

Summary
Intragenomic conflicts are fueled by rapidly evolving selfish genetic elements, which induce selective pressures to innovate opposing repressive mechanisms. This is patently manifest in sex-ratio (SR) meiotic drive systems, in which distorter and suppressor factors bias and restore equal transmission of X and Y sperm. Here, we reveal that multiple SR suppressors in D. simulans (Nmy and Tmy) encode related hairpin RNA (hpRNAs), which generate endo-siRNAs that repress the paralogous distorters Dox and MDox. All components in this drive network are recently-evolved and largely testis-restricted. To connect SR hpRNA function to the RNAi pathway, we generated D. simulans null mutants of Dcr-2 and AGO2. Strikingly, these core RNAi knockouts massively derepress Dox and MDox, and are in fact completely male sterile and exhibit highly defective spermatogenesis. Altogether, our data reveal how the adaptive capacity of hpRNAs is critically deployed to restrict selfish gonadal genetic systems that can exterminate a species.
eTOC blurb
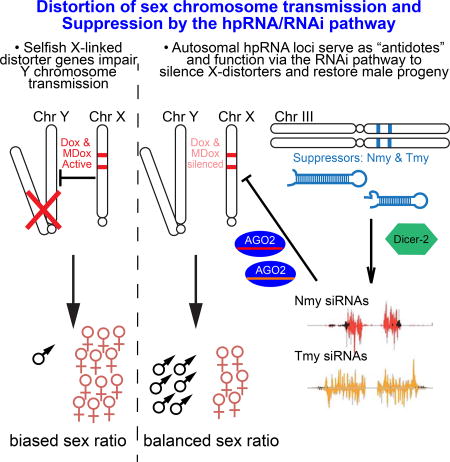
Lin and Hu et al. reveal a critical biological usage of RNAi in Drosophila simulans to tame meiotic drive systems. Multiple autosomal hairpin-siRNA loci are deployed to suppress X-encoded distorter loci that bias progeny sex ratio. These loci are rapidly evolving and testis-restricted, and such intragenomic conflicts may fuel speciation.
Introduction
RNA interference (RNAi) has long been recognized as a versatile experimental technique, but its endogenous biological utilities have been less tangible. This topic is in principle more accessible in invertebrates, several of which express diverse endogenous siRNAs (endo-siRNAs) via dedicated RNAi machinery that is distinct from the related miRNA pathway (Okamura and Lai, 2008). However, while RNAi mutants in nematodes and flies are compromised at defending viruses (Lu et al., 2005; Wang et al., 2006) and/or transposable elements (Chung et al., 2008; Czech et al., 2008; Ghildiyal et al., 2008; Kawamura et al., 2008), and affected by certain extreme environmental perturbations (Lucchetta et al., 2009), RNAi mutants generally exhibit few overt phenotypes under non-sensitized conditions.
We recently uncovered the biological logic of the Drosophila hairpin RNA (hpRNA) pathway (Czech et al., 2008; Okamura et al., 2008), in which inverted repeat transcripts preferentially generate endo-siRNAs in the testis and repress specific highly complementary mRNAs (Wen et al., 2015). Although all known hpRNAs are recently evolved, we observed clear evidence for siRNA:target co-evolution, indicating adaptive properties of this regulatory network. While ovaries detectably express hpRNAs and endo-siRNAs, RNAi mutants have relatively little consequence in females. Instead, genetic ablation of RNAi causes spermatogenesis defects and male subfertility (Wen et al., 2015). Nevertheless, D. melanogaster RNAi mutant males are fertile, suggesting this species can formally cope without siRNAs, at least within the laboratory setting.
In searching for other manifestations of the hpRNA pathway, we investigated the Winters sex ratio (SR) system of D. simulans. This meiotic drive system is absent from D. melanogaster and was born within D. simulans subclade species that diverged ~240,000 years ago (Tao et al., 2007a; Tao et al., 2007b). Despite its recent de novo appearance, Winters SR factors have profound activities. The Distorter on X (Dox) promotes X chromosome transmission by suppressing Y-bearing sperm (Tao et al., 2007a), a patently undesirable "wildtype" gene activity that must be silenced in order to maintain the D. simulans species. An antidote is encoded by autosomal Not much yang (Nmy), to which an inverted repeat with sequence similarity to Dox was mapped (Tao et al., 2007b). Signatures consistent with positive selection on Winters factors have been detected within D. simulans populations, indicating the system is actively evolving under an "arms race" scenario (Kingan et al., 2010). While the relationship of Dox and Nmy was evocative of homology-dependent silencing, there is currently no evidence (a) for molecular species constituting the active output of Nmy, (b) that Nmy directly or indirectly regulates expression of Dox, (c) whether Winters factors are truly distinct from other SR systems, or (d) that the RNAi pathway participates in SR control.
In this study, we provide first molecular evidence that hpRNA-siRNAs are functional mediators of son protection in the Winters SR system. Moreover, we reveal that a second, previously uncloned sex ratio suppressor in this species, known as the Durham SR system, involves a previously unknown hpRNA-siRNA locus termed Tmy. Although defined as genetically separable SR systems, we show that Nmy and Tmy are paralogous and have partially overlapping capacity to suppress both Dox as well as its progenitor locus MDox. To demonstrate a connection to the RNAi pathway, we employed CRISPR/Cas9 to engineer dcr-2 and ago2 null mutants in this non-model fruitfly species. Remarkably, these exhibit profound, testis-specific phenotypes that are much more severe than their well-studied D. melanogaster counterparts, in that they are completely male sterile due to profound defects in spermatogenesis progression, and harbor massive synergistic derepression of Dox and MDox transcripts, consistent with loss of collaborative suppression by Nmy and Tmy hpRNAs.
Altogether, these data demonstrate unanticipated complexity of sex distorting factors and hpRNA suppressing loci in D. simulans, all of which are rapidly evolving and none of which exist in D. melanogaster. Thus, RNAi is a key pathway that resolves intragenomic conflict that ensures species survival, and fulfills roles in adaptive gonadal gene regulation that are more commonly attributed to the piRNA pathway. This highlights the need to explore a wider range of species to appreciate more fully the evolving functions of germline small RNA pathways.
Results
The SR suppressor Nmy is a D. simulans hpRNA locus
We examined three nmy alleles SR12-2-7, nmy[5-2] and exf (Tao et al., 2007b; Yasuno et al., 2013), and validated that homozygous males are fertile, but sire extremely female-biased broods at 18°C (Figure 1A–B). Thus, loss of this individual locus is sufficient to lead to near population extinction, and these stocks require constant attention for maintenance since balancer chromosomes are not available in D. simulans. With this phenotypic confirmation, we sought evidence that Nmy might encode an hpRNA. D. simulans RNA-seq data indicated the presence of a ~2.8kb spliced transcript at the Nmy locus that is dominantly expressed in testis compared to other tissues (Figure 1C). Notably, analysis of testis small RNA data revealed that abundant small RNAs emanate specifically from the Nmy hairpin arms (Figure 1C). Although most small RNAs map to both strands of the highly self-complementary hairpin duplex, we could ascertain the transcribed strand based on the predominance of uniquely-mapping RNAseq reads (Figure 1C).
Figure 1.

The D. simulans sex ratio suppressor Nmy encodes a hairpin RNA. (A) Fertility assay and (B) sex ratio bias of progeny from D. simulans males of control w[XD1] and three independent nmy mutant alleles. Data are represented as mean ± SEM. (C) Genomics of the Nmy locus. Panel summarizes RNA-seq and small RNA data that define the primary, spliced, testis-biased Nmy transcript, and small RNAs that emanate specifically from the hairpin arms. These are siRNAs since they are mostly 21 nt (compare colored reads to "all" small RNAs in gray), and Nmy reads are missing in nmy mutants (red). Note that small RNAs map to both strands, but RNA-seq data define a single strand of transcription. A subset of Nmy reads cross-map to another autosomal location that we call Tmy (see Figure 2). Most of the cross-mapping reads (black) disappear in nmy[12-2-7], but a subset remain, indicating that they derive from Tmy. RNA-seq BigWig tracks represent normalized RNA-seq coverage. Small RNA data was normalized per million miRNAs. (D) Size distribution of Nmy and Tmy small RNAs. Small RNAs from w[XD1] testis were mapped to the Tmy and Nmy loci, and divided into reads that mapped to one of these loci or that mapped to both loci. All classes of reads showed a 21 nt bias, indicative of siRNA biogenesis. (E) Expression of Nmy and Tmy small RNAs across tissues plotted as reads per million (RPM). See also Figures S1 and S2.
Nmy small RNAs were predominantly ~21 nt RNA in length (Figure 1D), and were expressed at two orders of magnitude higher levels in testis than in ovaries, embryos or heads (Figure 1E), which are characteristics of D. melanogaster hpRNAs (Czech et al., 2008; Okamura et al., 2008). The exf mutant deletes most of the Nmy transcript including all of the hairpin, and thus abrogates possible functions of long and short RNAs. However, SR12-2-7 only removes a portion of the right hairpin arm and thus is expected to maintain the primary transcript (Figure 1C). Notably, sequencing from SR12-2-7 revealed nearly complete loss of all Nmy small RNAs, providing strong evidence that small RNAs are the functional products of Nmy (Figure 1C).
The SR suppressor Tmy encodes a newly-recognized D. simulans hpRNA
Curiously, we noticed that some Nmy small RNAs mapped to another autosomal location. This region contains a gap in the initial release D. simulans mosaic genome R1.3, and is missing in the w[501] R2.02 genome (Figure S1), due to assembly artifacts involving these repetitive regions. The genome assembly challenges are further emphasized by the fact that Nmy itself is gapped and placed on the wrong chromosome in R1.3, and remains gapped although in the correct location in R2.02 (Figure S1). Notably, the additional autosomal hit of Nmy-matching small RNAs resides within a <100kb interval to which another SR suppressor, Too much yin (Tmy), was delimited by introgressions of megabase D. mauritiana genomic segments into D. simulans (Figure 2A) over 17 years ago (Tao et al., 2001). The causal locus is unknown, and the original introgression lines are no longer extant; Tmy was also inferred to cooperate with another factor in the local vicinity to suppress hybrid sterility (Tao et al., 2001).
Figure 2.

The D. simulans sex ratio suppressor Tmy encodes an unannotated hpRNA related to Nmy. (A) Genetic map indicating examples of previously characterized introgression lines of D. mauritiana material into D. simulans that exhibit sex ratio (SR) and subfertility defects [summarized from reference (Tao et al., 2001)], which define a minimal genomic interval termed tmy. (B) This region is misassembled in publicly available genome builds (see Figure S1), but mapping to a PacBio contig reveals this region contains a bidirectionally transcribed inverted repeat (hpRNA) that is an abundant source of small RNAs. These data correspond to small RNA reads mapped from D. simulans testis SRR902009. Raw sRNA mapping density shown along a 14Mb PacBio contig, with sRNA peak corresponding to Tmy hpRNA highlighted in a yellow box and the Nmy hpRNA is shown in a blue box. PacBio contig resolves Nmy and Tmy hpRNA loci, which are in the same genomic vicinity separated by ~2 Mb on chromosome arm 3R. (C) Genomics of the Tmy region reveals an alternatively spliced transcript that is preferentially expressed in testis. Due to identical inverted repeat sequence at Tmy, mate-pairs from paired-end RNA-seq data occasionally gets misassigned to a non-proximal repeat arm. Therefore, RNA-seq data was mapped as unpaired, which identifies bidirectional transcription distinguished by uniquely mapping reads at the loop region. RNA-seq BigWig tracks represent normalized RNA-seq coverage. The Tmy hairpin yields abundant siRNAs that derive from this locus ("Tmy-only" reads, orange), with a subset of reads that cross-map to Nmy ("Tmy/Nmy-shared reads", black). Comparison of small RNA data from parallel library constructions from w[XD1] and nmy[12-2-7] testis confirm that Tmy siRNAs are maintained in the absence of Nmy. Small RNA data was normalized per million miRNAs. See also Figures S1 and S2.
Fortunately, PacBio sequencing data (A. Larracuente and J.J. Emerson, personal communication) now resolves the Tmy genomic region. Excitingly, it proved to harbor another long inverted repeat that generates small RNAs from its hairpin arms (Figure 2B). This duplex is much more self-complementary than Nmy, making it more difficult to assign the transcribed strand; the inverted repeat region is ~3.1Kb in length with almost identical sequence at the repeat arms and includes a fragmented portion of DNAREP1 (85 bp) and INVADER2 (274 bp) transposable element (TE) sequences. In between these two TE sequences, there is a 206 bp satellite DNA sequence and all three repeat sequences are identical in both arms of the inverted repeat (Figure 2B). Available RNAseq and small RNA data suggest that the spliced primary Tmy transcript is preferentially expressed in testis and does not include the distal flanking repeat elements. Nearly identical sequences of the inverted repeat arms pose a challenge to map paired-end RNA-seq data in this region, as occasionally mate-pair reads get misassigned to a nonproximal inverted repeat arm. Therefore, we treated the reads as unpaired sequences and found vast majority of reads map to both strands (Figure 2B). However, we recovered distinctive RNA-seq reads from both strands of the hairpin loop region at nearly equal frequency, providing evidence that both Tmy genomic strands are transcribed (Figure 2B).
Although we were led to this locus based on a subset of shared siRNAs, the bulk of small RNAs generated by Nmy and Tmy do not cross-map. In fact, Tmy generates a much higher-expressed population of unique siRNAs than does Nmy (Figure 1D–E) and nmy mutants maintain expression of abundant Tmy-specific small RNAs (Figure 2B). As with Nmy, all the Tmy-specific small RNAs are predominantly 21-nt in length and are strongly testis-biased (Figure 1D–E). Altogether, these data support the notion that SR phenotypes in tmy introgression lines (Tao et al., 2001) were due to loss of a newly-recognized bidirectional hpRNA, which we refer to as Tmy.
Remarkably, a single PacBio contig reveals that Nmy and Tmy are separated by ~2Mb (Figure 2A), which is reminiscent of the proximity of several homologous D. melanogaster hpRNAs into clusters (Wen et al., 2015). The ancestry of Nmy and Tmy hairpins is further indicated by their extensive homology (Figure 3A), even though the regions that can generate identical siRNAs are limited (Figure S2). Therefore, even though these loci were genetically defined by separable functions in SR control, we hypothesize that they may have overlapping activities.
Figure 3.

Biochemical evidence that Nmy and Tmy hpRNAs generate functional siRNAs. (A) The Tmy and Nmy hpRNAs share homologous segments. (B) Nmy generates 3'-modified small RNAs, that are insensitive to β-elimination. For comparison, the bantam miRNA is sensitive to this treatment while a known siRNA (hp-CG4068B) from hp-CG4068 is not. A mutant expression construct cloned from the nmy allele SR12-2-7 does not generate small RNAs. (C) Small RNA products of UAS-Nmy and UAS-Tmy can largely be distinguished using appropriate probe sets. All probes are listed in Table S2. (D) Nmy and Tmy small RNAs preferentially co-IP with the siRNA effector AGO2, while bantam miRNA associates with AGO1.
Nmy and Tmy have overlapping capacity to regulate Dox and MDox targets
Our molecular genetic evidence strongly supports the notion that Nmy and Tmy are hpRNA-siRNA loci (Figure 3A). To provide direct biochemical validation for this, we generated inducible (UAS) expression constructs bearing wildtype Nmy and mutant nmy isolated from the SR12-2-7 strain. We assayed them with ub-Gal4 activator in S2 cells from D. melanogaster, a non-cognate species that does not encode either of these loci. Northern blotting demonstrated production of small RNAs only from wildtype Nmy (Figure 3B). Moreover, ß-elimination increased the mobility of bantam miRNA, but not that of hp-CG4068 and Nmy small RNAs, indicating their 3' methylation as expected for siRNAs. We also generated a UAS-Tmy construct, and identified probes that verified largely distinct small RNA output of Nmy and Tmy hpRNAs (Figure 3C). Using these probes, we conducted Argonaute immunoprecipitation tests that showed Nmy and Tmy small RNAs load preferentially into AGO2, in contrast to the miRNA bantam that associates preferentially with AGO1 (Figure 3D). Thus, Nmy and Tmy are genuine endo-siRNA loci.
We used RNA-seq data to define the Dox transcription unit, which is preferentially expressed in testis compared to several other tissues (Figure S3). Both Nmy and Tmy hairpins share antisense homology to Dox (Figure 4A). However, Tmy exhibits a substantial complementary segment to the 5' region of Dox that is not shared by Nmy (Figure 4A), consistent with the interpretation that it provides an additional activity in SR control. We observe that abundant Nmy/Tmy siRNAs map perfectly to Dox (Figure S3), indicating that it is under direct control by these hpRNAs. Dox is related to an apparent progenitor locus MDox (Tao et al., 2007a; Tao et al., 2007b), which is not known to participate in SR distortion but is also located on the X chromosome (Figure S3). RNA-seq data shows that MDox is also testis-biased and also targeted by Nmy/Tmy, since siRNAs derived from these hpRNAs map perfectly to MDox (Figure 4B). We show the overall homology relationships of the SR hpRNA/target network (Figure 3E–F), detailed alignments (Figure S2), and illustrate the region of highest antisense complementarity between Nmy and Tmy hairpin arms and the transcribed strands of Dox and MDox, with exemplar Nmy-specific, Tmy-specific, and shared siRNAs indicated (Figure 4C). In this figure we emphasize perfect mappers, but note certain siRNAs that map with limited mismatches (e.g. the "blue" Tmy-specific siRNAs, Figure 4C); we address these in a following analysis.
Figure 4.

Nmy and Tmy hpRNAs have partially overlapping capacity to target a network of sex ratio distorter genes. (A–B) Homology relationships amongst complementary arms of Tmy/Nmy with the transcribed strands of Dox (A) and MDox (B). Note that Nmy and Tmy bear segments of antisense co-targeting to both Dox and MDox, but that Tmy has additional extensive complementarity to the 5' region of Dox that is not shared with Nmy (A). The relative numbers of Nmy-derived siRNAs that map perfectly to Dox and MDox is higher than with Tmy, suggesting it is a primary endogenous suppressor of these factors. Still, substantial perfect targeting by Tmy-derived siRNAs to Dox and MDox remains in nmy[12-2-7] testis, consistent with the notion that Tmy plays an overlapping role in their suppression. (C) Alignment of illustrative hairpin arm regions of Tmy and Nmy that exhibit extensive antisense complementarity to the Dox and MDox target genes. Selected individual siRNAs that map perfectly to Tmy (yellow), Nmy (pink) or both hpRNAs (orange) are indicated. (D) Both Tmy and Nmy hpRNAs suppress both Dox and MDox sensors, while mutant nmy and tmy constructs and non-cognate hpRNA1 construct lack activity on these sensors. Unpaired t-tests were used to calculate statistical significance. See also Figures S3 and S4.
Despite substantial homologies across this hpRNA/target network, we note higher antisense matching between Nmy and the Dox open reading frame (light orange and purple regions, Figure 4A). Correspondingly, many more Nmy-specific than Tmy-specific siRNAs cross-map to the Dox coding sequence, suggesting that Nmy might be a dominant regulator of Dox. We could confirm that these derive from Nmy, since they mostly disappeared when sequencing small RNAs from nmy[12-2-7]. Nevertheless, we observe that siRNAs still map to Dox in nmy mutants, and these derive from Tmy. These obviously include all Tmy-specific siRNAs, including those from the segment complementary to the 5' UTR region of Dox that is not shared by Nmy. Interestingly, we can also distinguish a population of Tmy/Nmy-shared siRNAs that cross-map to the 5' UTR region of Dox (colored as dark orange in the model picture of Figure 4A). Their provenance in wildtype would be ambiguous, but comparison to nmy mutant small RNAs allows us to assign that ~19% of apparent Tmy/Nmy-shared siRNAs that cross-map to this region actually derive from Tmy. Altogether, these data provide molecular evidence that Tmy is a likely co-regulator of Dox. Similar analyses indicate that Tmy likely coregulates MDox (Figure 4B).
To account for targeting more fully, we examined whether there were siRNAs that were complementary to Dox or MDox splice junctions. Indeed, we could identify a population of such reads, and depict their alignments in Figure S4. Because the duplex hpRNA duplex sequence is similar to the target sequence, we recovered Nmy/Tmy siRNAs that map both sense and antisense to Dox/MDox splice junctions of exons 1/2 and exons 2/3 (Figure S4). Also, since full siRNA:target pairing is not needed for AGO2-mediated target regulation (Haley and Zamore, 2004), the impact of Nmy/Tmy regulation on Dox/MDox may extend beyond perfect matching (see Figure 4C). Although the rules for siRNA targeting are not fully known, we implemented relaxed mapping with up to 3 mismatches, with the proviso of perfect pairing in an extended seed region that is continuous with the cleavage site (from siRNA nts 2–13). Under these conditions, we observed a moderate increase in Nmy/Tmy siRNAs with antisense matching to Dox/MDox (Figure S4); however, by far the strong majority of siRNAs match perfectly to these targets.
To provide experimental support for these regulatory relationships, we tested sufficiency of Nmy and Tmy hpRNA expression constructs for their ability to suppress luciferase sensors bearing the co-targeted regions of Dox or MDox in S2 cells. We compared these to a nmy[12-2-7] mutant construct or a tmy mutant bearing only one hairpin arm, as well as to empty or non-cognate (hpRNA1) constructs. We observed specific repression of both targets by both wildtype D. simulans hpRNAs, while mutant nmy and tmy constructs were non-functional (Figure 4D). Overall, these findings support the notion that a network of siRNA-generating loci mediate SR control in D. simulans, working to suppress multiple distorter loci.
We sought in vivo evidence for hpRNA-mediated target suppression. Although Dox and MDox share substantial homology, we were able to design unique primer pairs that amplify specific transcript amplicons (Figure S2). Using biologically replicate testis samples from the three independent alleles of nmy, we observed strong upregulation of both Dox (~10 fold, Figure 5A) and MDox (3–8 fold, Figure 5B). This constitutes the first direct evidence for endogenous deregulation of either target loci in nmy hpRNA mutants, conditions associated with strong depletion of male progeny (Figure 1A).
Figure 5.

Nmy and Tmy hpRNAs suppress SR distorters via a testis-directed RNAi pathway in D. simulans. (A–B) Testis qPCR tests of Dox (A) and MDox (B) show strong derepression in testis of three independent nmy alleles, and massive derepression in null alleles of RNAi factors dcr-2 and ago2. Data are represented as mean ± SD. (C–D) Strategy to generate deletion alleles of D. simulans dcr-2 and ago2 by replacing their coding regions with 3xP3-DsRed. (E–F) Validation of DsRed+ dcr-2 and ago2 mutants, with individual fly PCR demonstration of on-target knockin of the donor cassette using both left (P1/P2) and right arm (P3/P4) amplicons, and absence of dcr-2 or ago2 amplicon and presence of control amplicon. (G) Null alleles of dcr-2 and ago2 show only mild effects on female fertility; dcr-2-1 and dcr-2-2 are different deletions of dcr-2 (see also Figures S5 and S6). Data are represented as mean ± SEM. (H) Null alleles of dcr-2 and ago2 are absolutely male sterile. Data are represented as mean ± SEM.
D. simulans core RNAi pathway mutants exhibit profound male-specific defects
A rigorous connection of Nmy and Tmy functions to the RNAi pathway requires in vivo genetic evidence. We exploited CRISPR-Cas9 engineering to delete the core RNAi factors dcr-2 and ago2 and replace them with 3xP3-DsRed (Figure 5C–D); we used two donors to delete different regions of dcr-2 (Figure S5). For all experiments, we recovered multiple founders that expressed DsRed and passed this through the germline, and were validated as bearing on-target gene replacement by hybrid PCRs on both flanks and deleted the target genes (Figures S5–6). Because D. simulans lacks balancer chromosomes, and eye fluorescence could be used to track flies bearing a replacement allele (Figure 5E–F) but was not fully reliable to distinguish heterozygotes from homozygotes, we confirmed mutants based on individual fly genotyping for experiments (Figure 5E–F). These analyses showed that RNAi is dispensable for viability in D. simulans, as is the case in D. melanogaster.
These RNAi mutants maintained female fertility that was only slightly lower than that of control w[XD1] background used for CRISPR mutagenesis (Figure 5G). In sharp contrast, males of multiple independent alleles of all three D. simulans dcr-2 and ago2 mutants assayed were completely sterile (Figure 5H). Thus, RNAi is of greater overt phenotypic impact in this non-model fruitfly than in D. melanogaster. Moreover, we note that D. melanogaster mutants of the central primary piRNA effector P element-induced wimpy testis (piwi) are male fertile (Ku et al., 2016). Therefore, in terms of species propagation, RNAi is more critical for D. simulans males than primary piRNAs are for D. melanogaster males.
We performed cytological analyses to understand the basis of male infertility in D. simulans RNAi mutants, and to compare this to nmy mutants, which are fertile but sex-biased (Figure 1A). Indeed, we observed severe disruptions in spermatogenesis that were phenocopied by both dcr-2 mutants as well as ago2 mutants, indicating common biological consequences to loss of RNAi function in vivo (Figure 6A–D).
Figure 6.

Abnormal spermiogenesis in hpRNA/RNAi mutants. (A–H) Whole testes stained for DNA (blue), histones (red) and alpha-tubulin (green), showing that the normal pattern of spermatogenesis in control (A) is present in nmy (B) but severely disrupted in RNAi mutants dcr-2 (C) and ago2 (D). Scale bar: 100µm. (E–H) The apical tip of the testes containing the stem cell hub in wildtype (E) appears normal in nmy[5-2] (F), dcr-2 (G) and ago2 (H) mutants. Scale bar: 50µm. (I–L) Whole testis stained for F-actin to highlight individualization complexes (IC) that separate spermatids into individual cells to form wastebags (WB). These actin structures found in control (I) and nmy (J) are completely absent in dcr-2 (K) and ago2 (L) mutants. Scale bar: 50µm. (M–P) Closeups of post-meiotic spermatid nuclei. (M) In control testes, cysts of 64 interconnected spermatid nuclei after the histone-to-protamine transition are visible (arrows). (N) Closeup of nmy cysts that have completed histone-to-protamine transition. Two types of nuclei are visible, ones with normal canoe-stage spermatids and others with abnormal leaf shapes (arrowheads) that likely carry the Y chromosome. In dcr-2 (O) and ago2 (P) mutants, cysts are disorganized, spermatid nuclei are abnormally shaped and scattered in the testis, and histones remain associated. Scale bars: 10µm; note scale of panel (N) is enlarged relative to (M,O,P). (Q–T) Seminal vesicles. In control males (Q), the seminal vesicle is filled with sperm as revealed by the presence of needle-shape nuclei. (R) nmy exhibits reduced seminal vesicle contents. In contrast, seminal vesicles in dcr-2 (S) and ago2 (T) mutants are empty and only the round nuclei of the seminal vesicles walls are seen, indicating that no mature sperm are generated. Scale bar: 10µm. See also Figure S7.
We investigate the basis of defective spermatogenesis in detail. We first analyzed the testis apical tip where the stem cell niche resides; this appeared normal in nmy and RNAi mutants (Figure 6E–H). However, while the rest of the testis was normal in nmy, RNAi mutants exhibited severe disorganization. As normal spermatid cysts mature to generate individual sperm, they extrude their cytoplasm in a coordinated fashion via actin-rich cones that are collectively referred to as individualization complexes (ICs), eventually forming wastebags (WBs, Figure 6I). While both structures are present in nmy (Figure 6J), no ICs or WBs can be observed in any of the RNAi mutants, and only scattered actin cones can be seen (Figure 6K–L).
Closer examination of spermatid nuclei was informative. After wildtype meiosis, the cysts of 64 spermatid nuclei that mature in synchrony are visible (Figure 6M). In nmy mutants, we observed substantial post-meiotic nuclei with aberrant morphology that likely reflect Y-bearing spermatids (Figure 6N, arrowheads). The phenotype of RNAi mutants was more severe, as they exhibit aberrant meiotic divisions which produce spermatid nuclei that are abnormally shaped, frequently connected by chromatin threads and scattered. Moreover, histones remained associated, implying failure of protamine replacement (Figure 6O–P). In the end, nmy mutants exhibit a moderate reduction of mature sperm in the seminal vesicle, but dcr-2 and ago2 mutants fail to generate any mature sperm (Figure 6Q–T). We observed a similar suite of phenotypes in the other dcr-2 deletion allele, but not in heterozygotes (Figure S7). Thus, loss of RNAi is much more severe than loss of the single hpRNA Nmy.
The complete spermatogenesis defect and male sterility of D. simulans RNAi mutants prevented us from assessing SR bias in their progeny, and potentially hints at broader intragenomic conflicts that involve Tmy [i.e. hybrid sterility (Tao et al., 2001)]. We explored different strategies to obtain RNAi mutant progeny (e.g. by altering temperature or by aging) but these did not bypass sterility under any conditions tested; progeny of dcr-2/ago2 double heterozygotes also exhibited normal sex ratio. Nevertheless, we observed a profound and common molecular phenotype that links RNAi mutants to nmy mutants with respect to SR suppression. In particular, qPCR analysis revealed massive derepression of Dox (~50 fold) and MDox (~15–20 fold) transcripts in both dcr-2 and ago2 mutant testes, levels that were much greater than any nmy mutant background (Figure 5A–B). These data affirm that MDox is likely a functional distorter gene, which has not previously been suggested. More importantly, they strongly support the scenario that multiple hpRNAs (i.e., Nmy and Tmy) provide overlapping defense against both distorter loci during SR control.
Overall, our molecular analysis of the Winters and Durham systems reveal striking biological utilizations and functional necessities for RNAi in the control of intragenomic conflict, especially given (1) how rapidly evolving these sex ratio distorter systems are, and (2) how potent the consequences of loss of hpRNA-mediated control are in terms of population extinction.
Discussion
This study provides critical linkages amongst the RNAi pathway, hpRNA biogenesis and function, and suppression of SR bias. The evolutionary behavior of SR systems conforms to a "Red Queen" effect (Figure 7A), in which a seemingly static outcome (equal transmission of X and Y sperm) actually involves intense, opposing and rapidly-evolving, genetic programs (Figure 7B). Our studies provide striking evidence that endogenous RNAi is a central molecular pathway that resolves SR distortion and may potentially have impact on hybrid sterility. We provide molecular and genetic evidence of a potential hierarchy, in that Dox is a prime direct target of Nmy based on the observation that it supplies the majority of targeting siRNAs. Nevertheless, Tmy provides a secondary defense, since Tmy exhibits extensive complementarity to Dox that is non-overlapping with Nmy, Tmy siRNAs maintain targeting to Dox even in nmy mutants, Tmy can directly repress Dox in sensor assays, and most importantly, RNAi mutants exhibit strongly elevated Dox transcripts in testis, consistent with co-targeting endogenous action of both hpRNAs on Dox. Moreover, we show these principles to be true for MDox, strongly implying this locus as a functional distorter. Overall, we reveal that multiple evolutionary nascent hpRNAs (Nmy/Tmy) are individually required for species preservation via suppression of Winters and Durham SR (Dox/MDox) distorters in D. simulans (Figure 7B).
Figure 7.

Continuous cycles of sex ratio distortion and suppression via the hpRNA/RNAi pathway mediate balanced sex ratio. (A) A John Tenniel illustration from Lewis Carroll's "Through the Looking-Glass", where the Red Queen says to Alice, "Now, here, you see, it takes all the running you can do, to keep in the same place". Leigh Van Valen proposed this as a metaphor for a rapidly evolving genomic conflict, or arms race scenario, where the net outcome is to maintain the status quo. (B) In the case of this study, the seemingly static outcome equal male and female progeny is driven by an intense battle between the sex chromosomes, in which the X continually attempts to gain advantage over the Y via de novo meiotic X-linked distorter factors (e.g., Dox and MDox). The dramatic loss of Y transmission creates a strong pressure to innovate suppressors, which we have shown to be hpRNA loci that function via the RNAi pathway (e.g. Nmy and Tmy). In general, all known Drosophila hpRNAs are evolutionarily young, and consistent with this model, their action is only needed as long as the distorters are active. Once the distorter is inactivated, there is no selective pressure to maintain the hpRNA. Note that multiple cycles of distortion and suppression may occur in parallel.
In the future, further dissection of the genetic contributions of the individual hpRNAs and distorters will shed light on their relative contributions to Winters and Durham SR systems, and the extent to which they are distinct systems, or partially overlapping as indicated by our studies. This will be a challenge in a non-model fly that lacks the genetic tools available in D. melanogaster, but will be important to understand how these newly emerging factors are endowed with such powerful activities. For example, compelling hypotheses to test are whether specific deletions of the Tmy hairpin alone can recapitulate SR bias, whether Tmy exhibits derepression of other distorter factors, and whether nmy/tmy double mutant might exhibit only SR, or may prove to recapitulate sterility found in RNAi mutants. Thus far, Nmy and Tmy loci have proven recalcitrant to repeated attempts for CRISPR/Cas9 targeting, and it is not clear whether something about their repeat structure affects this endeavor. Moreover, the close linkage of these loci will be a challenge for any efforts to generate recombinants. Still, it will be worthwhile to pursue the generation of new genetic tools.
Remarkably, there is a third SR distortion system in D. simulans ("Paris"). While it is genetically complex, it was recently shown to depend on HP1D2, a recently-evolved paralog of the piRNA factor Rhino (Helleu et al., 2016). Thus, there are apparently molecular linkages of sex-ratio systems with small RNA systems, on both driving and suppressing sides. Although the mechanism of Paris SR remains to be determined, HP1D2 protein localizes to the heterochromatic Y chromosome, which provides a connection to the observation that driving Paris alleles prevent segregation of Y chromatids during meiosis II (Helleu et al., 2016). On the other hand, the defect in Winters sex-ratio appears to be post-meiotic (Tao et al., 2007a; Tao et al., 2007b), indicating mechanistic diversity in how depletion of male sperm might be achieved by de novo genes.
On the other hand, the sister species D. melanogaster appears to lack SR systems, testament to the extremely rapid rise and fall of SR systems during evolution. The adaptive properties of hpRNAs make them ideal genetic elements to tame such selfish meiotic drive elements, which are theorized to be under constant cycles of emergence, suppression and disappearance (Jaenike, 2008; Willis, 2009). There is mounting evidence that genetic systems that manifest in SR defects are often associated with sterility (Phadnis and Orr, 2009; Zhang et al., 2015). Our findings support the notion that the success or failure to resolve intragenomic conflicts using RNAi would intimately be connected to speciation.
While these roles were unexpected in light of the fact that metazoan RNAi biology has been so challenging to appreciate, the situation would have been different had a species only slightly diverged from D. melanogaster initially been selected as a genetic model. This parallels the inference that RNAi might have been recognized earlier, had certain budding yeasts other than S. cerevisiae been studied earlier (Drinnenberg et al., 2009). Indeed, functional studies across a broader phylogeny will be necessary to appreciate the evolving requirements of small RNA regulation, beyond standard model organisms (Sarkies et al., 2015). The availability of our D. simulans RNAi mutants opens the door to molecular identification of novel selfish genetic elements that induce SR bias and/or hybrid sterility, in both the Winters and Durham systems.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eric Lai (laie@mskcc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Drosophila simulans
w[XD1] was obtained from Bestgene, Inc. and used as the control strain for all the analyses in the paper. For nmy mutants, SR12-2-7 and nmy[5-2] were provided by Yun Tao and e exf p by Masatoshi Yamamoto. RNAi mutants, dcr-2 and ago2 are generated as described below in the main STAR Methods text. All stocks were maintained at 18°C or 25°C, as described.
Cell lines
We acquired S2R+ from DGRC and the cell were maintained at 25°C in Schneider’s medium containing 10% FBS.
METHOD DETAILS
Fertility and sex ratio tests
To obtain males for the assay, bottles were set up at 25°C to collect eggs of indicated genotypes for 2 days and transferred to 18°C until adult flies emerged. 5-dayold individual males were mated to 5 w[XD1] virgins and 5-day-old individual females were mated to 3 w[XD1] males. All vials were raised in 18°C and flipped every other day for 2 weeks. Progeny from 7 vials were counted for data analysis.
Small RNA library preparation
For small RNA analysis, we extracted RNA from testes and accessory glands of 7-day-old D. simulans w[XD1] and nmy mutants using Trizol (Invitrogen). 1 µg of total RNA were used to prepare small RNA libraries largely as previously described (Lee and Yi, 2014). Adenylation of 3’ linker was performed in a 40 µL reaction at 65°C for 1 hr containing 200 pmol 3’ linker, 1× 5’ DNA adenylation reaction buffer, 100 nM ATP and 200 pmol Mth RNA ligase and the reaction is terminated by heated to 85°C for 5 min. Adenylated 3’ linker was then precipitated using ethanol and was used for 3’ ligation reaction containing 10% PEG8000, 1× RNA ligase buffer, 20 µM adenylated 3’ linker and 100 U T4 RNA Ligase 2 truncated K227Q. The 3’ ligation reaction was performed at 4°C for overnight and the product were purified using 15% Urea-PAGE gel. The small RNA-3’ linker hybrid was then subjected to 5’ ligation reaction at 37°C for 4 hr containing 20% PEG8000, 1× RNA ligase buffer, 1 mM ATP, 10 µM RNA oligo, 20 U RNaseOUT and 5 U T4 RNA ligase 1. cDNA synthesis reaction was then proceeded immediately by adding following components to the ligated product: 2 µl 5× RT buffer, 0.75 µl 100 mM DTT, 1 µl 1 µM Illumina RT Primer, and 0.5 µl 10 mM dNTPs. The RT mix was incubated at 65µC for 5 min and cooled to room temperature and transfer on to ice. 0.5 µL of superscript III RT enzyme and 0.5 µL RNase OUT were added to the RT mix and the reaction was carried out at 50°C for 1 hr. cDNA lib raries were amplified using 15 cycles of PCR with forward and Illumina index reverse primers and the amplified libraries were purified by 8% non-denaturing acrylamide gel. Purified libraries were sequenced on HiSeq2500 using SR50 at the New York Genome Center. Oligo sequences and reagents used for library preparation are provided in Table S2 and Key Resources Table, respectively.
Small RNA analysis
We aggregated small RNA libraries data mentioned above with other public datasets (Table S1). sRNA reads generated in this study were processed as follows: Raw sequence reads were adapter trimmed using fastx toolkit (fastx_clipper -a TGGAATTCTCGGGT –Q 33 -i library_name.fastq. After filtering <15 nt reads using a custom shell script, we mapped the data to both D. simulans r2.02 from Flybase and PacBio contigs using the Bowtie2 software (very-sensitive-local; -D 20 -R 3 -N 0 -L 20 -i S,1,0.50). We mapped >15nt sRNAs to both r2.02 and PacBio assembly and normalized them per million mapped miRNAs from the respective libraries. The resulting small RNA alignments in SAM format were converted to BED for downstream processing using the BEDops software. To identify small RNA mappings with mismatches at nucleotide positions 1 and > 13 (no mismatches allowed in the seed region 2–13nt), small RNA alignment SAM file from bowtie mapping (with -v3 –best –strata options) was filtered using a custom shell script and converted to BED format for downstream processing and visualization. For public small RNA datasets described in this study, the data was mapped using Bowtie (with –v0 –best –strata options) to Pacbio assembly and the reads were normalized to million reads in their respective libraries.
Total RNA library preparation
We extracted RNA from testes (dissected free of accessory glands) of 7-day-old D. simulans w[XD1] flies using Trizol (Invitrogen). Illumina Truseq Total RNA library Prep Kit LT was used to make stranded RNA-seq libraries from 650 ng of total RNA. Manufacturer’s protocol was followed except for using 8 cycles of PCR to amplify the final library instead of the recommended 15 cycles, to minimize artifacts caused by PCR amplification. All samples were pooled together using the barcoded adapters provided by the manufacturer and the sequencing was performed by HiSeq2500 instrument using PE75 at the New York Genome Center.
RNA-Seq analysis
We supplemented our RNA-seq data with analysis of publicly available D. simulans RNA-seq data from various tissues (Table S1). The data was mapped to both D. simulans r2.02 and PacBio contigs using hisat2 aligner with default parameters. For annotated gene models available from FlyBase release r2.02, we obtained strand-specific mapped read counts using the featureCounts software (Liao et al., 2014). The read counts were normalized to FPKM using the DEseq2 package in R (Love et al., 2014). As genomic sequences of hpRNAs nmy and tmy were not assembled properly in r2.02, we quantified their expression using PacBio mappings. To visualize RNA-seq mapping in the IGV genome browser, we converted the sequence alignments in bam format to UCSC BigWig format using the BamToWig.py tool in the RSeqQC package (Wang et al., 2012).
Generation of hpRNA expression constructs
Nmy and nmy (mutant) expression constructs were cloned from w[XD1] and 12-2-7 strains, respectively, into the 3' UTR site of pUAST-DsRed. For Tmy construct, the left arm and right arm fragments were amplified separately from w[XD1] genomic DNA, and then three-way ligation was performed to clone both arms into the 3' UTR site of pUAST-DsRed. To avoid recombination, the pUAST-DsRed-Tmy construct was transformed into NEB stable competent cells. The primers used for cloning are listed in Table S2.
β-elimination test
To analyze small RNA 3' termini, S2R+ cells were transfected with UAS-Tmy, UAS-Nmy or UAS-nmy constructs using Effectene transfection reagent (QIAGEN) for 3 days and total RNAs were extracted using Trizol. RNAs were then treated with sodium periodate in borax/boric-acid buffer followed by sodium hydroxide treatment, and Northern blotting analysis as described (Okamura et al., 2008). Probes for blotting are listed in Table S2.
Argonaute immunoprecipitations
To analyze Argonaute loading, we transfected hpRNA constructs into FLAG-HAAGO2 stable S2R+ cell using Effectene transfection reagent (QIAGEN) for 3 days. We then followed the IP protocol described in (Okamura et al., 2009). Briefly, after washing with PBS, 107 cells were lysed with 500 µL of Ago IP buffer (30 mM HEPES pH7.5, 150 mM KOAc, 2 mM Mg(OAc)2, 5 mM DTT, 0.1% NP40, 1× Protease Inhibitor(Roche) and 20U RNaseOUT). Cleared cell lysates were first incubated with Flag-M2-conjugated beads and supernatant from the first IP was then used for AGO1-IP. Beads were washed five times using lysis buffer. Bound RNA was extracted from beads using Trizol, and was analyzed by small RNA Northern blotting describing below.
Small RNA Northern blotting
RNA was run on 15% urea acrylamide gel (SequaGel UreaGel System, National Diagnostics) in 1× TBE buffer, and transferred to GeneScreen Plus membrane (Perkin Elmer) in 0.5× TBE buffer for 1 hr. After UV-crosslinking, the membrane was incubated with prehyb buffer for 1 hr and was hybridized with gamma-P32 labeled probes for overnight at 50°C. Probe sequences are listed in Table S2.
Luciferase sensor assays
Dox and MDox sequences were amplified from w[XD1] into psiCHECK luciferase sensors. All the primers used for cloning are listed in Table S2. Other hpRNA constructs were described previously (Okamura et al., 2008). To perform sensor assays, 105 S2 cells were seeded per well of 96-well plate and transfected with 25 ng UAS-DsRed or UAS-DsRed hairpin constructs, 50 ng psiCHECK sensors and 12.5 ng Ub-Gal4. Luciferases were measured 3 days after transfection using Dual Glo luciferase assay system (Promega) and Cytation5 (BioTek). The fold repression values were normalized to empty psiCHECK and UAS-DsRed group. Individual tests were done in quadruplicate and the averaged values from three biological replicate samples were subjected to statistical analysis.
CRISPR/Cas9 Mutagenesis of D. simulans
sgRNAs were designed using http://flycrispr.molbio.wisc.edu/, and were designed to delete most of the target gene and replace it with a 3xP3:DsRed marker. sgRNA oligo pairs were phosphorylated by T4 PNK (NEB), annealed and ligated into BbsI digested pDCC6 vector using T4 DNA ligase (NEB). Homology arms of ago2 and dcr-2 were amplified from genomic DNA and cloned into pHD-DsRed-attp; two different donor arm combinations were used for dcr-2 to generate dcr-2-1 and dcr-2-2 alleles. All primers used to clone sgRNAs, validate sgRNA target sites, and clone donor constructs are listed in Table S2.
100 ng/µL of pDCC6-sgRNA constructs and 100 ng/µL donor plasmid were mixed for injection into D. simulans w[XD1] embryos (BestGene Inc, Chino Hills). Flies were cultured on standard cornmeal food at 25°C. To score for germline mutations, G0 adult flies were crossed to w[XD1] flies and F1 progeny were screened for DsRed+ eyes under a Leica fluorescent microscope. Primers used to screen for on-target hybrid PCR products on the left and right flanks at ago2 and dcr-2 are listed in Table S2, as are primer pairs to validate deletion of internal gene segments at each locus in homozygous mutants.
qPCR
Because D. simulans lacks balancer chromosomes, and the 3xP3:DsRed marker was not completely reliable for identifying homozygous mutants, we found it was necessary to genotype flies to confirm mutants prior to pooling RNA samples for further analysis. Therefore, we collected 7-day-old putative dcr-2 or ago2 mutant males based on DsRed eye intensity, and dissected their reproductive systems (testis and accessory gland) into PBS. We genotyped their respective thoraces, and prepared total RNA from combined, confirmed, mutant reproductive systems using Trizol (Invitrogen) and DNase treatment using TURBO DNA-free™ kit (Ambion). cDNA was synthesized using SuperScript III First-Strand Synthesis System with random primers (Invitrogen). qPCR reactions were performed with CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad). Primers for Dox, MDox and control Rpl32 are listed in Table S2.
Testis cytology
For each mutant genotype, 0–4 day old DsRed positive males were dissected and the mutant testes were recognized with the empty seminal vesicle and disrupted spermatogenesis. Whole-mount testes were stained as described (Dubruille and Loppin, 2015). Briefly, testes were dissected in PBS-T (1× PBS with 0.15% Triton), fixed in 4% formaldehyde 1× PBS for 20 min, washed 3 times in PBS-T before 4°C overnight incubation in primary antibodies. Primary antibodies were mouse IgG2a monoclonal anti-pan-histone, clone F152 (1:1000; Millipore), mouse IgG1 anti-alpha-tubulin clone DM1A (1:1000; Sigma-Aldrich). Then, they were washed 3 times for 20 min in PBS-T prior to incubation with secondary antibodies for 2–3 hours at room temperature. We used Alexa Fluor-coupled goat anti-mouse IGg1 and IgG2a (1:1,000; Jackson ImmunoResearch). Samples were then washed 3 times for 20 min in PBS-T before mounting.
For F-actin staining, testes were incubated in phalloidin diluted in 1×-PBS (1:100) and incubated at room temperature for 30 min. Testes were then washed again and incubated for 30 min at 37°C in 2mg/mL RNase A, rin sed and mounted in mounting medium containing 5 µg/mL propidium iodide (Sigma-Aldrich) to stain DNA. For other stainings, DNA was stained by directly mounting tissues in mounting medium (Dako) containing 10µg/mL DAPI (Sigma-Aldrich).
Images were acquired on a LSM800 confocal microscope (Carl Zeiss) using the Zen software in .czi format then converted to .tiff using Zen Digital Imaging or FIJI software.
Quantification and statistical analysis
To evaluate statistical significance for sensor assays and qPCR tests, we used unpaired Student’s t-test to calculate p-values. The error bars shown in fertility assay represent standard error (SEM), and standard deviation (SD) in sensor assays and qPCR tests.
Data and software availability
All of the raw small RNA and RNA-seq data reported in this study were deposited in the NCBI Short Read Archive (SRA) portal under the BioProject ID PRJNA477366.
Supplementary Material
Highlights.
Selfish sex ratio (SR) distorters impede son transmission, and must be silenced
Multiple SR suppressors in D. simulans correspond to hairpin RNA-siRNA loci
Knockout of RNAi pathway derepresses SR drivers and disrupts spermatogenesis
Rapidly evolving intragenomic conflicts and suppression by RNAi may underlie speciation
Acknowledgments
We are grateful to Yun Tao for discussion, and members of the simulans clade PacBio sequencing consortium (J. J. Emerson, Amanda Larracuente, Colin Meiklejohn and Kristi Montooth) for access to D. simulans PacBio data in the Tmy region. We thank Zhigang Jin, Lijuan Kan, Alex Flynt, and Rui Gao for helping to prepare D. simulans samples and Nicolas Robine for initial analysis of small RNA data. We thank Masatoshi Yamamoto and San Diego Drosophila Stock Center for fly stocks. Work in BL’s group was supported by a grant to RD from the Agence National de la Recherche (ANR-16-CE12-0006-01). Work in ECL’s group was supported by the National Institutes of Health (R01-NS083833 and R01-GM083300), BSF-2015398, and MSK Core Grant P30-CA008748.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
C.J.L. and F.H. performed the molecular and genetic analyses; F.H. performed the D. simulans CRISPR mutagenesis. R.D. analyzed testis cytology in mutants. J.V. and J.W. performed computational analyses. P.S. generated constructs and initiated the project. B.L. and E.C.L. supervised analysis and interpreted results, and E.C.L. wrote the manuscript with input from co-authors.
Declaration of Interests
The authors declare no competing interests.
References
- Chung WJ, Okamura K, Martin R, Lai EC. Endogenous RNA interference provides a somatic defense against Drosophila transposons. Current Biology. 2008;18:795–802. doi: 10.1016/j.cub.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech B, Malone CD, Zhou R, Stark A, Schlingeheyde C, Dus M, Perrimon N, Kellis M, Wohlschlegel J, Sachidanandam R, et al. An endogenous siRNA pathway in Drosophila. Nature. 2008;453:798–802. doi: 10.1038/nature07007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drinnenberg IA, Weinberg DE, Xie KT, Mower JP, Wolfe KH, Fink GR, Bartel DP. RNAi in budding yeast. Science. 2009;326:544–550. doi: 10.1126/science.1176945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubruille R, Loppin B. Protection of Drosophila chromosome ends through minimal telomere capping. Journal of cell science. 2015;128:1969–1981. doi: 10.1242/jcs.167825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghildiyal M, Seitz H, Horwich MD, Li C, Du T, Lee S, Xu J, Kittler EL, Zapp ML, Weng Z, et al. Endogenous siRNAs Derived from Transposons and mRNAs in Drosophila Somatic Cells. Science. 2008;320:1077–1081. doi: 10.1126/science.1157396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley B, Zamore PD. Kinetic analysis of the RNAi enzyme complex. Nature structural & molecular biology. 2004;11:599–606. doi: 10.1038/nsmb780. [DOI] [PubMed] [Google Scholar]
- Helleu Q, Gerard PR, Dubruille R, Ogereau D, Prud'homme B, Loppin B, Montchamp-Moreau C. Rapid evolution of a Y-chromosome heterochromatin protein underlies sex chromosome meiotic drive. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:4110–4115. doi: 10.1073/pnas.1519332113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenike J. X chromosome drive. Curr Biol. 2008;18:R508–511. doi: 10.1016/j.cub.2008.03.051. [DOI] [PubMed] [Google Scholar]
- Kawamura Y, Saito K, Kin T, Ono Y, Asai K, Sunohara T, Okada T, Siomi MC, Siomi H. Drosophila endogenous small RNAs bind to Argonaute2 in somatic cells. Nature. 2008;453:793–797. doi: 10.1038/nature06938. [DOI] [PubMed] [Google Scholar]
- Kingan SB, Garrigan D, Hartl DL. Recurrent selection on the Winters sex-ratio genes in Drosophila simulans. Genetics. 2010;184:253–265. doi: 10.1534/genetics.109.109587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku HY, Gangaraju VK, Qi H, Liu N, Lin H. Tudor-SN Interacts with Piwi Antagonistically in Regulating Spermatogenesis but Synergistically in Silencing Transposons in Drosophila. PLoS genetics. 2016;12:e1005813. doi: 10.1371/journal.pgen.1005813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Yi R. Highly efficient ligation of small RNA molecules for microRNA quantitation by high-throughput sequencing. Journal of visualized experiments : JoVE. 2014:e52095. doi: 10.3791/52095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Maduro M, Li F, Li HW, Broitman-Maduro G, Li WX, Ding SW. Animal virus replication and RNAi-mediated antiviral silencing in Caenorhabditis elegans. Nature. 2005;436:1040–1043. doi: 10.1038/nature03870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchetta EM, Carthew RW, Ismagilov RF. The endo-siRNA pathway is essential for robust development of the Drosophila embryo. PloS one. 2009;4:e7576. doi: 10.1371/journal.pone.0007576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Chung W-J, Ruby JG, Guo H, Bartel DP, Lai EC. The Drosophila hairpin RNA pathway generates endogenous short interfering RNAs. Nature. 2008;453:803–806. doi: 10.1038/nature07015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Lai EC. Endogenous small interfering RNAs in animals. Nature reviews Molecular cell biology. 2008;9:673–678. doi: 10.1038/nrm2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Liu N, Lai EC. Distinct mechanisms for microRNA strand selection by Drosophila Argonautes. Molecular cell. 2009;36:431–444. doi: 10.1016/j.molcel.2009.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phadnis N, Orr HA. A single gene causes both male sterility and segregation distortion in Drosophila hybrids. Science. 2009;323:376–379. doi: 10.1126/science.1163934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkies P, Selkirk ME, Jones JT, Blok V, Boothby T, Goldstein B, Hanelt B, Ardila-Garcia A, Fast NM, Schiffer PM, et al. Ancient and novel small RNA pathways compensate for the loss of piRNAs in multiple independent nematode lineages. PLoS biology. 2015;13:e1002061. doi: 10.1371/journal.pbio.1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Araripe L, Kingan SB, Ke Y, Xiao H, Hartl DL. A sex-ratio meiotic drive system in Drosophila simulans. II: an X-linked distorter. PLoS biology. 2007a;5:e293. doi: 10.1371/journal.pbio.0050293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Hartl DL, Laurie CC. Sex-ratio segregation distortion associated with reproductive isolation in Drosophila. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:13183–13188. doi: 10.1073/pnas.231478798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Masly JP, Araripe L, Ke Y, Hartl DL. A sex-ratio meiotic drive system in Drosophila simulans. I: an autosomal suppressor. PLoS biology. 2007b;5:e292. doi: 10.1371/journal.pbio.0050292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28:2184–2185. doi: 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- Wang XH, Aliyari R, Li WX, Li HW, Kim K, Carthew R, Atkinson P, Ding SW. RNA interference directs innate immunity against viruses in adult Drosophila. Science. 2006;312:452–454. doi: 10.1126/science.1125694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J, Duan H, Bejarano F, Okamura K, Fabian L, Brill JA, Bortolamiol-Becet D, Martin R, Ruby JG, Lai EC. Adaptive regulation of testis gene expression and control of male fertility by the Drosophila harpin RNA pathway. Molecular cell. 2015;57:165–178. doi: 10.1016/j.molcel.2014.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis JH. Genetics. Origin of species in overdrive. Science. 2009;323:350–351. doi: 10.1126/science.1169442. [DOI] [PubMed] [Google Scholar]
- Yasuno Y, Inoue YH, Yamamoto MT. Elimination of Y chromosome-bearing spermatids during spermiogenesis in an autosomal sex-ratio mutant of Drosophila simulans. Genes & genetic systems. 2013;88:113–126. doi: 10.1266/ggs.88.113. [DOI] [PubMed] [Google Scholar]
- Zhang L, Sun T, Woldesellassie F, Xiao H, Tao Y. Sex ratio meiotic drive as a plausible evolutionary mechanism for hybrid male sterility. PLoS genetics. 2015;11:e1005073. doi: 10.1371/journal.pgen.1005073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.