

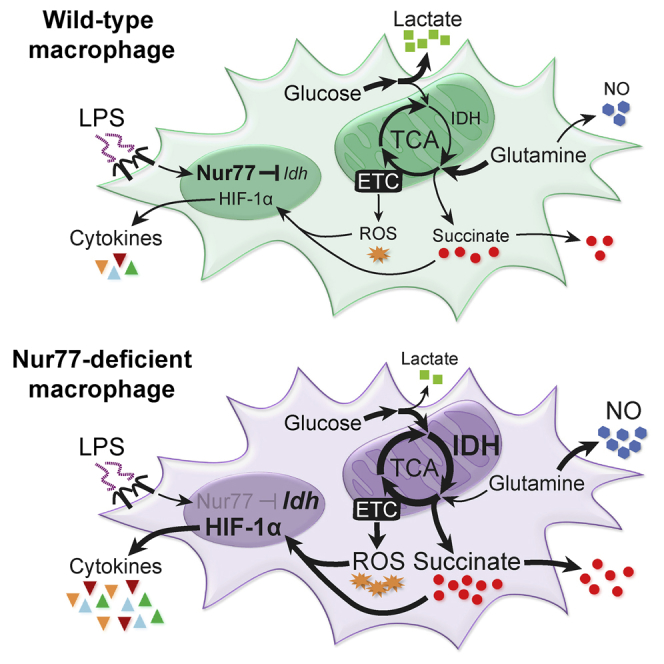
Summary
Activation of macrophages by inflammatory stimuli induces reprogramming of mitochondrial metabolism to support the production of pro-inflammatory cytokines and nitric oxide. Hallmarks of this metabolic rewiring are downregulation of α-ketoglutarate formation by isocitrate dehydrogenase (IDH) and accumulation of glutamine-derived succinate, which enhances the inflammatory response via the activity of succinate dehydrogenase (SDH). Here, we identify the nuclear receptor Nur77 (Nr4a1) as a key upstream transcriptional regulator of this pro-inflammatory metabolic switch in macrophages. Nur77-deficient macrophages fail to downregulate IDH expression and accumulate higher levels of succinate and other TCA cycle-derived metabolites in response to inflammatory stimulation in a glutamine-independent manner. Consequently, these macrophages produce more nitric oxide and pro-inflammatory cytokines in an SDH-dependent manner. In vivo, bone marrow Nur77 deficiency exacerbates atherosclerosis development and leads to increased circulating succinate levels. In summary, Nur77 induces an anti-inflammatory metabolic state in macrophages that protects against chronic inflammatory diseases such as atherosclerosis.
Keywords: nuclear receptor, Nur77, Nr4a1, macrophage, genome-wide profiling, inflammation, succinate dehydrogenase, immunometabolism, atherosclerosis
Graphical Abstract
Highlights
-
•
Genome-wide profiling indicates that Nur77 regulates macrophage mitochondrial metabolism
-
•
Nur77 inhibits IDH expression and TCA cycle activity in inflammatory macrophages
-
•
Nur77-deficient macrophages produce more nitric oxide and cytokines via SDH
-
•
Nur77 deficiency increases circulating succinate levels and atherosclerosis in vivo
Koenis et al. show that nuclear receptor Nur77 regulates the reprogramming of mitochondrial metabolism in inflammatory macrophages. Nur77-deficient macrophages accumulate higher levels of succinate and produce more pro-inflammatory cytokines and nitric oxide in a succinate dehydrogenase-dependent manner. In vivo, bone marrow Nur77 deficiency exacerbates atherosclerosis and increases circulating succinate levels.
Introduction
Macrophages are an essential part of the innate immune system and form the first line of defense against invading pathogens. However, chronic inflammatory diseases such as atherosclerosis are driven in large part by a prolonged and excessive macrophage-mediated inflammatory response, ultimately leading to the loss of local homeostasis and increased tissue damage (Moore and Tabas, 2011). Inhibiting excessive macrophage-mediated inflammation is therefore of considerable therapeutic interest, and a clinical trial studying the effects of pro-inflammatory cytokine interleukin (IL)-1β neutralization on cardiovascular morbidity and mortality has recently reported positive results (Ridker et al., 2017).
During the inflammatory response, macrophages produce immune effector molecules such as nitric oxide and cytokines to neutralize pathogens and recruit additional immune cells to the site of inflammation. Efficient production of such effector molecules by pro-inflammatory macrophages is dependent on several alterations in their cellular metabolism (Ryan and O’Neill, 2017). The three major metabolic hallmarks of pro-inflammatory macrophages include (1) suppressed mitochondrial oxidative phosphorylation (OXPHOS) and a dependence on increased glycolysis for energy; (2) a break in the tricarboxylic acid (TCA) cycle at isocitrate dehydrogenase (IDH), followed by an increase in glutamine-mediated anaplerosis; and (3) an accumulation of glutamine-derived succinate, which enhances reactive oxygen species (ROS) production, expression of the pro-inflammatory transcription factor hypoxiainducible factor (HIF)-1α, and, ultimately, pro-inflammatory cytokine production via succinate dehydrogenase (SDH) (Jha et al., 2015, Mills et al., 2016, Ryan and O’Neill, 2017).
Nur77 (Nr4a1) is a nuclear receptor that functions as a transcriptional activator or repressor, depending on coregulator protein recruitment and post-translational modifications (Kurakula et al., 2014). In human and mouse macrophages, Nur77 expression is induced by inflammatory stimuli such as lipopolysaccharide (LPS) and tumor necrosis factor (TNF)-α via nuclear factor kappa B (NF-κB) (Bonta et al., 2006, Pei et al., 2005). In turn, Nur77 has been shown to restrict the macrophage inflammatory response via the inhibition of NF-κB signaling (Bonta et al., 2006, Li et al., 2015a). Moreover, Nur77 deficiency exacerbates a variety of inflammation-driven pathologies, such as atherosclerosis (Hamers et al., 2012, Hanna et al., 2012, Hu et al., 2014) and LPS-induced sepsis (Li et al., 2015a). However, several observations suggest that the anti-inflammatory effects of Nur77 are not wholly mediated by the inhibition of NF-κB signaling. Nur77-deficient (Nur77-KO) macrophages produce higher levels of several pro-inflammatory cytokines, with the notable exception of TNF-α, the expression and secretion of which are unaffected (Hamers et al., 2012, Hanna et al., 2012). This lack of responsiveness is not consistent with NF-κB inhibition, but it is reminiscent of the anti-inflammatory effects of SDH inhibition, which also fails to attenuate TNF-α production (Lampropoulou et al., 2016, Mills et al., 2016). In addition, activation of NF-κB signaling is a rapid process, with nuclear translocation of NF-κB and target gene transcription starting within minutes of inflammatory stimulation. Detectable levels of nuclear Nur77 protein take several hours to accumulate, however (Pei et al., 2005). This temporal separation suggests that Nur77 cannot exert its anti-inflammatory effects until after the early, NF-κB-driven stage of the inflammatory response is well under way.
In this study, we examined the genome-wide Nur77 binding site profile and the Nur77-regulated transcriptome in macrophages. These experiments implicated Nur77 in the regulation of macrophage mitochondrial metabolism, and follow-up experiments showed that Nur77 is responsible for rewiring TCA cycle metabolism in pro-inflammatory macrophages. Furthermore, we show that a lack of Nur77-mediated metabolic changes in Nur77-deficient macrophages leads to excessive succinate production and SDH-driven nitric oxide and cytokine expression in vitro, and exacerbated atherosclerosis progression, circulating succinate accumulation, and lesion HIF-1α protein levels in vivo.
Results
Genome-wide Binding Site and Transcriptome Profiling Implicate Nur77 in the Regulation of Macrophage Mitochondrial Metabolism
To assess whether the anti-inflammatory effects of Nur77 are mediated by its activity as a transcription factor, we set out to characterize the target genes and genome-wide binding site profile of this nuclear receptor. We engineered RAW264.7 macrophage cell lines with the stable integration of a doxycycline (Dox)-inducible expression cassette that encoded either hemagglutinin (HA)-tagged Nur77 (HA-NUR77) or GFP as a control (Figure S1A). Dox treatment of these cells resulted in the strong expression of nuclear HA-NUR77 protein (Figures S1B and S1C) and induction of the established Nur77 target gene Eno3 in a time- and concentration-dependent manner (Figure S1D). These stable RAW264.7 cell lines were then used for genome-wide Nur77 binding site and transcriptome profiling by chromatin immunoprecipitation sequencing (ChIP-seq) and RNA-sequencing (RNA-seq) (Figure 1A).
Figure 1.

Genome-wide Binding Site and Transcriptome Profiling Implicate Nur77 in the Regulation of Macrophage Mitochondrial Metabolism
(A) ChIP-seq and RNA-seq experimental setup in RAW264.7 with inducible overexpression of HA-NUR77 or GFP.
(B) Venn diagram of Nur77 binding sites in ChIP-seq dataset. Binding sites present in at least 2 of 3 ChIP replicates were used for further analyses.
(C) Genomic distribution of 19,164 Nur77 binding sites in RAW264.7.
(D) ChIP-seq signal for RAW264.7 input and Nur77 ChIP samples in 100 kb window around 19,164 Nur77 binding sites.
(E) Top enriched de novo transcription factor (TF) motifs within 200 bp of 19,164 Nur77 binding sites.
(F) ChIP-seq signal for Nur77, PU.1, and H3K4me1 in indicated cell types in 5 kb window around 19,164 Nur77 binding sites in RAW264.7.
(G) Hierarchical clustering and Z score heatmap of Nur77-regulated genes as determined by RNA-seq.
(H) Top enriched pathways for genes (i) down- and (iii) upregulated by Nur77 overexpression in RNA-seq dataset or Nur77 binding sites linked to (ii) down- and (iv) upregulated genes in ChIP-seq dataset. Asterisks indicate metabolic pathways.
See also Figure S1.
Using ChIP-seq, we identified 19,164 high-confidence Nur77 binding sites (Figure 1B). These binding sites were primarily found near gene-coding regions of the genome (Figures 1C and S1E). Inspection of the raw ChIP-seq data showed a significantly higher signal in the three Nur77 ChIP replicates than in the input control (Figure 1D), and we successfully validated Nur77 binding sites from the ChIP-seq dataset by ChIP-qPCR for Eno3, Cx3cr1, and Pfkfb3 loci (Figure S1F).
Analysis of the ChIP-seq dataset showed enrichment of sequence motifs for Nur77 and the macrophage lineage-specifying transcription factors PU.1 and CEBPB (Figure 1E). Analysis of publically available PU.1 and H3K4me1 ChIP-seq datasets showed strong PU.1 binding and enrichment of H3K4me1—a histone modification associated with enhancer regions—around RAW264.7 Nur77 binding sites in bone marrow-derived macrophage (BMDM), but not in bone marrow, spleen, liver, or heart datasets (Figure 1F). These results suggest that Nur77 binds at genomic loci that are physiologically relevant to macrophages in our stable RAW264.7 cell lines.
Hierarchical clustering of the Nur77 RNA-seq dataset revealed good reproducibility of the biological replicates of this experiment, allowing us to identify 2,611 genes downregulated and 2,750 genes upregulated by Nur77 overexpression in RAW264.7 macrophages (Figure 1G; Table S1). We validated Nur77-mediated transcriptional regulation of three target genes identified in the RNA-seq dataset (Cx3cr1, Eno3, and Pfkfb3) by RT-qPCR (Figure S1G). We next linked our Nur77 ChIP-seq and RNA-seq datasets using the BETA software package (Wang et al., 2013) to identify genes under the direct control of Nur77. Pathway analyses of Nur77-regulated genes with associated Nur77 binding sites identified several processes known to be regulated by Nur77, such as mitogen-activated protein kinase (MAPK) and cytokine signaling (Li et al., 2015a), but also several pathways hitherto not known to involve Nur77 in macrophages, most notably several metabolic pathways (Figure 1H).
Nur77 Is Required for Normal Mitochondrial Activity in Naive Macrophages
Our genome-wide profiling experiments showed the enrichment of metabolic pathways. We therefore set out to assess the role of Nur77 in macrophage metabolism. Nur77 knockdown in RAW264.7 macrophages resulted in a lower basal, ATP synthase-coupled, and maximal (uncoupled) oxygen consumption rate (OCR), a measure of mitochondrial OXPHOS (Figures 2A and S2A). Similar results were obtained using BMDMs from wild-type (WT) and Nur77-KO mice (Figure 2B).
Figure 2.

Nur77 Is Required for Normal Mitochondrial Activity in Naive Macrophages
(A and B) Oxygen consumption rate (OCR) after sequential injection of glucose (Glc), oligomycin (OM), carbonyl cyanide 4-trifluoromethoxyphenylhydrazone (FCCP), and antimycin plus rotenone (AA+Rot) in RAW264.7 upon knock down of Nur77 (shNur77) versus control (shCtrl) (A), or in WT and Nur77-KO BMDMs left untreated or stimulated with LPS for 24 hr (B). Bar graphs show OXPHOS parameters derived from OCR values as illustrated in Figure S2C.
(C) Extracellular acidification rate (ECAR) in WT and Nur77-KO BMDMs left untreated or stimulated with LPS for 24 hr. Bar graphs show parameters of glycolysis from ECAR values, as illustrated in Figure S2E.
(D) Relative intracellular ATP in WT and Nur77-KO BMDMs left untreated or stimulated with LPS for 24 hr, followed by vehicle (−) or 5 μM OM (+) for 2 min.
(E–G) Mitochondrial abundance in untreated or 24 hr LPS-stimulated WT and Nur77-KO BMDMs determined by ratio of mitochondrial (mt)DNA to nuclear (n)DNA (E), flow cytometry (fluorescence-activated cell sorting [FACS]) for MitoTracker Green FM staining (F), or live-cell imaging of mitochondria visualized with MitoTracker Green FM (G). MFI, median fluorescence intensity. Micrographs on the left show representative MitoTracker stained cells. Scale bar indicates 20 μm.
Data are shown as means ± SEMs (n = 6 for [A]–[C]; n = 3 for [D], [F], and [G]; n = 106–136 cells for [E]). p values were calculated using two-tailed Student’s t test ([A]–[C], [E]–[G]) or two-way ANOVA with Tukey’s post hoc test (D). n.s., non-significant. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
See also Figure S2.
In response to inflammatory stimuli, macrophages switch from OXPHOS to glycolysis as their main source of energy (Ryan and O’Neill, 2017). Stimulating BMDMs with LPS induced expression of Nur77 in WT but not in Nur77-KO BMDMs (Figure S2B) and caused a significant reduction in OXPHOS in both cell populations (Figures 2B and S2C). Both proton leak (the proportion of oxygen consumption without concomitant ATP generation) and coupling efficiency (the proportion of oxygen consumption used for ATP generation) were significantly lower in Nur77-KO BMDMs compared to WT (Figures 2B and S2C). One possible explanation for this paradoxical decrease in both parameters is that LPS-activated Nur77-KO BMDMs instead suffer from increased electron leak, an unintended exit of electrons from the electron transport chain (ETC) that leads to ROS production.
Both WT and Nur77-KO BMDMs also massively increased glycolytic lactate production in response to LPS, but Nur77-KO BMDMs did so to a lesser extent (Figures 2C and S2E). Similar results were found using Nur77-depleted RAW264.7 macrophages (Figures S2D and S2E). Because several glycolytic genes were upregulated by Nur77 in our RAW264.7 transcriptome profiling experiment (Figure S1G), we hypothesized that the reduced expression of these genes could be responsible for the observed reduction in glycolytic lactate production in Nur77-depleted cells. However, while Nur77 overexpression in RAW264.7 macrophages increased the expression of two crucially important glycolytic enzymes (Pfkfb3 and Pfkp), the expression of these genes was unchanged in Nur77-KO BMDMs (Figures S2F and S2G).
Cells use OXPHOS and glycolysis to produce energy in the form of ATP. Consistent with their reduced OXPHOS, Nur77-KO BMDMs had significantly lower intracellular ATP levels than did WT BMDMs both before and after LPS stimulation (Figure 2D). The differences in ATP levels between WT and Nur77-KO BMDMs disappeared upon treatment with oligomycin, a mitochondrial ATP synthase inhibitor, showing that non-mitochondrial (glycolytic) ATP production is not affected in Nur77-KO BMDMs (Figure 2D).
A possible explanation for the reduced mitochondrial activity in Nur77-KO BMDMs would be a reduced number of mitochondria. We quantified mitochondrial abundance in WT and Nur77-KO BMDMs by measuring the ratio of mitochondrial to nuclear DNA (Figure 2E) and the intensity of a mitochondria-specific fluorescent probe using flow cytometry (Figure 2F) or live-cell fluorescence microscopy (Figure 2G). These three experimental approaches showed that unstimulated Nur77-KO BMDMs had significantly lower numbers of mitochondria than WT BMDMs, explaining their reduced OXPHOS rates. This difference in mitochondrial abundance disappeared after treating cells with LPS, however (Figure 2E). Reduced numbers of mitochondria may be caused by increased degradation (mitophagy). We found that Nur77-KO BMDMs had numbers of lysosomes similar to WT BMDMs (Figure S2H) but that these lysosomes co-localized more extensively with mitochondria (Figure S2I; Video S1)—a measure of mitophagy (Dolman et al., 2013). Similar to mitochondrial abundance, this difference in mitophagy between WT and Nur77-KO BMDMs disappeared after stimulating cells with LPS (Figure S2I).
Cells were co-stained with MitoTracker Green FM (green) and LysoTracker Red DND-99 (red) and imaged over a 7-minute period. Exposure was increased for Nur77-KO panel to better visualize mitochondrial-lysosomal dynamics. Arrows indicate MitoTracker Green FM/LysoTracker Red DND-99 co-localization events.
Nur77-Deficient Macrophages Fail to Downregulate IDH Expression and Accumulate Higher Levels of Succinate during Inflammation
In addition to mitochondrial copy number, OXPHOS rates are regulated by the availability of TCA cycle-derived ETC substrates. We therefore decided to investigate a possible role for Nur77 in the regulation of the TCA cycle.
Mitochondrial IDH catalyzes a rate-limiting step of the TCA cycle and macrophages downregulate the expression and activity of this enzyme in response to inflammatory stimuli (Jha et al., 2015). We hypothesized that Nur77 mediates the downregulation of IDH expression during inflammation because we observed Nur77-mediated inhibition of a subset of TCA cycle genes—including several IDH isoforms—in our RAW264.7 RNA-seq dataset (Figure 3A). Furthermore, in our Nur77 ChIP-seq dataset, we found that there was a general enrichment of Nur77 binding near the transcriptional start sites (TSSs) of TCA cycle enzyme genes (Figure 3B), including near the TSSs of several mitochondrial IDH genes (Figure 3C). Nur77-KO BMDMs consistently expressed higher levels of several mitochondrial IDH isoforms (Idh2, Idh3b, Idh3g) both before and after LPS stimulation, while expression of cytosolic IDH (Idh1) and several other TCA cycle enzyme genes was largely unchanged (Figures 3D and S3A). Nur77-dependent regulation of IDH-encoding genes was further confirmed by the overexpression of human Nur77 in Nur77-KO BMDMs, which resulted in the significantly reduced expression of mitochondrial IDH isoforms Idh2, Idh3b, and Idh3g, but not the cytosolic IDH isoform Idh1 (Figure S3B).
Figure 3.

Nur77-Deficient Macrophages Fail to Downregulate IDH Expression and Accumulate Higher Levels of Succinate during Inflammation
(A) Hierarchical clustering and expression heatmap of TCA cycle enzyme-encoding genes in RAW264.7 after inducible overexpression of GFP or HA-NUR77, as determined by RNA-seq.
(B) Mean Nur77 ChIP-seq signal in RAW264.7 in 20 kb window around transcription start sites (TSSs) of TCA cycle genes shown in (A).
(C) Genome browser view of Nur77 ChIP-seq signal around isocitrate dehydrogenase (IDH) gene loci in RAW264.7 macrophages.
(D and E) Gene expression of cytosolic and mitochondrial IDH isoforms (D) and succinate levels (E) in WT and Nur77-KO BMDMs left untreated or stimulated with LPS for the times indicated.
For (D) and (E), data are shown as means ± SEMs (n = 3). p values were calculated using two-tailed Student’s t test. n.s., non-significant. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
See also Figure S3.
We hypothesized that a failure to downregulate IDH expression in response to LPS could lead to aberrantly high TCA cycle activity in pro-inflammatory Nur77-KO BMDMs. To test this hypothesis, we measured the abundance of succinate, a pro-inflammatory TCA cycle intermediate formed downstream of IDH that accumulates during inflammation (Ryan and O’Neill, 2017). Consistent with their increased IDH expression, pro-inflammatory Nur77-KO BMDMs expressed higher levels of intracellular succinate and increased secretion of this metabolite into the extracellular environment (Figure 3E).
Pro-inflammatory Nur77-Deficient Macrophages Show Enhanced TCA Cycle Activity
We next investigated possible Nur77-induced changes in TCA cycle metabolism by performing metabolomic profiling of WT and Nur77-KO BMDMs using liquid chromatography-mass spectrometry (LC-MS). We found that WT and Nur77-KO BMDMs were metabolically similar at baseline but altered their metabolism in different ways in response to LPS (Figure 4A; Table S2). Random forest analysis showed that the differences between WT and Nur77-KO BMDMs were primarily caused by an altered abundance of α-ketoglutarate, glutamate, and succinate—three closely connected metabolites, the first of which is produced by IDH (Figure 4B). Similarly, separate random forest analyses of LPS-induced changes in WT and Nur77-KO BMDMs showed that WT cells primarily changed their levels of succinate, α-ketoglutarate, and glycolysis-related metabolites in response to LPS, while Nur77-KO cells primarily changed their abundance of succinate and several amino acids, but notably not α-ketoglutarate (Figure 4B). A closer look at the metabolomics dataset revealed that Nur77-KO BMDMs express significantly higher levels of IDH-derived α-ketoglutarate and downstream TCA cycle intermediates in response to LPS, while producing lower levels of lactate, possibly because of the increased demand for pyruvate entry into the TCA cycle (Figure 4C). In addition, Nur77-KO BMDMs had a significantly lower (iso)citrate-to-α-ketoglutarate ratio after LPS stimulation, which is indicative of increased IDH activity in these cells (Figure 4D).
Figure 4.

Pro-inflammatory Nur77-Deficient Macrophages Show Enhanced TCA Cycle Activity
(A and B) Principal-component analysis (PCA) (A) and random forest variable importance measures (B) of intracellular metabolite levels from untreated (n = 4 per group) or 24 hr LPS-treated (n = 6 per group) WT and Nur77-KO BMDMs, as determined by LC-MS (metabolomics).
(C) TCA cycle schematic summarizing metabolomics. Circles indicate metabolites, arrowheads indicate enzymes. Node colors indicate relative metabolite abundance, as shown in the lower left corner. Insets connected by dotted lines show relative metabolite abundance for untreated (−) and 24 hr LPS-treated groups. 2-HG, 2-hydroxyglutarate; IDH, isocitrate dehydrogenase; SDH, succinate dehydrogenase.
(D) Ratio of (iso)citrate to α-ketoglutarate abundance in WT and Nur77-KO BMDMs from metabolomics.
(E) Succinate secretion by WT and Nur77-KO BMDMs cultured in glutamine-containing (2 mM) or glutamine-free (0 mM) medium and either left untreated (−) or stimulated with LPS for 48 hr (+).
(F and G) Ratio of reduced (NADH) to oxidized (NAD+) nicotinamide adenine dinucleotide (F) and total 2-hydroxyglutarate (l- and d- forms) abundance (G) in WT and Nur77-KO BMDMs from metabolomics.
For (C)–(G), data are shown as means ± SEMs (n = 3 for [E]; n = 4–6 for [C], [D], [F], and [G]). p values were calculated using two-tailed Student’s t test (B, C, F, and H–J) or two-way ANOVA with Tukey’s post hoc test (E). n.s., non-significant. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
See also Figure S4.
We wondered what functional consequences high IDH activity in pro-inflammatory Nur77-KO macrophages could have. Downregulation of IDH in pro-inflammatory macrophages results in a TCA cycle break that is partially alleviated by the enhanced conversion of glutamine to α-ketoglutarate (anaplerosis), which is then converted to succinate (Jha et al., 2015). We therefore speculated that enhanced IDH activity in LPS-stimulated Nur77-KO BMDMs may lead to a reduced dependence on glutamine for succinate production. LPS-induced succinate accumulation was completely inhibited when WT BMDMs were cultured in glutamine-free medium, while succinate levels were unaffected by glutamine availability in Nur77-KO BMDMs (Figure 4E).
Taken together, these results strongly suggest there is increased IDH and TCA cycle activity in pro-inflammatory Nur77-KO macrophages. This idea was further supported by a significantly higher NADH:NAD+ ratio in pro-inflammatory Nur77-KO BMDMs (Figure 4F), which is consistent with the enhanced reduction of NAD+ by an overly active TCA cycle and reduced consumption of NADH by the ETC as a result of suppressed OXPHOS. Similarly, Nur77-KO BMDMs—but not WT BMDMs—accumulated 2-hydroxyglutarate (l- and d- forms) in response to LPS (Figures 4B and 4G). l-2-Hydroxyglutarate is a dead-end metabolite that is used by cells to regenerate NAD+ in response to rising α-ketoglutarate and NADH levels. Similar to succinate, it can stabilize the pro-inflammatory transcription factor HIF-1α (Oldham et al., 2015). In summary, these results demonstrate that Nur77 deficiency leads to increased TCA cycle activity and pro-inflammatory metabolite production in activated macrophages.
Nur77 Deficiency Enhances SDH Activity in Pro-inflammatory Macrophages
Nur77-KO BMDMs accumulate higher levels of succinate in response to LPS. Increased succinate levels could also be caused by a decrease in downstream succinate oxidation by SDH, however, especially because Nur77-KO BMDMs showed an increased abundance of the SDH-inhibitory metabolite itaconate and Irg1, the enzyme responsible for its production (Figures S4A and S4B) (Lampropoulou et al., 2016). Pro-inflammatory Nur77-KO BMDMs also produced more SDH-derived fumarate (Figure 4C). We therefore sought to clarify whether SDH activity is altered in Nur77-KO BMDMs. We measured oxygen consumption of cells treated with compounds that force mitochondrial respiration to occur exclusively via SDH (Salabei et al., 2014) and found that Nur77-KO BMDMs had a higher SDH-mediated respiration after LPS stimulation (Figure 5A). We also measured the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to formazan, a common assay for SDH activity (Van den Bossche et al., 2016), which showed that Nur77-KO BMDMs had higher levels of SDH activity after LPS stimulation. Treating cells with the SDH inhibitor dimethyl malonate (DMM) reduced SDH activity to levels comparable to those of WT (Figure 5B). Increased SDH activity in Nur77-KO BMDMs was not caused by a higher overall viability of these cells, however, because intracellular ATP levels, a measure of cell viability, were higher in WT cells (Figure S5A). Finally, a similar increase in SDH activity was observed in Nur77-depleted RAW264.7 macrophages (Figure S5B).
Figure 5.

Nur77 Deficiency Increases SDH Activity, Mitochondrial Membrane Potential, ROS Production, and SDH-Induced HIF-1α Stabilization in Pro-inflammatory Macrophages
(A) OCR in WT and Nur77-KO BMDMs left untreated or stimulated with LPS for 24 hr, followed by sequential injections of digitonin (Dig.), succinate (Succ.), rotenone (Rot.), and ADP in the first, OM in the second, and AA in the third injection. Bar graph shows SDH activity derived by dividing OCR at (a) by that at (b).
(B) SDH activity determined by MTT assay in WT and Nur77-KO BMDMs left untreated, stimulated with LPS for times indicated, or treated with SDH inhibitor DMM for 24 hr (SDHi).
(C) Flow cytometry dot plots of Δψm-insensitive MitoTracker Green FM (x axis) and Δψm-sensitive MitoTracker Red CM-H2Xros (y axis) fluorescence in WT and Nur77-KO BMDMs left untreated or stimulated with LPS for the times indicated. Cells in quadrant Q2 were considered to have hyperpolarized mitochondria.
(D) Δψm determined by flow cytometry in WT and Nur77-KO BMDMs left untreated or stimulated with LPS for the times indicated and stained with MitoTracker Red CM-H2Xros.
(E–G) Mitochondrial reactive oxygen species (mROS) (E) and total ROS production (F and G) determined by flow cytometry in WT and Nur77-KO BMDMs left untreated or stimulated with LPS for 48 hr. (G) Western blot for HIF-1α protein in WT and Nur77-KO BMDMs left untreated or stimulated with LPS for 24 hr with and without SDH inhibitor NPA pre-treatment (SDHi). β-Actin was used as loading control. Numbers on the right: molecular weight in kDa; numbers at the bottom: fold change (FC) in β-actin-normalized HIF-1α band intensity relative to unstimulated WT. The blot shown is representative of two independent experiments.
For (A), (B), and (D)–(F), data are shown as means ± SEMs (n = 4 for [A] and [D]; n = 8 for [B], n = 3 for [E] and [F]). p values were calculated using two-tailed Student’s t test. n.s., non-significant. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
See also Figure S5.
We hypothesized that Nur77 altered SDH activity by regulating the expression of SDH-encoding genes, because all four SDH subunits were downregulated by Nur77 overexpression in the RAW264.7 RNA-seq dataset (Figure 3A; Table S1). However, gene expression of all SDH subunits and protein expression of the SDH subunit A were unchanged in Nur77-KO BMDMs (Figures S3A and S5C). Taken together, these results show that elevated levels of succinate in pro-inflammatory Nur77-KO BMDMs are most likely caused by an increase in its production by upstream enzymes of the TCA cycle and not by a decrease in its conversion by SDH.
Nur77 Deficiency Increases Mitochondrial Membrane Potential, ROS Production, and SDH-Mediated HIF-1α Stabilization in Pro-inflammatory Macrophages
Succinate accumulates in activated macrophages and potentiates the inflammatory response by increasing SDH-facilitated mitochondrial ROS production, which in turn leads to stabilization of the pro-inflammatory transcription factor HIF-1α (Mills et al., 2016). Mitochondrial ROS is primarily produced by complex I of the ETC, which couples oxidation of TCA cycle-derived NADH with reduction of coenzyme Q (CoQ). ROS production by complex I is highest under two conditions: (1) when the NADH:NAD+ ratio is high (i.e., too much input), or (2) when the proton-motive force (Δp) is high, the rate of OXPHOS is low, and the CoQ pool is highly reduced (i.e., less potential output) (Murphy, 2009). Pro-inflammatory macrophages fulfill all three parameters of the second condition because they have low rates of OXPHOS, high rates of succinate-driven reduction of CoQ via SDH, and elevated mitochondrial membrane potential (Δψm, a component of Δp) (Ryan and O’Neill, 2017).
Because Nur77-KO BMDMs showed elevated levels of succinate, increased SDH activity, and a relatively higher NADH:NAD+ ratio, we hypothesized that these cells would suffer from exacerbated SDH-driven increases in Δψm and ROS production. We found that a larger proportion of Nur77-KO BMDMs had hyperpolarized mitochondria compared to WT BMDMs after LPS stimulation (Figures 5C and S5D), which also translated into higher median accumulation of the same Δψm-sensitive probe in the mitochondria of Nur77-KO BMDMs (Figure 5D). In addition, Nur77-KO BMDMs produced higher levels of both mitochondrial and whole-cell (total) ROS in response to LPS (Figures 5E and 5F). Similar results were obtained using Nur77-depleted RAW264.7 macrophages (Figure S5E). The increased ROS levels observed in Nur77-KO BMDMs were not caused by defective ROS scavenging, however, because there were no differences in the ratio of oxidized to reduced glutathione between WT and Nur77-KO BMDMs (Figure S5F), while the total pool of this endogenous antioxidant was actually increased in Nur77-KO BMDMs (Figure S5G). Finally, in line with their enhanced ROS, 2-hydroxyglutarate, and succinate production, Nur77-KO BMDMs expressed higher levels of HIF-1α protein after LPS stimulation, while pre-treating cells with the SDH inhibitor nitropropionic acid (NPA) completely abrogated this effect (Figure 5G). Taken together, these results show that Nur77 deficiency leads to increased mitochondrial membrane hyperpolarization, ROS production, and SDH-mediated HIF-1α stabilization in response to LPS.
Pro-inflammatory Nur77-KO Macrophages Produce More Cytokines in an SDH-Dependent Manner
In pro-inflammatory macrophages, SDH-mediated stabilization of HIF-1α leads to the increased production of several pro-inflammatory cytokines, with the notable exception of TNF-α (Lampropoulou et al., 2016, Mills et al., 2016). Consistent with our previous results, Nur77-KO BMDMs expressed higher levels of several genes encoding SDH-regulated pro-inflammatory cytokines, including Il1b, Il6, Il12a, Il12b, and Il18 (Figures 6A and 6D). Meanwhile, expression of the anti-inflammatory cytokine Il10 was decreased, while expression of pro-inflammatory Tnf was unchanged in Nur77-KO BMDMs (Figure 6A). Overexpression of human Nur77 in Nur77-KO BMDMs led to significantly reduced expression levels of Il1b and Il6, but not Tnf (Figure S6A). Correspondingly, protein expression of the cytokines IL-6, IL-12p70, and IL-1β were higher in LPS-treated Nur77-KO BMDMs, while IL-10 protein was lower and TNF-α protein was unchanged (Figures 6B and 6C). Treating cells with the SDH inhibitor NPA normalized the protein expression of all of the above-mentioned cytokines between WT and Nur77-KO BMDMs (Figures 6B and 6C). Nur77-KO BMDMs also induced the expression of the succinate receptor Sucnr1 (GPR91) and the pro-inflammatory cytokines Il18 and interferon (IFN)-γ in response to LPS stimulation, whereas WT BMDMs did not (Figures 6D and 6E). Taken together, these results show that the increased production of pro-inflammatory cytokines in Nur77-KO BMDMs is mainly the result of increased SDH activity in these cells.
Figure 6.

Nur77-Deficient Macrophages Produce More Pro-inflammatory Cytokines and NO in an SDH-Dependent Manner
(A and B) Cytokine gene expression (A) and protein secretion (B) in WT and Nur77-KO BMDMs left untreated or stimulated with LPS for times indicated with and without SDH inhibitor NPA pre-treatment (SDHi).
(C) Western blot for pro-IL-1β protein in WT and Nur77-KO BMDMs left untreated or stimulated with LPS for 24 hr with and without NPA pre-treatment (SDHi). α-Tubulin was used as loading control. Numbers on the right: molecular weight in kDa. Numbers at the bottom: fold change in α-tubulin-normalized pro-IL-1β band intensity relative to unstimulated WT. Blot shown is representative of two independent experiments.
(D and E) Il18 and Sucnr1 gene expression (D) and IFN-γ secretion (E) in WT and Nur77-KO BMDMs left untreated or stimulated with LPS for the times indicated.
(F) Schematic of TCA and NO cycles connected by the aspartate-argininosuccinate shunt. Circles indicate metabolites; arrowheads indicate enzymes. Node colors indicate metabolite abundance from metabolomics, as shown in the upper right corner. AST, aspartate aminotransferase; ALT, alanine aminotransferase; iNOS, inducible NO synthase; SDH, succinate dehydrogenase. Dashed line indicates α-ketoglutarate-consuming, glutamate-forming reaction of ALT.
(G and H) NO secretion by WT and Nur77-KO BMDMs left untreated (−) or stimulated with LPS for 48 hr with and without NPA pre-treatment (SDHi) (G), or cultured in glutamine-free (−) or 2 mM glutamine (l-glut)-containing (+) medium and left untreated (−) or stimulated with LPS for 48 hr (+) (H).
For (A), (B), (D), (E), (G), and (H), data are shown as means ± SEMs (n = 3). p values were calculated using two-tailed Student’s t test (A, B, D, E, and G) or two-way ANOVA with Tukey’s post hoc test (H). n.s., non-significant. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
See also Figure S6.
Altered TCA Cycle Metabolism Enhances Nitric Oxide Production in Pro-inflammatory Nur77-KO Macrophages
In addition to cytokines, pro-inflammatory macrophages produce large amounts of nitric oxide (NO), a molecule used to both attack pathogens and remodel intracellular metabolism (Van den Bossche et al., 2016). Nur77-KO BMDMs secreted larger amounts of NO in response to LPS (Figure S6B), even though both gene and protein expressions of inducible NO synthase (iNOS)—the enzyme responsible for LPS-induced NO production in mouse macrophages—were essentially unchanged between WT and Nur77-KO BMDMs (Figures S6C and S6D).
These observations led us to hypothesize that Nur77-KO BMDMs produce larger amounts of NO because of their altered TCA cycle metabolism. NO production in macrophages is supported by the aspartate-argininosuccinate shunt, which connects the TCA and NO cycles via aspartate aminotransferase (AST; Got2) (Jha et al., 2015). AST consumes TCA cycle-derived oxaloacetate and glutamine-derived glutamate to produce aspartate, which is then fed into the NO cycle (Figure 6F). Consistent with this model, reducing oxaloacetate availability by blocking TCA cycle activity at SDH decreased NO production in Nur77-KO BMDMs to levels comparable to those of WT BMDMs (Figure 6G). Furthermore, steady-state levels of glutamine, glutamate, and aspartate were found to be higher in LPS-treated Nur77-KO BMDMs (Figures 4C and S6E), potentially because of the reduced need by these cells to use glutamine and glutamate for TCA cycle anaplerosis. Finally, culturing cells in glutamine-free medium severely hampered the ability of WT BMDMs to produce NO in response to LPS, while NO production in Nur77-KO BMDMs was unaffected (Figure 6H). In summary, these results show that increased NO production in Nur77-KO BMDMs is primarily caused by their highly active TCA cycle metabolism.
Bone Marrow Nur77 Deficiency Exacerbates Western-type Diet-Induced Atherosclerotic Lesion Development, Circulating Succinate Accumulation, and Lesion HIF-1α Protein In Vivo
Given the enhanced SDH-driven inflammatory response in Nur77-KO BMDMs, we hypothesized that Nur77 deficiency leads to exacerbated inflammation in vivo. A number of studies by our group and others have shown that Nur77 has a protective effect in atherosclerosis (Hamers et al., 2012, Hanna et al., 2012, Hu et al., 2014), a chronic inflammatory disease driven in large part by macrophage-mediated inflammation (Moore and Tabas, 2011). However, others have reported that a lack of Nur77 does not affect atherosclerosis development, and it was suggested by the authors that this discrepancy may be the result of mouse stock-specific differences in genomic background (Chao et al., 2013). We therefore decided to re-evaluate the role of Nur77 in atherosclerosis using a newly developed Nur77-deficient mouse line created with the cytomegalovirus (CMV) promoter-driven Cre-Lox system. Bone marrow from these Nur77-deficient mice, which did not express Nur77 (Figure S7A), or from WT littermates was transplanted into Lldr−/− mice, followed by a Western-type diet (WTD) feeding to induce atherosclerotic lesion formation. After 14 weeks of WTD, mice that received a Nur77-KO bone marrow transplant (KO-tp) developed significantly larger atherosclerotic lesions than did mice that received WT bone marrow (WT-tp) (Figure 7A). In addition, lesions in KO-tp mice had significantly larger necrotic areas (Figure 7A), a hallmark of a more advanced disease stage (Moore and Tabas, 2011). Further analysis of atherosclerotic lesion composition showed that collagen and macrophage content was unchanged, while smooth muscle cell, T cell, and apoptotic cell counts were increased in KO-tp mice (Figure S7B). We found that blood inflammatory cytokine and succinate levels were increased in response to 14 weeks of WTD and that the accumulation of circulating IL-6, IFN-γ, and succinate was further increased in KO-tp mice (Figures 7B and 7C). Consistent with increased levels of circulating succinate and inflammatory cytokines, we found that HIF-1α protein was more highly expressed in atherosclerotic lesions of KO-tp mice (Figure 7D).
Figure 7.

Bone Marrow Nur77 Deficiency Exacerbates Western-type Diet-Induced Atherosclerotic Lesion Development, Circulating Succinate Accumulation, and Lesion HIF-1α Protein In Vivo
(A) Representative micrographs of H&E-stained atherosclerotic lesions in Ldlr−/− mice that received WT (WT-tp) or Nur77-deficient (KO-tp) bone marrow and were subsequently fed a Western-type diet (WTD) for 14 weeks. Scale bar indicates 1 mm. Scatterplots on the right: quantifications of atherosclerotic lesion size (left) and lesional necrotic area (right).
(B and C) Cytokine protein (B) and succinate (C) levels in blood plasma of mice described in (A).
(D) Representative micrographs of HIF-1α immunohistochemical staining in atherosclerotic lesions from mice described in (A). Scale bar indicates 0.25 mm. Scatterplot on the right: quantification of lesion HIF-1α staining.
For (A), (C), and (D), each filled circle (WT-tp) or hollow square (KO-tp) indicates one mouse, and horizontal bars indicate means ± SEMs. For (B), data are shown as means ± SEMs (n = 10). p values were calculated using two-tailed Student’s t test (A and D) or two-way ANOVA (B and C). n.s., non-significant. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Discussion
Rewiring of TCA cycle metabolism and activation of various metabolic shunt pathways is required for the efficient production of inflammatory effector molecules by activated macrophages (Ryan and O’Neill, 2017). The upstream regulators responsible for these inflammation-induced metabolic adaptations are not known, however. Here, we show that the anti-inflammatory activity of Nur77 in macrophages is primarily mediated by its ability to alter mitochondrial metabolism, in addition to its previously reported function as a repressor of NF-κB signaling. Furthermore, our results show that even though changes in TCA cycle metabolism are induced by inflammatory stimuli, they actually serve to limit the inflammatory response in macrophages.
The first step in the resolution of inflammation is a timely termination of pro-inflammatory signaling (Serhan and Savill, 2005). It is therefore particularly interesting that Nur77-KO macrophages possess the ability to produce pro-inflammatory molecules such as succinate and NO under conditions of glutamine restriction, a metabolite whose importance in promoting endotoxin tolerance in macrophages has recently appreciated (Liu et al., 2017). Although the exact cause of their glutamine independence is not clear, we hypothesize that Nur77-KO macrophages can maintain succinate and NO production in glutamine-restricted conditions because of their larger reservoir of IDH-derived α-ketoglutarate. This metabolite can be converted to either succinate via the TCA cycle or to glutamate—and subsequently aspartate—via alanine aminotransferase (ALT), the mitochondrial isoform of which (Gpt2) works mainly in the α-ketoglutarate-consuming, glutamate-forming direction (Gubern et al., 1990).
Two recent publications have reported that Nur77 silencing or pharmacological inhibition leads to increased gene expression of cytoplasmic IDH (Idh1) in various cancer cell lines (Hedrick et al., 2015, Lee et al., 2014). Although the exact cause of the discrepancy in Idh1 regulation between these studies and our RAW264.7 RNA-seq dataset showing reduced Idh1 expression in response to Nur77 overexpression was not studied in more detail, we speculate that it may be the result of cell-type-specific Idh1 regulation or differences between Nur77 overexpression and inhibition or silencing. It should be noted, however, that these studies do not report on the effects of Nur77 modulation on mitochondrial IDH (Idh2, Idh3) expression, which we found to be significantly lower in Nur77-KO macrophages.
Nur77-KO macrophages express higher levels of the pro-inflammatory cytokines IL-18 and IFN-γ in response to LPS, while WT macrophages do not. Such aberrant cytokine expression could be further exacerbated by increased autocrine signaling because IFN-γ stimulates the production of IL-12 by macrophages (Hayes et al., 1995), while efficient production of IFN-γ by these cells in turn is said to occur only in response to combined stimulation with IL-12 and IL-18 (Munder et al., 1998). All three of these cytokines are expressed more highly in Nur77-KO macrophages, suggesting the activation of an autocrine feedback loop that is inhibited in WT macrophages under the conditions tested here. A similar autocrine signaling loop involving extracellular succinate and the succinate receptor GPR91 could also occur, as activation of GPR91 by succinate enhances the inflammatory response of macrophages (Littlewood-Evans et al., 2016). Nur77-KO macrophages secrete more succinate and express higher levels of the GPR91-encoding gene Sucnr1. Because GPR91 senses extracellular succinate but does not internalize it, future research could focus on the relative contribution of intracellular versus extracellular succinate to the macrophage inflammatory response.
In vivo we found that Nur77 deficiency aggravates progression of the chronic inflammatory disease atherosclerosis and leads to increased circulating succinate levels, most likely because of the enhanced succinate and cytokine production of Nur77-KO macrophages. The potential consequences of increased circulating succinate levels should not be underestimated, as activation of the ubiquitously expressed succinate receptor GPR91 has been implicated in a wide variety of inflammation-driven diseases in both mice and humans (Ariza et al., 2012). Circulating succinate can therefore be thought of as a danger signal, the elevation of which could pave the way for a plethora of other inflammation-driven pathologies. Aged Nur77-deficient mice suffer from spontaneous, insult-free systemic inflammation (Li et al., 2015b), and Nur77 deficiency is detrimental in a variety of other inflammation-associated diseases, such as LPS-induced sepsis (Li et al., 2015a), diabetes (Chao et al., 2009), and myocardial infarction (Hilgendorf et al., 2014). Succinate signaling and metabolism have been shown to aggravate the progression of all three of these diseases (Chouchani et al., 2014, van Diepen et al., 2017, Mills et al., 2016), suggesting the involvement of Nur77-based restriction of succinate signaling in these diseases.
In conclusion, our work shows that Nur77 limits inflammation in pro-inflammatory macrophages by regulating TCA cycle remodeling. As a negative regulator of both NF-κB signaling and SDH-driven inflammation in macrophages, Nur77 is proposed as an attractive therapeutic target for the treatment of atherosclerosis and other chronic inflammatory diseases.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-HA tag | Diagenode | Cat# C15200190; RRID: AB_2737029 |
| Rabbit anti-Nur77 | Santa Cruz | Cat# sc-5569; RRID: AB_653373 |
| Mouse anti-α-Tubulin | Cedarlane | Cat# CLT9002; RRID: AB_10060319 |
| Rabbit anti-β-Actin | Cell Signaling Technology | Cat# 4967L; RRID: AB_330288 |
| Rabbit anti-mouse IL-1β | Cell Signaling Technology | Cat# 12507S; RRID: AB_2721117 |
| Rabbit anti-HIF-1α | Novus Biologicals | Cat# NB100-449; RRID: AB_10001045 |
| Rabbit anti-iNOS | Abcam | Cat# ab3523; RRID: AB_303872 |
| Mouse anti-SDHA | Abcam | Cat# ab14715; RRID: AB_301433 |
| Rat anti-mouse MAC-3 | PharMingen | Cat# 553322; RRID: AB_394780 |
| Rabbit anti-CD3 | Labvision | Cat# RM-9107; RRID: AB_149921 |
| Mouse anti-Smooth Muscle Actin (SMA) | DAKO | Cat# M0851; RRID: AB_2223500 |
| Bacterial and Virus Strains | ||
| Lentiviral pINDUCER20b-HA-NUR77 and pINDUCER20b-GFP | This paper | N/A |
| Lentiviral short hairpin (sh)RNA targeting mouse Nur77 (shNur77) or control (shCtrl) | (Bonta et al., 2006) | N/A |
| Lentiviral expression vector for CMV promoter-drive expression of human NUR77 or backbone control | (Bonta et al., 2006) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dynabeads® Protein G for Immunoprecipitation | Novex | Cat# 10003D |
| Di(N-succinimidyl) glutarate | Sigma-Aldrich | Cat# 80424; CAS: 79642-50-5 |
| Lipopolysaccharides (LPS) from Escherichia coli O55:B5 | Sigma-Aldrich | Cat# 62326 |
| Doxycycline hyclate | Sigma-Aldrich | Cat# D9891; CAS: 24390-14-5 |
| Oligomycin A | Sigma-Aldrich | Cat# 75351; CAS: 579-13-5 |
| Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) | Sigma-Aldrich | Cat# C2920; CAS: 370-86-5 |
| Rotenone | Sigma-Aldrich | Cat# R8875; CAS: 83-79-4 |
| Antimycin A | Sigma-Aldrich | Cat# A8674; CAS: 1397-94-0 |
| Digitonin | Sigma-Aldrich | Cat# D141; CAS: 11024-24-1 |
| MitoTracker Green FM | Molecular Probes | Cat# M7514; CAS: 201860-17-5 |
| MitoTracker Red CM-H2Xros | Molecular Probes | Cat# M7513; CAS: 167095-08-1 |
| LysoTracker Red DND-99 | Molecular Probes | Cat# L7528; CAS: 231946-72-8 |
| MitoSOX Red Mitochondrial Superoxide Indicator | Molecular Probes | Cat# M36008 |
| 2′,7′-Dichlorofluorescin diacetate (DCFDA) | Sigma-Aldrich | Cat# D6883; CAS: 4091-99-0 |
| Dimethyl malonate | Sigma-Aldrich | Cat# 136441; CAS: 108-59-8 |
| 3-Nitropropionic acid | Sigma-Aldrich | Cat# N5636; CAS: 504-88-1 |
| Hoechst 33258, Pentahydrate (bis-Benzimide) | Invitrogen | Cat# H3569; CAS: 23491-45-4 |
| Critical Commercial Assays | ||
| TruSeq Stranded mRNA Library Prep Kit | Illumina | Cat# 15031047 |
| Succinate Assay Kit (Colorimetric) | Abcam | Cat# ab204718 |
| Cytometric Bead Array (CBA) Mouse Inflammation Kit | BD Biosciences | Cat# 552364 |
| iScript cDNA synthesis kit | BioRad | Cat# 1708891 |
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat# G7570 |
| Bright-DAB | ImmunoLogic | Cat# BS04-500 |
| In Situ Cell Death Detection Kit, Fluorescein | Roche | Cat# 11684795910 |
| Deposited Data | ||
| Raw and analyzed Nur77 ChIP-seq and RNA-seq data | This paper | GEO: GSE102394 |
| Experimental Models: Cell Lines | ||
| Mouse: RAW264.7 | ATCC | TIB-71 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Nur77-deficient: Tg(CMV-cre)1Cgn/ Nr4a1tm1Pcn | This paper | N/A |
| Mouse: Nur77fl/fl: Nr4a1tm1Pcn | (Sekiya et al., 2013) | MGI: 5546594 |
| Mouse: CMV:cre: B6.C-Tg(CMV-cre)1Cgn/J | Charles River | JAX: 006054 |
| Mouse: Ldlr−/−: B6.129S7-Ldlrtm1Her/J | The Jackson Laboratory | JAX: 002207 |
| Oligonucleotides | ||
| Primers for mtDNA/nDNA quantification, ChIP-qPCR, and RT-qPCR: see Table S3. | This paper | N/A |
| Recombinant DNA | ||
| pINDUCER20b-HA-NUR77 and pINDUCER20b-GFP | This paper; based on (Meerbrey et al., 2011) | N/A |
| p156RRL-sinPPT-CMV-GFP-PRE-shNur77 and p156RRL-sinPPT-CMV-GFP-PRE-shCtrl | (Bonta et al., 2006) | N/A |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism/; RRID: SCR_002798 |
| Burrow-Wheeler Aligner 0.7.10 | (Li and Durbin, 2009) | http://bio-bwa.sourceforge.net/ |
| TopHat 2.1 | (Trapnell et al., 2009) | http://ccb.jhu.edu/software/tophat/index.shtml |
| HTSeq 0.6.1 | (Anders et al., 2015) | https://pypi.python.org/pypi/HTSeq |
| DESeq2 3.5 | (Love et al., 2014) | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| MACS 1.4 | (Zhang et al., 2008) | http://liulab.dfci.harvard.edu/MACS/ |
| DFilter 1.5 | (Kumar et al., 2013) | http://collaborations.gis.a-star.edu.sg/∼cmb6/kumarv1/dfilter/ |
| UCSC Genome Browser | (Kent et al., 2002) | http://genome.ucsc.edu/ |
| seqMINER 1.3.3e | (Ye et al., 2011) | http://bips.u-strasbg.fr/ |
| HOMER 4.9 | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/ |
| BETA 1.0.0 | (Wang et al., 2013) | http://cistrome.org/BETA/ |
| GREAT 3.0.0 | (McLean et al., 2010) | http://bejerano.stanford.edu/great/public/html/index.php |
| Metascape 3.0 | (Tripathi et al., 2015) | http://metascape.org |
| ImageJ 1.50 | NIH | https://imagej.nih.gov/ij/index.html; RRID: SCR_003070 |
| Leica QWin V3 | Leica | N/A |
| LinRegPCR 2015.3 | http://www.hartfaalcentrum.nl/index.php?main=files&fileName=LinRegPCR.zip&description=LinRegPCR:%20qPCR%20data%20analysis&sub=LinRegPCR | |
| CytExpert 2.0 | Beckman Coulter | https://www.beckman.com/coulter-flow-cytometry/software/cytexpert-software |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Carlie J.M. de Vries (c.j.devries@amc.nl).
Experimental Model and Subject Details
Mouse strains
All animal housing, care, and procedures were approved by the institutional Animal Experimental Committee for Animal Welfare in accordance with Directive 2010/63/EU of the European Parliament. Whole-body Nur77 knockout mice were generated by crossing mice overexpressing Cre recombinase under control of the human cytomegalovirus (CMV) promoter with mice containing a loxP-flanked Nur77 (Nr4a1) locus (Sekiya et al., 2013). Nur77 deletion was confirmed by genotyping PCR and mice were subsequently crossed to generate Nur77-deficient mice that no longer express Cre recombinase (Nur77-KO). These Nur77-KO mice and their wild-type (WT) littermates were used for experiments.
Bone marrow transplantation and atherosclerosis progression
Bone marrow cells were isolated from femurs and tibias of male 10-week-old WT or Nur77-KO mice and 1 × 107 cells/mouse were injected intravenously into female 10-week-old γ-irradiated Ldlr−/− recipient mice. After a 6-week recovery period, mice were fed a Western-type diet (Harlan) containing 42% calories from fat for 14 weeks. Mice were euthanized via ketamine/xylazine overdose (238 mg/kg ketamine, 102 mg/kg xylazine i.p. injection) and orbital bleeding. Hearts were excised, cut below the atrial tips, fixed with 4% formalin, embedded in paraffin, and sectioned to 7 μm. Aortic root lesion size and necrotic areas were assessed using hematoxylin and eosin-stained sections. Immunohistochemistry was performed using antibodies directed against HIF-1α, or macrophage (MAC-3), smooth muscle cell (smooth muscle actin; SMA), or T cell (CD3) specific markers and appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies, followed by detection with Bright-DAB. Apoptotic cells were assessed by TUNEL assay using the In Situ Cell Death Detection Kit, Fluorescein according to manufacturer’s instructions. Quantification was performed using ImageJ and Leica QWin V3 software.
Bone marrow-derived macrophage (BMDM) differentiation and cell culture
Bone marrow cells were isolated from femurs and tibias of male or female 6-8 weeks old WT and Nur77-KO mice and differentiated to macrophages by culturing in RPMI medium supplemented with 10% fetal calf serum, 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine, and 15% L929-conditioned medium for 7 days. After differentiation, cells were lifted, counted, and re-seeded for in vitro experiments at 0.6 × 106 BMDMs per mL. In all experiments involving LPS-stimulated BMDMs, a concentration of 100 ng/mL lipopolysaccharide (LPS) was used. In experiments involving concomitant SDH inhibition, BMDMs were pre-treated with 500 μM 3-nitropropionic acid (NPA) for 30 min prior to LPS stimulation.
RAW264.7 macrophage cell culture
The mouse macrophage RAW264.7 cell line was cultured in DMEM medium supplemented with 10% fetal calf serum, 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM L-glutamine. Low passage (< 15 passages) cells were used for all experiments. For experiments involving RAW264.7 stable cell lines with doxycycline (Dox)-inducible overexpression, cells were treated with 1 μg/mL Dox for 18 hr.
Method Details
Lentiviral transductions
Vector construction, transduction, and stable cell line generation for doxycycline-inducible HA-NUR77 expression in RAW264.7 macrophages
The lentiviral vectors pINDUCER20b-GFP and pINDUCER20b-HA-NUR77 encoding Dox-induced overexpression of either eGFP or HA-tagged NUR77 were constructed by cloning the coding-domain sequences of either eGFP or human Nur77 into the pENTR4-HA vector using the restriction enzymes KpnI and BamHI and subsequently recombining these plasmids into the pINDUCER20b destination vector by Gateway LR reaction. pINDUCER20b is a modified version of pINDUCER20 that contains a Blasticidin resistance cassette instead of Neomycin resistance. Lentiviral particles were produced in HEK293T cells by co-transfecting third generation lentiviral packaging vectors and lentiviral vector containing the gene-of-interest. Supernatants were collected over the course of 72 hr and filtered through a 0.45 μm filter. Lentiviral particles were concentrated by ultracentrifugation for 2 hr at 110,000 x g. Low-passage RAW264.7 cells were transduced for 24 hr at a multiplicity of infection (MOI) of 100 in the presence of 2 μg/mL polybrene. Transduced cells were selected for with 10 mg/mL Blasticidin (Sigma) starting 48 hr after transduction. After 5-7 days, resistant polyclonal colonies were further expanded and Dox-inducible expression of GFP or HA-NUR77 was assessed by RT-qPCR, western blotting, and immunofluorescence microscopy.
Lentiviral short hairpin RNA-mediated Nur77 knockdown in RAW264.7 macrophages and NUR77 overexpression in BMDMs
Lentiviral particles containing expression vectors for short hairpin (sh)RNA targeting mouse Nur77 (shNur77), scrambled shRNA control (shCtrl), human NUR77 overexpression, or backbone control (Mock) were previously described (Bonta et al., 2006) and produced in HEK293T cells as detailed above. BMDMs or RAW264.7 macrophages were transduced for 24 hr with lentiviral particles at a MOI of 10 in the presence of 2 μg/mL polybrene. Cells were subsequently cultured for an additional 48 hr after which GFP expression was verified by fluorescence microscopy as a measure of transduction efficiency. Cells were re-seeded for experiments and knockdown of endogenous Nur77 (Nr4a1) or overexpression of human NUR77 (NR4A1) was confirmed using RT-qPCR.
RNA sequencing (RNA-seq)
RAW264.7 stable cell lines with Dox-inducible overexpression of GFP or HA-NUR77 were seeded in triplicate in 10-cm culture dishes at 1 × 106 cells per plate. The next day, cells were stimulated with 1 μg/mL Dox for 18 hr, after which RNA was extracted using TRIreagent (Sigma) according to the manufacturer’s instructions. The RNA pellet was dried and dissolved in an appropriate volume of nuclease-free ultrapure water and quantified using Nanodrop UV-VIS Spectrophotometer (Thermo Fisher). RNA was further purified using the MinElute Cleanup Kit (QIAGEN) according to the manufacturer’s instructions. Quality and quantity of the isolated RNA was assessed using a 2100 Bioanalyzer with a Nano chip (Agilent). Strand-specific libraries were generated using the TruSeq Stranded mRNA Library Prep Kit (Illumina). Polyadenylated RNA from 1000 ng intact total RNA was purified using oligo-dT beads. Following purification, the RNA was fragmented, random primed, and reverse transcribed using SuperScript II Reverse Transcriptase (Invitrogen). Second strand synthesis was performed using Polymerase I and RNase H with replacement of dTTP for dUTP. The generated cDNA fragments were 3′ end adenylated and ligated to Illumina Paired-end sequencing adapters and subsequently amplified by 12 cycles of PCR. The libraries were analyzed on a 2100 Bioanalyzer using a 7500 chip (Agilent), diluted, and pooled in equimolar ratio into a 13-plex, 10 nM sequencing pool. Libraries were sequenced on an Illumina HiSeq 2500 using V4 chemistry in single-end 65 bp reads.
Chromatin immunoprecipitation sequencing (ChIP-seq)
RAW264.7 stable cell line with Dox-inducible overexpression of HA-NUR77 was seeded in eighteen 15-cm culture dishes at 6 × 106 cells per plate. The next day, cells were stimulated with 1 μg/mL Dox for 18 hr, after which cells were washed with PBS and dual crosslinked directly in culture plates with 2 mM Di(N-succinimidyl) glutarate (Sigma) in PBS for 45 min at room temperature, followed by 1% formaldehyde (Sigma) for 15 min at room temperature. Crosslinking reactions were quenched by addition of glycine to 0.125 M final concentration and incubation for 5 min at room temperature. Crosslinked cells were washed with ice-cold PBS and collected by scraping in ice-cold PBS followed by centrifugation for 5 min at 500 x g. Cell pellets were resuspended in buffer L1A (10 mM HEPES-KOH pH 7.9, 85 mM KCl, 1 mM EDTA). Cell membranes were lysed by diluting samples 1:1 with buffer L1B (L1A plus 1% IGEPAL CA-630) and incubating 10 min on ice. Cells were subsequently centrifuged 5 min at 700 x g and pellets were resuspended in buffer L2 (50 mM Tris-HCl pH 7.4, 1% SDS, 10 mM EDTA). Nuclei were ruptured and chromatin was sonicated into approximately 200 bp fragments using a Bioruptor Pico (Diagenode) for 20 cycles of 30 s on and 30 s off. Fragment length was confirmed by running an aliquot of reverse crosslinked chromatin on a 1.2% agarose gel. Sonicated chromatin samples were centrifuged for 10 min at 16,000 x g to pellet cell debris. Supernatants were transferred to DNA LoBind tubes (Sigma) and diluted 1:1.5 with dilution buffer (20 mM Tris-HCl pH 7.4, 100 mM NaCl, 2 mM EDTA, 0.5% Triton X-100). Per ChIP reaction, 30 μL Protein G Dynabeads (Novex) were washed twice with 0.5% bovine serum albumin (BSA) in PBS and incubated with 2 μg anti-HA antibody (Diagenode) for 3 hr at 4°C. Dynabead-antibody complexes were added to each diluted chromatin sample (18 ChIPs total) and incubated overnight at 4°C to immunoprecipitate chromatin-linked HA-NUR77 protein. Immunoprecipitates were washed four times with wash buffer 1 (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 2 mM EDTA), four times with wash buffer 2 (10 mM Tris-HCl pH 7.4, 250 mM LiCl, 1% IGEPAL CA-630, 0.7% sodium deoxycholate, 1 mM EDTA), twice with wash buffer 3 (10 mM Tris-HCl pH 7.4, 0.2% Triton X-100, 1 mM EDTA), and twice with wash buffer 4 (10 mM Tris-HCl pH 7.4, 50 mM NaCl, 1 mM EDTA). Samples were transferred to fresh DNA LoBind tubes after the first and second to last wash steps to reduce background. Chromatin was eluted from Dynabeads by incubating in elution buffer (10 mM Tris-HCl pH 7.4, 2% SDS, 1 mM EDTA) for 20 min at 37°C. Crosslinks were reversed by addition of NaCl to a final concentration of 300 mM and incubating overnight at 65°C. DNA was purified by incubating samples with 0.33 mg/mL RNase A for 1 hr at 37°C, followed by 0.5 mg/mL Proteinase K for 1 hr at 50°C, and extraction of DNA by phenol:chloroform:isoamylalcohol precipitation. Precipitated DNA was dissolved in ultrapure water and DNA from 6 separate ChIP reactions was pooled to create one biological replicate, resulting in three biological replicates total. ChIP DNA samples were then sequenced on an Illumina HiSeq 2500 in single-end 50 bp reads.
Quantitative PCR (qPCR)
Reverse transcription (RT)-qPCR
Total RNA from cultured cells was isolated using TRIreagent (Sigma). Reverse transcription (RT-)PCR was performed on 1 μg RNA using iScript cDNA Synthesis Kit (BioRad). qPCR was carried out using SensiFAST SYBR No-ROX Kit (Bioline) on a LightCycler 480 II PCR platform (Roche). Quantification cycles (Cq), primer set amplification efficiencies, and transcript starting concentrations (N0) were calculated using LinRegPCR. Target gene expression was normalized by dividing by the geometric mean of Actb and Rplp0 housekeeping gene expression. Sequences of the primers used for RT-qPCR can be found in Table S3.
ChIP-qPCR
The RAW264.7 stable cell line with Dox-inducible overexpression of HA-NUR77 was seeded in quadruplicate in 15-cm culture dishes at 6 × 106 cells per plate. The next day, cells were stimulated with 1 μg/mL Dox for 18 hr and chromatin was crosslinked, extracted, and immunoprecipitated using anti-HA antibody as described above. ChIP and 1% input DNA samples were quantified by qPCR as described above. Percentage of input was calculated using the following formula: primer efficiencyCq(input-6.644) – Cq(ChIP). ChIP-qPCR results are shown as fold change in percentage of input for target regions over the mean of two non-target regions. Sequences of the primers used for ChIP-qPCR can be found in Table S3.
SDS-PAGE and western blotting
Cultured cells were washed with PBS and lysed in RIPA Lysis and Extraction Buffer (Thermo Fisher) or nuclear and cytosolic fractions were made using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher). Protein samples were heated for 5 min at 96°C and resolved on 10% SDS-PAGE gels and transferred onto nitrocellulose membranes using the semi-dry Trans-Blot Turbo Transfer System (BioRad). Membranes were blocked for 1 hr at room temperature with Tris-buffered saline containing 0.05% Tween-20 and 5% (w/v) dried milk. Membranes were incubated overnight at 4°C with primary antibody, followed by the appropriate HRP-conjugated secondary antibody for 1 hr at room temperature. After washing, blots were developed using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher) and visualized on the ImageQuant LAS 4000 (GE Healthcare).
Immunofluorescence microscopy
RAW264.7 stable cell lines were cultured on uncoated glass coverslips, stimulated with 1 μg/mL Dox for 24 hr, and fixed with 4% (w/v) paraformaldehyde in PBS. Cells were subsequently permeabilized with 0.5% (v/v) Triton X-100 and HA-NUR77 expression was detected using rabbit anti-Nur77 (Santa Cruz) and Alexa Fluor 488-conjugated goat anti-rabbit (Molecular Probes) antibodies. Nuclei were visualized using Hoechst 33258 (Invitrogen).
Oxygen consumption and lactate production (Seahorse) assays
Oxygen consumption and lactate production were simultaneously measured in a Seahorse XF96 Analyzer (Agilent) using an established protocol (Van den Bossche et al., 2015). BMDMs were seeded at 0.5 × 105 cells/well in XF96-well plates (Agilent) and left unstimulated or stimulated with 100 ng/mL LPS for 24 h. Wells without cells were included as a background control. Sensor plates were calibrated overnight in a CO2-free incubator at 37°C using 200 μL/well XF calibrant solution (Agilent). After LPS stimulation, culture medium was replaced with glucose-free DMEM medium supplemented with 2 mM L-glutamine and cells were incubated 1 hr in a CO2-free incubator at 37°C. Injection ports were loaded with 10x injection mixes to obtain a final concentration in each well of 25 mM glucose after the first injection, 1.5 μM oligomycin after the second injection, 1 mM sodium pyruvate plus 1.5 μM FCCP after the third injection, and 1.25 μM rotenone plus 2.5 μM antimycin A after the fourth injection. After the run, supernatants were carefully aspirated and protein was isolated using RIPA Lysis and Extraction Buffer (Thermo Fisher). OCR and ECAR values were normalized to total protein content of each well as determined by DC Protein Assay (BioRad). Parameters of OXPHOS and glycolysis were calculated from OCR and ECAR values as shown in Figures S2C and S2E.
Determination of ATP levels
BMDMs were seeded at 0.6 × 106 cells/mL in 96-well plates and left unstimulated or stimulated with 100 ng/mL LPS for times indicated. After LPS stimulation, cells were treated with 5 μM oligomycin and total ATP levels were immediately measured using the CellTiter-Glo Luminescent Cell Viability Assay (Promega). Luminescence values were recorded using a GloMax Multi Microplate Multimode Reader (Promega).
Determination of mitochondrial and lysosomal abundance and co-localization
Mitochondrial DNA
BMDMs were seeded at 0.6 × 106 cells/mL in 12-well plates. The next day, total DNA was extracted from BMDMs using a DNeasy Blood & Tissue Kit (QIAGEN) and 25 ng of DNA was used for qPCR as described above using primers for mitochondrial DNA or nuclear DNA as an internal control. Primer sequences are shown in Table S3.
Flow cytometry
BMDMs were seeded at 0.6 × 106 cells/mL in non-tissue culture treated 48-well plates. The next day, culture medium was removed and cells were stained with 100 nM MitoTracker Green FM in HBSS/+Ca/+Mg for 30 min at 37°C and 5% CO2 in the dark or left unstained as a control. Cells were subsequently washed with PBS, lifted using ice-cold PBS containing 5 mM EDTA, and transferred to 96-well FACS plates. Fluorescence was measured using a Beckman Coulter CytoFLEX flow cytometer and data were analyzed using CytExpert software.
Wide-field microscopy
BMDMs were seeded at 0.5 × 105 cells/well in uncoated Nunc Lab-Tek chambered 1.0 borosilicated coverglass plates (Thermo Scientific). The next day, cells were stained with 100 nM MitoTracker Green FM, 75 nM LysoTracker Red DND-99, and/or 2 μg/mL Hoechst 33258 in HBSS/+Ca/+Mg for 30 min at 37°C to visualize mitochondria, lysosomes, and nuclei, respectively. Cells were subsequently washed with complete medium and directly imaged using a widefield microscope (NIKON Eclipse TI) equipped with a 60x 1.49 NA Apo TIRF (oil) objective and an Andor Zyla 4.2 plus sCMOS camera. An Okolab cage incubator and humidified CO2 gas chamber set to 37°C and 5% CO2 were used during the imaging process. For quantification of mitochondrial and lysosomal abundance, the mean fluorescence intensity (MFI) of MitoTracker Green FM or LysoTracker Red DND-99 fluorphores was determined in regions of interest demarcating individual cells in the original recorded data using ImageJ. Calculation of the Pearson correlation coefficient (Pearson’s r) for co-localization of MitoTracker Green FM and LysoTracker Red DND-99 signal was performed using ImageJ with the “Colocalization threshold” plugin. For display purposes, all images were similarly enhanced using the unsharp mask filter and window/level settings in ImageJ.
Determination of intracellular metabolite abundance by LC-MS (metabolomics)
BMDMs were seeded at 0.6 × 106 cells/mL in 6-well plates and left unstimulated or stimulated with 100 ng/mL LPS for 24 h. Cells were washed three times with ice-cold 0.9% NaCl and metabolism was quenched by adding 250 μL ice-cold methanol followed by 250 μL ice-cold ultrapure water. Cells were collected by scraping and each well was rinsed with an additional 250 μL ice-cold methanol followed by 250 μL ice-cold ultrapure water. The entire homogenate (1 mL total volume) was pooled and transferred to a 2 mL tube and 5 μM of internal standards were added to each sample: D3-aspartic acid, D3-serine, D5-glutamine, D3-glutamate, 13C3-pyruvate, 13C6-isoleucine, 13C6-glucose, 13C6-fructose-1,6-biphosphate, 13C6-glucose-6-phosphate, adenosine-15N5-monophosphate and guanosine-15N5-monophosphate. 1 mL of chloroform was added to each sample, followed by thorough mixing and centrifugation for 10 min at 14.000 rpm at 4°C. The “polar” top layer was transferred to a new 1.5 mL tube and dried in a vacuum concentrator. Dried samples were dissolved in 100 μL methanol/water (6/4; v/v). Metabolite abundance was measured using an ultra-high-pressure liquid chromatography system (Thermo Scientific) coupled to a Thermo Q Exactive (Plus) Orbitrap mass spectrometer (Thermo Scientific). The autosampler was held at 10°C during the runs and 5 μL of sample was injected on the analytical column. The chromatographic separation was established using a SeQuant ZIC-cHILIC column (PEEK 100 × 2.1mm, 3.0μm particle size; Merck) and kept at 15°C. The flow rate was set to 0.250 mL/min. The mobile phase was composed of (A) 9/1 acetonitrile/water with 5 mM ammonium acetate; pH 6.8 and (B) 1/9 acetonitrile/water with 5 mM ammonium acetate; pH 6.8, respectively. The LC gradient program was as follows: beginning with 100% (A) hold 0-3 min; ramping 3-20 min to 36% (A); ramping from 20-24 min to 20% (A); hold from 24-27 min at 20% (A); ramping from 27-28 min to 100% (A); and re-equilibrate from 28-35 min with 100% (A). The MS data were acquired in negative mode at full scan range at 140.000 resolution. Data were analyzed using Xcalibur software (Thermo Scientific) and normalized to total protein content in each well as determined by DC Protein Assay (BioRad).
SDH activity assays
SDH-mediated respiration
SDH-mediated respiration was measured in plasma membrane-permeabilized BMDMs using an established protocol (Salabei et al., 2014). BMDMs were seeded at 0.5 × 105 cells/well in XF96-well plates (Agilent) and left unstimulated or stimulated with 100 ng/mL LPS for 24 h. Oxygen consumption was measured using a Seahorse XF96 Analyzer (Agilent) as described above with the following modifications: after LPS stimulation, culture medium was replaced with MAS buffer (220 mM mannitol, 70 mM sucrose, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA, pH 7.4) and injection ports were loaded with 10x injection mixes to obtain a final concentration in each well of 10 mM succinate, 1 mM ADP, 30 μg/mL digitonin, and 1.25 μM rotenone after the first injection, 1.5 μM oligomycin after the second injection, and 2.5 μM antimycin A after the third injection. After the run, supernatants were carefully aspirated and protein was isolated using RIPA Lysis and Extraction Buffer (Thermo Fisher). OCR values were normalized to total protein content of each well as determined by DC Protein Assay (BioRad).
MTT assay
BMDMs were seeded at 0.6 × 106 cells/mL in 96-well plates and left unstimulated or stimulated with 100 ng/mL LPS for times indicated. Succinate dehydrogenase activity was assessed by yellow 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction to purple formazan. Cells were incubated with 1 mg/mL MTT (Sigma) for 1 hr at 37°C and 5% CO2 and subsequently lysed with 4.4 mM HCl and 0.1% IGEPAL CA-630 in isopropanol. Absorbance of soluble formazan was measured spectrophotometrically at 590 nm and background signal measured at 650 nm was subtracted from this value for each sample. Cells treated 24 hr with 10 mM of the SDH inhibitor dimethyl malonate (DMM) were included as a negative control.
Determination of mitochondrial membrane potential and reactive oxygen species (ROS) production by flow cytometry
BMDMs were seeded at 0.6 × 106 cells/mL in non-tissue culture treated 48-well plates and left unstimulated or stimulated with 100 ng/mL LPS for times indicated. After LPS stimulation, culture medium was removed and cells were stained with 200 nM MitoTracker Red CM-H2Xros plus 100 nM MitoTracker Green FM in RPMI medium (for mitochondrial membrane potential experiments), 5 μM MitoSOX in HBSS/+Ca/+Mg (for mitochondrial ROS), or 10 μM DCFDA in HBSS/+Ca/+Mg (for total ROS) for 30 min at 37°C and 5% CO2 in the dark. Unstained and single stained wells were included as a control. After staining, cells were washed with PBS, lifted using ice-cold PBS containing 5 mM EDTA, and transferred to 96-well FACS plates. Fluorescence was measured using a Beckman Coulter CytoFLEX flow cytometer and data were analyzed using CytExpert software.
Determination of nitric oxide (NO), cytokine, and succinate secretion
BMDMs were seeded at 0.6 × 106 cells/mL in 12-well plates and left unstimulated or stimulated with 100 ng/mL LPS for times indicated. Where indicated, cells were pre-treated with 500 μM of the SDH inhibitor NPA for 30 min or cultured in glutamine-free RPMI medium for at least 3 h prior to LPS stimulation. After LPS stimulation, culture medium was harvested and used for subsequent experiments. Nitrite levels, a measure of NO production, were determined using the Griess Reagent System (Promega). Cytokine levels in cell culture supernatants and mouse blood plasma samples were determined using the Cytometric Bead Array (CBA) Mouse Inflammation Kit. Succinate levels were determined using the Colorimetric Succinate Assay Kit (Abcam), with extracellular succinate being measured in culture medium and intracellular succinate being measured in Succinate Assay Buffer cell lysates according to the manufacturer’s instructions. For all assays, a standard curve was created to determine absolute quantities of nitrite, cytokines, or succinate.
Quantification and Statistical Analysis
Statistical analysis
Statistical analyses were performed using GraphPad Prism software. Data are reported as mean ± SEM. Number of replicates (n) and statistical tests used are described in the figure legends, with a p value < 0.05 being considered significant and levels of significance being indicated as follows: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. In general, a two-tailed unpaired Student’s t test was used when comparing two groups, and a two-way ANOVA was used when comparing interactions between genotype (WT or Nur77-KO) and stimulus (untreated or LPS-stimulated). For in vitro experiments, n = number of biological replicates; for in vivo experiments, n = number of animals. Normal distribution of data was confirmed using the D’Agostino & Pearson omnibus normality test.
Analysis of RNA-seq data
Raw sequencing reads were filtered for low complexity or repetitive sequences and aligned to the Mus musculus genome (assembly mm10, Dec. 2011) using TopHat (Trapnell et al., 2009). The number of uniquely mapping read counts per gene was determined using HTSeq (Anders et al., 2015). Differential expression analysis between GFP and HA-NUR77 overexpressing cells was performed using DESeq2 (Love et al., 2014), wherein genes with a false discovery rate (FDR)-adjusted p value < 0.05 and a log2(fold-change) < −0.3 or > 0.3 were considered to be differentially regulated. DESeq2 output can be found in Table S1. Pathway analysis on differentially regulated genes was performed using Metascape (Tripathi et al., 2015).
Analysis of ChIP-seq data
Raw sequencing reads were aligned to the Mus musculus genome (assembly mm9, July 2007) using Burrow-Wheeler Aligner (Li and Durbin, 2009). Reads that aligned poorly or mapped to multiple locations were discarded and only reads with a mapping quality score (MAPQ) > 20 were used for peak calling. RAW264.7 input sequencing data were obtained from the National Center for Biotechnology Information (NCBI) Gene Expression omnibus (GEO) database (GEO: GSM1232949, GSM1559473). High-confidence Nur77 binding sites were identified by comparing aligned input and Nur77 ChIP-seq data using two peak calling algorithms, MACS (Zhang et al., 2008) and DFilter (Kumar et al., 2013). For MACS, default settings were used except for a p value cut-off of 10−7. For DFilter, the settings bs = 50, ks = 30, refine, and nonzero were used with a p value cut-off of 10−6. Only those binding sites identified by both peak callers and present in at least two of the three biological replicates were used for further analyses.
A heatmap visualizing the raw ChIP-seq signal around Nur77 binding sites was created using seqMINER (Ye et al., 2011). H3K4me1 ChIP-seq datasets used for this analysis were obtained from the GEO database (GEO: GSE29218). Determination of genomic distribution of Nur77 binding sites and pathway analysis were performed using GREAT (McLean et al., 2010). De novo motif analysis on sequences underlying Nur77 binding sites was performed using HOMER (Heinz et al., 2010). For integration of RNA-seq and ChIP-seq datasets, genomic coordinates in the ChIP-seq dataset were converted to Mus musculus assembly mm10 using the UCSC Genome Browser liftOver tool (Kent et al., 2002). Prediction of direct Nur77 target genes based on both datasets was performed using BETA (Wang et al., 2013).
Data and Software Availability
Data resources
The accession number for the raw and processed ChIP-seq and RNA-seq data reported in this paper is GEO: GSE102394.
Acknowledgments
We thank Jessica Nelson and Marten Hoeksema for discussion and technical assistance. This research was supported by a grant from the Netherlands CardioVascular Research Initiative (CVON2011-19 “GENIUS”). N.Z. is supported by a European Research Council Consolidator grant (617376). E.K. is supported by a Dutch Diabetes Foundation grant (2009.60.003). J.V.d.B. is supported by a ZonMW (the Netherlands Organisation for Health Research and Development) VENI grant (91615052) and Netherlands Heart Foundation Junior Postdoc (2013T003) and Netherlands Heart Foundation Senior Postdoc (2017T048) grants. S.H. is supported by a ZonMW VIDI grant (016.156.327).
Author Contributions
D.S.K., E.K., and C.J.d.V. conceived the study and experiments. D.S.K., I.J.E.-v.G., L.M., P.B.v.L., M.v.W., M.V., and S.H. performed the experiments. D.S.K, Y.K., L.W., and W.Z. performed and analyzed the high throughput sequencing (HTSeq) experiments. D.S.K. and C.J.d.V. wrote the manuscript. J.V.d.B., D.S., N.Z., W.Z., and E.K. provided expertise and feedback.
Declaration of Interests
The authors declare no competing interests.
Published: August 21, 2018
Footnotes
Supplemental Information includes seven figures, three tables, and one video and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.07.065.
Supplemental Information
References
- Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariza A.C., Deen P.M.T., Robben J.H. The succinate receptor as a novel therapeutic target for oxidative and metabolic stress-related conditions. Front. Endocrinol. (Lausanne) 2012;3:22. doi: 10.3389/fendo.2012.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonta P.I., van Tiel C.M., Vos M., Pols T.W.H., van Thienen J.V., Ferreira V., Arkenbout E.K., Seppen J., Spek C.A., van der Poll T. Nuclear receptors Nur77, Nurr1, and NOR-1 expressed in atherosclerotic lesion macrophages reduce lipid loading and inflammatory responses. Arterioscler. Thromb. Vasc. Biol. 2006;26:2288–2294. doi: 10.1161/01.ATV.0000238346.84458.5d. [DOI] [PubMed] [Google Scholar]
- Chao L.C., Wroblewski K., Zhang Z., Pei L., Vergnes L., Ilkayeva O.R., Ding S.Y., Reue K., Watt M.J., Newgard C.B. Insulin resistance and altered systemic glucose metabolism in mice lacking Nur77. Diabetes. 2009;58:2788–2796. doi: 10.2337/db09-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao L.C., Soto E., Hong C., Ito A., Pei L., Chawla A., Conneely O.M., Tangirala R.K., Evans R.M., Tontonoz P. Bone marrow NR4A expression is not a dominant factor in the development of atherosclerosis or macrophage polarization in mice. J. Lipid Res. 2013;54:806–815. doi: 10.1194/jlr.M034157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani E.T., Pell V.R., Gaude E., Aksentijević D., Sundier S.Y., Robb E.L., Logan A., Nadtochiy S.M., Ord E.N.J., Smith A.C. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolman N.J., Chambers K.M., Mandavilli B., Batchelor R.H., Janes M.S. Tools and techniques to measure mitophagy using fluorescence microscopy. Autophagy. 2013;9:1653–1662. doi: 10.4161/auto.24001. [DOI] [PubMed] [Google Scholar]
- Gubern G., Imperial S., Busquets M., Cortés A. Partial characterization of the alanine aminotransferase isoenzymes from human liver. Biochem. Soc. Trans. 1990;18:1288–1289. doi: 10.1042/bst0181288. [DOI] [PubMed] [Google Scholar]
- Hamers A.A.J., Vos M., Rassam F., Marinković G., Kurakula K., van Gorp P.J., de Winther M.P.J., Gijbels M.J.J., de Waard V., de Vries C.J.M. Bone marrow-specific deficiency of nuclear receptor Nur77 enhances atherosclerosis. Circ. Res. 2012;110:428–438. doi: 10.1161/CIRCRESAHA.111.260760. [DOI] [PubMed] [Google Scholar]
- Hanna R.N., Shaked I., Hubbeling H.G., Punt J.A., Wu R., Herrley E., Zaugg C., Pei H., Geissmann F., Ley K., Hedrick C.C. NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ. Res. 2012;110:416–427. doi: 10.1161/CIRCRESAHA.111.253377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes M.P., Wang J., Norcross M.A. Regulation of interleukin-12 expression in human monocytes: selective priming by interferon-gamma of lipopolysaccharide-inducible p35 and p40 genes. Blood. 1995;86:646–650. [PubMed] [Google Scholar]
- Hedrick E., Lee S.O., Kim G., Abdelrahim M., Jin U.H., Safe S., Abudayyeh A. Nuclear receptor 4A1 (NR4A1) as a drug target for renal cell adenocarcinoma. PLoS One. 2015;10:e0128308. doi: 10.1371/journal.pone.0128308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgendorf I., Gerhardt L.M.S., Tan T.C., Winter C., Holderried T.A.W., Chousterman B.G., Iwamoto Y., Liao R., Zirlik A., Scherer-Crosbie M. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ. Res. 2014;114:1611–1622. doi: 10.1161/CIRCRESAHA.114.303204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.W., Zhang P., Yang J.Y., Huang J.L., Ma X., Li S.F., Zhao J.Y., Hu Y.R., Wang Y.C., Gao J.J. Nur77 decreases atherosclerosis progression in apoE(-/-) mice fed a high-fat/high-cholesterol diet. PLoS One. 2014;9:e87313. doi: 10.1371/journal.pone.0087313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha A.K., Huang S.C.C., Sergushichev A., Lampropoulou V., Ivanova Y., Loginicheva E., Chmielewski K., Stewart K.M., Ashall J., Everts B. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42:419–430. doi: 10.1016/j.immuni.2015.02.005. [DOI] [PubMed] [Google Scholar]
- Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V., Muratani M., Rayan N.A., Kraus P., Lufkin T., Ng H.H., Prabhakar S. Uniform, optimal signal processing of mapped deep-sequencing data. Nat. Biotechnol. 2013;31:615–622. doi: 10.1038/nbt.2596. [DOI] [PubMed] [Google Scholar]
- Kurakula K., Koenis D.S., van Tiel C.M., de Vries C.J.M. NR4A nuclear receptors are orphans but not lonesome. Biochim. Biophys. Acta. 2014;1843:2543–2555. doi: 10.1016/j.bbamcr.2014.06.010. [DOI] [PubMed] [Google Scholar]
- Lampropoulou V., Sergushichev A., Bambouskova M., Nair S., Vincent E.E., Loginicheva E., Cervantes-Barragan L., Ma X., Huang S.C.C., Griss T. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 2016;24:158–166. doi: 10.1016/j.cmet.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.O., Jin U.H., Kang J.H., Kim S.B., Guthrie A.S., Sreevalsan S., Lee J.S., Safe S. The orphan nuclear receptor NR4A1 (Nur77) regulates oxidative and endoplasmic reticulum stress in pancreatic cancer cells. Mol. Cancer Res. 2014;12:527–538. doi: 10.1158/1541-7786.MCR-13-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Liu Y., Chen H.Z., Li F.W., Wu J.F., Zhang H.K., He J.P., Xing Y.Z., Chen Y., Wang W.J. Impeding the interaction between Nur77 and p38 reduces LPS-induced inflammation. Nat. Chem. Biol. 2015;11:339–346. doi: 10.1038/nchembio.1788. [DOI] [PubMed] [Google Scholar]
- Li X.-M., Lu X.-X., Xu Q., Wang J.-R., Zhang S., Guo P.-D., Li J.-M., Wu H. Nur77 deficiency leads to systemic inflammation in elderly mice. J. Inflamm. (Lond.) 2015;12:40. doi: 10.1186/s12950-015-0085-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlewood-Evans A., Sarret S., Apfel V., Loesle P., Dawson J., Zhang J., Muller A., Tigani B., Kneuer R., Patel S. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J. Exp. Med. 2016;213:1655–1662. doi: 10.1084/jem.20160061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.-S., Wang H., Li X., Chao T., Teav T., Christen S., Di Conza G., Cheng W.-C., Chou C.-H., Vavakova M. α-Ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017;18:985–994. doi: 10.1038/ni.3796. [DOI] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerbrey K.L., Hu G., Kessler J.D., Roarty K., Li M.Z., Fang J.E., Herschkowitz J.I., Burrows A.E., Ciccia A., Sun T. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc. Natl. Acad. Sci. USA. 2011;108:3665–3670. doi: 10.1073/pnas.1019736108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills E.L., Kelly B., Logan A., Costa A.S.H., Varma M., Bryant C.E., Tourlomousis P., Däbritz J.H.M., Gottlieb E., Latorre I. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. 2016;167:457–470.e13. doi: 10.1016/j.cell.2016.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore K.J., Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munder M., Mallo M., Eichmann K., Modolell M. Murine macrophages secrete interferon gamma upon combined stimulation with interleukin (IL)-12 and IL-18: a novel pathway of autocrine macrophage activation. J. Exp. Med. 1998;187:2103–2108. doi: 10.1084/jem.187.12.2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham W.M., Clish C.B., Yang Y., Loscalzo J. Hypoxia-mediated increases in L-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metab. 2015;22:291–303. doi: 10.1016/j.cmet.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei L., Castrillo A., Chen M., Hoffmann A., Tontonoz P. Induction of NR4A orphan nuclear receptor expression in macrophages in response to inflammatory stimuli. J. Biol. Chem. 2005;280:29256–29262. doi: 10.1074/jbc.M502606200. [DOI] [PubMed] [Google Scholar]
- Ridker P.M., Everett B.M., Thuren T., MacFadyen J.G., Chang W.H., Ballantyne C., Fonseca F., Nicolau J., Koenig W., Anker S.D., CANTOS Trial Group Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- Ryan D.G., O’Neill L.A.J. Krebs cycle rewired for macrophage and dendritic cell effector functions. FEBS Lett. 2017;591:2992–3006. doi: 10.1002/1873-3468.12744. [DOI] [PubMed] [Google Scholar]
- Salabei J.K., Gibb A.A., Hill B.G. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat. Protoc. 2014;9:421–438. doi: 10.1038/nprot.2014.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya T., Kashiwagi I., Yoshida R., Fukaya T., Morita R., Kimura A., Ichinose H., Metzger D., Chambon P., Yoshimura A. Nr4a receptors are essential for thymic regulatory T cell development and immune homeostasis. Nat. Immunol. 2013;14:230–237. doi: 10.1038/ni.2520. [DOI] [PubMed] [Google Scholar]
- Serhan C.N., Savill J. Resolution of inflammation: the beginning programs the end. Nat. Immunol. 2005;6:1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi S., Pohl M.O., Zhou Y., Rodriguez-Frandsen A., Wang G., Stein D.A., Moulton H.M., DeJesus P., Che J., Mulder L.C.F. Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe. 2015;18:723–735. doi: 10.1016/j.chom.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bossche J., Baardman J., de Winther M.P.J. Metabolic characterization of polarized M1 and M2 bone marrow-derived macrophages using real-time extracellular flux analysis. J. Vis. Exp. 2015 doi: 10.3791/53424. Published online November 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bossche J., Baardman J., Otto N.A., van der Velden S., Neele A.E., van den Berg S.M., Luque-Martin R., Chen H.J., Boshuizen M.C.S., Ahmed M. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. 2016;17:684–696. doi: 10.1016/j.celrep.2016.09.008. [DOI] [PubMed] [Google Scholar]
- van Diepen J.A., Robben J.H., Hooiveld G.J., Carmone C., Alsady M., Boutens L., Bekkenkamp-Grovenstein M., Hijmans A., Engelke U.F.H., Wevers R.A. SUCNR1-mediated chemotaxis of macrophages aggravates obesity-induced inflammation and diabetes. Diabetologia. 2017;60:1304–1313. doi: 10.1007/s00125-017-4261-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Sun H., Ma J., Zang C., Wang C., Wang J., Tang Q., Meyer C.A., Zhang Y., Liu X.S. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat. Protoc. 2013;8:2502–2515. doi: 10.1038/nprot.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye T., Krebs A.R., Choukrallah M.-A., Keime C., Plewniak F., Davidson I., Tora L. seqMINER: an integrated ChIP-seq data interpretation platform. Nucleic Acids Res. 2011;39:e35. doi: 10.1093/nar/gkq1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cells were co-stained with MitoTracker Green FM (green) and LysoTracker Red DND-99 (red) and imaged over a 7-minute period. Exposure was increased for Nur77-KO panel to better visualize mitochondrial-lysosomal dynamics. Arrows indicate MitoTracker Green FM/LysoTracker Red DND-99 co-localization events.