Hewitt NJ, Albrecht U, Andersen ME, Ansari N, Bhattacharya S, Bode JG, Bolleyn J, Borner C, Bottger J, Braeuning A, Budinsky RA, Burkhardt B, Cameron NR, Camussi G, Cho CS, Choi YJ, Rowlands JC, Dahmen U, Damm G, Dirsch O, Donato MT, Dong J, Dooley S, Drasdo D, Eakins R, Ferreira KS, Fonsato V, Fraczek J, Gebhardt R, Gibson A, Glanemann M, Goldring CEP, Gomez-Lechon MJ, Groothuis GMM, Gustavsson L, Guyot C, Hallifax D, Hammad S, Hayward A, Haussinger D, Hellerbrand C, Hewitt P, Hoehme S, Holzhutter HG, Houston JB, Hrach J, Ito K, Jaeschke H, Keitel V, Kelm JM, Park BK, Kordes C, Kullak-Ublick GA, LeCluyse EL, Lu P, Luebke-Wheeler J, Lutz A, Maltman DJ, Matz-Soja M, McMullen P, Merfort I, Messner S, Meyer C, Mwinyi J, Naisbitt DJ, Nussler AK, Olinga P, Pampaloni F, Pi JB, Pluta L, Przyborski SA, Ramachandran A, Rogiers V, Rowe C, Schelcher C, Schmich K, Schwarz M, Singh B, Stelzer EHK, Stieger B, Stober R, Sugiyama Y, Tetta C, Thasler WE, Vanhaecke T, Vinken M, Weiss TS, Widera A, Woods CG, Xu JJ, Yarborough KM and Hengstler JG, Arch. Toxicol, 2013, 87, 1315–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yum K, Hong SG, Healy KE and Lee LP, Biotechnology journal, 2014, 9, 16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marx U, Andersson TB, Bahinski A, Beilmann M, Beken S, Cassee FR, Cirit M, Daneshian M, Fitzpatrick S, Frey O, Gaertner C, Giese C, Griffith L, Hartung T, Heringa MB, Hoeng J, de Jong WH, Kojima H, Kuehnl J, Leist M, Luch A, Maschmeyer I, Sakharov D, Sips AJAM, Steger-Hartmann T, Tagle DA, Tonevitsky A, Tralau T, Tsyb S, van de Stolpe A, Vandebriel R, Vulto P, Wang JF, Wiest J, Rodenburg M and Roth A, Altex-Altern Anim Ex, 2016, 33, 272–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shuler ML, Lab on a chip, 2017, 17, 2345–2346. [DOI] [PubMed] [Google Scholar]
- 5.Zhang B and Radisic M, Lab on a chip, 2017, 17, 2395–2420. [DOI] [PubMed] [Google Scholar]
- 6.Clark AM, Ma B, Taylor DL, Griffith L and Wells A, Exp Biol Med (Maywood), 2016, 241, 1639–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Low LA and Tagle DA, Expert Review of Precision Medicine and Drug Development, 2018, DOI: 10.1080/23808993.2018.1456333, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beckwitt CH, Clark AM, Wheeler S, Taylor DL, Stolz DB, Griffith L and Wells A, Exp. Cell Res, 2018, 363, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vernetti LA, Vogt A, Gough A and Taylor DL, Clin Liver Dis, 2017, 21, 197–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colnot S and Perret C, in Molecular Pathology of Liver Diseases, ed. Monga SSP, Springer US, Boston, MA, 2011, DOI: 10.1007/978-1-4419-7107-4_2, pp. 7–16. [DOI] [Google Scholar]
- 11.Materne EM, Tonevitsky AG and Marx U, Lab on a chip, 2013, 13, 3481–3495. [DOI] [PubMed] [Google Scholar]
- 12.Soto-Gutierrez A, Gough A, Vernetti LA, Taylor DL and Monga SP, Exp Biol Med (Maywood), 2017, 242, 1605–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wisse E, Braet F, Luo D, De Zanger R, Jans D, Crabbe E and Vermoesen A, Toxicologic pathology, 1996, 24, 100–111. [DOI] [PubMed] [Google Scholar]
- 14.Kajikawa K, Yasui W, Sumiyoshi H, Yoshida K, Nakayama H, Ayhan A, Yokozaki H, Ito H and Tahara E, Virchows Archiv. A, Pathological anatomy and histopathology, 1991, 418, 27–32. [DOI] [PubMed] [Google Scholar]
- 15.Asrih M and Jornayvaz FR, Mol. Cell. Endocrinol, 2015, 418 Pt 1, 55–65. [DOI] [PubMed] [Google Scholar]
- 16.Satapathy SK and Sanyal AJ, Semin Liver Dis, 2015, 35, 221–235. [DOI] [PubMed] [Google Scholar]
- 17.Meex RCR and Watt MJ, Nature reviews. Endocrinology, 2017, 13, 509–520. [DOI] [PubMed] [Google Scholar]
- 18.Jungermann K and Kietzmann T, Hepatology (Baltimore, Md, 2000, 31, 255–260. [DOI] [PubMed] [Google Scholar]
- 19.Gebhardt R and Matz-Soja M, World journal of gastroenterology : WJG, 2014, 20, 8491–8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olson H, Betton G, Robinson D, Thomas K, Monro A, Kolaja G, Lilly P, Sanders J, Sipes G, Bracken W, Dorato M, Van Deun K, Smith P, Berger B and Heller A, Regulatory toxicology and pharmacology : RTP, 2000, 32, 56–67. [DOI] [PubMed] [Google Scholar]
- 21.Starokozhko V and Groothuis GM, Expert opinion on drug metabolism & toxicology, 2017, 13, 125–128. [DOI] [PubMed] [Google Scholar]
- 22.Rennert K, Steinborn S, Groger M, Ungerbock B, Jank AM, Ehgartner J, Nietzsche S, Dinger J, Kiehntopf M, Funke H, Peters FT, Lupp A, Gartner C, Mayr T, Bauer M, Huber O and Mosig AS, Biomaterials, 2015, 71, 119–131. [DOI] [PubMed] [Google Scholar]
- 23.Gori M, Simonelli MC, Giannitelli SM, Businaro L, Trombetta M and Rainer A, Plos One, 2016, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jellali R, Bricks T, Jacques S, Fleury MJ, Paullier P, Merlier F and Leclerc E, Biopharm Drug Dispos, 2016, 37, 264–275. [DOI] [PubMed] [Google Scholar]
- 25.Wagner I, Materne EM, Brincker S, Sussbier U, Fradrich C, Busek M, Sonntag F, Sakharov DA, Trushkin EV, Tonevitsky AG, Lauster R and Marx U, Lab Chip, 2013, 13, 3538–3547. [DOI] [PubMed] [Google Scholar]
- 26.Lee H and Cho DW, Lab on a chip, 2016, 16, 2618–2625. [DOI] [PubMed] [Google Scholar]
- 27.Senutovitch N, Vernetti L, Boltz R, DeBiasio R, Gough A and Taylor DL, Exp Biol Med (Maywood), 2015, 240, 795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vernetti LA, Senutovitch N, Boltz R, DeBiasio R, Shun TY, Gough A and Taylor DL, Exp Biol Med (Maywood), 2016, 241, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vernetti L, Gough A, Baetz N, Blutt S, Broughman JR, Brown JA, Foulke-Abel J, Hasan N, In J, Kelly E, Kovbasnjuk O, Repper J, Senutovitch N, Stabb J, Yeung C, Zachos NC, Donowitz M, Estes M, Himmelfarb J, Truskey G, Wikswo JP and Taylor DL, Scientific reports, 2017, 7, 42296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee-Montiel FT, George SM, Gough AH, Sharma AD, Wu J, DeBiasio R, Vernetti LA and Taylor DL, Exp Biol Med (Maywood), 2017, 242, 1617–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Domansky K, Inman W, Serdy J, Dash A, Lim MH and Griffith LG, Lab Chip, 2010, 10, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kostadinova R, Boess F, Applegate D, Suter L, Weiser T, Singer T, Naughton B and Roth A, Toxicol. Appl. Pharmacol, 2013, 268, 1–16. [DOI] [PubMed] [Google Scholar]
- 33.Yoon No D, Lee KH, Lee J and Lee SH, Lab Chip, 2015, 15, 3822–3837. [DOI] [PubMed] [Google Scholar]
- 34.Ramachandran SD, Schirmer K, Munst B, Heinz S, Ghafoory S, Wolfl S, Simon-Keller K, Marx A, Oie CI, Ebert MP, Walles H, Braspenning J and Breitkopf-Heinlein K, Plos One, 2015, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weng YS, Chang SF, Shih MC, Tseng SH and Lai CH, Advanced materials, 2017, 29. [DOI] [PubMed] [Google Scholar]
- 36.Ho CT, Lin RZ, Chang WY, Chang HY and Liu CH, Lab on a chip, 2006, 6, 724–734. [DOI] [PubMed] [Google Scholar]
- 37.Schutte J, Hagmeyer B, Holzner F, Kubon M, Werner S, Freudigmann C, Benz K, Bottger J, Gebhardt R, Becker H and Stelzle M, Biomed. Microdevices, 2011, 13, 493–501. [DOI] [PubMed] [Google Scholar]
- 38.Sarkar U, Rivera-Burgos D, Large EM, Hughes DJ, Ravindra KC, Dyer RL, Ebrahimkhani MR, Wishnok JS, Griffith LG and Tannenbaum SR, Drug metabolism and disposition: the biological fate of chemicals, 2015, 43, 1091–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Esch MB, Prot JM, Wang YI, Miller P, Llamas-Vidales JR, Naughton BA, Applegate DR and Shuler ML, Lab on a chip, 2015, 15, 2269–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Esch MB, Ueno H, Applegate DR and Shuler ML, Lab on a chip, 2016, 16, 2719–2729. [DOI] [PubMed] [Google Scholar]
- 41.Bavli D, Prill S, Ezra E, Levy G, Cohen M, Vinken M, Vanfleteren J, Jaeger M and Nahmias Y, Proceedings of the National Academy of Sciences of the United States of America, 2016, 113, E2231–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prodanov L, Jindal R, Bale SS, Hegde M, McCarty WJ, Golberg I, Bhushan A, Yarmush ML and Usta OB, Biotechnology and bioengineering, 2016, 113, 241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang YB, Rawat S, Duchemin N, Bouchard M and Noh M, Micromachines-Basel, 2017, 8. [Google Scholar]
- 44.Zhang MY, Lee PJ, Hung PJ, Johnson T, Lee LP and Mofrad MR, Biomedical microdevices, 2008, 10, 117–121. [DOI] [PubMed] [Google Scholar]
- 45.Goral VN, Hsieh YC, Petzold ON, Clark JS, Yuen PK and Faris RA, Lab on a chip, 2010, 10, 3380–3386. [DOI] [PubMed] [Google Scholar]
- 46.Clark AM, Wheeler SE, Taylor DP, Pillai VC, Young CL, Prantil-Baun R, Nguyen T, Stolz DB, Borenstein JT, Lauffenburger DA, Venkataramanan R, Griffith LG and Wells A, Exp Biol Med (Maywood), 2014, DOI: 10.1177/1535370214532596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhushan A, Senutovitch N, Bale SS, McCarty WJ, Hegde M, Jindal R, Golberg I, Berk Usta O, Yarmush ML, Vernetti L, Gough A, Bakan A, Shun TY, DeBiasio R and Lansing Taylor D, Stem cell research & therapy, 2013, 4 Suppl 1, S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Elvevold KH, Nedredal GI, Revhaug A and Smedsrod B, Comparative hepatology, 2004, 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Knolle PA and Wohlleber D, Cellular & molecular immunology, 2016, 13, 347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Samasara Sciences, HUMAN LIVER-DERIVED ENDOTHELIAL CELLS (hLEC), http://www.samsarasciences.com/wp-content/uploads/2017/03/SAMSARA-hLEC-GENERAL-INFORMATION_FINAL_2017.pdf, (accessed June 1, 2018).
- 51.Nahmias Y, Berthiaume F and Yarmush ML, Advances in biochemical engineering/biotechnology, 2007, 103, 309–329. [DOI] [PubMed] [Google Scholar]
- 52.Ochs CJ, Kasuya J, Pavesi A and Kamm RD, Lab Chip, 2014, 14, 459–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baumann RP, Penketh PG, Seow HA, Shyam K and Sartorelli AC, Radiat Res, 2008, 170, 651–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Balis UJ, Behnia K, Dwarakanath B, Bhatia SN, Sullivan SJ, Yarmush ML and Toner M, Metab. Eng, 1999, 1, 49–62. [DOI] [PubMed] [Google Scholar]
- 55.Stephenne X, Najimi M, Ngoc DK, Smets F, Hue L, Guigas B and Sokal EM, Cell transplantation, 2007, 16, 409–419. [DOI] [PubMed] [Google Scholar]
- 56.Dunn JC, Yarmush ML, Koebe HG and Tompkins RG, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 1989, 3, 174–177. [DOI] [PubMed] [Google Scholar]
- 57.Kono Y, Yang S and Roberts EA, In vitro cellular & developmental biology. Animal, 1997, 33, 467–472. [DOI] [PubMed] [Google Scholar]
- 58.Kaplowitz N and DeLeve L, Drug-Induced Liver Disease, Academic Press, Boston, Third edn, 2013. [Google Scholar]
- 59.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P and Cardona A, Nature methods, 2012, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smit JW, Meijer CJ, Decary F and Feltkamp-Vroom TM, J. Immunol. Methods, 1974, 6, 93–98. [DOI] [PubMed] [Google Scholar]
- 61.Martins-Green M, Petreaca M and Yao M, Methods in enzymology, 2008, 443, 137–153. [DOI] [PubMed] [Google Scholar]
- 62.Tinsley JH, Wu MH, Ma W, Taulman AC and Yuan SY, The Journal of biological chemistry, 1999, 274, 24930–24934. [DOI] [PubMed] [Google Scholar]
- 63.Duffy DC, McDonald JC, Schueller OJ and Whitesides GM, Analytical chemistry, 1998, 70, 4974–4984. [DOI] [PubMed] [Google Scholar]
- 64.Bhatia SN and Ingber DE, Nat. Biotechnol, 2014, 32, 760–772. [DOI] [PubMed] [Google Scholar]
- 65.Wang JD, Douville NJ, Takayama S and ElSayed M, Ann Biomed Eng, 2012, 40, 1862–1873. [DOI] [PubMed] [Google Scholar]
- 66.Halldorsson S, Lucumi E, Gomez-Sjoberg R and Fleming RM, Biosensors & bioelectronics, 2015, 63, 218–231. [DOI] [PubMed] [Google Scholar]
- 67.van Meer BJ, de Vries H, Firth KSA, van Weerd J, Tertoolen LGJ, Karperien HBJ, Jonkheijm P, Denning C, AP IJ and Mummery CL, Biochemical and biophysical research communications, 2017, 482, 323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Domansky K, Sliz JD, Wen N, Hinojosa C, Thompson G, Fraser JP, Hamkins-Indik T, Hamilton GA, Levner D and Ingber DE, Microfluid Nanofluid, 2017, 21. [Google Scholar]
- 69.Vernetti L, Gough A, Baetz N, Blutt S, Broughman JR, Brown JA, Foulke-Abel J, Hasan N, In J, Kelly E, Kovbasnjuk O, Repper J, Senutovitch N, Stabb J, Yeung C, Zachos NC, Donowitz M, Estes M, Himmelfarb J, Truskey G, Wikswo JP and Taylor DL, Scientific reports, 2017, 7, 44517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu C and Xia Y, Chemical Society reviews, 2017, 46, 7668–7682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lalor PF, Lai WK, Curbishley SM, Shetty S and Adams DH, World J Gastroentero, 2006, 12, 5429–5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Braet F and Wisse E, Comparative hepatology, 2002, 1, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martinez I, Nedredal GI, Oie CI, Warren A, Johansen O, Le Couteur DG and Smedsrod B, Comparative hepatology, 2008, 7, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huebert RC, Jagavelu K, Liebl AF, Huang BQ, Splinter PL, LaRusso NF, Urrutia RA and Shah VH, Laboratory investigation; a journal of technical methods and pathology, 2010, 90, 1770–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McConnell M and Iwakiri Y, Hepatol Int, 2018, 12, 11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Knolle PA and Gerken G, Immunological reviews, 2000, 174, 21–34. [DOI] [PubMed] [Google Scholar]
- 77.McCuskey RS, Anatomical record, 2008, 291, 661–671. [DOI] [PubMed] [Google Scholar]
- 78.Gebhardt R, Pharmacology & therapeutics, 1992, 53, 275–354. [DOI] [PubMed] [Google Scholar]
- 79.Halpern KB, Shenhav R, Matcovitch-Natan O, Toth B, Lemze D, Golan M, Massasa EE, Baydatch S, Landen S, Moor AE, Brandis A, Giladi A, Avihail AS, David E, Amit I and Itzkovitz S, Nature, 2017, 542, 352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thomas PC, Raghavan SR and Forry SP, Analytical chemistry, 2011, 83, 8821–8824. [DOI] [PubMed] [Google Scholar]
- 81.Brennan MD, Rexius-Hall ML, Elgass LJ and Eddington DT, Lab Chip, 2014, 14, 4305–4318. [DOI] [PubMed] [Google Scholar]
- 82.Allen JW and Bhatia SN, Biotechnol. Bioeng, 2003, 82, 253–262. [DOI] [PubMed] [Google Scholar]
- 83.Allen JW, Khetani SR and Bhatia SN, Toxicol. Sci, 2005, 84, 110–119. [DOI] [PubMed] [Google Scholar]
- 84.Gough A, Vernetti L, Bergenthal L, Shun TY and Taylor DL, Appl In Vitro Toxicol, 2016, 2, 103–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Koo TK and Li MY, J Chiropr Med, 2016, 15, 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kullak-Ublick GA, Stieger B and Meier PJ, Gastroenterology, 2004, 126, 322–342. [DOI] [PubMed] [Google Scholar]
- 87.Lee KS, Lee SJ, Park HJ, Chung JP, Han KH, Chon CY, Lee SI and Moon YM, Yonsei medical journal, 2001, 42, 1–8. [DOI] [PubMed] [Google Scholar]
- 88.Washington K, Wright K, Shyr Y, Hunter EB, Olson S and Raiford DS, Human pathology, 2000, 31, 822–828. [DOI] [PubMed] [Google Scholar]
- 89.Breitkopf K, Haas S, Wiercinska E, Singer MV and Dooley S, Alcoholism, clinical and experimental research, 2005, 29, 121s–131s. [DOI] [PubMed] [Google Scholar]
- 90.Dooley S, Delvoux B, Streckert M, Bonzel L, Stopa M, ten Dijke P and Gressner AM, FEBS letters, 2001, 502, 4–10. [DOI] [PubMed] [Google Scholar]
- 91.Gant TW, Rao DN, Mason RP and Cohen GM, Chemico-biological interactions, 1988, 65, 157–173. [DOI] [PubMed] [Google Scholar]
- 92.Kadenbach B, Biochimica et biophysica acta, 2003, 1604, 77–94. [DOI] [PubMed] [Google Scholar]
- 93.McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC and Jaeschke H, J. Clin. Invest, 2012, 122, 1574–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fisher K, Vuppalanchi R and Saxena R, Archives of pathology & laboratory medicine, 2015, 139, 876–887. [DOI] [PubMed] [Google Scholar]
- 95.Strauss O, Phillips A, Ruggiero K, Bartlett A and Dunbar PR, Scientific reports, 2017, 7, 44356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Satchell SC and Braet F, American journal of physiology. Renal physiology, 2009, 296, F947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Watson DE, Hunziker R and Wikswo JP, Exp. Biol. Med, 2017, 242, 1559–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Borysiak MD, Bielawski KS, Sniadecki NJ, Jenkel CF, Vogt BD and Posner JD, Lab Chip, 2013, 13, 2773–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wikswo JP, Curtis EL, Eagleton ZE, Evans BC, Kole A, Hofmeister LH and Matloff WJ, Lab Chip, 2013, 13, 3496–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.