

Abstract
The epigenetic mark 5-hydroxymethylcytosine (5-hmC) is a distinct product of active DNA demethylation that is linked to gene regulation, development, and disease. In particular, 5-hmC levels dramatically decline in many cancers, potentially serving as an epigenetic biomarker. The noise associated with next-generation 5-hmC sequencing hinders reliable analysis of low 5-hmC containing tissues such as blood and malignant tumors. Additionally, genome-wide 5-hmC profiles generated by short-read sequencing are limited in providing long-range epigenetic information relevant to highly variable genomic regions, such as the 3.7 Mbp disease-related Human Leukocyte Antigen (HLA) region. We present a long-read, highly sensitive single-molecule mapping technology that generates hybrid genetic/epigenetic profiles of native chromosomal DNA. The genome-wide distribution of 5-hmC in human peripheral blood cells correlates well with 5-hmC DNA immunoprecipitation (hMeDIP) sequencing. However, the long single-molecule read-length of 100 kbp to 1 Mbp produces 5-hmC profiles across variable genomic regions that failed to show up in the sequencing data. In addition, optical 5-hmC mapping shows a strong correlation between the 5-hmC density in gene bodies and the corresponding level of gene expression. The single-molecule concept provides information on the distribution and coexistence of 5-hmC signals at multiple genomic loci on the same genomic DNA molecule, revealing long-range correlations and cell-to-cell epigenetic variation.
Keywords: 5-hydroxymethylcytosine, methylation, single-molecule, optical mapping, epigenetics, fluorescence microscopy, nanotechnology, nanochannels
The characterization and profiling of new DNA epigenetic modifications has been the focus of many studies in recent years. 5-Hydroxymethylcytosine (5-hmC), the first in a chain of chemical oxidation products catalyzed by the Ten-11 translocation (TET) family of dioxygenases during active DNA demethylation, has garnered special attention since its discovery in mammalian cells in 2009.1,2 5-hmC was first thought to be a transient state; however, increasing evidence suggests that this modification plays a role in the regulation of gene expression, affecting development and cell differentiation.3−7 Although its mechanism of action is not fully resolved, it is considered a key player in several processes, including binding of transcription factors and regulators, altering chromatin structure, and modulating alternative splicing, presumably by influencing the recruitment and binding of associated proteins.8−12 Several studies have linked 5-hmC with neurological disorders, stress response, and aging.13−16 Additionally, depletion in global 5-hmC levels has been observed in various pathological conditions, including several types of cancer.17−24 Furthermore, recent reports showed characteristic 5-hmC profiles in the cell-free circulating DNA of cancer patients.25−27 Such profiles may provide information regarding tumor type and stage, making 5-hmC a potential biomarker for early diagnosis and response to therapy. The increasing interest in 5-hmC has served as a catalyst for the development of several techniques for profiling its distribution on a genome-wide scale. Currently accepted methods include single-base resolution methods such as oxidative bisulfite sequencing (oxBS-seq)28 and TET-assisted bisulfite sequencing (TAB-seq),29 as well as lower resolution affinity-based enrichment methods such as 5-hmC DNA immunoprecipitation (hMeDIP),4,30 5-hmC selective chemical labeling (hMe-Seal),31 and nano-hmC-Seal.32 The main limitation of all of these techniques is their reliance on sequencing by synthesis (SBS), also frequently termed next generation sequencing (NGS), which requires pooling and fragmentation of genomic DNA. As such, the epigenetic profile reported by these techniques is the average distribution of an entire population of cells, where cell-to-cell variation is lost, and with it the ability to detect small subpopulations. Recently reported single-cell 5-hmC sequencing may potentially capture 5-hmC variation,33 but its low genomic sampling rate and reliance on NGS lead to difficulties in the analysis of long-range correlations at a reasonable cost.34 Third generation sequencing approaches, including single-molecule, real-time (SMRT) sequencing (Pacific Biosciences), and nanopore sequencing (Oxford Nanopore Technologies) have demonstrated the potential ability to detect chemical modifications directly on single, long DNA molecules.35−39 However, extensive development of these applications is still necessary before they are used for whole-genome epigenetic profiling. A significant challenge arises when profiling tissues that exhibit ultralow levels of 5-hmC, such as human blood (0.001–0.005%). In such experiments, the real 5-hmC signal drops to the inherent noise level of the above-mentioned techniques. For affinity approaches, a relatively constant rate of false positives is dictated by nonspecific capture of DNA, and for oxBS-seq and TAB-seq, nonideal chemical conversion results in false readout. Specifically, gold-standard TAB-seq relies on enzymatic conversion that at best reaches 99% efficiency. In the case of blood, the result is an equivalent number of true and false 5-hmC signals, undermining the ability to extract informative data.
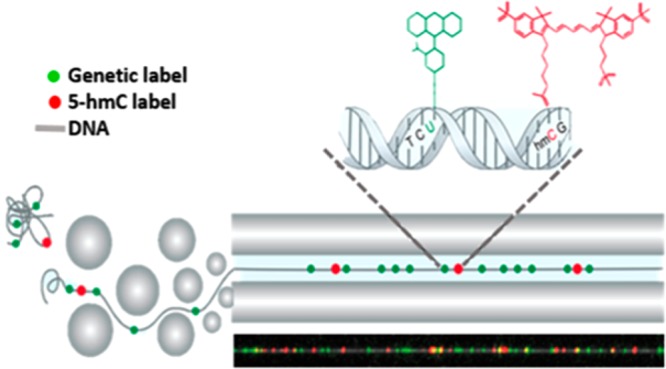
Here we present optical 5-hmC mapping, a single-molecule mapping approach for studying the genomic distribution of 5-hmC. We apply our method to human peripheral blood mononuclear cells (PBMCs), emphasizing the high sensitivity of this single-molecule approach. Our method integrates into genome mapping technology, commercialized by BioNano Genomics Inc., which relies on extending fluorescently labeled DNA molecules in nanochannel arrays.40,41 Fluorescence microscopy allows simultaneous detection of genetic and epigenetic information on the same molecule, by labeling each feature with a different color. By using color as a contrast mechanism, this method can be extended further to detect several epigenetic observables simultaneously, allowing the study of modification coexistence.42,43 In this study, genetic and epigenetic marks on individual DNA molecules are labeled simultaneously in two different colors. The genetic barcode is generated by enzymatically labeling DNA in a specific sequence motif, and an additional epigenetic information layer is produced by labeling 5-hmC through a specific chemo-enzymatic reaction.44 By aligning the genetic labels to a reference, the genomic positions of 5-hmC labels are mapped to obtain a genome-wide profile of epigenetic modifications (Figure 1).
Figure 1.

Optical 5-hmC mapping experimental scheme. (A) Left: Scanning electron microscope (SEM) image of a silicon nanochannel array. Right: Stretched DNA molecules (gray) fluorescently labeled in two colors. Green: Sequence specific genetic barcode. Red: 5-hmC labels. (B) Fluorescently labeled molecules are extended in nanochannel arrays by electrophoresis. (C) Fluorescence intensity of genetic labels (green) and 5-hmC labels (red) along a single molecule. (D) Genetic labels (green) are used to align a digital representation of the molecule in part C (yellow) to an in silico generated reference (gray) of chromosome 5, highlighting large structural variations such as the 7 kbp deletion in the midright part of the molecule, denoted by the diagonal alignment marks. 5-hmC labels (red) are mapped on the basis of genetic alignment.
We compare the 5-hmC patterns detected by our optical mapping approach in PBMCs to hMeDIP-seq results generated for the same sample. We show that optical mapping displays higher sensitivity and lower background noise and that global patterns correlate well with previously reported results regarding the distribution of 5-hmC near regulatory elements and its enrichment in highly expressed genes. Furthermore, we show the correlation between 5-hmC density in gene bodies and their corresponding expression levels over a large dynamic range. Finally, we demonstrate the potential strength of long-read optical mapping in characterizing epigenetic patterns in variable genomic regions, which currently pose a challenge for NGS technology.
Results and Discussion
We developed optical 5-hmC mapping on high-molecular-weight DNA extracted from fresh human PBMCs. A nick-translation reaction with the nicking enzyme Nt.BspQI was performed in order to incorporate a green fluorophore into the DNA, producing a sequence specific labeling pattern for genome mapping. A second layer of information was obtained by performing a chemo-enzymatic reaction to specifically label 5-hmC with a red fluorophore. This simultaneous labeling scheme enables the positioning of 5-hmC sites according to the genetic labels, yielding single-molecule whole-genome 5-hmC maps.
Efficiency of 5-hmC Labeling
Efficient labeling of 5-hmC is critical for obtaining meaningful information from individual molecules. In order to evaluate the efficiency of 5-hmC labeling, we performed two separate nick-labeling reactions on purified lambda phage DNA. The reactions contained either fluorescent nucleotides, for assessment of nick-labeling efficiency, or 5-hmC nucleotides, which were then fluorescently labeled according to our 5-hmC labeling scheme and used for assessment of 5-hmC labeling efficiency. DNA from both reactions was combined, driven into nanochannels, and imaged together on an Irys instrument (Figure 2A). The lambda phage genome contains 10 expected Nt.BspQI nicking sites, with two sites that cannot be separated due to the optical resolution limit. Labeling efficiency was calculated by comparing the number of detected spots to the number of expected nicking sites.
Figure 2.

Assessment of 5-hmC labeling efficiency. Lambda DNA was nicked with Nt.BspQI (nine expected labeling spots) and labeled with either 5-hmC or fluorescent dUTP. 5-hmC was labeled according to our labeling scheme, and the samples were mixed and imaged together in order to evaluate the labeling efficiency. (A) Representative field of view showing a mixed population of green (nicking) and red (5-hmC) labeled molecules. (B) Histograms showing the number of labels per molecule for 5-hmC labeling (top) and nicking (bottom).
Figure 2B shows the number of detected labels per molecule in the red channel (5-hmC labeling) and in the green channel (nick-labeling). Nick-labeling efficiency was calculated by dividing the average number of green labels per molecule in each scan by the total number of expected labels. 5-hmC labeling efficiency was calculated by dividing the average number of red labels per molecule by the average number of green labels per molecule in each scan. A total of 20,520 scanned images were analyzed to determine labeling efficiency. Accordingly, the nicking efficiency was determined as 85 ± 2%, and the 5-hmC labeling efficiency as 82 ± 3%.
Quantification of 5-hmC Sites per Detected Label by Measuring Photobleaching Steps
Due to the diffraction limit, multiple 5-hmC sites on the same 1 kb region will result in multiple close-by fluorescent labels that generate a single fluorescent spot. The number of fluorophores detected in each isolated 5-hmC spot has a crucial role in determining the actual density of 5-hmC. It was therefore important that we validate the characteristic amount of fluorophores, and therefore the number of 5-hmC sites, per isolated fluorescent spot. In order to assess the number of fluorophores, we monitored the photobleaching process which fluorophores undergo when exposed to intense laser excitation (see Methods). Quantifying the amount of photobleaching steps enabled us to accurately determine the number of 5-hmC sites per detected spot (Figures S1–S3). Approximately 1000 fluorescent spots along the DNA were analyzed in order to construct the distribution of 5-hmC clusters. The full distribution allowed us to correct for this effect as part of the calibration procedure. Effectively, 1.45 fluorophores are present in each fluorescent spot and this correction factor was taken into account for global quantification. Since over 90% of the analyzed 5-hmC spots contained one to two 5-hmC labels, our method can reliably quantify 5-hmC in genomic DNA from blood, without considering the fluorescence intensity of the spots. Using such global quantification, we have recently shown that our labeling scheme can detect a decrease in the global level of 5-hmC in blood and colon tumor cells vs normal cells.21
Generation of Genome-Wide 5-hmC Profiles
To recover the genome-wide distribution of 5-hmC, we combined our labeling scheme with conventional genome mapping by generating a sequence specific fluorescent barcode in addition to the epigenetic marks. This dual-color labeling provides information on the amount as well as the location and distribution of 5-hmC sites along the genome.
Labeled DNA was stretched and imaged on nanochannel array chips, and the positions of genetic and epigenetic labels were automatically detected. Molecule images and genome browser compatible molecule tracks were generated with Irys Extract45 (Figure 1A,C). A total of 992,030 long molecules (>150 kb) were aligned to the reference (human hg19), and 587,583 that passed our confidence criteria (see Methods) were used for downstream analysis, generating a median genomic coverage of 46X. In order to verify the 5-hmC density obtained from optical mapping, we performed LC-MS/MS measurements on DNA extracted from the same PBMCs sample (see Supporting Information, Figure S4). The percentage of 5-hmC in DNA extracted from PBMCs was calculated as 0.0035%. This is in agreement with the mean 5-hmC density measured for the optical mapping experiment (0.0029%).
In order to validate our optical mapping results, we also performed two independent hMeDIP-seq experiments on the same sample. On the basis of the correlation between the genomic 5-hmC profiles of these two experiments (Pearson correlation coefficient = 0.7), we were able to merge the sequencing reads from both data sets, in order to enhance the 5-hmC signal. The extremely low levels of 5-hmC in blood,46 together with nonspecific pulldown inherent in this type of experiments,47,48 resulted in low signal-to-noise ratio for the sequencing data. Thus, a minimal threshold for 5-hmC reads was set for downstream analysis (Supporting Information, Figures S5 and S6). We note that it is likely that using more sensitive enrichment techniques for sequencing may have resulted in better sequencing signal and coverage, with overall better correlation with our optical mapping results. However, at the time of experiments, hMeDIP-Seq was a more established benchmark for comparison.
Global Optical Mapping Patterns of 5-hmC Correlate with Sequencing Results
We first wanted to verify that the epigenetic patterns we observe using our optical mapping approach correlate with the patterns observed in the sequencing results. To this end, we examined the global distribution of 5-hmC in both data sets near several regulatory elements (Figure 3A–C). Both methods display distinct patterns near transcription start sites (TSS), enhancers marked by the histone modification H3K4me1, and active enhancers marked by the histone modification H3K27Ac, as previously reported for embryonic stem cells (ESCs), brain, and blood.49−51 In all cases, the patterns detected by both methods are highly correlated. However, global examination of both data sets on a genome-wide scale (Figure 3) establishes the advantages of the optical mapping approach. While the sequencing results show finer features due to the higher resolution of hMeDIP (∼200 bp) compared to optical mapping (∼1500 bp), the optical data displays superior signal-to-noise ratio, allowing us to reliably detect low levels of 5-hmC. This high sensitivity is likely due to the low false positive and false negative rates of our labeling scheme (Figure 1). Moreover, while antibody-based methods are biased toward heavily modified regions,48 the high sensitivity of optical detection, combined with the single-molecule aspect of this approach, allows the detection of rare, isolated 5-hmC residues occurring only in a small subset of cells. This is clearly seen in the additional signals present in the optical mapping track in Figure 3D.
Figure 3.

Global correlation between 5-hmC profiles produced by optical 5-hmC mapping and hMeDIP-seq. (A) Coverage as a function of distance from TSS. (B) Coverage as a function of distance from H3K4me1 histone modification peaks. (C) Coverage as a function of distance from H3K27Ac histone modification peaks. (D) Comparison of coverage produced by both methods in a representative 500 kbp region from chromosome 1. hMeDIP-seq results are presented in 1 kbp resolution.
5-hmC Levels in Gene Bodies Correlate with Gene Expression
Following reports regarding 5-hmC enrichment in the bodies of highly expressed genes,49−51 we examined the correlation between gene expression and 5-hmC levels for both optical mapping and hMeDIP-seq results (Figure 4). Although for both methods higher levels of 5-hmC are observed in highly expressed genes, optical 5-hmC mapping presents a much larger dynamic range. The high sensitivity and specificity of the fluorescent labeling utilized in optical 5-hmC mapping enable a clear distinction between the 5-hmC levels in gene bodies of low expressed and unexpressed genes. These two groups are almost indistinguishable in the hMeDIP-seq results. These results suggest that 5-hmC levels may be used to infer gene expression without directly quantifying RNA levels.
Figure 4.

5-hmC coverage across gene bodies, in correlation with gene expression level. Gene lengths were normalized to 15 kbp, and 3 kbp was added to each gene upstream of the TSS and downstream of the TES. Left: optical mapping data. Right: hMeDIP-seq data.
Optical Mapping “Long Reads” Allow the Characterization of Highly Variable Regions
The relatively low resolution of optical-based detection is undoubtedly the main drawback of this approach. Nevertheless, one of the main advantages of genetic optical mapping is the ability to use long-range information encoded in long DNA molecules to characterize highly variable genomic regions.52,53 These “long reads” extend beyond the variable region and may be anchored to the reference based on reliable alignment to conserved flanking regions, maintaining contiguity along the variable region itself.
The Human Leukocyte Antigen (HLA) region, located on chromosome arm 6p21.3, is one of the most polymorphic regions in the human genome.54 This 3.6 Mb region has been associated with more than 100 diseases, including diabetes, psoriasis, and asthma, and several alleles of HLA genes have been linked to hypersensitivity to specific drugs.55,56 Therefore, information regarding the epigenetic landscape of this region may have clinical importance.
Examining the region around the HLA-A gene, one of three major histocompatibility complex (MHC) class I cell surface receptors, we found that single molecules can indeed be aligned to the reference with high confidence around this locus. This resulted in the detection of high levels of 5-hmC in this region, information that was not detected by hMeDIP-seq, as there were no sequencing reads aligned to this gene (Figure 5A). The highly variable nature of this locus is known to pose a challenge to NGS sequencing technologies. In contrast, the alignment of long optical reads to the reference is not hampered by the variable region, since it occupies only a small portion of the detected molecule. Consequently, using optical 5-hmC mapping, we are able to directly detect 5-hmC modifications in the HLA region without the amplification or specific targeting methods currently needed for short-read-based experiments.57,58
Figure 5.

Epigenetic characterization of variable regions by optical 5-hmC mapping “long reads”. Light blue: hMeDIP-seq. Black: optical 5-hmC mapping. Blue: gene symbol. (A) 5-hmC coverage of 23 kbp around the HLA-A gene. (B) 5-hmC coverage of 111 kbp containing the histone gene cluster. (C) 5-hmC coverage of 103 kbp around TLR7 and TLR8-AS1. (D) 5-hmC coverage of 98 kbp containing the TLR cluster located on chromosome 4.
Similarly, our optical mapping results could profile the epigenetic patterns of other variable regions that were not well represented in the sequencing results. For example, we were able to detect the 5-hmC distribution in the histone gene cluster located on chromosome arm 1q21, which harbors variant genes as well as duplicated regions (Figure 5B).59−61 The sequencing signal was very low in this ∼100 kbp cluster, with no detected signals in histone gene bodies. Correlation between methylation in histone genes and disease has been previously reported,62,63 but a thorough investigation is required to establish the importance of 5-hmC density in these genes. The advantages of optical mapping in variable regions are further exemplified by the highly polymorphic toll-like receptor (TLR) gene clusters located on chromosome arms 4p14 and Xp22.2.64 TLR genes presented modest 5-hmC density in the optical map, presumably since only a small subset of PBMCs express these genes. In the sequencing results, the sparse 5-hmC content is practically undetected (Figure 5C and D).
In recent years, genetic optical mapping has proven to be invaluable for the characterization of variable and repetitive regions inaccessible to NGS technologies, as well as for completing assemblies of various species.65,66 In this work, we demonstrate the ability to add a second, epigenetic layer of information to long individual DNA molecules. The combination of existing optical genome mapping technology with a 5-hmC-specific labeling reaction creates a genome-wide profile of this epigenetic mark in human PBMCs.
We demonstrate that optical 5-hmC mapping produces an epigenetic map that correlates well with the epigenetic map produced by hMeDIP-seq, which relies on gold-standard NGS technologies. Despite the resolution limit of optical detection, optical 5-hmC mapping proves to be superior to hMeDIP-seq in sensitivity and specificity, resulting in a high signal-to-noise ratio and broad dynamic range. The recently presented nano-hmC-Seal32 capture approach provides highly sensitive genome wide profiling of 5-hmC using as low as ∼1000 cells. Although still limited by the short-read nature of NGS, this approach holds promise for accurate 5-hmC profiling where very small amounts of DNA are available. TAB-seq, another gold standard technique for genome-wide 5-hmC profiling, can detect the epigenetic mark with high resolution and high efficiency.29 Nevertheless, it is limited to tissues expressing relatively high levels of 5-hmC. This is due to the conversion rate of 5-methylcytosine (5-mC) to 5-carboxycytosine (5-CaC) by the TET enzyme. Even in the case of 99% conversion efficiency, 1% of nonconverted methylated C will be detected as 5-hmC. This false positive rate of TAB-seq is on the same order as the real 5-hmC content in tissues exhibiting extremely low levels of 5-hmC such as blood. With an average signal-to-noise ratio of 1, TAB-seq is limited in its ability to characterize the distribution of 5-hmC in PBMCs.
Our labeling and imaging technique highlights single 5-hmC sites with no false positives and a minor false negative rate. These attributes enable the detection of extremely low levels of epigenetic modifications. This is especially important for cancer related studies, where 5-hmC levels decline drastically with disease progression.17 Additionally, utilizing the long-range information encoded in optical “long reads” enables the epigenetic characterization of variable and repetitive regions known to pose a challenge to NGS technologies.
Conclusions
We present a method for whole-genome, single-molecule, epigenetic mapping of 5-hmC. The method offers extremely long reads and high sensitivity that allow the characterization of variable or complex genomic regions. The presented concept is not limited to the measurement of 5-hmC or to the measurement of a single epigenetic feature. This fluorescence-based technique relies on color to distinguish different genomic features. Only the number of specific labeling chemistries and the spectral properties of the optical measurement system limit the number of markers that can be detected simultaneously. Thus, further development of this technique may enable the simultaneous measurement of 5-hmC and 5-mC, giving a more comprehensive genomic profile of the cell population as a whole and of cell-to-cell variability in particular.
Methods
Human Subjects
The healthy donor sample used in this study was collected with informed consent for research use and approved by the Tel-Aviv University and Meir Medical Center ethical review boards, in accordance with the declaration of Helsinki.
Measuring the Labeling Efficiency of 5-hmC
For the nicking reaction, 900 ng of lambda phage DNA (New England Biolabs) was digested with 30 units of Nt.BspQI nicking enzyme (New England Biolabs) for 2 h at 50 °C in the presence of 3 μL of 10× buffer 3.1 (New England Biolabs) and ultrapure water to a total volume of 30 μL. Next, nicked DNA was incubated for 1 h at 72 °C with 15 units of Taq DNA polymerase (New England Biolabs), supplemented with 600 nM of dATP, dGTP, dCTP (Sigma) and atto-532-dUTP (Jena Bioscience), or dATP, dGTP, dTTP (Sigma) and 5hmdCTP (Zymo Research), in the presence of 4.5 μL of 10× thermopol buffer (New England Biolabs) and ultrapure water to a total reaction volume of 45 μL. Following labeling, DNA was repaired for 30 min at 45 °C with 12 units of Taq DNA ligase (New England Biolabs) in the presence of 1.5 μL of 10× thermopol buffer, 1 mM NAD+ (New England Biolabs), and ultrapure water to a total reaction volume of 60 μL. DNA that was labeled with 5hmdCTP nucleotides was further used for 5-hmC labeling. A 300 ng portion of nicked DNA was incubated overnight at 37 °C with 30 units of T4 β-glucosyltransferase (New England Biolabs), in the presence of 4.5 μL of 10× buffer 4 (New England Biolabs) and 200 mM homemade UDP-glucose-azide67 in a total reaction volume of 45 μL. Next, dibenzocyclooctyl (DBCO)-Cy5 (Jena Bioscience) was added to a final concentration of 620 μM and the reaction was incubated overnight at 37 °C. For purification of 5-hmC-labeled DNA from an excess of fluorescent dyes, plugs were generated by adding agarose to the DNA (see the “High-Molecular-Weight DNA Extraction” section) to a final agarose concentration of 0.8% and were then washed extensively with TE (pH 8) on a horizontal shaker. Plugs were melted and purified by drop dialysis using a 0.1 μm dialysis membrane (Millipore) floated on TE (pH 8). DNA from both labeling reactions was stained with YOYO-1 staining solution (BioNano Genomics) according to the manufacturer’s instructions and gently mixed together with wide bore tips until homogeneous. DNA concentration was measured by a Qubit HS dsDNA assay. Labeled DNA was loaded in nanochannels and imaged on an Irys system (BioNano Genomic Inc.).
Images were processed by the IrysView software package (version 2.3, BioNano Genomics) to detect individual molecules and positions of labels along each molecule. In accordance with the length of the lambda phage genome (48.5 kbp), only molecules that were 40–60 kbp in length were used for downstream analysis. Additionally, only labels with SNR ≥ 2.75 were considered. For each molecule, the number of 5-hmC labels (red channel) or the number of fluorescent nucleotides (green channel) were counted (in-house software). Molecules labeled in both colors were considered noise and discarded from the data set.
High-Molecular-Weight DNA Extraction
Human peripheral blood mononuclear cells (PBMCs) were isolated from peripheral blood of a healthy donor by density gradient centrifugation using Ficoll Paque Plus (GE Healthcare) according to the manufacturer’s instructions. PBMCs were trapped in agarose plugs to protect DNA from shearing during the labeling process.68 Samples were prepared according to the IrysPrep Plug Lysis Long DNA Isolation Protocol (Bionano Genomics Inc.) with slight modifications. Briefly, 1 × 106 cells were washed twice with PBS, resuspended in cell suspension buffer (CHEF mammalian DNA extraction kit, Bio-Rad), and incubated at 43 °C for 10 min. 2% low melting agarose (CleanCut agarose, Bio-Rad) was melted at 70 °C followed by incubation at 43 °C for 10 min. Melted agarose was added to the resuspended cells at a final concentration of 0.7% and mixed gently. The mixture was immediately cast into a plug mold, and plugs were incubated at 4 °C until solidified. Plugs were incubated twice (2 h of incubation followed by an overnight incubation) at 50 °C with 167 μL of freshly prepared proteinase K (Qiagen) in 2.5 mL of lysis buffer (BioNano Genomics Inc.) with occasional shaking. Next, plugs were incubated with 50 μL of RNase (Qiagen) in 2.5 mL of TE (10 mM Tris, pH 8, 1 mM EDTA) for 1 h at 37 °C with occasional shaking. Plugs were washed three times by adding 10 mL of wash buffer (10 mM Tris, pH 8, 50 mM EDTA), manually shaking for 10 s, and discarding the wash buffer before adding the next wash. Plugs were then washed four times by adding 10 mL of wash buffer and shaking for 15 min on a horizontal platform mixer at 180 rpm at room temperature. Following washes, plugs were stored at 4 °C in wash buffer or used for labeling. In order to extract high-molecular-weight DNA, plugs were washed three times in TE (pH 8) and were melted for 2 min at 70 °C, followed by 5 min of incubation at 43 °C. Next, 0.4 units of Gelase (Epicenter) were added and the mixture was incubated for 45 min. High-molecular-weight DNA was purified by drop dialysis using a 0.1 μm dialysis membrane (Millipore) floated on TE (pH 8). Viscous DNA was gently pipetted and incubated at room temperature overnight in order to achieve homogeneity. DNA concentration was determined using Qubit BR dsDNS assay (Thermo Fisher Scientific).
Genetic/Epigenetic Labeling and Data Collection
Genetic labeling was performed using either the commercial IrysPrep NLRS assay (Bionano Genomics Inc.) or an in-house developed alternative protocol (see “Measuring the Labeling Efficiency of 5-hmC” section). A 900 ng portion of high-molecular-weight DNA from PBMCs was subjected to a nick-translation reaction with Nt.BspQI for nicking and atto-532-dUTP for labeling. For labeling of 5-hmC,44 nick-labeled DNA was incubated overnight at 37 °C with 90 units of T4 β-glucosyltransferase (New England Biolabs), in the presence of 13.5 μL of 10× buffer 4 (New England Biolabs), 200 mM UDP-glucose-azide,67 and ultrapure water (135 μL total reaction volume). Next, dibenzocyclooctyl (DBCO)-Cy5 (Jena Bioscience) was added to a final concentration of 620 μM and the reaction was incubated overnight at 37 °C. For purification of labeled DNA from excess fluorescent dye, plugs were prepared by adding agarose to the DNA as described above to a final agarose concentration of 0.8%. Plugs were then washed extensively with TE (pH 8) on a horizontal shaker. Plugs were melted and purified by drop dialysis as described, and DNA was stained with YOYO-1 staining solution (BioNano Genomics) according to the manufacturer’s instructions with the addition of 25 mM Tris (pH 8) and 30 mM NaCl. DNA concentration was measured by the Qubit HS dsDNA assay (Thermo Fisher Scientific). For evaluation of photobleaching steps, labeled DNA was stretched on glass coverslips and imaged. For optical mapping experiments, labeled DNA was loaded into nanochannel-array Irys Chips and imaged on an Irys system (BioNano Genomics Inc.).
Evaluation of 5-hmC Sites per Detected Fluorescent Spot
Due to the diffraction-limited resolution of optical mapping, a single fluorescent spot may contain more than a single 5-hmC site. We used single-molecule photobleaching to assess the distribution of sites per spot in the studied DNA sample, in order to correct for any bias caused by this potential underestimation. DNA molecules that were labeled with Cy5 to indicate 5-hmC locations and their backbone stained with YOYO-1 were stretched on modified coverslips prepared as previously described44 with minor modifications. In short, 22 × 22 mm2 glass coverslips were cleaned for 7 h to overnight by incubation in a freshly made 2:1 (v/v) mixture of 70% nitric acid and 37% hydrochloric acid. After extensive washing with ultrapure water (18 MΩ) and then with ethanol 96%, coverslips were dried under a stream of nitrogen. Dry coverslips were immersed in a premixed silane solution containing 750 μL of N-trimethoxysilylpropyl-N,N,N-trimethylammonium chloride and 200 μL of vinyltrimethoxysilane in 300 mL of ultrapure water and incubated overnight at 65 °C. After incubation, coverslips were thoroughly washed with ultrapure water and ethanol and stored at 4 °C in ethanol. The silane solution was freshly made and thoroughly mixed before the coverslips were introduced into the mixture. Stored coverslips were normally used within 2 weeks. Prelabeled and stained DNA was diluted to a final concentration of 0.25 ng/μL in TE (pH 8) and 0.2 M DTT. DNA molecules were stretched on the silanized glass coverslips by placing 6 μL of diluted DNA between a dry silanized coverslip and a nontreated microscope slide.
The extended DNA molecules were imaged on an Olympus
IX81 microscope adapted for laser-fluorescence microscopy. A 637 nm
CW laser diode (Coherent, OBIS 637LX) and a 473 nm CW laser (OEM,
200 mW) were used as excitation sources for the 5-hmC Cy5 labels and
YOYO-1 intercalating backbone staining dye, respectively. Excitation
light was focused on the back aperture of a 100× oil immersion
objective (Olympus, UPlanSApo 100×/N.A 1.4) with an average excitation
power density of 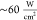 in the sample plane. Sample fluorescence
was collected by the same objective and imaged through a polychroic
beamsplitter (Chroma, ZT473/532/637 rpc-xt890) and emission filter
(Semrock, 679/41 Brightline) onto an electron multiplying charge coupled
device (EMCCD) camera (Andor, iXon Ultra 897) with single-molecule
detection sensitivity. All measurements were carried out at room temperature
under ambient conditions. Upon excitation, 5-hmC sites were detected
as fluorescent diffraction limited spots in the field of view (80 ×
80 μm2). Time lapse movies (500 frames, 20 ms/frame)
were acquired for over 100 different fields of view, containing 1000
individual isolated observable 5-hmC marker sites. For each time lapse
movie, individual labels were automatically localized by 2D Gaussian
fitting and manually validated to be isolated fluorescent marks consisting
of a single Gaussian spot and colocalized with a DNA molecule. After
localization, time traces of fluorescence intensity were calculated
for each marker. The intensity was calculated by the average intensity
of a 3 × 3 pixel2 window around the center of each
fitted Gaussian. In order to account for the uneven excitation intensity,
a local background subtraction was performed for each marker by taking
the average intensity of a 7 × 7 pixel2 contour window
around the fitted Gaussian.
in the sample plane. Sample fluorescence
was collected by the same objective and imaged through a polychroic
beamsplitter (Chroma, ZT473/532/637 rpc-xt890) and emission filter
(Semrock, 679/41 Brightline) onto an electron multiplying charge coupled
device (EMCCD) camera (Andor, iXon Ultra 897) with single-molecule
detection sensitivity. All measurements were carried out at room temperature
under ambient conditions. Upon excitation, 5-hmC sites were detected
as fluorescent diffraction limited spots in the field of view (80 ×
80 μm2). Time lapse movies (500 frames, 20 ms/frame)
were acquired for over 100 different fields of view, containing 1000
individual isolated observable 5-hmC marker sites. For each time lapse
movie, individual labels were automatically localized by 2D Gaussian
fitting and manually validated to be isolated fluorescent marks consisting
of a single Gaussian spot and colocalized with a DNA molecule. After
localization, time traces of fluorescence intensity were calculated
for each marker. The intensity was calculated by the average intensity
of a 3 × 3 pixel2 window around the center of each
fitted Gaussian. In order to account for the uneven excitation intensity,
a local background subtraction was performed for each marker by taking
the average intensity of a 7 × 7 pixel2 contour window
around the fitted Gaussian.
The number of photobleaching steps was manually quantified according to the number of intensity drops in each time trace. All data analysis in this section was carried out by a custom-made MATLAB program.
Optical Mapping and Data Analysis
Two optical epigenome mapping experiments of PBMCs were performed separately, and their results were combined for downstream analysis.
Raw images were processed by the IrysView software package (version 2.3, BioNano Genomics Inc.) to detect individual molecules and positions of genetic and epigenetic labels along each molecule. Only labels with SNR ≥ 2.75 were considered. The positions of genetic labels were used to align molecules longer than 150 kbp to an in silico generated map of expected Nt.BspQI nicking sites in the hg19 human reference (IrysView), and only molecules with an alignment confidence equal or higher than 12 (P ≤ 10–12) were used for downstream analysis. In order to minimize ambiguous alignments of the overlaid 5-hmC pattern, we filtered out molecules that were not aligned to the reference for over 60% of their total length. Genomic positions of epigenetic labels were then extracted by interpolation, on the basis of the genetic alignment results (in-house script). 500 bp were added to either side of each label position, in order to account for optical resolution, and genomic coverage of both 5-hmC and aligned molecules was calculated using BEDTools (version 2.25.0)69 genomecov. Regions represented by less than 19 molecules were discarded for downstream analysis, and molecule coverage was normalized by dividing the coverage in each genomic position by the maximum coverage. Normalized 5-hmC coverage was then calculated by dividing the number of detected 5-hmC labels in each genomic position by the normalized molecule coverage in the same position.
hMeDIP-seq Library Preparation
DNA extracted from PBMCs was used to prepare two sequencing libraries by Epigentek Inc. Each library included one hMeDIP sample and an input sample. hMeDIP experiments were performed with 5-hmC monoclonal antibody, using 1.5–4 μg of DNA. Sample libraries were sequenced on a HiSeq 2500 (Illumina Inc.).
Sequencing Alignment and Data Analysis
Sequencing reads were aligned to the hg19 human reference using Bowtie 2 (version 2.2.6)70 with default parameters. Following alignment, reads with MAPQ less than 30 were filtered with SAMtools,71 and the remaining reads were de-duplicated with Picard (https://broadinstitute.github.io/picard/) to eliminate PCR duplication bias. Reads from the pull-down data set were extended in the 3′ direction to a total length of 200 bp, in order to account for insert size. Genomic coverage of 5-hmC and input DNA was calculated using BEDTools genomecov. Due to a low signal-to-noise ratio in the pull-down experiment, the signal extraction scaling (SES)72 method was used to estimate a minimal signal level. Briefly, the genome was divided into 1 kbp nonoverlapping windows and the number of 5-hmC and input reads in each window was counted. Windows were then sorted in increasing order on the basis of 5-hmC counts, and the cumulative sum in each window was calculated and normalized to the cumulative sum of the complete data set. The absolute value of the difference between these normalized sums was calculated for each window, and the minimal signal level was defined as the 5-hmC counts value in the window where this difference reached a maximum. On the basis of this calculation, all regions where the 5-hmC signal was lower than 7 were set to zero. Additionally, ENCODE-defined signal artifact regions in the hg19 human genome reference73 (https://sites.google.com/site/anshulkundaje/projects/blacklists) were discarded.
For visual assessment of the correlation between the hMeDIP-seq results and the optical mapping results, sequencing coverage resolution was lowered to 1 kbp using a running average calculation. This was accomplished by dividing the genome into overlapping 1 kbp windows with a 1 bp shift and calculating the mean 5-hmC coverage in each window. The calculated mean was then set as the 5-hmC value in the midpoint genomic position of each window.
Analysis of Global 5-hmC Distribution across Genomic Features
Transcription start sites (TSS) were defined according to RefSeq annotation for hg19 downloaded from the UCSC genome browser database. H3K4me1 and H3K27Ac ChIP-seq data for PBMCs was downloaded as alignment results from the Roadmap Epigenomics Project (GEO accession numbers GSM1127143 and GSM1127145, respectively) and converted into genomic coverage using BEDTools. Enhancer regions were defined by examining coverage percentiles (>99% percentile for H3K4me1 and >99.5% for H3K27Ac), and regions closer than 100 bp apart were merged. 5-hmC counts in each region were calculated using BEDTools by dividing regions into equally sized bins (90 bp for H3K4me1, 30 bp for H3K27Ac, and 20 bp for TSS), computing the mean 5-hmC signal in each bin, and summing the signal across all bins of equal distance from the region midpoint. Normalization of values to a 0–1 range was performed separately for each data set.
Analysis of 5-hmC Correlation to Gene Expression
Expression data of protein-coding genes for PBMCs was downloaded from the Roadmap Epigenomics Project as RPKM values (http://egg2.wustl.edu/roadmap/data/byDataType/rna/expression/). Genes were classified into three groups: high expression (5987 genes, log10(RPKM) ≥ 10), low expression (5741 genes, 1 ≤ log10(RPKM) < 10), and no expression (8074 genes, log10(RPKM) < 1). Mean 5-hmC values along genes were calculated using deepTools74 computeMatrix in scale regions mode. Each gene was scaled to 15 kbp and divided into 300 bp bins. The mean 5-hmC score in each bin was calculated, and all scores in the same bin were summed and normalized to the number of genes in the group.
Acknowledgments
This study was supported by the European Research Council starter grant for financial support (Grant No. 337830), the BeyondSeq consortium (EC program 63489), the I-Core program of the Israel Science Foundation (Grant No. 1902/12), the Marie Curie Career Integration grant (Grant No. 322249), and the Gertner fellowship. We thank Zohar Shipony and Prof. Amos Tanay for assistance with hMeDIP data analysis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.8b03023.
Mass spectrometry, additional experimental details, and computational analysis (PDF)
Author Contributions
T.G. and H.S. contributed equally to this work. Y.E. and Y.M. conceived the study and supervised the project. H.S., T.G., Y.M., and Y.E. designed the study. T.G. and Y.M. performed optical 5-hmC mapping experiments. H.S. performed optical mapping, and hMeDIP-seq data analysis. G.N. and J.J. performed photobleaching measurements. T.S. performed mass spectrometry measurements. R.A., M.L.-S., and L.H. performed supporting measurements and data analysis. N.A. supervised sample collection and ethics. H.S., Y.M., and Y.E. wrote the manuscript. All authors edited the manuscript.
The authors declare no competing financial interest.
Notes
Optical mapping cmap and xmap files have been uploaded as NCBI supplementary files: SUPPF_0000002688, SUPPF_0000002687. Sequencing data was uploaded to GEO (accession number GSE115454). In-house analysis scripts are available on GitHub: https://github.com/ebensteinLab/Irys-data-analysis.
Supplementary Material
References
- Tahiliani M.; Koh K. P.; Shen Y.; Pastor W. A.; Bandukwala H.; Brudno Y.; Agarwal S.; Iyer L. M.; Liu D. R.; Aravind L.; Rao A. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science (Washington, DC, U. S.) 2009, 324, 930–935. 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriaucionis S.; Heintz N. The Nuclear DNA Base 5-Hydroxymethylcytosine Is Present in Purkinje Neurons and the Brain. Science (Washington, DC, U. S.) 2009, 324, 929–930. 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficz G.; Branco M. R.; Seisenberger S.; Santos F.; Krueger F.; Hore T. a; Marques C. J.; Andrews S.; Reik W. Dynamic Regulation of 5-Hydroxymethylcytosine in Mouse ES Cells and during Differentiation. Nature 2011, 473, 398–402. 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- Stroud H.; Feng S.; Morey Kinney S.; Pradhan S.; Jacobsen S. E. 5-Hydroxymethylcytosine Is Associated with Enhancers and Gene Bodies in Human Embryonic Stem Cells. Genome Biol. 2011, 12, R54. 10.1186/gb-2011-12-6-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wossidlo M.; Nakamura T.; Lepikhov K.; Marques C. J.; Zakhartchenko V.; Boiani M.; Arand J.; Nakano T.; Reik W.; Walter J. 5-Hydroxymethylcytosine in the Mammalian Zygote Is Linked with Epigenetic Reprogramming. Nat. Commun. 2011, 2, 241. 10.1038/ncomms1240. [DOI] [PubMed] [Google Scholar]
- Bachman M.; Uribe-Lewis S.; Yang X.; Williams M.; Murrell A.; Balasubramanian S. 5-Hydroxymethylcytosine Is a Predominantly Stable DNA Modification. Nat. Chem. 2014, 6, 1049–1055. 10.1038/nchem.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulwach K. E.; Li X.; Li Y.; Song C.-X.; Wu H.; Dai Q.; Irier H.; Upadhyay A. K.; Gearing M.; Levey A. I.; Vasanthakumar A.; Godley L. A.; Chang Q.; Cheng X.; He C.; Jin P. 5-hmC–mediated Epigenetic Dynamics during Postnatal Neurodevelopment and Aging. Nat. Neurosci. 2011, 14, 1607–1616. 10.1038/nn.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T.-L.; Chen J.; Wan H.; Tang B.; Tian W.; Liao L.; Qiu Z. Regulation of mRNA Splicing by MeCP2 via Epigenetic Modifications in the Brain. Sci. Rep. 2017, 7, 42790. 10.1038/srep42790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruijt C. G.; Gnerlich F.; Smits A. H.; Pfaffeneder T.; Jansen P. W. T. C.; Bauer C.; Münzel M.; Wagner M.; Müller M.; Khan F.; Eberl H. C.; Mensinga A.; Brinkman A. B.; Lephikov K.; Müller U.; Walter J.; Boelens R.; van Ingen H.; Leonhardt H.; Carell T.; et al. Dynamic Readers for 5-(Hydroxy)Methylcytosine and Its Oxidized Derivatives. Cell 2013, 152, 1146–1159. 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- Shi D.-Q.; Ali I.; Tang J.; Yang W.-C. New Insights into 5hmC DNA Modification: Generation, Distribution and Function. Front. Genet. 2017, 8, 100. 10.3389/fgene.2017.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan D.; Perner J.; Carrillo de Santa Pau E.; Marsili S.; Ochoa D.; Chung H.-R.; Vingron M.; Rico D.; Valencia A. Epigenomic Co-Localization and Co-Evolution Reveal a Key Role for 5hmC as a Communication Hub in the Chromatin Network of ESCs. Cell Rep. 2016, 14, 1246–1257. 10.1016/j.celrep.2016.01.008. [DOI] [PubMed] [Google Scholar]
- Marina R. J.; Sturgill D.; Bailly M. A.; Thenoz M.; Varma G.; Prigge M. F.; Nanan K. K.; Shukla S.; Haque N.; Oberdoerffer S. TET-Catalyzed Oxidation of Intragenic 5-Methylcytosine Regulates CTCF-Dependent Alternative Splicing. EMBO J. 2016, 35, 335–355. 10.15252/embj.201593235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López V.; Fernández A. F.; Fraga M. F. The Role of 5-Hydroxymethylcytosine in Development, Aging and Age-Related Diseases. Ageing Res. Rev. 2017, 37, 28–38. 10.1016/j.arr.2017.05.002. [DOI] [PubMed] [Google Scholar]
- Madrid A.; Papale L. A.; Alisch R. S. New Hope: The Emerging Role of 5-Hydroxymethylcytosine in Mental Health and Disease. Epigenomics 2016, 8, 981–991. 10.2217/epi-2016-0020. [DOI] [PubMed] [Google Scholar]
- Condliffe D.; Wong A.; Troakes C.; Proitsi P.; Patel Y.; Chouliaras L.; Fernandes C.; Cooper J.; Lovestone S.; Schalkwyk L.; Mill J.; Lunnon K. Cross-Region Reduction in 5-Hydroxymethylcytosine in Alzheimer’s Disease Brain. Neurobiol. Aging 2014, 35, 1850–1854. 10.1016/j.neurobiolaging.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack L. M.; Dick A. L. W.; Provençal N. Epigenetic Mechanisms Involved in the Effects of Stress Exposure: Focus on 5-Hydroxymethylcytosine. Environ. Epigenet. 2016, 2, dvw016. 10.1093/eep/dvw016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Liu Y.; Bai F.; Zhang J.-Y.; Ma S.-H.; Liu J.; Xu Z.-D.; Zhu H.-G.; Ling Z.-Q.; Ye D.; Guan K.-L.; Xiong Y. Tumor Development Is Associated with Decrease of TET Gene Expression and 5-Methylcytosine Hydroxylation. Oncogene 2013, 32, 663–669. 10.1038/onc.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Liu M. Distribution of 5-Hydroxymethylcytosine in Different Human Tissues. J. Nucleic Acids 2011, 2011, 870726. 10.4061/2011/870726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner M. C.; Chaux A.; Meeker A. K.; Esopi D. M.; Gerber J.; Pellakuru L. G.; Toubaji A.; Argani P.; Iacobuzio-Donahue C.; Nelson W. G.; Netto G. J.; De Marzo A. M.; Yegnasubramanian S. Global 5-Hydroxymethylcytosine Content Is Significantly Reduced in Tissue Stem/progenitor Cell Compartments and in Human Cancers. Oncotarget 2011, 2, 627–637. 10.18632/oncotarget.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M.; Huang Y.; Jankowska A. M.; Pape U. J.; Tahiliani M.; Bandukwala H. S.; An J.; Lamperti E. D.; Koh K. P.; Ganetzky R.; Liu X. S.; Aravind L.; Agarwal S.; Maciejewski J. P.; Rao A. Impaired Hydroxylation of 5-Methylcytosine in Myeloid Cancers with Mutant TET2. Nature 2010, 468, 839–843. 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilat N.; Tabachnik T.; Shwartz A.; Shahal T.; Torchinsky D.; Michaeli Y.; Nifker G.; Zirkin S.; Ebenstein Y. Single-Molecule Quantification of 5-Hydroxymethylcytosine for Diagnosis of Blood and Colon Cancers. Clin. Epigenet. 2017, 9, 70. 10.1186/s13148-017-0368-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslanyan M. G.; van Rooij A.; Koorenhof-Scheele T. N.; Massop M.; Carell T.; Boezeman J. B.; Marie J.-P.; Halkes C. J. M.; de Witte T. M.; Huls G.; Suciu S.; Wevers R.; van der Reijden B. A.; Jansen J. H. Aberrant 5-Hydroxymethylcytosine Levels Correlate With Poor Overall Survival In Acute Myeloid Leukemia. Blood 2013, 122, 1261. [Google Scholar]
- LIAO Y.; GU J.; WU Y.; LONG X.; GE D.; XU J.; DING J. Low Level of 5-Hydroxymethylcytosine Predicts Poor Prognosis in Non-Small Cell Lung Cancer. Oncol. Lett. 2016, 11, 3753–3760. 10.3892/ol.2016.4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeze L. I.; Aslanyan M. G.; van Rooij A.; Koorenhof-Scheele T. N.; Massop M.; Carell T.; Boezeman J. B.; Marie J.-P.; Halkes C. J. M.; de Witte T.; Huls G.; Suciu S.; Wevers R. A.; van der Reijden B. A.; Jansen J. H. EORTC Leukemia Group and GIMEMA. Characterization of Acute Myeloid Leukemia Based on Levels of Global Hydroxymethylation. Blood 2014, 124, 1110–1118. 10.1182/blood-2013-08-518514. [DOI] [PubMed] [Google Scholar]
- Song C.-X.; Yin S.; Ma L.; Wheeler A.; Chen Y.; Zhang Y.; Liu B.; Xiong J.; Zhang W.; Hu J.; Zhou Z.; Dong B.; Tian Z.; Jeffrey S. S.; Chua M.-S.; So S.; Li W.; Wei Y.; Diao J.; Xie D.; et al. 5-Hydroxymethylcytosine Signatures in Cell-Free DNA Provide Information about Tumor Types and Stages. Cell Res. 2017, 27, 1231–1242. 10.1038/cr.2017.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X.; Sun B.; Chen C.; Gao C.; Zhang J.; Lu X.; Wang L.; Li X.; Xing Y.; Liu R.; Han X.; Qi Z.; Zhang X.; He C.; Han D.; Yang Y.-G.; Kan Q. Circulating Tumor DNA 5-Hydroxymethylcytosine as a Novel Diagnostic Biomarker for Esophageal Cancer. Cell Res. 2018, 28, 597–600. 10.1038/s41422-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Zhang X.; Lu X.; You L.; Song Y.; Luo Z.; Zhang J.; Nie J.; Zheng W.; Xu D.; Wang Y.; Dong Y.; Yu S.; Hong J.; Shi J.; Hao H.; Luo F.; Hua L.; Wang P.; Qian X.; et al. 5-Hydroxymethylcytosine Signatures in Circulating Cell-Free DNA as Diagnostic Biomarkers for Human Cancers. Cell Res. 2017, 27, 1243–1257. 10.1038/cr.2017.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth M. J.; Branco M. R.; Ficz G.; Oxley D.; Krueger F.; Reik W.; Balasubramanian S. Quantitative Sequencing of 5-Methylcytosine and 5-Hydroxymethylcytosine at Single-Base Resolution. Science (Washington, DC, U. S.) 2012, 336, 934–937. 10.1126/science.1220671. [DOI] [PubMed] [Google Scholar]
- Yu M.; Hon G. C.; Szulwach K. E.; Song C. X.; Zhang L.; Kim A.; Li X.; Dai Q.; Shen Y.; Park B.; Min J. H.; Jin P.; Ren B.; He C. Base-Resolution Analysis of 5-Hydroxymethylcytosine in the Mammalian Genome. Cell 2012, 149, 1368–1380. 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S.-G.; Wu X.; Li A. X.; Pfeifer G. P. Genomic Mapping of 5-Hydroxymethylcytosine in the Human Brain. Nucleic Acids Res. 2011, 39, 5015–5024. 10.1093/nar/gkr120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C.-X.; Szulwach K. E.; Fu Y.; Dai Q.; Yi C.; Li X.; Li Y.; Chen C.-H.; Zhang W.; Jian X.; Wang J.; Zhang L.; Looney T. J.; Zhang B.; Godley L. A.; Hicks L. M.; Lahn B. T.; Jin P.; He C. Selective Chemical Labeling Reveals the Genome-Wide Distribution of 5-Hydroxymethylcytosine. Nat. Biotechnol. 2011, 29, 68–72. 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D.; Lu X.; Shih A. H.; Nie J.; You Q.; Xu M. M.; Melnick A. M.; Levine R. L.; He C. A Highly Sensitive and Robust Method for Genome-Wide 5hmC Profiling of Rare Cell Populations. Mol. Cell 2016, 63, 711–719. 10.1016/j.molcel.2016.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooijman D.; Dey S. S.; Boisset J.-C.; Crosetto N.; van Oudenaarden A. Single-Cell 5hmC Sequencing Reveals Chromosome-Wide Cell-to-Cell Variability and Enables Lineage Reconstruction. Nat. Biotechnol. 2016, 34, 852–856. 10.1038/nbt.3598. [DOI] [PubMed] [Google Scholar]
- Treangen T. J.; Salzberg S. L. Repetitive DNA and next-Generation Sequencing: Computational Challenges and Solutions. Nat. Rev. Genet. 2012, 13, 36–46. 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C.-X.; Clark T. A.; Lu X.-Y.; Kislyuk A.; Dai Q.; Turner S. W.; He C.; Korlach J. Sensitive and Specific Single-Molecule Sequencing of 5-Hydroxymethylcytosine. Nat. Methods 2012, 9, 75–77. 10.1038/nmeth.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flusberg B. A.; Webster D. R.; Lee J. H.; Travers K. J.; Olivares E. C.; Clark T. A.; Korlach J.; Turner S. W. Direct Detection of DNA Methylation during Single-Molecule, Real-Time Sequencing. Nat. Methods 2010, 7, 461–465. 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand A. C.; Jain M.; Eizenga J. M.; Musselman-Brown A.; Olsen H. E.; Akeson M.; Paten B. Mapping DNA Methylation with High-Throughput Nanopore Sequencing. Nat. Methods 2017, 14, 411–413. 10.1038/nmeth.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson J. T.; Workman R. E.; Zuzarte P. C.; David M.; Dursi L. J.; Timp W. Detecting DNA Cytosine Methylation Using Nanopore Sequencing. Nat. Methods 2017, 14, 407–410. 10.1038/nmeth.4184. [DOI] [PubMed] [Google Scholar]
- Stoiber M. H.; Quick J.; Egan R.; Lee J. E.; Celniker S. E.; Neely R.; Loman N.; Pennacchio L.; Brown J. B.. De Novo Identification of DNA Modifications Enabled by Genome-Guided Nanopore Signal Processing. 2016, bioRxiv. https://doi.org/10.1101/094672. [Google Scholar]
- Lam E. T.; Hastie A.; Lin C.; Ehrlich D.; Das S. K.; Austin M. D.; Deshpande P.; Cao H.; Nagarajan N.; Xiao M.; Kwok P.-Y. Genome Mapping on Nanochannel Arrays for Structural Variation Analysis and Sequence Assembly. Nat. Biotechnol. 2012, 30, 771–776. 10.1038/nbt.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy-Sakin M.; Ebenstein Y. Beyond Sequencing: Optical Mapping of DNA in the Age of Nanotechnology and Nanoscopy. Curr. Opin. Biotechnol. 2013, 24, 690–698. 10.1016/j.copbio.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Song C.-X.; Diao J.; Brunger A. T.; Quake S. R. Simultaneous Single-Molecule Epigenetic Imaging of DNA Methylation and Hydroxymethylation. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 4338–4343. 10.1073/pnas.1600223113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zirkin S.; Fishman S.; Sharim H.; Michaeli Y.; Don J.; Ebenstein Y. Lighting up Individual DNA Damage Sites by in Vitro Repair Synthesis. J. Am. Chem. Soc. 2014, 136, 7771–7776. 10.1021/ja503677n. [DOI] [PubMed] [Google Scholar]
- Michaeli Y.; Shahal T.; Torchinsky D.; Grunwald A.; Hoch R.; Ebenstein Y. Optical Detection of Epigenetic Marks: Sensitive Quantification and Direct Imaging of Individual Hydroxymethylcytosine Bases. Chem. Commun. (Cambridge, U. K.) 2013, 49, 8599–8601. 10.1039/c3cc42543f. [DOI] [PubMed] [Google Scholar]
- Arielly R.; Ebenstein Y. Irys Extract. Bioinformatics 2018, 34, 134. 10.1093/bioinformatics/btx437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscarlet M.; Tessier A.; Provost S.; Mollica L.; Busque L. Human Blood Cell Levels of 5-Hydroxymethylcytosine (5hmC) Decline with Age, Partly Related to Acquired Mutations in TET2. Exp. Hematol. 2016, 44, 1072–1084. 10.1016/j.exphem.2016.07.009. [DOI] [PubMed] [Google Scholar]
- Thomson J. P.; Hunter J. M.; Nestor C. E.; Dunican D. S.; Terranova R.; Moggs J. G.; Meehan R. R. Comparative Analysis of Affinity-Based 5-Hydroxymethylation Enrichment Techniques. Nucleic Acids Res. 2013, 41, e206. 10.1093/nar/gkt1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C.-X.; Yi C.; He C. Mapping Recently Identified Nucleotide Variants in the Genome and Transcriptome. Nat. Biotechnol. 2012, 30, 1107–1116. 10.1038/nbt.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulwach K. E.; Li X.; Li Y.; Song C. X.; Han J. W.; Kim S. S.; Namburi S.; Hermetz K.; Kim J. J.; Rudd M. K.; Yoon Y. S.; Ren B.; He C.; Jin P. Integrating 5-Hydroxymethylcytosine into the Epigenomic Landscape of Human Embryonic Stem Cells. PLoS Genet. 2011, 7, e1002154. 10.1371/journal.pgen.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L.; Li X.; Yan L.; Tan Y.; Li R.; Zhao Y.; Wang Y.; Xie J.; Zhang Y.; Song C.; Yu M.; Liu X.; Zhu P.; Li X.; Hou Y.; Guo H.; Wu X.; He C.; Li R.; Tang F.; et al. Whole-Genome Analysis of 5-Hydroxymethylcytosine and 5-Methylcytosine at Base Resolution in the Human Brain. Genome Biol. 2014, 15, R49. 10.1186/gb-2014-15-3-r49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekpli X.; Urbanucci A.; Hashim A.; Vågbø C. B.; Lyle R.; Kringen M. K.; Staff A. C.; Dybedal I.; Mills I. G.; Klungland A.; Staerk J. Changes of 5-Hydroxymethylcytosine Distribution during Myeloid and Lymphoid Differentiation of CD34+ Cells. Epigenet. Chromatin 2016, 9, 21. 10.1186/s13072-016-0070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaratlerdsiri W.; Chan E. K. F.; Petersen D. C.; Yang C.; Croucher P. I.; Bornman M. S. R.; Sheth P.; Hayes V. M. Next Generation Mapping Reveals Novel Large Genomic Rearrangements in Prostate Cancer. Oncotarget 2017, 8, 23588–23602. 10.18632/oncotarget.15802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak A. C. Y.; Lai Y. Y. Y.; Lam E. T.; Kwok T.-P.; Leung A. K. Y.; Poon A.; Mostovoy Y.; Hastie A. R.; Stedman W.; Anantharaman T.; Andrews W.; Zhou X.; Pang A. W. C.; Dai H.; Chu C.; Lin C.; Wu J. J. K.; Li C. M. L.; Li J.-W.; Yim A. K. Genome-Wide Structural Variation Detection by Genome Mapping on Nanochannel Arrays. Genetics 2016, 202, 351. 10.1534/genetics.115.183483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandiedonck C.; Knight J. C. The Human Major Histocompatibility Complex As a Paradigm in Genomics Reseaarch. Briefings Funct. Genomics Proteomics 2009, 8, 379–394. 10.1093/bfgp/elp010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiina T.; Inoko H.; Kulski J. K. An Update of the HLA Genomic Region, Locus Information and Disease Associations: 2004. Tissue Antigens 2004, 64, 631–649. 10.1111/j.1399-0039.2004.00327.x. [DOI] [PubMed] [Google Scholar]
- Hetherington S.; Hughes A. R.; Mosteller M.; Shortino D.; Baker K. L.; Spreen W.; Lai E.; Davies K.; Handley A.; Dow D. J.; Fling M. E.; Stocum M.; Bowman C.; Thurmond L. M.; Roses A. D. Genetic Variations in HLA-B Region and Hypersensitivity Reactions to Abacavir. Lancet 2002, 359, 1121–1122. 10.1016/S0140-6736(02)08158-8. [DOI] [PubMed] [Google Scholar]
- Carapito R.; Radosavljevic M.; Bahram S. Next-Generation Sequencing of the HLA Locus: Methods and Impacts on HLA Typing, Population Genetics and Disease Association Studies. Hum. Immunol. 2016, 77, 1016–1023. 10.1016/j.humimm.2016.04.002. [DOI] [PubMed] [Google Scholar]
- Hosomichi K.; Shiina T.; Tajima A.; Inoue I. The Impact of next-Generation Sequencing Technologies on HLA Research. J. Hum. Genet. 2015, 60, 665–673. 10.1038/jhg.2015.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bönisch C.; Hake S. B. Histone H2A Variants in Nucleosomes and Chromatin: More or Less Stable?. Nucleic Acids Res. 2012, 40, 10719–10741. 10.1093/nar/gks865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braastad C. D.; Hovhannisyan H.; van Wijnen A. J.; Stein J. L.; Stein G. S. Functional Characterization of a Human Histone Gene Cluster Duplication. Gene 2004, 342, 35–40. 10.1016/j.gene.2004.07.036. [DOI] [PubMed] [Google Scholar]
- Talbert P. B.; Henikoff S. Histone Variants — Ancient Wrap Artists of the Epigenome. Nat. Rev. Mol. Cell Biol. 2010, 11, 264–275. 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- Sapienza C.; Lee J.; Powell J.; Erinle O.; Yafai F.; Reichert J.; Siraj E. S.; Madaio M. DNA Methylation Profiling Identifies Epigenetic Differences between Diabetes Patients with ESRD and Diabetes Patients without Nephropathy. Epigenetics 2011, 6, 20–28. 10.4161/epi.6.1.13362. [DOI] [PubMed] [Google Scholar]
- Zhai J.-M.; Yin X.-Y.; Hou X.; Hao X.-Y.; Cai J.-P.; Liang L.-J.; Zhang L.-J. Analysis of the Genome-Wide DNA Methylation Profile of Side Population Cells in Hepatocellular Carcinoma. Dig. Dis. Sci. 2013, 58, 1934–1947. 10.1007/s10620-013-2663-4. [DOI] [PubMed] [Google Scholar]
- Netea M. G.; Wijmenga C.; O’Neill L. A. J. Genetic Variation in Toll-like Receptors and Disease Susceptibility. Nat. Immunol. 2012, 13, 535–542. 10.1038/ni.2284. [DOI] [PubMed] [Google Scholar]
- Jiao Y.; Peluso P.; Shi J.; Liang T.; Stitzer M. C.; Wang B.; Campbell M. S.; Stein J. C.; Wei X.; Chin C.; Guill K.; Regulski M.; Kumari S.; Olson A.; Gent J.; Schneider K. L.; Wolfgruber T. K.; May M. R.; Springer N. M.; Antoniou E. Improved Maize Reference Genome with Single-Molecule Technologies. Nature 2017, 546, 524–527. 10.1038/nature22971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton M.; Sebra R.; Pang A. W. C.; Ummat A.; Franzen O.; Rausch T.; Stütz A. M.; Stedman W.; Anantharaman T.; Hastie A.; Dai H.; Fritz M. H.-Y.; Cao H.; Cohain A.; Deikus G.; Durrett R. E.; Blanchard S. C.; Altman R.; Chin C.-S.; Guo Y.; et al. Assembly and Diploid Architecture of an Individual Human Genome via Single-Molecule Technologies. Nat. Methods 2015, 12, 780–786. 10.1038/nmeth.3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nifker G.; Levy-Sakin M.; Berkov-Zrihen Y.; Shahal T.; Gabrieli T.; Fridman M.; Ebenstein Y. One-Pot Chemoenzymatic Cascade for Labeling of the Epigenetic Marker 5-Hydroxymethylcytosine. ChemBioChem 2015, 16, 1857–1860. 10.1002/cbic.201500329. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Zhang Y.; Scheuring C. F.; Wu C.-C.; Dong J. J.; Zhang H.-B. Preparation of Megabase-Sized DNA from a Variety of Organisms Using the Nuclei Method for Advanced Genomics Research. Nat. Protoc. 2012, 7, 467–478. 10.1038/nprot.2011.455. [DOI] [PubMed] [Google Scholar]
- Quinlan A. R.; Hall I. M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B.; Salzberg S. L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Handsaker B.; Wysoker A.; Fennell T.; Ruan J.; Homer N.; Marth G.; Abecasis G.; Durbin R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz A.; Park K.; Lim D. A.; Song J. S. Normalization, Bias Correction, and Peak Calling for ChIP-Seq. Stat. Appl. Genet. Mol. Biol. 2012, 10.1515/1544-6115.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham I.; Kundaje A.; Aldred S. F.; Collins P. J.; Davis C. A.; Doyle F.; Epstein C. B.; Frietze S.; Harrow J.; Kaul R.; Khatun J.; Lajoie B. R.; Landt S. G.; Lee B.-K.; Pauli F.; Rosenbloom K. R.; Sabo P.; Safi A.; Sanyal A.; Shoresh N.; et al. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F.; Ryan D. P.; Grüning B.; Bhardwaj V.; Kilpert F.; Richter A. S.; Heyne S.; Dündar F.; Manke T. deepTools2: A next Generation Web Server for Deep-Sequencing Data Analysis. Nucleic Acids Res. 2016, 44, W160–W165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.