

Abstract
Purpose
It was first suggested more than 40 years ago that heterozygous carriers for the human autosomal recessive disorder Ataxia-Telangiectasia (A-T) might also be at increased risk for cancer. Subsequent studies have identified the responsible gene, Ataxia-Telangiectasia Mutated (ATM), characterized genetic variation at this locus in A-T and a variety of different cancers, and described the functions of the ATM protein with respect to cellular DNA damage responses. However, an overall model of how ATM contributes to cancer risk, and in particular, the role of DNA damage in this process, remains lacking. This review considers these questions in the context of contralateral breast cancer (CBC).
Conclusions
Heterozygous carriers of loss of function mutations in ATM that are A-T causing, are at increased risk of breast cancer. However, examination of a range of genetic variants, both rare and common, across multiple cancers, suggests that ATM may have additional effects on cancer risk that are allele dependent. In the case of CBC, selected common alleles at ATM are associated with a reduced incidence of CBC, while other rare and predicted deleterious variants may act jointly with radiation exposure to increase risk. Further studies that characterize germline and somatic ATM mutations in breast cancer and relate the detected genetic changes to functional outcomes, particularly with regard to radiation responses, are needed to gain a complete picture of the complex relationship between ATM, radiation and breast cancer.
Keywords: ATM, breast cancer, Ataxia-Telangiectasia, radiation therapy, allelic heterogeneity
Introduction
The Ataxia-Telangiectasia Mutated (ATM) gene, which is mutated in the autosomal recessive genetic disorder Ataxia-Telangiectasia (A-T) (Savitsky et al. 1995), encodes a master regulator of the cellular response to DNA double strand breaks, whether these result from endogenous agents such as reactive oxygen species or external agents such as ionizing radiation. Involved in both the sensing and signaling of the presence of DNA double strand breaks, ATM mediates critical cellular decisions regarding the response to genotoxic insults, which may include activation of cell cycle checkpoint machinery, application of DNA repair, or initiation of apoptosis (Lavin 2008).
The ATM gene and protein
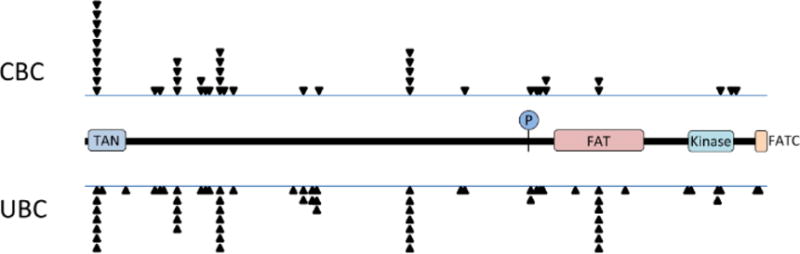
In humans, the ATM gene maps to chromosome 11q22.3 and includes 69 exons of which 65 are coding (Gatti et al. 1988, Uziel et al. 1996). The predominant ATM transcript is more than 13,000 nucleotides in length and encodes a 3056 amino acid protein of approximately 370 kD in size. ATM is a member of the phosphatidylinositol 3-kinase-related protein kinase (PIKK) family which includes other proteins, such as ATR and DNA-PK, that are also involved in DNA damage responses (Bakkenist and Kastan 2004). There are a limited number of conserved domains mapped in ATM of which the most important is the C-terminal serine/threonine kinase domain which mediates much of the signaling function of ATM. The kinase domain is flanked by FAT and FATC domains indicating sequence similarity with the related proteins FRAP and TRRAP (Bosotti et al. 2000). At the N terminal end of ATM is a TAN domain involved in telomere length maintenance (Seidel et al. 2008) and an array of repeated HEAT motifs (Andrade and Bork 1995) that may facilitate the formation of large protein complexes.
ATM exists in cells as an inactive dimer. In the presence of DNA double-strand breaks, ATM transitions to an active monomeric form, a transition that is rapid, occurring in less than a minute from break induction, and coincident with a trans-autophosphorylation event on serine 1981 (S1981) (Bakkenist and Kastan 2003). While S1981 phosphorylation is often used as a marker of ATM activation, there are other non-canonical activation pathways for ATM, responding to stimuli such as chromatin relaxation (Bakkenist and Kastan 2003, Kanu and Behrens 2007), reactive oxygen species (Guo et al. 2010) or replication stress (Stiff et al. 2006, Yajima et al. 2009), which can result in the production of active ATM monomers without the characteristic phosphorylation on S1981, or active, but disulfide linked, ATM dimers. Site specific mutagenesis studies of serine 1987 in murine Atm, the analogous position to S1981 in humans, phosphorylation at this site is not essential for Atm activation (Pellegrini et al. 2006). ChIP studies in human cells suggest that the primary role of S1981 phosphorylation may be to promote retention of activated ATM monomers at the sites of DNA double strand breaks (Berkovich et al. 2008). Upon activation, ATM phosphorylates hundreds of substrates (Matsuoka et al. 2007), preferentially targeting serine and threonine residues when followed by a glutamine. These substrates include many of the key proteins involved in sensing, signaling and repairing DNA double-strand breaks as well as other proteins with roles in diverse cellular processes from metabolism to transcriptional regulation.
ATM mutations and disease
ATM was originally identified based upon its mutation in A-T, a disorder characterized by progressive cerebellar ataxia, oculocutaneous telangiectasias, oculomotor apraxia, imunodeficiency, elevated serum alpha fetoprotein, hypersensitivity to ionizing radiation and a high incidence of cancer, primarily of lymphoid origin (Gatti et al. 1991). Cells from A-T patients, upon exposure to ionizing radiation or radiomimetic chemicals, display increased numbers of DNA double strand breaks and impaired survival in clonogenic assays (Taylor et al. 1975). A-T results from bi-allelic deleterious mutations in ATM that, in most cases, result in the production of low or undetectable levels of ATM protein. Of the 1345 mutations reported from A-T patients listed in the ATM Mutation Database (www.lovd.nl/ATM) greater than 80% are predicted to truncate the protein with no obvious clustering in specific regions of the gene.
A-T patients have a high incidence of lymphoid cancers typically developing in the first two decades of life. Older patients have an elevated risk for solid tumors including breast and gastric cancers. It was first recognized 40 years ago that female first degree relatives of A-T patients had an elevated incidence of breast cancer (Swift et al. 1990, Swift et al. 1987, Swift et al. 1976). Subsequent studies of high risk breast cancer families have revealed statistically significant associations between heterozygosity for loss of function mutations in the ATM gene (i.e. mutations that are usually A-T causing) and breast cancer (Renwick et al. 2006). More recently, similar associations with germline ATM mutations have also been observed in familial gastric and pancreatic cancers (Helgason et al. 2015, Roberts et al. 2012). Given that the products of a number of genes already implicated in breast cancer risk such as BRCA1, BRCA2, TP53, PALB2, CHEK2 and NBN are either directly phosphorylated by ATM, or are dependent on ATM function for their radiation-induced phosphorylation, a role for ATM mutations in breast cancer risk, particularly in radiation exposed individuals, is highly plausible.
Somatic mutations in the ATM gene also occur in cancer, and are particularly characteristic of certain lymphoid cancers such as mantle cell lymphoma and T pro-lymphocytic leukemia where bi-allelic de novo ATM mutations are observed (Schaffner et al. 2000, Stankovic et al. 2001, Stilgenbauer et al. 1997, Stoppa-Lyonnet et al. 2000, Stoppa-Lyonnet et al. 1998, Vorechovsky et al. 1997). The COSMIC database (http://cancer.sanger.ac.uk/cosmic) indicates that ATM is mutated in a wide array of cancers and tissues (Forbes et al. 2015). However, the distribution of mutations is strongly biased towards missense substitutions (approximately 67% of all mutations) and thus distinct from the distribution of germline mutations reported in A-T patients where truncating mutations predominate.
The discordance between the predominant loss of function mutations detected in A-T patients and A-T carriers who develop breast cancer and the predominantly missense somatic mutations observed in some cancers is noteworthy. Given that certain lymphoid cancers are associated with biallelic mutations in ATM yet A-T carriers are not at increased risk for these cancers (Stankovic, et al. 2001, Stoppa-Lyonnet, et al. 1998, Vorechovsky, et al. 1997), it seems likely that the mutations contributing to these disorders are functionally distinct from those that contribute to A-T. We have previously proposed a model for ATM action in cancer that assumed that truncating mutations in the ATM gene were functionally distinct from missense mutations (Gatti et al. 1999). Several newer studies provide suggestive support for such a model. A missense mutation, c.7271T>G, in ATM has been described that results in an atypically mild form of A-T but carries significant risk of breast cancer, comparable to that for BRCA2 (Bernstein et al. 2006, Stankovic et al. 1998). In the mouse, homozygous truncating mutations that mimic mutations observed in human A-T patients are viable, but missense substitutions at critical residues for the kinase activity of Atm lead to genomic instability and embryonic lethality (Daniel et al. 2012, Yamamoto et al. 2012), demonstrating that at least selected missense substitutions can have distinct phenotypic effects from null mutations.
Contralateral breast cancer
Breast cancer patients are 2–5 times more likely to develop contralateral breast cancer (CBC) than are women without breast cancer to develop an initial breast cancer (Bernstein et al. 1992). Women at higher risk for CBC include those with a first primary at an early age (Chen et al. 1999, Li et al. 2003), lobular histology in a first primary (Bernstein, et al. 1992, Claus et al. 2003, Donovan 1990, Horn and Thompson 1988, Horn et al. 1987, Prior and Waterhouse 1978), and/or a family history of breast cancer (Bernstein, et al. 1992, Donovan 1990, Horn, et al. 1987, Kelsey et al. 1993, Peto and Mack 2000, Prior and Waterhouse 1978). The risk of developing CBC can be modified by therapies for the first primary. Women treated with radiation therapy have an increased risk of CBC: risk is proportional to increasing dose and inversely related to age at exposure. Multiple studies have demonstrated that BRCA1 and BRCA2 mutation carriers have substantially greater risk of CBC than non-carriers (Ansquer et al. 1998, Begg et al. 2008, Gershoni-Baruch et al. 2000, Malone et al. 2010, Pierce et al. 2000, Robson et al. 1998, Robson et al. 1999, Steinmann et al. 2001, Verhoog et al. 1999, Verhoog et al. 2000, Verhoog et al. 1998). However, the vast majority (90–95%) of all breast cancer cases are not carriers of BRCA1 or BRCA2 mutations (Campeau et al. 2008). Finally, other risk factors for UBC have also been shown to affect the risk of CBC, including reproductive factors (Largent et al. 2007), menarche age (Largent, et al. 2007), body size (Brooks et al. 2012, Li et al. 2009), alcohol consumption (Knight et al. 2009, Li, et al. 2009), and smoking (Knight, et al. 2009). In a recent study, reduction of mammographic density (MD), one of the strongest risk factors for UBC, was shown to relate to a significantly reduced risk of CBC (Li, et al. 2009). The context of CBC creates a unique opportunity to explore the role of ATM in breast cancer risk both because of the ability to incorporate well-defined information on radiation exposure from treatment and that any mutation that is associated with breast, cancer but rare in the general population, will be more prevalent in a sample of breast cancer patients.
The Women’s Environmental, Cancer, and Radiation Epidemiology (WECARE) Study
The WECARE Study is an international, population-based, case-control study designed to identify the genetic and environmental factors contributing to asynchronous CBC. The study population consists of 708 case subjects, women with CBC, and 1399 control subjects, women with unilateral breast cancer (UBC). Two controls were individually matched to each case on age at diagnosis, length of follow up, registry reporting region, and race and/or ethnicity. For statistical efficiency, controls were further counter-matched on radiation exposure (Bernstein et al. 2004). All women in the WECARE Study were interviewed and complete medical treatment history information was collected. For all women who received radiation therapy for their first primary breast cancer, the radiation dose to the contralateral breast was reconstructed from treatment records and radiation measurements via simulations using patient-specific phantoms (Stovall et al. 2008). Blood samples were obtained from all women and the extracted DNA was used to complete full mutation screening of the ATM (Bernstein et al. 2003), BRCA1 and BRCA2 genes (Borg et al. 2010, Malone, et al. 2010), partial screening for PALB2 (Antoniou et al. 2014, Tischkowitz et al. 2012) and CHEK2 (Mellemkjaer et al. 2008) and a genome-wide association study (manuscript in preparation).
Radiation therapy and CBC risk in the WECARE Study
Results from the WECARE Study, as well as reports from others, indicate that women receiving radiation therapy for a first primary breast cancer have an increased risk of developing CBC (Bernstein, et al. 1992, Bertelsen et al. 2008, Boice et al. 1992, Gao et al. 2003, Hooning et al. 2008, Stovall, et al. 2008). Among WECARE CBC cases, the mean cumulative absorbed radiation dose to the quadrant of the contralateral breast where the second tumor developed was 1.1 Gy (range=0.02–6.2 Gy), corresponding to an average absorbed dose per fraction of 44 mGy. Women less than 40 years of age with a greater than 1.0 Gy absorbed dose to the tumor specific quadrant of the contralateral breast had a significantly increased risk of CBC relative to those unexposed, (RR = 2.5, 95% CI 1.4–4.5). This risk was inversely related to age at first exposure and dose dependent. Across all cases, the most common tumor site for second cancers was the upper outer quadrant of the breast. However in women receiving radiation therapy for a first cancer there was a significant increase in tumors occurring in the inner and central quadrants (P = 0.03). This shift in tumor location is consistent with the gradient of radiation exposure across the contralateral breast where absorbed doses decrease from the inner region (Mean = 1.9 Gy, range 0.2–7.7) to the central (Mean = 1.2 Gy, range 0.1–3.8) to the outer (Mean = 0.8 Gy, range 0.1–2.5) region (Stovall, et al. 2008).
ATM and the risk of CBC
The WECARE Study provides a unique opportunity to examine the main effects of genetic variation at ATM on risk for CBC as well as the joint effects of ATM and radiation therapy on risk. Mutation screening of the ATM gene in all 2105 WECARE Study participants by a tiered approach using denaturing high performance liquid chromatography followed by sequencing, identified 240 distinct sequence variants, most of which (225 of 240) were rare, occurring in less than 1% of subjects (Concannon et al. 2008). Among the rare variants, A-T causing variants, defined as those that either had been previously identified in A-T patients or were predicted to result in protein truncation, were associated with a non-significant increase in the risk of CBC (RR = 2.0, 95% CI = 0.7–5.9). Rare missense variants were associated with a modest, but non-significant increase in risk (RR = 1.2, 95% CI = 0.8–1.7). Using the bioinformatic tools SIFT (Kumar et al. 2009) or PolyPhen (Adzhubei et al. 2010) to classify these missense variants into those likely to be tolerated or deleterious did not reveal any statistically significant main effects on CBC risk for either group. In contrast to these results obtained for rare ATM variants, carriers of common variants, overall, had a statistically significant reduction in risk of CBC (RR = 0.8, 95% CI = 0.6–0.9). Four common variants were individually associated with a significantly decreased risk of second primary breast cancer (c.1899-55T>G, RR = 0.5, 95% CI = 0.3–0.8; c.3161C>G, RR = 0.5, 95% CI = 0.3–0.9; c.5558A>T, RR = 0.2, 95% CI = 0.1–0.6; c.6348-54T>C RR = 0.2, 95% CI = 0.1–0.8).
Incorporating radiation dosimetry into the analyses of ATM genetic association with CBC in the WECARE Study revealed a more complex interaction. Women in the WECARE Study who carried any rare ATM missense variant predicted to be deleterious using either SIFT or PolyPhen, and who received radiation therapy for their first cancer, had a significantly elevated risk of CBC compared with unexposed women who carried the reference ATM sequence (< 1.0 Gy dose to the contralateral breast: RR = 2.8, 95% CI = 1.2 to 6.5; ≥1.0 Gy: RR = 3.3, 95% CI = 1.4 to 8.0) and, more importantly, compared with unexposed women who carried the same deleterious missense variant (< 1.0 Gy: RR = 5.3, 95% CI = 1.6 to 17.3; ≥1.0 Gy: RR = 5.8, 95% CI = 1.8 to 19.0). For this latter comparison, there was a statistically significant dose-response trend (excess relative risk (ERR)/Gy = 2.6, Ptrend = .044). The interaction of radiation and deleterious ATM variants was further explored by considering age at, and time since, first diagnosis. Among women who carried ATM missense variants that were predicted to be deleterious, the rate ratio for treated versus not treated with radiation therapy was greater for women younger than 45 years at diagnosis (RR = 10.4, 95% CI = 2.3 to 47.2) than for older women (RR = 2.4, 95% CI = 0.6 to 9.5). Similarly, among women who carried predicted deleterious ATM missense variants, there was a steeper radiation dose slope for those with a longer latency (≥ 5 years) compared with those with a shorter latency (ERR/Gy: 4.3, 95% CI = 0.1 to 20.7 vs. 1.9, 95% CI = −0.1 to 9.1; respectively).
Discussion
How do the data derived from the WECARE Study inform the evolving understanding of the role of ATM in breast cancer and cancer risk in general? Unlike studies that compare affected cases to unaffected controls, WECARE explores factors that influence the risk of developing a second cancer in patients already treated for a first. This model may not be as generalizable as a classical population-based case-control design for identifying variants that increase risk for breast cancer as a main effect, but does allow for analyses of how genetic risk factors, both rare and common, may interact with treatment to modify risk. In analyzing the WECARE Study data, results were stratified into the broad mutation categories truncating, missense, splicing and silent, as prior studies, summarized in this review, had suggested that different classes of mutation might have different phenotypic consequences. Missense mutations were then further classified using bioinformatic predictors of effects on protein function. Given the low prevalence of A-T in the population and the association between A-T heterozygosity and breast cancer risk, common and rare variants in ATM were analyzed separately.
Three conclusions can be drawn from the findings regarding ATM and CBC from the WECARE Study : [1] selected common variants in ATM may exert a protective effect, reducing the risk of developing CBC, [2] rare ATM missense variants classified as likely deleterious to protein function increase risk for CBC in women treated with radiation therapy for a first cancer in a dose dependent manner, and [3] the joint effect of ATM mutations and radiation therapy on CBC risk suggests that ATM responds to the low doses of radiation scattering from individual fractionated treatments.
Protective effects with regards to breast cancer for variants in ATM have not been previously reported. One possible mechanism for such effects would be that these variants alter the threshold for ATM activation, destabilizing inactive ATM dimers and/or increasing the pool of activated ATM present at any given time in a cell. Indeed, preliminary studies carried out in B cell lines established from WECARE Study subjects suggests that the level of ATM phosphorylated on S1981 normalized to overall cellular ATM levels is increased in carriers of putative protective variants in ATM (Concannon et al., unpublished observations). This would be a main effect of these variants, protecting carriers from environmental sources of cellular stress, and more readily detectable in the WECARE Study where CBC cases and UBC controls differ in their exposure to either radiation therapy or systemic chemotherapy.
The observation that rare, deleterious missense variants in ATM act jointly with radiation to increase risk of CBC while A-T causing mutations do not, is consistent with prior suggestions of allelic heterogeneity at ATM with regard to breast cancer risk. However, caution is warranted in the interpretation of these findings. Carriers of A-T causing mutation are infrequent; numbers of such subjects may have been too small in the WECARE Study to observe a significant effect on CBC risk for this category. In addition, classification of missense variants in the WECARE Study is based only on bioinformatic predictors. There are no functional data indicating deleterious effects on ATM, nor is the possibility that these variants might, in fact, be A-T causing, ruled out. However, the low cumulative scatter doses to the contralateral breast estimated from simulations, particularly when averaged over the number of fractions, provides epidemiological evidence that genetic variation at ATM does affect the response to low individual radiation doses in the range of 50mGy. There are conflicting reports on whether ATM is activated by such doses (Bakkenist and Kastan 2003, Enns et al. 2015, Nakamura et al. 2006, Ojima et al. 2008).
Summary
The relationship between ATM, radiation and breast cancer remains complex. Genetic variants in ATM may have a range of effects on breast cancer risk from protective to high risk. Heterozygous carriers of germline ATM mutations that are A-T causing have an increased risk of developing breast cancer. Other rare missense variants, which may not be A-T causing, can, nevertheless, interact with radiation exposure, even at low doses, and increase risk for breast cancer. Studies that combine ATM mutation detection in both germline and tumor tissue with detailed functional characterization of variants will be necessary to resolve the role of ATM in breast cancer and enable reliable risk prediction.
Figure 1.
Acknowledgments
The Women’s Environmental, Cancer and Epidemiology (WECARE) Study Collaborative Group includes Memorial Sloan Kettering Cancer Center (Coordinating Center) Investigators and Staff: Jonine L. Bernstein Ph.D. (WECARE Study P.I.); Marinela Capanu Ph.D.; Xiaolin Liang M.D.; Irene Orlow Ph.D.; Anne S. Reiner M.P.H.; Mark Robson, M.D.; Meghan Woods M.P.H. Collaborative Site Investigators: Leslie Bernstein Ph.D.; John D. Boice Jr. Sc.D.; Jennifer Brooks Ph.D.; Patrick Concannon Ph.D.; Dave V. Conti Ph.D.; David Duggan Ph.D.; Joanne W. Elena Ph.D., M.P.H.; Robert W. Haile Dr.P.H.; Esther M. John Ph.D.; Julia A. Knight Ph.D.; Charles F. Lynch M.D., Ph.D.; Kathleen E. Malone Ph.D.; Lene Mellemkjær Ph.D.; Jørgen H. Olsen M.D. DMSc.; Daniela Seminara Ph.D. M.P.H.; Roy E. Shore Ph.D., Dr.P.H.; Marilyn Stovall Ph.D.; Daniel O. Stram Ph.D.; Marc Tischkowitz M.D., Ph.D.; Duncan C. Thomas Ph.D. Collaborative Site Staff: Kristina Blackmore M.Sc.; Anh T. Diep; Judy Goldstein; Irene Harris B.S., C.M.D.; Rikke Langballe M.P.H.; Cecilia O’Brien; Susan Smith M.P.H.; Rita Weathers M.S.; Michele West Ph.D.
Funding
Patrick Concannon was supported by grants from the NIEHS under Grant R01 ES027121 and the Defense Threat Reduction Agency under Grant 1-16-1-0048. This work was also supported by grants from the National Institutes of Health (U01 CA83178, R01 CA97397, R01 CA129639, R01 CA114236, and P30 CA008748)
Footnotes
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature methods. 2010 Apr;7:248–249. doi: 10.1038/nmeth0410-248. Epub 2010/04/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade MA, Bork P. HEAT repeats in the Huntington’s disease protein. Nat Genet Oct. 1995;11:115–116. doi: 10.1038/ng1095-115. Epub 1995/10/01. [DOI] [PubMed] [Google Scholar]
- Ansquer Y, Gautier C, Fourquet A, Asselain B, Stoppa-Lyonnet D. Survival in early-onset BRCA1 breast-cancer patients. Institut Curie Breast Cancer Group. Lancet. 1998 Aug 15;352:541. doi: 10.1016/s0140-6736(05)79248-5. Epub 1998/08/26. [DOI] [PubMed] [Google Scholar]
- Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkas K, Roberts J, Lee A, Subramanian D, De Leeneer K, Fostira F, et al. Breast-cancer risk in families with mutations in PALB2. The New England journal of medicine. 2014 Aug 7;371:497–506. doi: 10.1056/NEJMoa1400382. Epub 2014/08/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003 Jan 30;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB. Initiating cellular stress responses. Cell. 2004 Jul 9;118:9–17. doi: 10.1016/j.cell.2004.06.023. Epub 2004/07/10. [DOI] [PubMed] [Google Scholar]
- Begg CB, Haile RW, Borg A, Malone KE, Concannon P, Thomas DC, Langholz B, Bernstein L, Olsen JH, Lynch CF, et al. Variation of breast cancer risk among BRCA1/2 carriers. Jama-J Am Med Assoc. 2008 Jan 9;299:194–201. doi: 10.1001/jama.2007.55-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkovich E, Monnat RJ, Kastan MB. Assessment of protein dynamics and DNA repair following generation of DNA double-strand breaks at defined genomic sites. Nat Protoc. 2008;3:915–922. doi: 10.1038/nprot.2008.54. 2008. [DOI] [PubMed] [Google Scholar]
- Bernstein JL, Langholz B, Haile RW, Bernstein L, Thomas DC, Stovall M, Malone KE, Lynch CF, Olsen JH, Anton-Culver H, et al. Study design: evaluating gene-environment interactions in the etiology of breast cancer - the WECARE study. Breast cancer research: BCR. 2004;6:R199–214. doi: 10.1186/bcr771. 2004. Epub 2004/04/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein JL, Teraoka S, Haile RW, Borresen-Dale AL, Rosenstein BS, Gatti RA, Diep AT, Jansen L, Atencio DR, Olsen JH, et al. Designing and implementing quality control for multi-center screening of mutations in the ATM gene among women with breast cancer. Hum Mutat. 2003 May;21:542–550. doi: 10.1002/humu.10206. [DOI] [PubMed] [Google Scholar]
- Bernstein JL, Teraoka S, Southey MC, Jenkins MA, Andrulis IL, Knight JA, John EM, Lapinski R, Wolitzer AL, Whittemore AS, et al. Population-based estimates of breast cancer risks associated with ATM gene variants c.7271T>G and c.1066-6T>G (IVS106T>G) from the Breast Cancer Family Registry. Hum Mutat. 2006 Nov;27:1122–1128. doi: 10.1002/humu.20415. Epub 2006/09/08. [DOI] [PubMed] [Google Scholar]
- Bernstein JL, Thompson WD, Risch N, Holford TR. Risk factors predicting the incidence of second primary breast cancer among women diagnosed with a first primary breast cancer. Am J Epidemiol. 1992;136:925–936. doi: 10.1093/oxfordjournals.aje.a116565. 10/15/1992. [DOI] [PubMed] [Google Scholar]
- Bertelsen L, Bernstein L, Olsen JH, Mellemkjaer L, Haile RW, Lynch CF, Malone KE, Anton-Culver H, Christensen J, Langholz B, et al. Effect of systemic adjuvant treatment on risk for contralateral breast cancer in the Women’s Environment, Cancer and Radiation Epidemiology Study. Journal of the National Cancer Institute. 2008 Jan 2;100:32–40. doi: 10.1093/jnci/djm267. Epub 2007/12/27. [DOI] [PubMed] [Google Scholar]
- Boice JD, Jr, Harvey EB, Blettner M, Stovall M, Flannery JT. Cancer in the contralateral breast after radiotherapy for breast cancer. The New England journal of medicine. 1992 Mar 19;326:781–785. doi: 10.1056/NEJM199203193261201. Epub 1992/03/29. [DOI] [PubMed] [Google Scholar]
- Borg A, Haile RW, Malone KE, Capanu M, Diep A, Torngren T, Teraoka S, Begg CB, Thomas DC, Concannon P, et al. Characterization of BRCA1 and BRCA2 deleterious mutations and variants of unknown clinical significance in unilateral and bilateral breast cancer: the WECARE study. Hum Mutat. 2010 Mar;31:E1200–1240. doi: 10.1002/humu.21202. Epub 2010/01/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosotti R, Isacchi A, Sonnhammer EL. FAT: a novel domain in PIK-related kinases. Trends in biochemical sciences. 2000 May;25:225–227. doi: 10.1016/s0968-0004(00)01563-2. Epub 2000/04/27. [DOI] [PubMed] [Google Scholar]
- Brooks JD, John EM, Mellemkjaer L, Reiner AS, Malone KE, Lynch CF, Figueiredo JC, Haile RW, Shore RE, Group WSC et al. Body mass index and risk of second primary breast cancer: the WECARE Study. Breast Cancer Res Treat. 2012 Jan;131:571–580. doi: 10.1007/s10549-011-1743-4. Epub 2011/09/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau PM, Foulkes WD, Tischkowitz MD. Hereditary breast cancer: new genetic developments, new therapeutic avenues. Human genetics. 2008 Aug;124:31–42. doi: 10.1007/s00439-008-0529-1. Epub 2008/06/26. [DOI] [PubMed] [Google Scholar]
- Chen Y, Thompson W, Semenciw R, Mao Y. Epidemiology of contralateral breast cancer. Cancer Epidemiol Biomarkers Prev. 1999;8:855–861. 10/1999. [PubMed] [Google Scholar]
- Claus EB, Stowe M, Carter D, Holford T. The risk of a contralateral breast cancer among women diagnosed with ductal and lobular breast carcinoma in situ: data from the Connecticut Tumor Registry. Breast. 2003 Dec;12:451–456. doi: 10.1016/s0960-9776(03)00152-8. Epub 2003/12/09. [DOI] [PubMed] [Google Scholar]
- Concannon P, Haile RW, Borresen-Dale AL, Rosenstein BS, Gatti RA, Teraoka SN, Diep TA, Jansen L, Atencio DP, Langholz B, et al. Variants in the ATM gene associated with a reduced risk of contralateral breast cancer. Cancer Res. 2008 Aug 15;68:6486–6491. doi: 10.1158/0008-5472.CAN-08-0134. Epub 2008/08/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel JA, Pellegrini M, Lee BS, Guo Z, Filsuf D, Belkina NV, You Z, Paull TT, Sleckman BP, Feigenbaum L, et al. Loss of ATM kinase activity leads to embryonic lethality in mice. The Journal of cell biology. 2012 Aug 06;198:295–304. doi: 10.1083/jcb.201204035. Epub 2012/08/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan AJ. Bilateral breast cancer. Surg Clin North Am. 1990;70:1141–1149. doi: 10.1016/s0039-6109(16)45235-7. 10/1990. [DOI] [PubMed] [Google Scholar]
- Enns L, Rasouli-Nia A, Hendzel M, Marples B, Weinfeld M. Association of ATM activation and DNA repair with induced radioresistance after low-dose irradiation. Radiation protection dosimetry. 2015 Sep;166:131–136. doi: 10.1093/rpd/ncv203. Epub 2015/04/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic acids research. 2015 Jan;43:D805–811. doi: 10.1093/nar/gku1075. Epub 2014/10/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Fisher SG, Emami B. Risk of second primary cancer in the contralateral breast in women treated for early-stage breast cancer: a population-based study. Int J Radiat Oncol Biol Phys. 2003;56:1038–1045. doi: 10.1016/s0360-3016(03)00203-7. 7/15/2003. [DOI] [PubMed] [Google Scholar]
- Gatti RA, Berkel I, Boder E, Braedt G, Charmley P, Concannon P, Ersoy F, Foroud T, Jaspers NGJ, Lange K, et al. Localization of an ataxia-telangiectasia gene to chromosome 11q22–23. Nature. 1988;336:577–580. doi: 10.1038/336577a0. 1988. Epub 1988/12/08. [DOI] [PubMed] [Google Scholar]
- Gatti RA, Boder E, Vinters HV, Sparkes RS, Norman A, Lange K. Ataxia-telangiectasia: an interdisciplinary approach to pathogenesis. Medicine (Baltimore) 1991;70:99–117. 3/1991. [PubMed] [Google Scholar]
- Gatti RA, Tward A, Concannon P. Cancer risk in ATM heterozygotes: a model of phenotypic and mechanistic differences between missense and truncating mutations. Mol Genet Metab. 1999;68:419–423. doi: 10.1006/mgme.1999.2942. 12/1999. [DOI] [PubMed] [Google Scholar]
- Gershoni-Baruch R, Dagan E, Fried G, Bruchim Bar-Sade R, Sverdlov-Shiri R, Zelicksson G, Friedman E. Significantly lower rates of BRCA1/BRCA2 founder mutations in Ashkenazi women with sporadic compared with familial early onset breast cancer. European journal of cancer. 2000 May;36:983–986. doi: 10.1016/s0959-8049(00)00045-9. Epub 2000/07/08. [DOI] [PubMed] [Google Scholar]
- Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010 Oct 22;330:517–521. doi: 10.1126/science.1192912. Epub 2010/10/23. [DOI] [PubMed] [Google Scholar]
- Helgason H, Rafnar T, Olafsdottir HS, Jonasson JG, Sigurdsson A, Stacey SN, Jonasdottir A, Tryggvadottir L, Alexiusdottir K, Haraldsson A, et al. Loss-of-function variants in ATM confer risk of gastric cancer. Nat Genet. 2015 Aug;47:906–910. doi: 10.1038/ng.3342. Epub 2015/06/23. [DOI] [PubMed] [Google Scholar]
- Hooning MJ, Aleman BM, Hauptmann M, Baaijens MH, Klijn JG, Noyon R, Stovall M, van Leeuwen FE. Roles of radiotherapy and chemotherapy in the development of contralateral breast cancer. J Clin Oncol. 2008;26:5561–5568. doi: 10.1200/JCO.2007.16.0192. 12/1/2008. [DOI] [PubMed] [Google Scholar]
- Horn PL, Thompson WD. Risk of contralateral breast cancer. Associations with histologic, clinical, and therapeutic factors. Cancer. 1988;62:412–424. doi: 10.1002/1097-0142(19880715)62:2<412::aid-cncr2820620228>3.0.co;2-3. 7/15/1988. [DOI] [PubMed] [Google Scholar]
- Horn PL, Thompson WD, Schwartz SM. Factors associated with the risk of second primary breast cancer: an analysis of data from the Connecticut Tumor Registry. J Chronic Dis. 1987;40:1003–1011. doi: 10.1016/0021-9681(87)90114-7. 1987. [DOI] [PubMed] [Google Scholar]
- Kanu N, Behrens A. ATMIN defines an NBS1-independent pathway of ATM signalling. EMBO J. 2007 Jun 20;26:2933–2941. doi: 10.1038/sj.emboj.7601733. Epub 2007/05/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsey JL, Gammon MD, John EM. Reproductive factors and breast cancer. Epidemiologic reviews. 1993;15:36–47. doi: 10.1093/oxfordjournals.epirev.a036115. Epub 1993/01/01. [DOI] [PubMed] [Google Scholar]
- Knight JA, Bernstein L, Largent J, Capanu M, Begg CB, Mellemkjaer L, Lynch CF, Malone KE, Reiner AS, Liang X, et al. Alcohol intake and cigarette smoking and risk of a contralateral breast cancer: The Women’s Environmental Cancer and Radiation Epidemiology Study. American journal of epidemiology. 2009 Apr 15;169:962–968. doi: 10.1093/aje/kwn422. Epub 2009/02/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. 2009. [DOI] [PubMed] [Google Scholar]
- Largent JA, Capanu M, Bernstein L, Langholz B, Mellemkaer L, Malone KE, Begg CB, Haile RW, Lynch CF, Anton-Culver H, et al. Reproductive history and risk of second primary breast cancer: the WECARE study. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2007 May;16:906–911. doi: 10.1158/1055-9965.EPI-06-1003. Epub 2007/05/18. [DOI] [PubMed] [Google Scholar]
- Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nature reviews Molecular cell biology. 2008 Oct;9:759–769. doi: 10.1038/nrm2514. Epub 2008/09/25. [DOI] [PubMed] [Google Scholar]
- Li CI, Daling JR, Porter PL, Tang MT, Malone KE. Relationship between potentially modifiable lifestyle factors and risk of second primary contralateral breast cancer among women diagnosed with estrogen receptor-positive invasive breast cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009 Nov 10;27:5312–5318. doi: 10.1200/JCO.2009.23.1597. Epub 2009/09/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CI, Malone KE, Porter PL, Daling JR. Epidemiologic and molecular risk factors for contralateral breast cancer among young women. British journal of cancer. 2003 Aug 4;89:513–518. doi: 10.1038/sj.bjc.6601042. Epub 2003/07/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone KE, Begg CB, Haile RW, Borg A, Concannon P, Tellhed L, Xue S, Teraoka S, Bernstein L, Capanu M, et al. Population-based study of the risk of second primary contralateral breast cancer associated with carrying a mutation in BRCA1 or BRCA2. J Clin Oncol. 2010;28:2404–2410. doi: 10.1200/JCO.2009.24.2495. 5/10/2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, III, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. 5/25/2007. [DOI] [PubMed] [Google Scholar]
- Mellemkjaer L, Dahl C, Olsen JH, Bertelsen L, Guldberg P, Christensen J, Borresen-Dale AL, Stovall M, Langholz B, Bernstein L, et al. Risk for contralateral breast cancer among carriers of the CHEK2*1100delC mutation in the WECARE Study. Br J Cancer. 2008;98:728–733. doi: 10.1038/sj.bjc.6604228. 2/26/2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H, Yasui Y, Saito N, Tachibana A, Komatsu K, Ishizaki K. DNA repair defect in AT cells and their hypersensitivity to low-dose-rate radiation. Radiation research. 2006 Mar;165:277–282. doi: 10.1667/rr3519.1. Epub 2006/02/24. [DOI] [PubMed] [Google Scholar]
- Ojima M, Ban N, Kai M. DNA double-strand breaks induced by very low X-ray doses are largely due to bystander effects. Radiation research. 2008 Sep;170:365–371. doi: 10.1667/RR1255.1. Epub 2008/09/04. [DOI] [PubMed] [Google Scholar]
- Pellegrini M, Celeste A, Difilippantonio S, Guo R, Wang W, Feigenbaum L, Nussenzweig A. Autophosphorylation at serine 1987 is dispensable for murine Atm activation in vivo. Nature. 2006;443:222–225. doi: 10.1038/nature05112. 9/14/2006. [DOI] [PubMed] [Google Scholar]
- Peto J, Mack TM. High constant incidence in twins and other relatives of women with breast cancer. Nat Genet. 2000 Dec;26:411–414. doi: 10.1038/82533. Epub 2000/12/02. [DOI] [PubMed] [Google Scholar]
- Pierce LJ, Strawderman M, Narod SA, Oliviotto I, Eisen A, Dawson L, Gaffney D, Solin LJ, Nixon A, Garber J, et al. Effect of radiotherapy after breast-conserving treatment in women with breast cancer and germline BRCA1/2 mutations. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2000 Oct 01;18:3360–3369. doi: 10.1200/JCO.2000.18.19.3360. Epub 2000/10/03. [DOI] [PubMed] [Google Scholar]
- Prior P, Waterhouse JA. Incidence of bilateral tumours in a population-based series of breast-cancer patients. I. Two approaches to an epidemiological analysis. Br J Cancer. 1978;37:620–634. doi: 10.1038/bjc.1978.92. 4/1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, North B, Jayatilake H, Barfoot R, Spanova K, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38:873–875. doi: 10.1038/ng1837. 8/2006. [DOI] [PubMed] [Google Scholar]
- Roberts NJ, Jiao Y, Yu J, Kopelovich L, Petersen GM, Bondy ML, Gallinger S, Schwartz AG, Syngal S, Cote ML, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer discovery. 2012 Jan;2:41–46. doi: 10.1158/2159-8290.CD-11-0194. Epub 2012/05/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson M, Gilewski T, Haas B, Levin D, Borgen P, Rajan P, Hirschaut Y, Pressman P, Rosen PP, Lesser ML, et al. BRCA-associated breast cancer in young women. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 1998 May;16:1642–1649. doi: 10.1200/JCO.1998.16.5.1642. Epub 1998/05/20. [DOI] [PubMed] [Google Scholar]
- Robson M, Levin D, Federici M, Satagopan J, Bogolminy F, Heerdt A, Borgen P, McCormick B, Hudis C, Norton L, et al. Breast conservation therapy for invasive breast cancer in Ashkenazi women with BRCA gene founder mutations. Journal of the National Cancer Institute. 1999 Dec 15;91:2112–2117. doi: 10.1093/jnci/91.24.2112. Epub 1999/12/22. [DOI] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995 Jun 23;268:1749–1753. doi: 10.1126/science.7792600. Epub 1995/06/23. [DOI] [PubMed] [Google Scholar]
- Schaffner C, Idler I, Stilgenbauer S, Dohner H, Lichter P. Mantle cell lymphoma is characterized by inactivation of the ATM gene. Proc Natl Acad Sci USA. 2000;97:2773–2778. doi: 10.1073/pnas.050400997. 3/14/2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel JJ, Anderson CM, Blackburn EH. A novel Tel1/ATM N-terminal motif, TAN, is essential for telomere length maintenance and a DNA damage response. Molecular and cellular biology. 2008 Sep;28:5736–5746. doi: 10.1128/MCB.00326-08. Epub 2008/07/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankovic T, Kidd AM, Sutcliffe A, McGuire GM, Robinson P, Weber P, Bedenham T, Bradwell AR, Easton DF, Lennox GG, et al. ATM mutations and phenotypes in ataxia- telangiectasia families in the British Isles: expression of mutant ATM and the risk of leukemia, lymphoma, and breast cancer. Am J Hum Genet. 1998;62:334–345. doi: 10.1086/301706. 2/1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankovic T, Taylor AM, Yuille MR, Vorechovsky I. Recurrent ATM mutations in T-PLL on diverse haplotypes: no support for their germline origin. Blood. 2001;97:1517–1518. doi: 10.1182/blood.v97.5.1517. 3/1/2001. [DOI] [PubMed] [Google Scholar]
- Steinmann D, Bremer M, Rades D, Skawran B, Siebrands C, Karstens JH, Dork T. Mutations of the BRCA1 and BRCA2 genes in patients with bilateral breast cancer. British journal of cancer. 2001 Sep 14;85:850–858. doi: 10.1054/bjoc.2001.2016. Epub 2001/09/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiff T, Walker SA, Cerosaletti K, Goodarzi AA, Petermann E, Concannon P, O’Driscoll M, Jeggo PA. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. EMBO J. 2006;25:5775–5782. doi: 10.1038/sj.emboj.7601446. 12/13/2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stilgenbauer S, Schaffner C, Litterst A, Liebisch P, Gilad S, Bar-Shira A, James MR, Lichter P, Döhner H. Biallelic mutations in the ATM gene in T-prolymphocytic leukemia. Nat Med. 1997;3:1155–1159. doi: 10.1038/nm1097-1155. 1997. [DOI] [PubMed] [Google Scholar]
- Stoppa-Lyonnet D, Lauge A, Sigaux F, Stern MH. No germline ATM mutation in a series of 16T-cell prolymphocytic leukemias. Blood. 2000;96:374–376. 7/1/2000. [PubMed] [Google Scholar]
- Stoppa-Lyonnet D, Soulier J, Lauge A, Dastot H, Garand R, Sigaux F, Stern MH. Inactivation of the ATM gene in T-cell prolymphocytic leukemias. Blood. 1998;91:3920–3926. 5/15/1998. [PubMed] [Google Scholar]
- Stovall M, Smith SA, Langholz BM, Boice JD, Jr, Shore RE, Andersson M, Buchholz TA, Capanu M, Bernstein L, Lynch CF, et al. Dose to the contralateral breast from radiotherapy and risk of second primary breast cancer in the WECARE study. Int J Radiat Oncol Biol Phys. 2008;72:1021–1030. doi: 10.1016/j.ijrobp.2008.02.040. 11/15/2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift M, Chase CL, Morrell D. Cancer predisposition of ataxia-telangiectasia heterozygotes. Cancer Genet Cytogenet. 1990;46:21–27. doi: 10.1016/0165-4608(90)90004-t. 1990. [DOI] [PubMed] [Google Scholar]
- Swift M, Reitnauer PJ, Morrell D, Chase CL. Breast and other cancers in families with ataxia-telangiectasia. N Engl J Med. 1987;316:1289–1294. doi: 10.1056/NEJM198705213162101. 1987. [DOI] [PubMed] [Google Scholar]
- Swift M, Sholman L, Perry M, Chase C. Malignant neoplasms in the families of patients with ataxia- telangiectasia. Cancer Res. 1976;36:209–215. 1/1976. [PubMed] [Google Scholar]
- Taylor AMR, Harnden DG, Arlett CF, Harcourt SA, Lehmann AR, Stevens S, Bridges BA. Ataxia-telangiectasia: a human mutation with abnormal radiation sensitivity. Nature. 1975;258:427–429. doi: 10.1038/258427a0. 1975. [DOI] [PubMed] [Google Scholar]
- Tischkowitz M, Capanu M, Sabbaghian N, Li L, Liang X, Vallee MP, Tavtigian SV, Concannon P, Foulkes WD, Bernstein L, et al. Rare germline mutations in PALB2 and breast cancer risk: a population-based study. Hum Mutat. 2012 Apr;33:674–680. doi: 10.1002/humu.22022. Epub 2012/01/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uziel T, Savitsky K, Platzer M, Ziv Y, Helblitz T, Nehls M, Boehm T, Rosenthal A, Shiloh Y, Rotman G. Genomic organization of the ATM gene. Genomics. 1996;33:317–320. doi: 10.1006/geno.1996.0201. 1996. [DOI] [PubMed] [Google Scholar]
- Verhoog LC, Brekelmans CT, Seynaeve C, Dahmen G, van Geel AN, Bartels CC, Tilanus-Linthorst MM, Wagner A, Devilee P, Halley DJ, et al. Survival in hereditary breast cancer associated with germline mutations of BRCA2. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 1999 Nov;17:3396–3402. doi: 10.1200/JCO.1999.17.11.3396. Epub 1999/11/05. [DOI] [PubMed] [Google Scholar]
- Verhoog LC, Brekelmans CT, Seynaeve C, Meijers-Heijboer EJ, Klijn JG. Contralateral breast cancer risk is influenced by the age at onset in BRCA1-associated breast cancer. British journal of cancer. 2000 Aug;83:384–386. doi: 10.1054/bjoc.2000.1239. Epub 2000/08/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoog LC, Brekelmans CT, Seynaeve C, van den Bosch LM, Dahmen G, van Geel AN, Tilanus-Linthorst MM, Bartels CC, Wagner A, van den Ouweland A, et al. Survival and tumour characteristics of breast-cancer patients with germline mutations of BRCA1. Lancet. 1998 Jan 31;351:316–321. doi: 10.1016/s0140-6736(97)07065-7. Epub 1998/07/04. [DOI] [PubMed] [Google Scholar]
- Vorechovsky I, Luo L, Dyer MJ, Catovsky D, Amlot PL, Yaxley JC, Foroni L, Hammarstrom L, Webster AD, Yuille MA. Clustering of missense mutations in the ataxia-telangiectasia gene in a sporadic T-cell leukaemia. Nat Genet. 1997;17:96–99. doi: 10.1038/ng0997-96. 9/1997. [DOI] [PubMed] [Google Scholar]
- Yajima H, Lee KJ, Zhang S, Kobayashi J, Chen BP. DNA double-strand break formation upon UV-induced replication stress activates ATM and DNA-PKcs kinases. Journal of molecular biology. 2009 Jan 23;385:800–810. doi: 10.1016/j.jmb.2008.11.036. Epub 2008/12/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Wang Y, Jiang W, Liu X, Dubois RL, Lin CS, Ludwig T, Bakkenist CJ, Zha S. Kinase-dead ATM protein causes genomic instability and early embryonic lethality in mice. The Journal of cell biology. 2012 Aug 06;198:305–313. doi: 10.1083/jcb.201204098. Epub 2012/08/08. [DOI] [PMC free article] [PubMed] [Google Scholar]