

Abstract
In recent decades, nanoparticles have shown significant promise as an oncology treatment modality. Responsive polymers represent a promising class of nanoparticles that can trigger delivery through the exploitation of a specific stimuli. Response to a stimulus is one of the most basic processes found in living systems. As such, the desire to engineer dynamic and functional materials is becoming more prevalent in an effort to achieve precise control over our environment. The combination of controlled radical polymerization and high yielding chemistry strategies provide an excellent basis for the development of the next generation of drug delivery systems. The versatility of polymer chemistries available enables the synthesis of increasingly complex architectures with enhanced delivery specificity and control over the desired properties to interface with biological systems. This tutorial review highlights recent developments in polymer-based approaches to internally responsive nanoparticles for oncology. Presented are concise overviews of the current challenges and opportunities in cancer nanomedicine, common polymer-based architectures, and the basis for internally triggered stimuli–response relationships commonly employed in oncology applications. Examples of the chemistry used in the design of environmentally labile nanomaterials are discussed, and we outline recent advances in creating advanced bioresponsive drug delivery architectures.
Keywords: biomaterials, biomedical applications, drug delivery systems, nanostructured polymers, stimuli-sensitive polymers
INTRODUCTION
The field of structured, advanced biomaterials has seen remarkable progress in the past decade. This progress has yielded novel materials to address problems in a number of applications including controlled drug delivery, biosensors, diagnostics, and regenerative tissue engineering.1–9 Research in cancer cell and molecular biology has also brought about significant advances in our fundamental understanding of tumor complexity, heterogeneity, and the biological barriers to treatment.10–16 Combining this understanding along with the development of new fabrication methods, polymer-based nanoparticles have emerged as a promising oncology treatment modality.
Advances in controlled radical polymerization have allowed for the design and synthesis of polymeric materials with unique and complicated architectures. Recently, the introduction of bioresponsive moieties into polymeric materials has been of particular interest because of the ability of the materials to respond to physiological changes in the body. The responsive properties serve to improve drug delivery properties, such as cellular uptake, drug release, or clearance.17,18 These materials can trigger delivery through a bond cleavage or conformational change in the presence of a specific stimuli, such as pH, enzyme activity, and redox-potential. The specific response of the polymer network may be controlled by incorporation of a variety of functional chemistries in chain side groups, branches, and cross-links. Increasingly complex architectures of these bioresponsive polymers show the potential to enhance delivery specificity.
This review highlights recent developments in polymer-based approaches to bioresponsive nanoparticles for oncology. Presented are concise overviews of the current challenges and opportunities in cancer nanomedicine, common polymer-based architectures, and the basis for internally triggered stimuli–response relationships commonly employed in designing systemically administer bioresponsive systems for oncology applications. Examples of the chemistry used in the design of environmentally labile nanomaterials are discussed. Finally, we outline recent advances in created advanced responsive drug delivery architectures. The scope is intended to cover systemically administered nanomaterials, and is not intended to include inorganic materials and externally-driven stimuli. For more comprehensive reviews on topics not included, the readers are pointed to recent work in literature.8,9,17,18
NANOMEDICINE IN CANCER THERAPY
The Centers for Disease Control and Prevention estimates that 14 million people are diagnosed with cancer globally each year and 8 million die from the disease.19 Current treatment plans for cancer include chemotherapy, radiation, surgical removal of the tumors, or, most often, a combination of these methods. However, traditional chemotherapies do not distinguish between healthy and cancerous cells and result in nonspecific biodistribution. Due to this, only a tiny fraction of the drug dosed reaches and kills cancer cells, meaning patients need to be given very high levels of the drug to be effective.
The high doses required and lack of specificity typically lead to substantial and debilitating side effects to healthy tissues. These dose-limiting toxicities require patients to wait for long periods for recovery between treatments. During this time, cancerous cells that have been exposed to the drug, but not killed, also have an opportunity to recover from the treatment and develop resistance. This resistance is often due to overexpression of efflux transporters such as plasma membrane P-glycoprotein (P-gp), where P-gp mediated resistance is capable of removing cytotoxic chemotherapeutics from cells before they are lethal.20
In recent years, nanoparticles have shown some promise for enhancing the systemic delivery of chemotherapeutic agents and solving some of the limitations that conventional administration presents. There has been growing interest in the field due to the potential for altering drug pharmacokinetics and biodistribution, where nanoparticle formulations have been shown to increase drug concentrations around a tumor by 100%–400% over systemic free drug administration.10,16,21
Ultimately, localizing and controlling drug release at the disease site would offer advantages over the current chemotherapeutic regimens because it limits the toxicity to healthy tissues and decreases the side effects for the patient. Further, the use of nanoparticles for intracellular targeted delivery could increase the therapeutic dose delivered to the cytosol. Recent literature has also shown evidence that nanoparticles are able to overcome acquired drug resistance because efflux pumps in the cell are inefficient at removing drug-nanoparticle complexes that have entered by receptor-mediated events.22 Overall, this can lead to an improved therapeutic margin and patient quality of life.
The following section provides a brief, fundamental overview of the current challenges and opportunities in cancer nanomedicine and how understanding the complexities of tumor biology can guide rational particle engineering and material design choices. More comprehensive reviews on the challenges and barriers faced were recently published by Wicki et al.,14 Blanco et al.,10 and Park (Figure 1).16
Figure 1.

(a) Nanoparticles for cancer therapy can be made from a variety of materials, be produced to have varied physicochemical properties, and include an array of targeting ligands on the surface. (b,c) The nanoparticle surface properties and targeting ligands greatly affect its interactions with serum proteins, and can be modified to increase blood circulation time and slow clearance mechanisms from the body. (c,d,f) Particle properties (such as size, geometry, surface charge, surface hydrophilicity, elasticity, etc.) can be modified to drive preferential biodistribution in tumors by increasing particle extravasation into the tumor microenvironment via the characteristic leaky vasculature in tumor vessels and increasing uptake and intracellular trafficking by tumor cells. (g) Particles can also be designed to control cargo release in response to intracellular biological stimuli. (Reproduced from Ref. 12, with permission from Nature Publishing Group.) [Color figure can be viewed at wileyonlinelibrary.com]
Barriers to Delivery in Tumors: Challenges and Opportunities
There are several major requirements for a nanoparticle to deliver its payload to a tumor. Effective nanoparticles need to: (i) be stable in the circulation without releasing drug prematurely (retain), (ii) have sufficient residence time during circulation and accumulate in the tumor efficiently (evade, target, and accumulate), and (iii) enable controlled release the cargo locally in the tumor tissue or inside the tumor cells (release).15,23
However, there are several significant biological barriers that the carriers need to overcome.15,16,24 Renal filtration or rapid renal clearance can occur if hydrodynamic diameter of the nanoparticle is less than 10 nm. However, if the diameter is more than 200 nm, the particles can be removed by the reticuloendothelial (RES) system through interaction with proteins and subsequent opsonization. This most often results in nanoparticle accumulation in the liver, spleen, and lungs. The particles may also be cleared from circulation in the body by nonspecific uptake by macrophages and/or lymphocytes. Even if a particle were to make it to the tumor tissue, the tortuosity and high interstitial pressure of the tumor tissue can lead to an outward convective fluid flow.
Extravasation.
Several nanoparticle properties (e.g., size, shape, surface chemistry) can be rationally designed to slow these clearance mechanisms and increase circulation time in the bloodstream.14,21,24 This aims to increase nanoparticle accumu lation in the areas in and around the tumor microenvironment via passage through blood vessel leaky vasculature, commonly referred to as passive targeting by the enhanced permeation and retention effect.
Tumors have larger pores or leaky vessels in which nanoparticles can enter into the tumor tissue and are retained due to poor lymphatic drainage. Accumulation of nanoparticles in the tumor microenvironment is based on blood circulation and extravasation, and increasing circulation time can increase the fraction of nanoparticles reaching a tumor. Particle size is one of the most commonly employed strategies for increasing circulation time to exploit the enhanced permeation and retention effect, as it is well known to be related to the rate of clearance.15,25 In general, particles 50–300 nm in diameter have longer circulation times, as compared with those having larger sizes which readily accumulate in the liver and spleen. However, rapid clearance occurs via kidney filtration typically occurs for particle sizes less than 10 nm.
The problem with many of these design strategies is that the conditions for prolonged circulation also reduce cellular uptake.14,26–28 A hydrophilic, neutral surface charge is better for avoiding interactions with nonspecific proteins and prolonging circulation, but a positively charged is better for increasing cellular uptake. Stealth coatings, such as poly(ethylene glycol), shield the particle surface charge and reduce opsonization by blood proteins and subsequent uptake by macrophages of the mononuclear phagocyte system. Yet, after localizing at the tumor site, the coating can become a hurdle to achieving an efficacious response by slowing drug release and interaction with the target cells.
Active Targeting.
Another design strategy includes active targeting, which is used to increase cellular uptake and tumor residence time. The nanoparticle surface can be decorated with a variety of motilities (e.g., antibodies, peptides, carbohydrates). These ligands then enable interaction between the carrier and target cells receptors (membrane-associated proteins) through specific ligand-receptor affinity and promotes cell uptake. This can be particularly useful when particles cannot penetrate deep into the tumor, as with small, poorly vascularized metastases.
The identity and density of the targeting moiety are important to consider as they have been shown to affect circulation time and stability in the bloodstream, cellular uptake, affinity, and extravasation.29 When selecting target receptor, an important aspect to consider is the cell surface receptor recycling rate, as it directly relates to endocytosis-mediated cellular uptake. Additionally, it is important to consider the level of overexpression on the cancerous cell as compared to other healthy cells. A general guideline in literature is that overexpression by cancer cells of at least threefold warrants investigation.
Clinically Approved Nanomedicines
In the last decade, remarkable advances have been seen in the fundamental understanding of cancer, the complexities of tumor biology, and the interactions between chemical structures and biological properties. Still, the promise of nanoparticles as a controlled, targeted treatment for cancer has yet to be fully realized.
Despite extensive research on nanoparticle systems, there are only seven approved for use in cancer therapy by the Food and Drug Administration in the United States or the European Medicines Agency in the European Union (Table I), with only a few recommended as the first line of treatment.10,16,30–32 Of the carriers, liposomes have been the most extensively studied and have had the most success, representing six of the seven of the approved nanomedicines. These formulations have long circulation times and the ability to extravasate to tumor sites, leading to improved safety and tolerability.10,16,30–32
Table I.
Nanomedicines Approved for Use in Cancer Therapy by the FDA in the United States and/or the European Medicines Agency (EMA) in the European Union
| Trade name (company) | Carrier type and drug | Approved indication | Approval year | Approval agency |
|---|---|---|---|---|
| Doxil/Caelyx (Janssen) | Liposomal doxorubicin (PEGylated) | Ovarian cancer (secondary to platinumbased therapies). HIV-associated Kaposi’s sarcoma (secondary to chemotherapy). Multiple myeloma (secondary) | 1995; 1996 | FDA; EMA |
| DaunoXome (Galen) | Liposomal daunorubicin (non-PEGylated) | HIV-associated Kaposi’s sarcoma (primary) | 1996 | FDA |
| Myocet (Teva UK) | Liposomal doxorubicin (non-PEGylated) | Treatment of metastatic breast cancer (primary) | 2000 | EMA |
| Abraxane (Celgene) | Albumin-particle bound paclitaxel | Advanced nonsmall cell lung cancer (if surgery or radiation is not an option). Metastatic breast cancer (secondary). Metastatic pancreatic cancer (primary) | 2005; 2008 | FDA; EMA |
| Marqibo (Spectrum) | Liposomal vincristine (non-PEGylated) | Philadelphia chromosome-negative acute lymphoblastic leukemia (tertiary) | 2012 | FDA |
| MEPACT (Millennium) | Liposomal mifamurtide (non-PEGylated) | Treatment for osteosarcoma (primary following surgery) | 2009 | EMA |
| Onivyde MM-398 (Merrimack) | Liposomal irinotecan (PEGylated) | Metastatic pancreatic cancer (secondary) | 2015 | FDA |
Adapted from Ref. 26, with permission from Wiley.
Nevertheless, the particles only offer modest efficacious patient responses with marginal improvements over conventional treatment.10 Obstacles such as low drug carrying capacities, premature release, nonspecific distribution, and inadequate cellular uptake still remain formidable challenges.10,16,30–32 A next generation nanoparticle rationally designed to address these material and biological obstacles would enable realization of efficacious, site-specific delivery.
Advantages of Polymeric Nanoparticles
Polymer-based approaches to design nanoparticles hold promise to yield key advantages towards addressing the limitations seen to date. To date, polymers have shown potential as effective carriers in the clinical development stages (Table II). They are a particularly attractive option due to their ease of manufacturing, high drug carrying capacity, controllable size distribution, long-term stability, tunable properties, and ease of surface functionalization.9,17,26 The versatility of polymer chemistries available can enable the synthesis of increasingly complex architectures with enhanced delivery specificity and control over the desired properties to interface with biological systems.
Table II.
Polymer-Based Nanomedicines under Clinical Evaluation for Use in Cancer Therapy
| Name (company) | Particle type and drug | Investigated indication | Clinical phase | Identifiera |
|---|---|---|---|---|
| AZD2811 (AstraZeneca with BIND Therapeutics) | Aurora B kinase inhibitor in BIND therapeutics polymer particle accurin platform | Advanced solid tumors | Ph I | NCT02579226 |
| BIND-014 (BIND Therapeutics) | PSMA targeted (via ACUPA) docetaxel PEG-PLGA or PLAPEG particle | Prostate, metastatic, nonsmall cell lung, cervical, head and neck, or KRAS positive lung cancers | Ph II; Ph II; Ph II; Ph II; Ph I | NCT02479178; NCT02283320; NCT01812746; NCT01792479; NCT01300533 |
| Cynviloq IG-001 (Sorrento) | Paclitaxel polymeric micelle nanoparticle | Breast cancer | Not provided | NCT02064829 |
| Genexol-PM (Samyang Biopharmaceuticals) | Paclitaxel polymeric micelle nanoparticle | Head and neck or breast cancer | Ph II; Ph II; Ph IV | NCT01689194; NCT02263495; NCT00912639 |
| NC-6004 Nanoplatin (Nanocarrier) | Polyamino acid, PEG, and cisplatin derivative micellar nanoparticle | Advanced solid tumors, lung, biliary, bladder, or pancreatic cancers | Ph I/II; Ph III | NCT02240238; NCT02043288 |
| NC-4016 DACH-Platin Micelle (Nanocarrier) | Polyamino acid, PEG, and oxaliplatin micellar nanoparticle | Advanced solid tumors or lymphomas | Ph I | NCT01999491 |
| NK105 (Nippon Kayaku) | Paclitaxel micelle | Breast cancer | Ph III | NCT01644890 |
| Docetaxel-PM DOPNP201 (Samyang Biopharmaceuticals) | Docetaxel micelle | Head and neck cancer and advanced solid tumors | Ph II; Ph I | NCT02639858; NCT02274610 |
| CriPec (Cristal Therapeutics) | Docetaxel micelles | Solid tumors | Ph I | NCT02442531 |
| CRLX101 (Cerulean) | Cyclodextrin based nanoparticlecamptothecin conjugate | Ovarian, renal cell, small cell lung, or rectal cancers | Ph II; Ph I/II; Ph I; Ph II; Ph II | NCT02187302; NCT02010567; NCT02389985; NCT01803269; NCT01652079 |
| CRLX301 (Cerulean) | Cyclodextrin based nanoparticle-docetaxel conjugate | Dose escalation study in advanced solid tumors | Ph I/II | NCT02380677 |
Adapted from Ref. 26, with permission from Wiley.
From ClinicalTrials.gov.
POLYMERIC NANOPARTICLE ARCHITECTURES
Polymer nanoparticles offer many significant advantages to the other common carriers, as detailed above. Common architectures extensively studied in cancer nanomedicine include: micelles, nanospheres, nanocapsules, dendrimers, and highly branched polymer networks (Figure 2).10,21,22,24 These are typi cally prepared either through the dispersion or crosslinking of preformed polymers or the direct polymerization of monomers.
Figure 2.

Schematic representative of common polymer nanoparticle architectures: (A) micelle (structure depending on packing parameter, p), (B) dendrimer, (C), hyperbranched polymer, (D) nanocapsule, and (E) nanosphere. [Color figure can be viewed at wileyonlinelibrary.com]
Nanoparticles can be synthesized using a variety of natural and synthetic polymers. Natural polymers, such as chitosan, alginate, dextran, and albumin, are appealing because they offer a high degree of biocompatibility, making them an ideal choice for biomedical applications.33 However, natural polymers have several disadvantages. They may lack sufficient mechanical strength or degrade too rapidly in vivo for a desired application, and natural polymers are often derived from methods that result in inconsistent production or potential contamination.4 In that regard, synthetic polymers may offer increased control over structure–property relationships.
The selection of a specific carrier matrix and architecture will depend highly on the intended cargo, preferred site of action, and mechanism of release.10,21,22,24 Therefore, those factors must be carefully considered early in the design process. Specifically, it is important to understand how by the cargo physico-chemical properties influence the drug-polymer interactions as it will determine the drug carrying capacity and release profile.
This section will provide a concise overview of common polymeric nanoparticle systems and highlight several benefits and challenges in their application for cancer therapy. Comprehensive reviews of all types of carriers (organic and inorganic) were recently published by Peer et al.,28 Wang et al.,21 and Wicki et al.14
Micelles
Micelles are simple self-assembled macromolecules formed in solution as aggregates of amphiphilic polymers.28,34 The di- or triblock polymers contain soluble and insoluble regions that molecularly assemble into nanoscale structures, with the insoluble region forming a semisolid core and the soluble segment forming a coronal layer on the surface.
The assembly is thermodynamically-driven, and the components are in equilibrium with the surrounding medium. To this degree the micelles are dynamic, and formation and stability are highly influenced by environmental conditions, including concentration, solvent, temperature, pH, and ionic strength.35 Micelles can be formed in many different shapes which are dependent on the packing parameter, which is a function of the hydrophilic head group area and the hydrophilic tail length and volume. Micellar structures can be spherical (p < 1/3), cylindrical or worm-like (1/3 < p < 1/2), vesicle or planar bilayers (1/2 < p < 1), and inverted micelles (p > 1).36
In systemic delivery for cancer, the hydrophobic region forms the core of the micelle, providing a favorable environment for solubilizing chemotherapeutic agents.28 The hydrophilic region then dominates the surface of the micelle, yielding desirable properties to prolong circulation by avoiding opsonization and subsequent clearance by the mononuclear phagocyte system/reticuloendothelial system. Micelles are typically 10–100 nm diameter, enabling them to extravasate through the leaky vasculature in tumor tissue.
Micellar structures carry several significant advantages such as a high drug loading capacity and controllable size through variations in the hydrophobic region. Most importantly, the drugs or agents may either be physically entrapped within the hydrophobic core of the micelle or covalently linked to the polymer prior to aggregate formation. This offers wide versatility in selection of chemistries.
Despite having many advantages, several challenges still exist for the field. While micelles are stable on shorter timescales, improving the long-term stability both in storage and in physiological conditions is still an active research area.35 Crosslinking after aggregate formation may offer significant promise in improving this issue.
Dendrimers and Hyberbranched Polymers
Dendrimers are symmetric, highly branched macromolecules with a well-defined molecular weight and high degree of monodispersity obtained through single-step polymerization reactions.37–39 A dendrimer has a globular architecture consisting of two types of structural units: the dendron units in the core of the particle and the terminal units on the molecular surface. The terminal unit symmetry and exact structure result from all bonds converging to a single focal point.
Hyberbranched polymers are also globular macromolecules with a densely branched, tree-like structure. In contrast to dendrimers, the branches stem from the focal core in irregular structures with imperfect branching. These systems contain three types of structural units: dendron units and linear units in the core, and terminal units on the surface. The single-step polymerization typically results in random branching and an unpredictable polydispersity, topology, and molecular size.37 Both dendrimers and hyberbranched systems typically result in structures less than 10 nm in diameter.39
Dendrimers and hyperbranched polymers offer several advantages and disadvantages.39 In both systems, the properties are driven by the functional groups present on the surface. These end groups also enable easy conjugation with ligands that improve targeting, stealth, compatibility, or membrane permeability. Additionally, the smaller size of dendrimers and branched polymers can lead to deeper penetration within tumors and enhanced efficacy. However, shortened half-lives are typically seen with these systems due to a more rapid clearance via kidney filtration.
The systems also necessitate covalent conjugation of the active component to the polymer itself, which can be advantageous in preventing premature release. However, it present a significant limitation as it requires the drug to contain functionalizable groups for conjugation or the addition of functionalizable groups to the agent, which can be difficult depending on the mechanism of action or required orientation to a binding pocket.38 While this may be overcome with careful analysis, it limits the ease of the approach and use as a platform technology.
Nanoparticles: Nanocapsules and Nanospheres
Polymeric nanoparticles can be classified as nanocapsule or nano-sphere architectures depending on the composition. Nanocapsules are considered a reservoir system as they are comprised of a spherical polymer membrane shell which separates a core or cavity comprised of an aqueous or oily environment.22 The active drug is contained within the core and diffuses through the membrane shell into the surrounding environment with an approximately constant release rate.1 In cancer nanomedicine, the core is commonly a hydrophobic liquid containing the active drug in a molecular dispersion.40 This enables a high degree of versatility as the core and shell compositions can be independently optimized for drug solubility and desired biological interactions, respectively. Additionally, a lower polymer content compared to other carriers may offer improved biocompatibility.
Nanospheres represent spherical particles comprised of a polymer matrix.41 The active agent can instead be distributed evenly throughout the matrix or absorbed near the particle surface. The depth of distribution is typically a function of the drug size, matrix porosity, drug-polymer interactions, and drug-solvent solubility.1 The active agent is released into the surrounding environment through diffusive mechanisms. The rate at which it is released is a function of the polymer composition, the ability of the surrounding fluid to imbibe the matrix, and the drug physico-chemical properties.1 When the matrix is formed from the polymerization of monomers, crosslinking offers high stability both in storage and in vivo. Additionally, targeting ligands or hydrophilic agents may be covalently bound to the outer surface of the polymer after synthesis in order to prolong circulation or enhance uptake in tumors.
BIORESPONSIVE POLYMERS
The nanoparticle systems detailed above have been studied extensively for controlling delivery through diffusion-driven mechanisms. In contrast, environmentally-responsive polymers represent a promising class of materials that can trigger delivery through the exploitation of a specific stimuli.4,21,24,42,43
Response to a stimulus is one of the most basic processes found in living systems. As such the desire to engineer dynamic and functional materials is becoming more prevalent in an effort to achieve precise control over our environment. At the simplest definition, responsive materials adapt to their environment and undergo a desired change. What makes responsive polymers unique is that each response has been carefully and purposefully engineered at the molecular level to communicate through complex structure–property relationships and produce a macroscopic functional behavior.
The response can take many forms, from a simple change in solubility to produce swelling or a complete degradation of the network structure (Figure 3). The degree of response can be controlled by the intensity of applied stimuli. These responsive components can also be joined together and built up via hierarchical system to create intelligent devices that simultaneously respond to multiple stimuli and perform complex functions.
Figure 3.

(A,B) The tumor extracellular and intracellular environments have several characteristic phenotypes, such as enzyme overexpression, lower pH, reactive oxygen species (ROS), and a reducing environment, that can be used as stimuli to trigger responsive polymers. (C) In the presence of the specific stimuli, the nanoparticles can be designed to release their cargo by degrading, swelling, shrinking, or disassociating. The stimuli can also be used to degrade a masking ligand and activate a change in the nanoparticle surface in order to increase cellular uptake (for example, switch surface charge or expose a ligand targeted to cell-surface receptors). [Color figure can be viewed at wileyonlinelibrary.com]
Stimuli can be divided into three main categories: chemical, physical, and biological. Chemical stimuli are categorized as those that stimulate intermolecular interactions such as pH, solvent, oxidation–reduction, hypoxia, and ionic strength. Biological stimuli typically include a variety of enzymatic reactions. Physical cues can include mechanical stress, temperature, light, and magnetic or electrical fields.
Polymers that respond to internal biological cues specific to a part of the body or disease phenotype, such as degradation or swelling in response to the acidic environment of the tumor extracellular matrix or intracellular endosome, are of great interest in developing advanced architectures in cancer therapy.44 Understanding the intricacies of a stimulus–response relationship is essential to achieve kinetic, thermodynamic, and spatial control in the final product.
This section will focus on providing the reader with a concise understanding of key internal stimuli localized to tumor tissues, specifically pH, enzyme expression, and redox potential. Later sections will offer chemistry and engineering considerations to enable the rational design of such advanced materials. More comprehensive reviews on responsive carriers and stimuli were recently published by Spencer et al.44 and Liechty and Peppas.17
pH-Responsive Systems
The tumor microenvironment is characterized as slightly acidic compared to physiological pH (pH 6.8–7.2) due to lactate secretion from aerobic glycolysis, and the intracellular early and late endosomes are characterized as pH 4.5–6.8.11 This abnormal phenotypic property can be exploited for controlled, stimuli-triggered drug delivery. pH-responsive polymers, also known as polyelectrolytes, contain moieties that are capable of accepting or donating protons in response to an environmental pH change.44 The optimal design would be inert and stable under normal physiological conditions (pH 7.4), and would be capable of a triggered response upon transition to more acidic conditions. The ultimate goal of the design is to provoke a response to specific conditions through either bond cleavage or solubility changes at the molecular level, leading to controlled degradation or a conformational change in structure.
In the case of bond cleavage, the polymers can be designed with acid-sensitive bonds, such as hydrazone, which allow for site-specific triggered hydrolysis. Typically, this cleavage results directly in the subsequent release of the cargo (through entrapment or covalent conjugation), or an increase in nanoparticle cell uptake via alteration in the particle surface charge or exposure of a targeting ligand.18,45,46
In the case of structural change, electrostatic interactions between charged polymer chains repulse each other, which cause the polymer chains to extend or collapse and show an alternation between hydrophobic and hydrophilic behavior to release the cargo. Polymers that bear charged groups can be divided into anionic or cationic, with the mechanism of response being the same for both.
Anionic polymers, also known as polyacids, are polymers containing acidic functional groups. Under acidic conditions (below the pKa), the functional groups are negatively charged and contribute the formation of complexes by engaging in hydrogen bonding with accepting groups in the polymer network. The functional groups release protons and cause the polymer to become soluble when the environmental pH value is higher than the pKa value. Common examples of acidic functional groups include carboxylic acids, sulfonic acids, boronic acids, and phosphonic acids, with carboxylic acid groups being the most extensively studied.9,44
Cationic polymers, also known as polybases, behave in an inverse manner to polyacids. The most common example of basic functional groups includes amino or amine groups (—NH3).9,44 In cationic polymers, the functional group will accept a proton under acidic conditions as with anionic polymers. However, the polymer chains become positively charged, hydrophilic, and expand due to coulomb repulsion when the environmental pH value is lower than the pKb value.
A polymer has amphoteric properties if it is comprised of both acidic and basic functional groups, typically in separate monomer units.9,44 Polybetaines are a subcategory of polyampholytes where each individual monomer carries both a positive and negative ion. Both distributions result in a more complex pH-dependent behavior than simple anionic or cationic polymers. Depending on the nature of the functional groups, a dual charge may exist throughout the entire pH range, resulting in a zero net charge of the polymer. If the polymer contains weak ionic functional groups, then it can acquire a net charge at certain pH values. One of the obvious reasons for interest in this property is that many biomacromolecules, in particular proteins, are amphoteric.
In order for nanoparticles to be suitable for triggered intracellular delivery, they must be able to release their cargo and provide a mechanism for endosomal escape and delivery to the site of action.17 Endocytosis transports nanoparticles into cells within vesicles and, depending on the mode of internalization, can either be recycled and exocytosed out of the cell or trafficked to other organelles. Entrapment of nanoparticles within those vesicles is undesirable. However, there is a natural pH gradient along the endocytic pathway from pH 6.0–6.5 in early endosomes to pH 4.5–5.5 in late endosomes and lysosomes.11 This characteristic can be leveraged to facilitate cytosolic delivery of the entrapped cargo. As the endosomes acidify, cationic polymers with a pKb near pH 5 to 7 can mediate their endosomal escape through the proton-sponge effect.17 Here, the polymer absorbs protons from its environment and induces osmotic swelling of the endosome, leading to rupture of the compartment membrane.
Redox-Responsive Systems
Redox stimulus occurs due to the change in oxidation state of redox sensitive groups.42,47,48 Reactive oxygen species and reactive nitrogen species are formed as natural byproducts of normal metabolism and play key roles in many native biological processes, including cell signaling, host innate immunity, apoptosis, and proliferation. Most commonly, reactive oxygen species of physiological significance include the superoxide anion and hydrogen peroxide, as well as their derived reactive species including the hydroxyl radical, peroxyl radicals, hypochlorous acid, and singlet oxygen. Reactive nitrogen species generally include nitric oxide and its derivatives (e.g., peroxynitrite, nitrogen dioxide).
Overproduction may be endogenously-induced or provoked by external stimuli (e.g., ionizing radiation, pollutants, tobacco, smoke). Importantly, the inflammatory responses associated with many diseases, such as cancer, cardiovascular disease, and neuro-degenerative diseases, are associated with abnormal enzyme activity that leads to high concentrations of oxidizing agents. When overproduced, the oxidative stress can damage key proteins, DNA, and RNA, resulting in oxidative deactivation of certain enzymes and of polyunsaturated fatty acids in lipids (lipid peroxidation).
Oxidation–reduction responsive materials incorporate similar design principles to pH-responsive systems.42 The ultimate goal of redox-responsive designs is to provoke a response to specific conditions through either bond cleavage or solubility changes at the molecular level, leading to controlled degradation or a conformational change in structure. The chemistry generally employed in the design of materials for biological applications utilize those with multiple oxidation states, such as disulfide linkages. A promising strategy for degradation is via disulfide linkers as they can be cleaved by reductive glutathione, which is present at significantly higher levels intracellularly (concentrations of 1–11 mM).49 By incorporating a disulfide crosslinker or conjugation, points of degradation can be imparted into the polymer network while retaining mechanical integrity and desired macroscale properties.
Enzyme-Responsive Systems
Metabolic dysregulation of intracellular and extracellular enzymes (such as matrix metalloproteinases, endopeptidases, phospoholipases, and glycosidases) has been observed in tumor tissues when compared to healthy tissues.17,44,50 In nanomateri als, the altered expression profile of enzymatic activity can be used to impart a highly specific mechanism for triggered response. One of the most common designs it to incorporate an enzyme-responsive bond that is designed only to degrade upon cellular uptake. This serves to both facilitate full drug release inside the cell and maximize therapeutic effect, and reduce long-term nanogel toxicity and facilitate clearance from the body after releasing the cargo.
Peptide linkers can provide extremely high specificity for intra-cellular and/or extracellular endopeptidases, and can be incorporated with high chemical and mechanical stability. For example, cathepsin B has been linked to tumor progression and found in malignant tumors, where the cells at the invasive edge express the highest activity.51 It cleaves several bonds including Leu-Leu, Ala-Leu, Arg-Arg, Phe-Arg, Phe-Lys, Ala-Phe-Lys, Gly-Phe-Leu-Gly (GFLG), Gly-Leu-Phe-Gly, and Ala-Leu-Ala-Leu.52 The tetrapeptide GFLG has been previously studied as a cleavable linker in doxorubicin (DOX) prodrugs and polymer conjugates, and has been tested in several clinical trials.53
CHEMISTRY FOR ENVIRONMENTALLY LABILE NANOMATERIALS
The most popular techniques for polymerization of bioresponsive materials include atom transfer radical polymerization (ATRP), reversible addition-fragmentation chain-transfer polymerization (RAFT), and ring opening polymerization (ROP). These techniques allow for the use of functional initiators commonly containing poly(ethylene glycol) (PEG) chains or reactive species for additional high yielding chemistries. In this section, the approaches to synthesizing polymeric materials that undergo irreversible cleavage of covalent bonds in response to acidic pH, proteolytic enzymes, or reducing conditions are highlighted. In this section, the chemistries are broken into the following categories:(i) functional initiators, (ii) side chain modification and chemical modifications, and (iii) functional monomers and crosslinkers.
Functional Initiators
One common method for the introduction of bioresponsive functionalities is through the initiator. A wide range of initiators is commercially available and has been used to incorporate PEG, azide, alkyne, N-hydroxysuccinimide, and many other functionalities. Recently, synthesis of custom initiators has emerged as a popular way to impart physiological responsiveness. The most common functionalities are disulfide linkages that are reduced intracellularly thought the tripeptide glutathione, peptide linkages that are cleaved in the presence of proteolytic enzymes, and linkages sensitive to acidic pH.
The synthesis of disulfide ATRP initiators was reported in the literature by Tsarevsky and Matyjaszewski.54,55 Specifically, the homobi-functional initiator bis[2-(2’-bromoisobutyryloxy)ethyl]disulfide (DSDMA) was synthesized by Steglich esterification using equimolar ratios of bis(2-hydroxyethyl) disulfide and 2-bromoisobutyric acid with dicyclohexylcarbodiimide (DCC) and 4-(dimethylamino)pydridine (4-DMAP) In this work, the authors reported the synthesis of linear methacrylate polymers with ATRP as well as cleavage of the disulfide to yield polymers of half the molecular weight. Utilizing the same technique, but a molar excess of bis(2-hydroxyethyl) disulfide, Sourkohi et al. synthesized a heterobifunctional initiator for ROP and ATRP. Column chromatography was utilized to separate the heterobifunctional initiator from the homo-bifuntional initiator and other products.56 The heterobifunctional initiator offers the advantage of polymerizing unique polymer blocks on either end of the initiator. With this methodology, in addition to reducing the molecular weight by up to a factor of two, intracellular reduction can yield separation of hydrophilic and hydrophobic polymer chains.
PEG terminated initiators have been widely used as a convenient method for increasing circulation half-life of polymers. Recently, PEG-based initiators have been synthesized to contain disulfide linkages between the PEG and CRP initiator. For example, a PEG 2000 with a nitro phenyl carbonate (NPC) terminal functionality was reacted with cystamine to synthesize PEG-SS-NH2 for ROP.57 Similarly, PEG-SS-NH2 was synthesized and coupled to CPADN with carbodiimide chemistry to give a reduction sensitive RAFT agent. In a similar report, NPC-PEG-NPC was reacted with cyst-amine and then CPADN to give CPADN-SS-PEG-SS-CPADN.48,58 In these reactions, the reduction of the disulfide will lead to the separation of the hydrophilic PEG segment from the other block.
Boyer et al. reported the synthesis of a heterotelechelic polymer containing α-azide and ω-dithiopyridine functionalities. The initiator was synthesized in multiple steps and is unique in that it allows for RAFT polymerization followed by conjugation via click chemistry and thiol-disulfide exchange (Figure 4).59 This approach allows for great versatility and conjugation in that targeting or stealth agents can be easily conjugated post polymerization.
Figure 4.

Synthesis route for a hetereotelechelic bioresponsive RAFT agent. (Reproduced from Ref. 59, with permission from American Chemical Society.)
The majority of literature on functional bioresponsive initiators has focused on disulfide bond containing materials because they are readily available and can be processed in a wide range of solvents and temperatures. Polymers sensitive to acidic pH are of specific interest because of the response to the pH of tumor microenvironments or endosomes. A homobifunctional hemiacetal initiator was synthesized from 2-bromoisobutyric acid and ethylene glycol divinyl ether.60 This initiator serves as an acid degradable analog to the disulfide homobifunctional initiators.
Polymers sensitive to proteolytic degradation are of interest for environmentally labile nanomaterials because their response is not limited to the intracellular reducing environment. Instead, the presence of elevated enzymes in certain cancerous tissues can lead to the cleavage of the peptide. For example, a RAFT agent with an enzymatically degradable peptide was synthesized in three steps, taking advantage of an N-hydroxysuccinimide functional chain transfer agent and maleimide thiol click chemistry (Figure 5).61,62 The resulting material, an enzymatically degradable RAFT agent, was designed to deshield the PEG layer in the presence of MMP-7, yielding an increase in surface charge to facilitate cell uptake.
Figure 5.

Synthesis route of chain transfer agent with MMP-7 degradable linkage to enable poly(ethylene glycol) deshielding. (Reproduced from Ref. 61, with permission from Wiley.)
The use of functional initiators is a promising method to synthesize bioresponsive polymers. Synthesizing polymers from functional initiators typically results in the incorporation of the responsive unit in the backbone of the polymer. Heterobifunctional disulfide initiators provide a much more powerful responsive mechanism because of the ability to synthesize polymers with two or more unique segments. In this case, molecular weight is cut by up to a factor of two and can lead to outcomes including (i) complete dissociation of the nanomaterials because of separation of hydrophilic and hydrophobic segments, and (ii) cleavage of PEG/other chains to reveal increased surface charge or targeting ligands. Extensions of these methodologies will continue to enhance the responsive materials properties of nanoscale drug delivery systems for cancer therapy (Table III).
Table III.
Recent Chemistries Utilized in Direct Polymerization with Functional Initiators to Impart Bioresponsive Properties
| Name and structure | Synthesis | Degradation | References |
|---|---|---|---|
| Disulfanediylbis(ethane-2,1-diyl) bis(2-bromo-2-methylpropanoate) |
ATRP | Backbone cleaved intracellularly into two identical chains | 54 |
| 2-((2-Hydroxyethyl)disulfaneyl)ethyl 2-bromo-2-methylpropanoate |
ATRP/ROP | Backbone cleaved intracellularly into two unique chains | 56 |
| 6-(2-((2-((2-Bromo-2-methylpropanoyl)oxy) ethyl)disulfaneyl)ethoxy)-6-oxohexyl 6-(1-(2-chloroethoxy)ethoxy)hexanoate |
ATRP/ROP | Backbone cleaved intracellularly into multiple unique chains | 63 |
| PEG-SS-CPADNa |
RAFT | Backbone cleaved intracellularly into PEG chain and polymer chain | 48 |
CTA-peptide-PEGb RAFT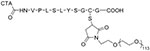
|
RAFT | Backbone cleaved extracellularly into PEG chain and polymer chain | 61 |
| 3-Azidopropyl 2-((((3-oxo-3-(2-(pyridin-2-yldisulfaneyl) ethoxy)propyl)thio)carbonothioyl)thio)propanoate |
RAFT | Backbone cleaved intracellularly into two unique chains | 59 |
| 2-nPEG methyl ether (2-((2-aminoethyl) disulfaneyl)ethyl)carbamate |
ROP | Backbone cleaved intracellularly into PEG chain and polymer chain | 57 |
CPADN, 4-cyanopentanoic acid dithionaphthalenoate.
CTA, chain transfer agent.
Side Chain Modifications
Chemical modifications of pendant functional groups have been a popular strategy for the synthesis of polymer-drug conjugates. Correspondingly, modifications of pendant functional groups with a bioresponsive functionality can result in polymeric drug delivery systems that can modulate materials properties in response to physiological stimuli. In this case, the cleavage can result in a hydrophobe to hydrophile transition or cleavage of a crosslink.
In 2003, Gillies and Frechet published an article detailing the synthesis of amphiphilic block copolymers via the attachment of hydrophobic groups susceptible to acid hydrolysis to one block of a diblock copolymer. Specifically, the conjugation of cyclic benzylidene acetals to poly(aspartic acid) was reported.64 In the initial report, conjugation was achieved through EDC chemistry. However, coupling efficiency was later improved though the use of the amine reactive 4-nitrophenyl carbonates to yield carbamates with near quantitative yield.64,65 The group demonstrated pH-dependent hydrolysis of the side chains and concomitant release of the hydrophobic small molecule, nile red. In this case, the mechanism of micelle disruption stems from the transition from a hydrophobic to polar side chain.
Ghosh et al. reported a method for disulfide incorporation into polymeric sidechains.66 Disulfide exchange is a convenient mechanism for the synthesis of reduction labile side chains, or crosslinking. The disulfide exchange is popular in bioconjugation reactions, and has been heavily utilized in protein and prodrug conjugation schemes with the heterobifunctional cross-linker succinimidyl 3-(2-pyridyldithio)propionate.67
Ryu et al.’s group demonstrated the incorporation of a pyridyldisulfide (PDS) functionality into the backbone of polymer using RAFT polymerization of a methacrylated pyridyldisulfide monomer.68,69 The addition of a deficient amount of dithiothreitol (DTT); resulted in the cleavage of a fraction of the PDS groups, which could then react with uncleaved PDS groups to form disulfide crosslinks. Further exploration of this system based on thioldisulfide exchange led to assemblies with multiple responsive side chain units through postpolymerization modifications. This strategy can further enhance the diversity of responsive side chains.70
Wei et al. also utilized disulfide exchange to form crosslinked micelles through side-chain modification. The authors synthesized the diblock copolymer PEG-b-PHPMA by RAFT polymerization and then esterified the hydroxyl group with lipoic acid to give the disulfide functionality.47 After esterification, the polymers assembled into micelles and were crosslinked through addition of DTT.
Li et al. utilized RAFT polymerization containing N-acryloxysuccinimide to form shell crosslinked micelles with diamines.71 The authors later utilized cystamine to yield covalent crosslinks labile under reducing conditions.72 This scheme allows for the construction of shell-crosslinked micelles with enhanced diversity as the shell block is only limited to monomers that can copolymerize with N-acryloxysuccinimide under appropriate conditions.
Sequential Chemistries.
Jin et al. reported they synthesis of a triblock copolymer containing an oxime linkage in the backbone. First, an oxime-tethered polycaprolactone (OPCL) was synthesized by oxime coupling polycondensation. The amino-oxy groups of the OPCL were then reacted with the alde-hyde terminal PEG in an oxime coupling ligation reaction to give the PEG-OPCL-PEG triblock copolymer.73 Compared to the amide linked control, the acid labile bond served to enhance the drug release from the micelles.
In 2014, Zhang et al. reported a terpolymer with both acetal and disulfide linkages in the polymeric backbone.63 In this work, the authors used a combination of ROP, ATRP, and click chemistry to synthesize the multiresponsive system. First, a polycaprolactone (PCL) block was reacted with ROP using the heterobifunctional ATRP/ROP initiator. An acetal linkage was made through reaction with 2-chloroethyl vinyl ether, 2-(dimethyl-amino)ethyl methacrylate (DMAEMA) was reacted via ATRP, and the terminal chlorine was converted to an azide with sodium azide. The disulfide and acetal functional diblock copolymer was then functionalized with a targeting ligand through copper catalyzed azide-alkyne cycloaddition. This multistep synthesis reflects the ability to generate complex intelligently responsive structures with CRP techniques and high yielding chemistry. The techniques in combination with insightful designs promise to maximize therapeutic of nanoparticles for drug delivery (Figure 6).
Figure 6.

Synthesis route of a reduction and pH dual responsive polymer with combination ATRP ROP initiator. (Reproduced from Ref. 63, with permission from Royal Society of Chemistry.)
Direct Polymerization of Labile Monomers or Crosslinkers
Perhaps one of the most well-characterized methods for incorporating bioresponsive side chains into polymeric materials is the work on peptide spacers into hydroxy propyl methacrylamide pioneered by Kopeček and colleagues starting in the 1970s.74 Spe cifically, they reported the synthesis of hydroxyl propyl methacrylamide monomers with enzymatically degradable side chains.75
Monomers and polymers with peptide spacers degradable under lysosomal conditions with p-nitroaniline end functionality were reported.76,77 The majority of their work focused on drug and protein conjugates. The strategies they utilized are very relevant to ongoing work into biologically responsive nanomaterials.
Kwon et al. developed an acetal containing acrylate monomer and crosslinker.78 In the scheme, an acetal containing diamine was synthesized as a precursor to the monomer and crosslinker and could serve as an acid labile analog for the amine reactive scheme reviewed earlier. The monomers were later reacted into polyethyleneimine for use for delivery of DNA or siRNA.79
Griset et al. synthesized methacrylate cyclic acetal monomers and crosslinker.80 The monomers were used in a UV-initiated miniemulsion polymerization to synthesize nanoparticles that underwent a dramatic volume expansion upon cellular uptake and exposure to acidic conditions (approximately pH 5). The acid labile nanoparticles loaded with paclitaxel (PTX) resulted in minimal release over 24 h at pH 7.4, while nearly 100% of the drug was released at 24 h at pH 5. With in vivo evaluation, the authors observed that injection of the PTX loaded, acid labile nanoparticles prevented tumor formation in a subcutaneous model of rapidly growing Lewis lung carcinoma tumors in C57Bl/6 female mice. In contrast, equivalent dose of free PTX, media alone, empty nanoparticles, and nonresponse nanoparticles did not prevent tumor formation.
Tang et al. reported the synthesis of an acid labile ortho-ester methacrylate monomer. The monomer was used directly in ATRP from a PEG macroinitiator yielding a diblock copolymer with an or tho-ester side chain.81 The diblock copolymer formed core–shell micelles in slightly alkaline water with a hydrodynamic diameter of 74 nm. In these acid responsive nanomaterials, upon exposure of the micelles to acid, the goal is the polymer to be rendered completely hydrophilic and to release its contents.
Du et al. have published a series of articles based upon the concept of polymer blocks with acid labile side chains. In one report, a triblock copolymer was synthesized by sequential RAFT polymerization from a PEG-macro chain transfer agent with the acid labile monomer 2,4,6-trimethoxybenzylidene-1,1,1-tris(hydroxymethyl) ethane methacrylate and acrylic acid.82 The authors reported the self-assembly of the triblock copolymers into chimeric polymerosomes. Hydrolysis of the acetal under slightly acidic conditions was designed to result in a water soluble triblock copolymer. Wu et al. also synthesized 2,4,6-trimethoxybenzylidene-pentaerythritol carbonate and mono-4-methoxybenzylidene-pen-taerythritol carbonate. The monomers were utilized in ROP with PEG initiators. In one example, the authors reported copolymerization with acryoyl carbonate, and the ability photo-crosslink the materials into a core-crosslinked micelle.83,84
Crosslinkers.
N,N′-bis(acryloyl)cystamine was used for the synthesis of disulfide labile nanoparticles of PEG and NIPAAm were reported as early as 2005.85 The authors reported the synthesis of temperature responsive nanoparticles with hydrodynamic diameters ranging from 60 to 200 nm. The authors further demonstrated the degradability of the particles as well as the ability to load doxorubicin and a fluorescent probe.
Dai et al. reported the synthesis of cationic start polymers via ATRP of DMAEMA with bis(acryloylcystamine) (BAC).86 The star polymers were synthesized by polymerization of the desired arm length of DMAEMA onto a difunctional ATRP initiator, followed injection of the crosslinker (BAC) at 4 h to form a reducible core. The resulting star polymers were 20–60 nm depending on arm length.
Tsarevsky and Matyjaszewski54 and Li et al.87 reported the synthesis and use of a methacrylate version of the disulfide cross-linker DSDMA and used it to form branched polymers or networks that were degradable under reducing conditions. Utilizing DSDMA, Wei et al.47 synthesized star polymers via RAFT polymerization with n-butyl methacrylate, 2-(dimethylamino)ethyl methacrylate, and poly[oligo(ethylene glyol) methacry-late] in a similar manner to Dai et al.
Recently, Hu et al. reported the use of a dual responsive cross-linking agent into a PCL-b-p(OEGMA-co-MAEMA) diblock copolymer.88 The crosslinking agent was first proposed by Rodriguez-Docampo and Otto et al.89 In the study, the authors used a combination of ROP and ATRP, and the crosslinking agent was responsive to both acid and reducing conditions in the final copolymer assembly (Table IV).
Table IV.
Recent Chemistries Utilized in the Direct Polymerization of Labile Monomers or Crosslinking Agents to Impart Bioresponsive Properties
| Name and structure | Incorporation | Type of response | References |
|---|---|---|---|
2-(2,4,6-Trimethoxyphenyl)-1,3-dioxan-5-amine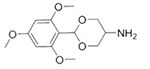
|
EDC coupling | Acid labile, hydrophobic to polar | 64 |
| (5-Methyl-2-(2,4,6-trimethoxyphenyl)-1,3-dioxan-5-yl) methyl (4-nitrophenyl) carbonate |
Amine reactive | Acid labile, hydrophobic to polar | 65 |
9-(2,4,6-Trimethoxyphenyl)-2,4,8,10-tetraoxaspiro [5.5]undecan-3-one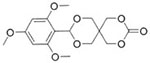
|
ROP monomer | Acid labile, hydrophobic to polar | 84 |
(5-Methyl-2-(2,4,6-trimethoxyphenyl)-1,3-dioxan-5-yl) methyl methacrylate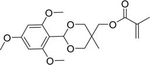
|
Monomer | Acid labile, hydrophobic to polar | 80 |
2-(Pyridin-2-yldisulfaneyl)ethyl methacrylate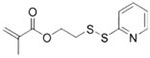
|
Monomer | SPDP, thiol-disulfide exchange for crosslinking or coupling | 66 |
2,5-Dioxopyrrolidin-1-yl acrylate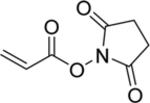
|
Monomer | Coupling with cystamine gives labile crosslinks | 71 |
|
N,N′-(Disulfanediylbis(ethane-2,1-diyl))diacrylamide |
Crosslinker | Intracellular cleavage of crosslinks | 90 |
|
N,N′-(7,7-Dimethyl-3,6,8,11-tetraoxatridecane-1,13- diyl)diacrylamide |
Crosslinker | Acid catalyzed hydrolysis of crosslinks | 78 |
| Disulfanediylbis(ethane-2,1-diyl) bis(2-methylacrylate) |
Crosslinker | Intracellular cleavage of crosslinks | 87 |
| 3,3′-Disulfanediyldi(propanehydrazide) |
Crosslinker | Intracellular cleavage of crosslinks via reduced and acid-catalyzed hydrolysis | 88 |
RECENT ADVANCES IN BIORESPONSIVE NANOSCALE POLYMERIC DRUG DELIVERY SYSTEMS
In the section above, three main methods for the incorporation of bioresponsive functionalities into polymer backbones were reviewed. Specifically, methods for functional initiators, postpolymerization chemical modifications, and direct polymerization of functional monomers were detailed. In each of methods, the goal was to incorporate acid labile, enzymatic labile, or reduction sensitive linkers in either the backbone, side chain, or crosslinks of the polymeric drug delivery system. The overall goal of incorporating the labile covalent bond sensitive to a specific physiological environment is to promote nanoparticle/drug uptake, penetration, release, or clearance. Here, some notable examples of nanoparticle drug delivery systems containing one or more bioresponsive moieties are highlighted.
Zhu et al. recently compared in vivo delivery of doxorubicin to U87MG glioma xenografts from PEG-SS-PCL with and without cRGD targeting PEG-PCL with cRGD targeting and free drug.91 The study demonstrated that the incorporation of the disulfide linkage did not significantly alter pharmacokinetics or biodistribution. A significant difference was observed in the distribution of doxorubicin to tumor tissue to healthy tissue with the targeting ligand. The reduction sensitive formulation with targeting demonstrated the greatest inhibition to tumor growth after 30 days.
In a similar study, the Zou et al. reported the delivery of DOX from disulfide crosslinked particles.92 In the study, ROP was used to synthesize diblock copolymer of PEG-b-PDTC in which the disulfide bond could participate in crosslinking. Micellar nanoparticles with and without cRGD functionality were assessed for biodistribution and efficacy in B16 melanoma-bearing C57BL/6 mice as compared to liposomal DOX formulations and PBS. Notably, the disulfide containing formulations resulted in the enhanced survivability compared to controls, with 100% survival at the highest drug loading after 43 days.
Ko et al. developed charge convertible nanoparticles containing disulfide crosslinks.93 The nanoparticles contained cationic polyethyleneimine, hydrophobic lithochloic acid, charge convertible 2,3-dimethylmaleic anhydride, and were crosslinked with succinimidyl 3-(2-pyridyldithio)propionate. The charge converting nanoparticles switched from a negative zeta potential (–11.4 mV) at pH 7.4 to cationic (112.4) at pH 6.5 compared to materials without the charge-converting group that were cationic at both pH 7.4 and 6.5 at 20.9 and 28.4 mV, respectively. The charge converting particles demonstrated higher accumulation tumor to liver accumulation ratio compared to the control.
Li et al. recently reported the design of a proximity activated targeting smart polymeric nanoparticle (PAT-SPN). An MMP-7 cleavable peptide was incorporated between a PEG block and mixed blocks of DMAEMA and mixed block of DMAEMA, propyl acrylic acid, and butyl methacrylate. In response to MMP-7 (also MMP-2 and MMP-9) the peptide linker degraded, shedding the PEG layer, which resulted in an increase in surface charge. A2.5-fold increase in internalization was observed in MDA-MB-231 breast cancer cells for MMP7 activated nanocarriers compared to the controls.61
Recently an in vivo assessment of stimuli-responsive clustered nanoparticles was reported. The assembly was based upon the self-assembly of dendrimers with platinum pro-drugs linked to PCL, PEG-PCL, and PCL. The reaction of anhydride and amino groups led to an acid labile amide bond. At physiological pH, the assemblies were around 100 nm in diameter, but upon exposure to acidic environments, PAMAM prodrugs approximately 5 nm in size were released enabling deeper tumor penetration. In vivo evaluation of the system in a cisplatin-resistant A549R human lung cancer demonstrated 95% inhibition of tumor growth compared to the PBS control. In comparison, free drug demonstrated 10% inhibition and pH-stable assemblies showed 60% inhibition.45
Liu and Thayumanavan recently investigated the pH sensitivity of nanogels synthesized with 18 unique acetal and ketal cross-linkers subdivided into six categories of structural variation. The comprehensive study of acid catalyzed hydrolysis demonstrated the ability to fine-tune degradation kinetics over 6 orders of magnitude. Notably the authors reported that the relative position of the ketal moiety and electron withdrawing amide resulted in a 2 order of magnitude differences in cleavage rates for 2-carbon 6-carbon linkers.94
CONCLUSIONS AND FUTURE PERSPECTIVE
In recent decades, nanoparticles have shown significant promise as an oncology treatment modality. Polymer-based carriers offer important advantages, and the field is rapidly progressing as evidenced by the extensive studies in the preclinical and clinical stages. Despite this, significant obstacles in overcoming the complexities of tumor biology still exist, preventing the full realization of cancer nanomedicine.
Responsive polymers represent a promising class of nanoparticles that can trigger delivery through the exploitation of a specific stimuli. Response to a stimulus is one of the most basic processes found in living systems. As such the desire to engineer dynamic and functional materials is becoming more prevalent in an effort to achieve precise control over our environment. The versatility of polymer chemistries available can enable the synthesis of novel systems with enhanced delivery specificity and control over the desired properties to interface with biological systems.
The initiators, monomers, crosslinkers, and conjugation chemistries highlighted in this review demonstrate the advancement in the design and synthesis of novel responsive polymer architectures for enhancing drug delivery to tumors. The utility of these chemistries either alone or in tandem will allow for the design and realization of increasingly complex architectures. However, the design of simple, elegant materials utilizing high yield synthesis or bioconjugation schemes with highly reproducible material and responsive properties will be beneficial for progressing materials into larger animal models and the clinic.
The combination of quality materials that respond to environmental triggers to change surface charge, size, or release drugs represent the new direction of polymeric nanocarriers in drug delivery. The utilization of these polymers to small molecule, short or long RNA, or proteins represents an incredible challenge. However, the combination of controlled radical polymerization and high yielding chemistry strategies outlined here provide an excellent basis for the development the next generation of drug delivery systems.
ACKNOWLEDGMENTS
The work was supported in part by a grant from the National Institutes of Health (R01-EB-022025) and the Cockrell Family Regents Chair. A.M.W. was supported by a National Science Foundation Graduate Research Fellowship (DGE-1610403), the S.E.S.H.A. Endowed Graduate Fellowship in Engineering, and the Philanthropic Educational Organization Scholar Award. D.S.S. was supported by a National Science Foundation Graduate Research Fellowship (DGE-1610403) and a Cockrell School of Engineering Doctoral Fellowship. Nicholas A. Peppas greets the publication of this special volume and thanks Wiley for their contributions the field, as he has been a member of the Editorial Board since July 1976 having been appointed by the legendary Editor Hermann Mark.
Biographies

Angela M. Wagner received her B.S. in Chemical Engineering at the Pennsylvania State University. She earned her M.S.E. and is currently working towards her Ph.D. in Chemical Engineering at the University of Texas at Austin, as a National Science Foundation Graduate Research Fellow under the guidance of Professor Peppas. She has nearly 7 years research experience in the pharmaceutical industry working on nanoparticles for gene therapy and solid dispersions for oral delivery. Her research interests include responsive polymers for drug delivery, immunotherapy, and biomedical applications.

David S. Spencer received his B.S. in Chemical Engineering at the University of Kentucky in 2013 and M.S.E. in Chemical Engineering from The University of Texas at Austin in 2016. He is currently working toward his Ph.D. in Chemical Engineering at The University of Texas at Austin, as a National Science Foundation Graduate Research Fellow under the guidance of Professor Peppas. His research interests include intelligent polymeric and inorganic nanomaterials for drug delivery and diagnostic applications.

Nicholas A. Peppas’ research spans a career of 40 years and has concentrated on the elucidation of transport phenomena (notably diffusion and convection problems) in biological and chemical problems with emphasis on the integration of mathematical models and engineering design with cellular processes. He is particularly recognized for his pioneering work on polymer networks and hydrogels. He received his Dipl. Eng. at the National Technical University of Athens in 1971, Greece, and received his Sc.D. at the Massachusetts Institute Technology in 1973. He was professor at Purdue University from 1976–2002, and has been the Cockrell Family Chaired Professor at the University of Texas at Austin since 2003. He is a member of several Academies such as the U.S. NAM, NAE, NAI, AAAS, the National Academy of France, the Academy of Athens, the Royal Academy of Spain, the Chinese Academy of Engineering, and the Academy of Medicine, Engineering and Sciences of Texas. He has been recognized with more than 120 national and international awards. He is the author of over 1450 refereed publications and 37 patents, and he has been cited over 107,000 times with an H-index of 152.
REFERENCES
- 1.Ratner BD; Hoffman AS; Schoen FJ Biomaterials Science: An Introduction to Materials in Medicine; Elsevier Academic Press: San Diego, CA, 2004. [Google Scholar]
- 2.Singh A; Peppas NA Adv. Mater 2014, 26, 6530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peppas NA; Hilt JZ; Bryne MW O. Pat 116,734–A3 (2006). [Google Scholar]
- 4.Peppas NA; Hilt JZ; Thomas JB Nanotechnology in Therapeutics: Current Technology and Applications; Horizon Bioscience: Oxford, UK, 2007. [Google Scholar]
- 5.Peppas NA; Van Blarcom DS J. Controlled Release 2016, 240, 142. [DOI] [PubMed] [Google Scholar]
- 6.Koetting MC; Peters JT; Steichen SD; Peppas NA Mater. Sci. Eng. R Rep 2015, 93, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knipe JM; Peppas NA Regen. Biomater 2014, 1, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caldorera-Moore ME; Liechty WB; Peppas NA Acc. Chem. Res 2011, 44, 1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liechty WB; Kryscio DR; Slaughter BV; Peppas NA Annu. Rev. Chem. Biomol. Eng 2010, 1, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blanco E; Shen H; Ferrari M Nat. Biotechnol 2015, 33, 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kato Y; Ozawa S; Miyamoto C; Maehata Y; Suzuki A; Maeda T; Baba Y Cancer Cell Int 2013, 13, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi J; Kantoff PW; Wooster R; Farokhzad OC Nat. Rev. Cancer 2017, 17, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nolen BM; Lokshin AE Mol. Diagn. Ther 2013, 17, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wicki A; Witzigmann D; Balasubramanian V; Huwyler JJ Controlled Release 2015, 200, 138. [DOI] [PubMed] [Google Scholar]
- 15.Bae YH; Park KJ Controlled Release 2011, 153, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park K ACS Nano 2013, 7, 7442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liechty WB; Peppas NA Eur. J. Pharm. Biopharm 2012, 80, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Du J; Lane LA; Nie SJ Controlled Release 2015, 219, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.http://www.cdc.gov/cancer/international/statistics.htm, 2015.
- 20.Kirtane AR; Kalscheuer SM; Panyam J Adv. Drug Delivery Rev 2013, 65, 1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang AZ; Langer R; Farokhzad OC Annu. Rev. Med 2012, 63, 185. [DOI] [PubMed] [Google Scholar]
- 22.Brigger I; Dubernet C; Couvreur P Adv. Drug Delivery Rev 2002, 54, 631. [DOI] [PubMed] [Google Scholar]
- 23.Mills JK; Needham D Expert Opin. Therap. Patents 1999, 9, 1499. [Google Scholar]
- 24.Steichen SD; Caldorera-Moore M; Peppas NA Eur. J. Pharm. Sci 2013, 48, 416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsumura Y; Maeda H Cancer Res 1986, 46, 6387. [PubMed] [Google Scholar]
- 26.Anselmo AC; Mitragotri S Bioeng. Transl. Med 2016, 1,10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bertrand N; Wu J; Xu X; Kamaly N; Farokhzad OC Adv. Drug Delivery Rev 2014, 66, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peer D; Karp JM; Hong S; Farokhzad OC; Margalit R; Langer R Nat. Nanotechnol 2007, 2, 751. [DOI] [PubMed] [Google Scholar]
- 29.Xu S; Olenyuk BZ; Okamoto CT; Hamm-Alvarez SF Adv. Drug Delivery Rev 2013, 65, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barenholz YJ Controlled Release 2012, 160, 117. [DOI] [PubMed] [Google Scholar]
- 31.Dawidczyk CM; Kim C; Park JH; Russell LM; Lee KH; Pomper MG; Searson PC J. Controlled Release 2014, 187, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gelderblom H; Verweij J; Nooter K; Sparreboom A Eur.J. Cancer 2001, 37, 1590. [DOI] [PubMed] [Google Scholar]
- 33.Dang JM; Leong KW Adv. Drug Delivery Rev 2006, 58, 487. [DOI] [PubMed] [Google Scholar]
- 34.Blanco E; Kessinger CW; Sumer BD; Gao J Exp. Biol. Med. (Maywood, NJ) 2009, 234, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Owen SC; Chan DPY; Shoichet MS Nano Today 2012, 7, 53. [Google Scholar]
- 36.Nagarajan R Langmuir 2002, 18, 31. [Google Scholar]
- 37.Fr echet JMJ; Hawker CJ; Gitsov I; Leon JW J. Macromol. Sci. Chem 1996, 33, 1399. [Google Scholar]
- 38.Kurniasih IN; Keilitz J; Haag R Chem. Soc. Rev 2015, 44, 4145. [DOI] [PubMed] [Google Scholar]
- 39.Inoue K Prog. Polym. Sci 2000, 25, 453. [Google Scholar]
- 40.Mora-Huertas CE; Fessi H; Elaissari A Int. J. Pharm 2010, 385, 113. [DOI] [PubMed] [Google Scholar]
- 41.Haley B; Frenkel E Urol. Oncol 2008, 26, 57. [DOI] [PubMed] [Google Scholar]
- 42.Kelley EG; Albert JNL; Sullivan MO; Epps TH Chem. Soc. Rev 2013, 42, 7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spencer DS; Puranik AS; Peppas NA Curr. Opin. Chem. Eng 2015, 7, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mura S; Nicolas J; Couvreur P Nat. Mater 2013, 12, 991. [DOI] [PubMed] [Google Scholar]
- 45.Li HJ; Du JZ; Du XJ; Xu CF; Sun CY; Wang HX; Cao ZT; Yang XZ; Zhu YH; Nie SM; Wang J Proc. Natl. Acad. Sci. U.S.A 2016, 113, 4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Romberg B; Hennink WE; Storm G Pharm. Res 2008, 25, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wei RR; Cheng L; Zheng M; Cheng R; Meng FH; Deng C; Zhong ZY Biomacromolecules 2012, 13, 2429. [DOI] [PubMed] [Google Scholar]
- 48.Zhang J; Wu L; Meng F; Wang Z; Deng C; Liu H; Zhong Z Langmuir 2012, 28, 2056. [DOI] [PubMed] [Google Scholar]
- 49.Baudouin-Cornu P; Lagniel G; Kumar C; Huang ME; Labarre JJ Biol. Chem 2012, 287, 4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nishikawa H; Ozaki Y; Nakanishi T; Blomgren K; Tada T; Arakawa A; Suzumori K Gynecol. Oncol 2004, 92, 881. [DOI] [PubMed] [Google Scholar]
- 51.Berquin IM; Sloane BF Adv. Exp. Med. Biol 1996, 389, 281. [DOI] [PubMed] [Google Scholar]
- 52.Zhong Y-J; Shao L-H; Li YA N. Int. J. Oncol 2013, 42, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Malugin A; Kopečkov a P; Kopeček JJ Controlled Release 2007, 124, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsarevsky NV; Matyjaszewski K Macromolecules 2005, 38, 3087. [Google Scholar]
- 55.Tsarevsky NV; Matyjaszewski K Macromolecules 2002, 35, 9009. [Google Scholar]
- 56.Sourkohi BK; Cunningham A; Zhang Q; Oh JK Bio-macromolecules 2011, 12, 3819. [Google Scholar]
- 57.Thambi T; Yoon HY; Kim K; Kwon IC; Yoo CK; Park JH Bioconjug. Chem 2011, 22, 1924. [DOI] [PubMed] [Google Scholar]
- 58.Zhu C; Zheng M; Meng F; Mickler FM; Ruthardt N; Zhu X; Zhong Z Biomacromolecules 2012, 13, 769. [DOI] [PubMed] [Google Scholar]
- 59.Boyer C; Liu J; Bulmus V; Davis TP; Barner-Kowollik C; Stenzel MH Macromolecules 2008, 41, 5641. [Google Scholar]
- 60.Rikkou-Kalourkoti M; Loizou E; Porcar L; Matyjaszewski K; Patrickios CS Polym. Chem 2012, 3, 105. [Google Scholar]
- 61.Li HM; Yu SS; Miteva M; Nelson CE; Werfel T; Giorgio TD; Duvall CL Adv. Funct. Mater 2013, 23, 3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H; Miteva M; Kirkbride KC; Cheng MJ; Nelson CE; Simpson EM; Gupta MK; Duvall CL; Giorgio TD Biomacromolecules 2015, 16, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang Y; He JL; Cao DL; Zhang MZ; Ni PH Polym. Chem 2014, 5, 5124. [Google Scholar]
- 64.Gillies ER; Frechet JM J. Chem. Commun 2003, 1640. [DOI] [PubMed] [Google Scholar]
- 65.Gillies ER; Jonsson TB; Frechet JM J. J. Am. Chem. Soc 2004, 126, 11936. [DOI] [PubMed] [Google Scholar]
- 66.Ghosh S; Basu S; Thayumanavan S Macromolecules 2006, 39, 5595. [Google Scholar]
- 67.Carlsson J; Drevin H; Axen R Biochem. J 1978, 173, 723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ryu JH; Chacko RT; Jiwpanich S; Bickerton S; Babu RP; Thayumanavan SJ Am. Chem. Soc 2010, 132, 17227. [DOI] [PubMed] [Google Scholar]
- 69.Ryu JH; Jiwpanich S; Chacko R; Bickerton S; Thayumanavan SJ Am. Chem. Soc 2010, 132, 8246. [DOI] [PubMed] [Google Scholar]
- 70.Liu XC; Hu D; Jiang ZW; Zhuang JM; Xu YS; Guo XH; Thayumanavan S Macromolecules 2016, 49, 6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li YT; Lokitz BS; McCormick CL Macromolecules 2006, 39, 81. [Google Scholar]
- 72.Li YT; Lokitz BS; Armes SP; McCormick CL Macromolecules 2006, 39, 2726. [Google Scholar]
- 73.Jin Y; Song L; Su Y; Zhu LJ; Pang Y; Qiu F; Tong GS; Yan DY; Zhu BS; Zhu XY Biomacromolecules 2011, 12, 3460. [DOI] [PubMed] [Google Scholar]
- 74.Ulbrich K; Subr V Adv. Drug Delivery Rev 2010, 62, 150. [DOI] [PubMed] [Google Scholar]
- 75.Drobnik J; Kopeček J; Labsky J; Rejmanova P; Exner J; Saudek V; Kalal J Makromolekul. Chem. Macromol. Chem. Phys 1976, 177, 2833. [Google Scholar]
- 76.Duncan R; Cable HC; Lloyd JB; Rejmanova P; Kopecek J Makromolekul. Chem. Macromol. Chem. Phys 1983, 184, 1997. [Google Scholar]
- 77.Rejmanova P; Kopecek J; Pohl J; Baudys M; Kostka V Makromolekul. Chem. Macromol. Chem. Phys 1983, 184, 2009. [Google Scholar]
- 78.Kwon YJ; Standley SM; Goodwin AP; Gillies ER; Frechet JM J. Mol. Pharm 2005, 2, 83. [DOI] [PubMed] [Google Scholar]
- 79.Shim MS; Kwon YJ Biomacromolecules 2008, 9, 444. [DOI] [PubMed] [Google Scholar]
- 80.Griset AP; Walpole J; Liu R; Gaffey A; Colson YL; Grinstaff MW J. Am. Chem. Soc 2009, 131, 2469. [DOI] [PubMed] [Google Scholar]
- 81.Tang RP; Ji WH; Panus D; Palumbo RN; Wang CJ Controlled Release 2011, 151, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Du YF; Chen W; Zheng M; Meng FH; Zhong ZY Biomaterials 2012, 33, 7291. [DOI] [PubMed] [Google Scholar]
- 83.Wu YL; Chen W; Meng FH; Wang ZJ; Cheng R; Deng C; Liu HY; Zhong ZY J. Controlled Release 2012, 164, 338. [DOI] [PubMed] [Google Scholar]
- 84.Chen W; Meng FH; Li F; Ji SJ; Zhong ZY Biomacromolecules 2009, 10, 1727. [DOI] [PubMed] [Google Scholar]
- 85.Zeng Y; Pitt WG J. Biomater. Sci. Polym. Ed 2005, 16, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dai FY; Sun P; Liu YJ; Liu WG Biomaterials 2010, 31, 559. [DOI] [PubMed] [Google Scholar]
- 87.Li YT; Armes SP Macromolecules 2005, 38, 8155. [Google Scholar]
- 88.Hu XL; Li H; Luo SZ; Liu T; Jiang YY; Liu SY Polym. Chem 2013, 4, 695. [Google Scholar]
- 89.Rodriguez-Docampo Z; Otto S Chem. Commun. (Camb) 2008, 42, 5301. [DOI] [PubMed] [Google Scholar]
- 90.Emilitri E; Ranucci E; Ferruti PJ Polym. Sci. Part A: Polym. Chem 2005, 43, 1404. [Google Scholar]
- 91.Zhu Y; Zhang J; Meng FH; Deng C; Cheng R; Feijen J; Zhong ZY J. Controlled Release 2016, 233, 29. [DOI] [PubMed] [Google Scholar]
- 92.Zou Y; Fang Y; Meng H; Meng FH; Deng C; Zhang J; Zhong ZY J. Controlled Release 2016, 244, 326. [DOI] [PubMed] [Google Scholar]
- 93.Ko H; Son S; Jeon J; Thambi T; Kwon S; Chae YS; Kang YM; Park JH J. Controlled Release 2016, 234, 68. [DOI] [PubMed] [Google Scholar]
- 94.Liu B; Thayumanavan SJ Am. Chem. Soc 2017, 139, 2306. [DOI] [PMC free article] [PubMed] [Google Scholar]