

Abstract
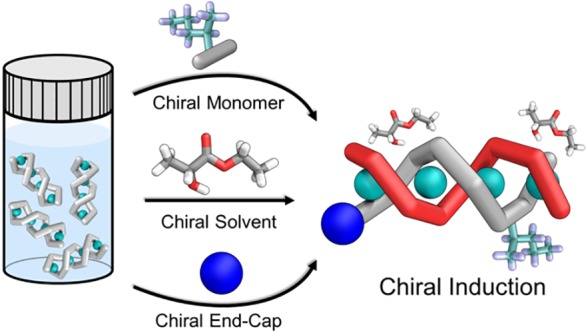
Self-assembled helical polymers hold great promise as new functional materials, where helical handedness controls useful properties such as circularly polarized light emission or electron spin. The technique of subcomponent self-assembly can generate helical polymers from readily prepared monomers. Here we present three distinct strategies for chiral induction in double-helical metallopolymers prepared via subcomponent self-assembly: (1) employing an enantiopure monomer, (2) polymerization in a chiral solvent, (3) using an enantiopure initiating group. Kinetic and thermodynamic models were developed to describe the polymer growth mechanisms and quantify the strength of chiral induction, respectively. We found the degree of chiral induction to vary as a function of polymer length. Ordered, rod-like aggregates more than 70 nm long were also observed in the solid state. Our findings provide a basis to choose the most suitable method of chiral induction based on length, regiochemical, and stereochemical requirements, allowing stereochemical control to be established in easily accessible ways.
Introduction
Chiral molecules interact differentially with circularly polarized light and electrons in different spin states, giving rise to useful properties1,2 and functions.3−7 Helical materials are employed throughout biological systems, serving as scaffolds8−10 for mechanical support and for the precise spatial arrangement of dipoles in ion channels.11,12 Recently, interest has grown in using helical structures for applications13−17 that include circularly polarized light emission,3,4,6,18−21 information storage,22 and in spintronics as electron-spin filters.1,5 Helical polymers23−28 are particularly useful in these contexts26 due to their modular structures and scalable methods of preparation.29,30 Achieving control over the helical handedness of these materials is necessary for their use.5,31,32
Encoding stereochemical information in the form of stereocenters appended to the monomer is an effective means of dictating helical handedness.33−35 However, this method requires the preparation of enantiomerically pure building blocks, the ease of which is specific to the choice of monomer. A more flexible approach would employ an additive36,37 that dictates the helical handedness of a polymer comprising either achiral or racemic monomer units.38−43 This approach has borne fruit44 for the helically folded polymers known as foldamers.44−46 These synthetic oligomers are formed of amide-linked achiral monomer units terminated with end-groups containing chiral moieties, which dictate the preferred foldamer helicity.38,47−49 Enantioenriched supramolecular polymers have also been formed using an enantiopure initiating species, whereby the chiral information imprinted at initiation propagates along the length of the polymer.39,41,50
Identifying the optimal approach to generate a helical polymer having a single handedness requires consideration of the strength51 of the helical bias and the mechanism52−54 of polymerization. The former can be probed using statistical mechanics,51,55 which has proved useful in describing the strength of chiral induction in supramolecular polymers.56 Complementarily, kinetic models have elucidated the polymerization mechanism52 for a variety of covalent polymers.57−59 The polymerization mechanisms of self-assembled polymers54,60−62 can be more difficult to unravel63−65 due to interplay between the dynamic interactions66,67 that knit such structures together.
The self-assembly of monomer units68 is an effective approach to obtaining functional helical polymers10,69 with a minimum of synthetic effort.31,70−76 Prior studies have demonstrated an approach to forming helical, self-assembled polymers based on subcomponent self-assembly.24,67 This method relies on the use of dynamic-covalent imine (C=N) bonds, whereby reversibility serves as an error-checking mechanism within the growing structure.24 The CuI-templated imine condensation of an amino-aldehyde monomer has thus afforded conjugated, double-helical metallopolymers, which exhibited a tunable white-light emission, dependent upon the length of the polymer.24
The dynamic-covalent imine linkages within polymers prepared via subcomponent self-assembly can define a pathway for electronic conjugation24 between monomer units. Such structures are of interest in the study of electronic delocalization in a helical77 environment.1,5 As yet, methods to induce a preferred helical handedness in these polymers24 have not been reported. Control over the helicity is critical for applications that include circularly polarized light emission and chirality-induced spin selectivity.1
Herein, three strategies to control helical handedness in subcomponent self-assembled two-stranded helical metallopolymers are presented and compared: (1) polymerization from an enantiopure monomer, (2) chiral induction from an enantiopure solvent, (3) use of an enantiopure initiating group. In this discussion, we interleave discussion of these three strategies with the development of a model that describes the growth mechanism of the polymer. This model quantifies the varying degrees of induction observed between the different chiral induction strategies.
Our model includes the different behaviors arising from the relative orientations of the two strands of the polymer helix, which can be controlled through the use of different initiating groups. The strength of chiral induction for each of the different methods was then evaluated using a statistical mechanics model. This model enables the selection of the most suitable method of chiral induction for a given application. Finally, electron microscopy revealed the presence of crystalline aggregates in thin films, with lengths exceeding 70 nm. The structure of these aggregates revealed the hierarchical self-assembly of polymer chains having the same handedness into ordered, homochiral bundles.
Results and Discussion
Mechanism of Polymerization
The origins of the helical biases can be understood in terms of the mechanistic model shown in Figure 1. Two key steps thus govern the polymerization process: first, initiation results in the formation of a P or M nucleus, P-P1 or M-P1, which dictates the screw-sense of the growing polymer. Second, polymerization yields P-Pn or M-Pn from a nucleus of either P or M helicity, respectively, maintaining the handedness of the nucleus from which it grew.
Figure 1.

Schematic of the “standard polymerization” model of monomer A around CuI. An extension to this model allows for a chiral amine initiator, C, to induce a helical bias (“amine induction”). The polymerization model is detailed in Supporting Information Section 6.
In the case of no applied bias toward forming one helical handedness, i.e., when monomer A formed polymer 1, the energetic barriers to nucleation of the M-P1 and P-P1 double helices were equal, as were the subsequent polymerization steps leading to M-Pn and P-Pn, respectively. Therefore, polymerization proceeded equally down the M and P pathways to afford a racemic mixture of polymers, reflected in a featureless CD spectrum (Figure 2). However, in the case of chiral monomer B, the steric requirements of the side-chain stereocenter rendered the barrier to forming M-P1 higher than for P-P1, leading to a higher population of P polymer helices.
Figure 2.
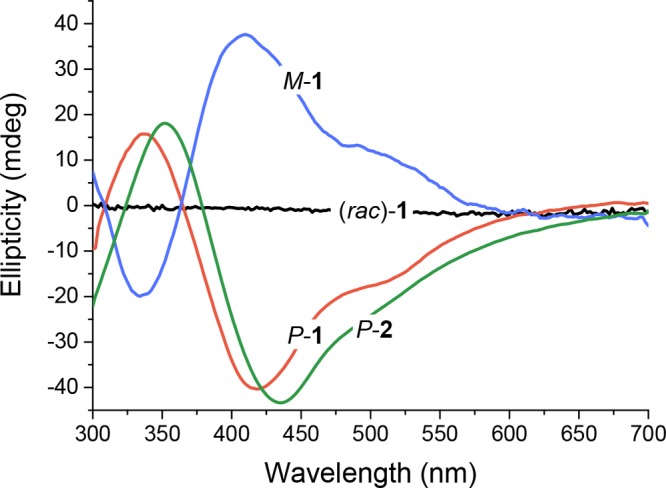
Circular dichroism (CD) spectra of polymers 1 and 2. The red-shift observed for P-2 with respect to P-1 is attributed to the greater length of P-2. Assembly of chiral monomer B with CuI produced enantioenriched polymer P-2. Racemic polymer 1 showed no CD signal. The corresponding UV–vis spectra are presented in Figure S2. Polymer chiral induction occurred when chiral chain-capping groups were employed, as discussed below. Chain caps of opposite handedness gave polymers with mirror-image CD spectra.
Polymerization from an Enantiopure Monomer
Polymer 1 self-assembled from monomer A (1 equiv) and CuINTf2 (0.5 equiv; Tf = O2SCF3) in dry degassed CH3CN (Scheme 1). The absence of a circular dichroism (CD) signal (Figure 2) indicated the presence of a racemic mixture of M and P helices.
Scheme 1. Subcomponent Self-Assembly of Double-Helical Metallopolymers 1, and 2 from Monomers A and B, Respectively.

Addition of monomer A to CuI gave racemic helical metallopolymer 1.
Introducing an enantiomerically pure initiating group, (R)-C or (S)-C, lent stereoselectivity to the polymerization reaction, yielding M-1 or P-1 preferentially.
Formation of helical polymers from enantiomerically pure monomer (S)-B also biased the reaction toward the formation of enantioenriched P-2.
Chiral monomer B, prepared as the S enantiomer as described in the Supporting Information Section 2, was designed to exhibit similar electronic properties to A while imparting asymmetry to the polymerization process, thus rendering one helical screw-sense more favorable to formation. CD bands for the resulting self-assembled polymer 2 were observed at 440 and 500 nm, which we assign to the π–π* and MLCT absorptions, respectively (Figure 2). We infer polymer 2 to have predominantly P-handedness, based upon the results of molecular mechanics modeling (Figure S1). The P helical preference for 2 is a consequence of how the stereocenters on adjacent chains interact at the periphery of the polymer chain. Our assignments obtained using different modes of stereochemical induction (see below) are self-consistent and consistent with stereochemical assignments for CuI helicates.78,79
Effects of Concentration
The strengths of chiroptical responses were compared across different experiments using the Kuhn dissymmetry factor,80,81gabs:
| 1 |
where AL (or AR) is the absorbance of left- (or right-) handed circularly polarized light at λmax in the CD spectra. The term (AL + AR)/2 corresponds to the unpolarized UV–vis absorption, rendering gabs independent of the sample concentration used in the measurement. The dissymmetry factor gabs is proportional to the enantiomeric excess (ee) of the system under study.82
The magnitude of gabs was used to probe the concentration dependence of polymerization (Figure 3). A series of samples containing chiral monomer B and CuINTf2 (0.5 equiv) in MeCN were prepared at concentrations between 0.002 and 42 mM. Below 0.2 mM, no CD signal was observed, consistent with the absence of helical polymer chains. Upon exceeding 0.2 mM, a CD signal started to appear. The gabs continued to increase before reaching a plateau at 6 mM, a concentration that we inferred to correspond to the integration of all free monomer units into chains of polymer 2.
Figure 3.
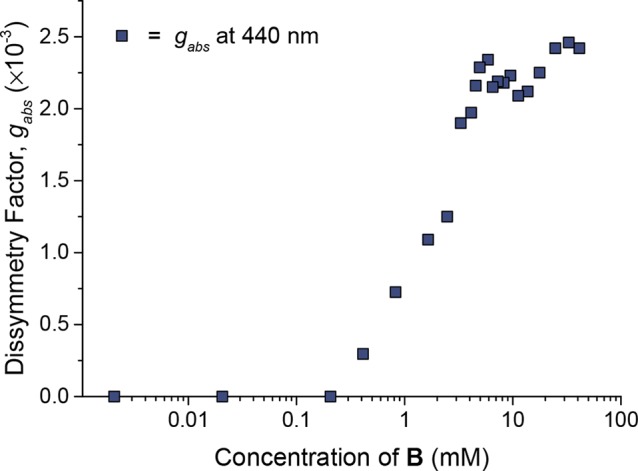
Plot of gabs (at 440 nm) against the concentration of chiral monomer B when mixed with CuI (0.5 equiv) in acetonitrile. No signal was observed below 0.2 mM. The increase in gabs between 0.2 and 6 mM is inferred to reflect an increase in the degree of polymerization, and the plateau above 3 mM to reflect a regime in which all monomer is incorporated into polymers.
Whereas 1H NMR at 0.1 mM revealed predominantly monomer B (Figure S3), at 20 mM concentration no signals corresponding to monomer B were observed, with broader signals assigned to polymer 2 growing in their place. The observed increase in gabs is thus correlated with the growth of longer helices in solution at the expense of free monomer: B absorbed at 440 nm (Figure S4), but did not show a CD signal (Figure S5). Thus, monomers began to self-assemble into helical polymers at 0.2 mM and were fully assembled above 6 mM. Subsequent experiments were thus carried out within the plateau regime (6–60 mM) of Figure 3, where polymers are inferred to be fully assembled.
Probing Excited-State Delocalization along the Polymer
Increasing proportions of end-capping groups yielded oligomers of decreasing lengths (Figures S6, S7).24 Oligomers of P-2 having different lengths were prepared from 18 mM monomer B initiated with p-toluidine E. Longer oligomers had red-shifted π–π* absorptions (Figures S8 and S9), consistent with an increase in conjugation length.83 The degree of red shifting reached a plateau after 15 repeat units, increasing minimally for longer polymers. This observation suggested that the excitons produced following visible light absorption could delocalize across ca. 15 polymer repeat units.
Plotting gabs against polymer length (Figure 4) provides a complementary means to gauge the degree of exciton delocalization. An increase in gabs was observed with increasing polymer length (Figure 4), also approaching a plateau after 15 repeat units. This observation thus indicates that the excited-state wave functions probed using CD delocalize across ca. 15 repeat units.24
Figure 4.
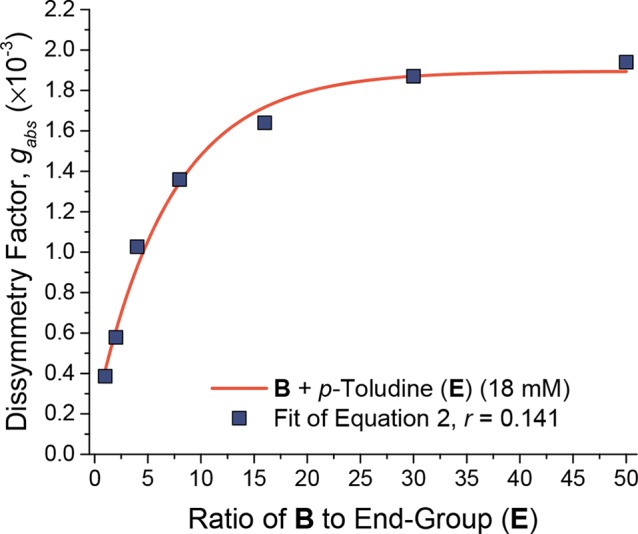
Plot of gabs as a function of the stoichiometric ratio of monomer B (at 18 mM in MeCN) to end-cap p-toluidine E and its fit to eq 2 (with least-squares R2 = 0.991).
The behavior of gabs with respect to polymer length was fitted to the empirical equation
| 2 |
where gmax is the maximum observed dissymmetry factor (we found a gmax of 1.95 × 10–3 in the case of polymer 2 initiated with p-toluidine, Figure 4), p is the ratio of monomer to initiating group, g1 is a parameter accounting for deviations of polymer length from the targeted polymer length based on the B to E ratio24 (here, g1 = 1.7 × 10–3), and r is an empirical constant, with a fitted value of 0.141 (at a monomer concentration of 18 mM), which describes the rise in gabs as a function of the number of repeat units in the polymer. The maximum gabs for a specific polymer length can thus be predicted using eq 2, so that it may be used subsequently in our kinetic model (see Supporting Information, Section 6).
Control of Polymer Handedness Using a Chiral Solvent
The nucleus of a helical polymer chain may absorb stereochemical information from the environment, without incorporating a chiral building block.84,85 The reaction of achiral monomer A with CuINTf2 in (S)-ethyl lactate ((S)-EL) thus yielded polymers biased toward M helical handedness (Figure 5a,b, Figure S11). The ability to perform self-assembly in (S)-EL is attractive because this solvent can be produced from renewable resources.86,87 In a control experiment, a racemic sample of polymer 1 (previously prepared in MeCN) was dissolved in (S)-EL. The corresponding CD spectrum (Figure 5b) was featureless. We thus infer the chiral solvent to imprint handedness during polymer formation, instead of preferentially solubilizing one handedness of 1 over the other.
Figure 5.

(a) Self-assembly of A around CuI in (S)-EL. Shorter oligomers were observed when more p-toluidine initiator (E) was added. (b) CD spectra of polymer 1 prepared in R vs S EL. Redissolving 1 that had been formed in MeCN in (S)-EL did not give rise to CD bands. (c) The gabs of polymer 1 incorporating only monomer A (no added E) correlated linearly with the ee of the EL solvent. The relative ee was calculated inferring that gabs = 2.54 × 10–3 corresponds to enantiopure 1 (see below). (d) Plots showing the increase of gabs with the ratio of monomer A to achiral initiating group E, with polymerization carried out in EL of different ee. The data were fitted to eq 2 using r = 0.141, and largest observed gabs values of 0.89 × 10–3, 0.32 × 10–3, and 0.10 × 10–3 were obtained in 100%, 40%, and 15% ee (S)-EL, respectively.
The chiral solvent effect can be understood in terms of the model of Figure 1, where the asymmetric solvent environment biases initiation by increasing the barrier to forming M-P1 relative to that of P-P1. Moreover, this bias was found to correlate linearly with the ee of the solvent (Figure 5c).
The relationship between gabs and the ratio of monomer A to initiating group E for polymer 1 assembled in (S)-EL of varying ee was probed using eq 2 (Figures 5d, S11, S12). The same value of r = 0.141 previously obtained for polymer 2 grown from chiral monomer B (Figure 4) gave excellent fits (Figure 5d). We thus infer that similar polymer lengths for a given ratio of monomer to initiator are obtained in MeCN and (S)-EL solvents. Equation 2 thus appears to have general application in correlating gabs with length in this class of helical polymer.
The gabs values of the polymers formed from achiral A in enantiopure (S)-EL are lower than those obtained using chiral monomer B in achiral solvent, implying a lower degree of enantiodifferentiation during polymerization in the chiral solvent. This weaker discrimination implies a smaller energetic difference between the activation barriers leading to M and P helices in the chiral solvent (S)-EL than with the chiral monomer B. We hypothesize that greater enantiodiscrimination might be achieved by using a solvent having stronger, specific interactions with the polymer nucleus formed during initiation.
Although the chiral solvent gave a lesser degree of enantiomeric enrichment, its use does not require the preparation of an enantiopure monomer. This method of chiral induction may thus be applicable to the stereoselective preparation of other chiral structures that are composed of achiral building blocks using subcomponent self-assembly.88
Free-Energy Penalty of Forming the Disfavored Helical Handedness
The magnitude of the screw-sense bias imparted by a given agent (i.e., a chiral solvent or monomer) can be gauged using a statistical mechanics model. This model estimates a free energy penalty, f1, measured in thermal energy units (kBT), incurred when a chiral subcomponent adopts an unfavorable helical handedness (Figure 6).51 The statistical mechanics model developed to quantify the strength of chiral induction in these polymers was predicated upon a static polymer structure, with no unwinding taking place following initial screw-sense determination. Since the handedness is determined at nucleation, our approach evaluated the additive contributions to the free energy of a “seed” complex P1 (Figure 1) composed of two ligands, which contribute independently. We then evaluate the Boltzmann distribution of stereochemical states, P or M, of this complex. A maximum gabs of 2.54 × 10–3 was observed in several different cases of chiral induction, as described in Supporting Information Section 7.2. We infer that this gabs corresponds to an ee of polymer 2 that approaches 100%, in these different cases where f1 ≫ kBT. Thus, we define the relative enantiomeric excess (rel ee) in each case as the maximum observed gabs divided by the maximum gabs (2.54 × 10–3) of enantiopure helix 2.
Figure 6.
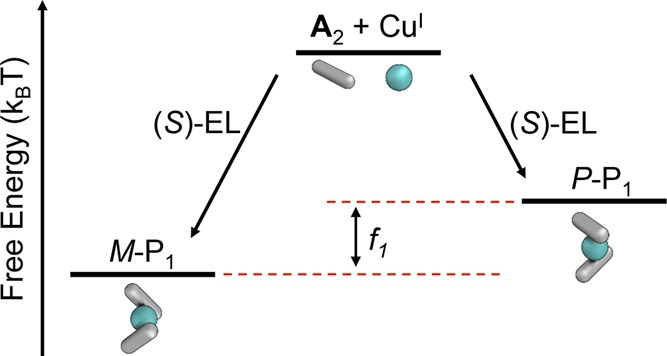
Our statistical mechanics model predicts the free energy penalty, f1, for forming a nucleus of the disfavored helical handedness using a given method of chiral induction (here the chiral solvent (S)-EL).
This maximum gabs value was measured from a sample that assembled at 20 mM monomer concentration, i.e., in the regime of complete polymerization (Figure 3). In order to simplify our model, we consider only end-cap to polymer ratios within the plateau region from Figure 4, thus where gabs is independent of length. We did not observe any sergeants-and-soldiers40 or majority-rules23 behavior over the course of this study; the use of a source of chiral information on less than 100% ee always resulted in a commensurate decrease in the polymer ee. Section 7 of the Supporting Information provides a complete discussion and derivation of the statistical model.
Initiation involving chiral monomer B incurred a greater free energy penalty than was observed when forming polymer 1 in (S)-EL: in the case of B, f1 was determined to be 2.15 kBT, whereas the weaker discrimination between P and M helices in the chiral solvent (S)-EL afforded an f1 of 0.36 kBT.
Stereochemical Amplification Using Chiral End-Cap C
Biasing the helical screw-sense using either chiral monomer B or chiral solvent (S)-EL required the use of synthetically challenging enantiopure monomers or a large excess of chiral solvent, respectively. A substoichiometric amount of an enantiopure chain end-capping group, in contrast, might allow a small amount of chiral signal to fix the handedness of the entire length of a helical polymer strand.33,41,89−91
When enantiopure amine C was employed to nucleate the growth of helical polymer 1 from A, the polymer was observed to take on a preferred helical handedness; using the other enantiomer of C gave rise to the opposite helical handedness (Figure 2). Racemic C yielded no helical bias (Figure S13). Our mechanistic model of polymerization was thus extended beyond “standard polymerization” to describe the functioning of this “amine induction” approach (Figure 1), providing an alternate pathway to forming a chiral helical nucleus, P/M-CP1.
Stereochemical amplification thus occurs during the polymerization of A from chiral initiating group C; subsequent polymer growth from this chiral nucleus maintains the initial helical handedness. The addition of enantiopure (R)-C to a racemic solution of preformed 1 thus did not result in any change in the helical handedness (Figure S14). This observation is consistent with our inference of a high energetic cost to the helical unwinding of polymer 1.
Effects of Regiochemistry on Chiral Induction
The two strands of double-helical polymer 1 can orient in either a parallel head-to-head (HH) or an antiparallel head-to-tail (HT) configuration (Figure 7a).24 When less-bulky amine C was employed to cap the aldehyde groups of A within 1, the HH regioisomer was obtained. We hypothesize that hydrogen bonds between the hydroxyl groups of B residues favor this conformation, in similar fashion to what was observed in the case of dicopper(I) helicates.78 Such HH polymers contain a free, uncapped diamine terminus and are living, since the polymer is capable of continuing to grow upon further introduction of monomer.
Figure 7.

(a) Chiral amine C afforded an excess of the HH regioisomer, whereas bulkier chiral amine F led predominantly to the HT regioisomer. The HH regioisomer possesses a free terminus capable of elongation, whereas HT has both termini capped, hindering further elongation. The degree of stereochemical induction was in all cases gauged using the dissymmetry factor gabs, which is proportional to ee, with a gabs of 2.54 × 10–3 corresponding to 100% ee (Supporting Information Section 7.2). (b) Plot of gabs against the monomer-to-chiral-inducer ratio (proportional to polymer length, Figure S24) for the HH system where all precursors were combined simultaneously. A decrease in induction was observed at an A/C ratio above 2. (c) gabs plotted against the rate of addition of A (18 equiv) and CuI (9 equiv) to (S)-C (1 equiv); slower rates of addition favored a greater degree of induction. (d) Plot of gabs against the ratio of monomer A to bulky chiral inducer (R)-F where all precursors were combined simultaneously. Two regimes were observed: (i) where an increase in the A/F ratio favored a greater degree of chiral induction and (ii) where a further increase in the A/F ratio reduced the degree of chiral induction, as a greater proportion of polymer chains initiate from achiral A as opposed to chiral F. (e) Plot of gabs (440 nm) against the rate of addition of A (18 equiv) and CuI (9 equiv) to F (1 equiv), affording an HT polymer, again showing two regimes: (i) where an increase in rate of addition disfavored termination through chain-capping by free F, affording an increase in induction; (ii) more rapid addition reduced enantioenrichment because proportionally fewer polymer chains nucleated from chiral F residues and more initiation took place via the racemic standard polymerization pathway (Figure 1).
HT regioisomers were observed to form in the presence of bulkier amines.24 In contrast with the HH configuration, in HT double helices the bulky amine groups also serve as terminators, halting polymer growth at the end where they are incorporated. We hypothesized that the regiochemistry of the polymer influences the degree of chiral induction due to the different growth characteristics of the HH and HT regioisomers.
Oligomers of 1 having different lengths were prepared by increasing the ratio of monomer A and CuI with respect to (S)-C (Figure 7a) at the same overall CuI concentration (Scheme S5, Figures S15, S16, S17). The strength of chiral induction, gauged using gabs, correlated linearly with the ee of the (S)-C employed (Figure S18).
Our inference that C led to HH regiochemistry was supported by 19F NMR of an experiment wherein 5-fluoropyridine carboxaldehyde was employed to terminate the ends of oligomers initiated using C (Figure S19).24 In the case of HH regiochemistry, polymerization proceeded unidirectionally from the nucleus CP1 (Figure 1), pausing once either A or CuI was consumed and ceasing upon the addition of a terminating monoaldehyde. Polymerization can occur in both the presence and absence of chiral amine C. Therefore, the strength of enantioenrichment was dictated by the imbalance in the rates of formation of M-CP1 and P-CP1, with only polymers formed from the C-initiated pathway contributing to enantioenrichment.
As shown in Figure 7b, gabs underwent a sharp initial increase for oligomers as the A/C ratio increased from 1 to 2. We attribute this observation to the intrinsic increase in gabs associated with initial oligomer growth (Figure 4). Further elongation brought about a decrease in gabs, however. As discussed above, a greater proportion of A relative to C is required to obtain longer polymers. As the proportion of C decreases, the number of polymer chains initiating from achiral A increased, leading to a decrease in the helical handedness bias reflected in gabs.
The individual processes shown in Figure 1 could not be picked apart through direct spectroscopic analysis due to overlap of the monomer and polymer bands in UV–vis and also because CD intensity varies as a function of polymer length (Figure 4). Instead, the degree of induction was determined by fitting dissymmetry factors (gabs) as a function of different starting stoichiometries using a series of first-order differential equations (Supporting Information Section 6). Our model operates under the assumption that the CD spectrum is the sum of individual species in solution92 that do not interact electronically to give rise to new CD transitions. Thus, the CD intensity, gauged using gabs, was used to measure the difference in population between P and M helices (Supporting Information Section 6.2). The populations of P and M helices, determined using our model, were substituted into eq 3, which relates the modeled enantiomeric excess to gabs and takes into account the dependence of gabs on polymer length:
| 3 |
where gmax, g1, and r are the same fit parameters used in eq 2; NM and NP correspond to the population fractions of M and P helices, respectively; q is the difference in absorption cross section for left- and right-handed circularly polarized light; and σT is the total absorption cross section for unpolarized light. A full derivation of eq 3 is given in Supporting Information Section 6. The fit shown in Figure 7b is in good agreement with the experimental results, reproducing the key features of a rise followed by a decrease in gabs.
Maximizing Chiral Amplification in an HH Polymer
Our kinetic model predicted stereoinduction to be maximized either when A reacted with (S)-C, so as to nucleate a helix with a preferred handedness, or when A adds to a growing polymer chain that has nucleated from (S)-C already. This set of circumstances occurred during the slow, simultaneous addition of A and CuI into a solution of (S)-C (Schemes S7, S20). This slow addition reduces the probability of nucleation from achiral A by minimizing the free concentration of A throughout polymerization. As shown in Figure 7c, progressively higher degrees of helicity induction were observed at slower rates of addition, reaching a plateau at 0.07 μmol min–1. Here the A-to-C ratio was selected to produce an 18-mer at a concentration within the plateau region of Figure 4. We infer that this plateau corresponds to a gabs wherein minimal contributions occurred from the standard polymerization mechanism; the likelihood of a polymer nucleating in the absence of a chiral inducer is minimized under these conditions.
Under such conditions of maximal stereoinduction, the difference between the observed gabs and the maximum gabs must arise from the intrinsic directing strength of the end-capping group, which renders M-CP1 and P-CP1 energetically inequivalent (Figure 6); our statistical mechanics model reported a free-energy penalty (f1) for (S)-C that disfavored growth of the M helical screw-sense by 0.74 kBT.
Polymer growth from chiral initiator C thus resulted in enantioenriched HH polymers, which exhibited living behavior in maintaining an uncapped terminus capable of further elongation. Slow addition of monomer to the chiral initiator exploited this living behavior of the HH regioisomer, resulting in stereochemical amplification from the initiator throughout the helical polymer chain.
Influence of Head-to-Tail Regiochemistry on Chiral Induction
Chiral amplification occurred under conditions favoring HT regiochemistry when the bulkier chiral amine F was employed.24 The HT orientation was evidenced by 19F NMR spectroscopy of a model oligomer (Figure S19) and is supported by molecular modeling, which showed extensive steric clash in an HH terminus incorporating two F residues (Figure S1).
In constrast to the living HH regioisomer, an HT strand terminated by bulky F residues at both ends may not readily elongate, requiring dissociation of one terminating group before another monomer unit may attach.
In order to account for the nonliving state of a doubly terminated HT oligomer, an off-pathway polymer termination step was incorporated into our kinetic model (Supporting Information Section 6). Oligomers of different lengths were prepared by rapidly adding varying proportions of A and CuI to (R)-F in CH3CN (Scheme S8, Figures S21, S22, S23). The response of gabs to the A-to-F ratio (A/F) showed two distinct regimes (Figure 7d). An initial increase in gabs was observed for A/F ratios below 15, followed by a decrease in gabs for A/F > 15. Our kinetic model also captures the key features of a sharp rise, followed by a gradual fall, with eq 3 fitting well the experimental data of Figure 7d.
Similarities were observed between the responses of gabs to changes in the A/F ratio (for the HT regioisomer) and the A/C ratio (for the HH regioisomer). For HH polymers (Figure 7b), the initial increase in gabs with respect to the increasing A/C ratio was attributed to the inherently higher gabs for longer compared to shorter oligomers (Figure 4). However, the increase in gabs extended to higher chiral amine/monomer ratios in the case of the HT regiosiomer (Figure 7d) due to polymer termination, as F residues capped oligomers at shorter lengths for the HT regioisomer, whereas C did not cap the living HH regioisomer (Figures S23, S24). Hence at all ratios of monomer to chiral amine, longer polymers were produced in the case of amine C, which generated HH regiochemistry and which acted as an initiator, as compared to amine F, which favored HT regiochemistry and could both initiate and terminate polymer strands (Figure S24).
Maximizing Chiral Amplification in an HT Polymer
The rate of addition of A and CuI to a solution of F was varied, while keeping the A/F ratio constant (Figure 7e). Slower addition led to the formation of mixtures of shorter, F-initiated oligomers together with longer polymers that contained no F residues (Figures 7e, S25, S26; Scheme S9). The higher effective concentration of F with respect to A promoted termination at the expense of polymer growth under conditions of slow addition. This set of circumstances resulted in more F residues terminating short oligomers, while the residual A and CuI polymerized independently (via the standard polymerization mechanism, Figure 1), thereby reducing the helical handedness bias. At higher addition rates, we infer the A/F ratio to increase during polymerization, as A was added more rapidly than it was consumed. This state favors initiation from achiral A monomers, reducing the ee. The highest degree of chiral induction achieved for F thus involved an intermediate addition rate of 6.52 μmol min–1, at which the racemic standard polymerization mechanism (Figure 1) proceeds to a minimal degree.
The maximum gabs obtained in this slow addition experiment corresponds to a free energy penalty (f1) for amine F forming the disfavored helical handedness of 2.00 kBT (Supporting Information Section 7). We infer the magnitude of f1 for F to be greater than the corresponding value for C (0.74 kBT) because the bulkier sterics of F enforce a greater free energy penalty for “incorrect” incorporation than for C.51
Chiral amplification in polymer 1 thus depended upon its regiochemistry. The living character of the HH regioisomer was capable of yielding long, enantiomerically enriched polymers at slower rates of polymer growth. In contrast, the capability of amine F to terminate the HT regioisomer gave rise to poorer length control and required careful tuning of the initation and termination processes to obtain a high degree of enantioenrichment. The living HH regioisomer thus appears most appealing for future applications, not least due to its potential to grow multiple blocks of a single handedness, following sequential addition of different monomer units.
Hierarchical Assembly into Rod-like Aggregates
Directing polymer aggregation is critical for translating the benefits of chiral control over individual molecules into bulk properties for use in applications, including circularly polarized light emission93,94 and chiral-induced spin selectivity of electrical current.95−97 Consequently, structures formed through the aggregation of metallopolymer 1 were explored.
A population of aggregates was observed across all samples using dynamic light scattering (Figure S27). We sought to elucidate the structure of these aggregates using electron microscopy. A solid sample of 1 was prepared by evaporation of 1 mL of a 10 mg mL–1 solution of polymer 1 in MeCN at 298 K over 3 days in a N2-filled glovebag, yielding a smooth film. The film was then ground to a powder using a mortar and pestle and imaged using transmission electron microscopy (TEM). The majority of the specimen consisted of a poorly ordered phase (Figure 8a), with some regions appearing ordered (Figure 8b,c). Energy dispersive spectroscopy of both the amorphous and ordered regions revealed an elemental composition consistent with polymer 1 (Figure S29). We infer the amorphous regions to consist of strands of polymer 1 that adopt a disordered arrangement.
Figure 8.

(a) TEM image indicating the presence of high-aspect-ratio rod-like aggregates scattered among an amorphous phase. (b) High-resolution TEM image of a single rod-like aggregate; a series of striations are observable along the length of the aggregate; the ends of the rod were well-defined, suggesting aggregation of polymers having similar lengths; shorter polymers are observed at the edge of the structure (indicated by a red line). (c) HRTEM micrograph of a portion of a rod-shaped aggregate of polymer 1 showing two sets of striations. One set runs along the length of the rods, while a secondary set was observed diagonally across the aggregate. The dimensions of these striations coincide with the experimental and calculated interstrand and inter-CuI spacing, respectively. (d) Schematic model of the aggregates where rods of 1 undergo close packing, in which each helix interdigitates into the grooves of its neighbor.
The ordered structures observed within the TEM samples were high in contrast, revealing a rod-like architecture. The structures were observed to have a mean length of approximately 70 nm and a range of 20 to 110 nm. Electron energy loss spectroscopy indicated that these structures contained copper in the +1 oxidation state (Figure S30).
Striations running parallel to the long axes of the rods exhibited a spacing of 0.55 nm (Figure 8b). This distance is in agreement with the expected hexagonal close-packed spacing (ca. 0.6 nm) calculated from the closest spacing between adjacent polymers (1.2 nm) in the single crystal structure of an analogous oligomer.24 A second set of striations were observed along the length of the rod, running diagonally (Figure 8c). The separation between these striations was 0.3 nm, consistent with the CuI–CuI distance in the crystal structure of a 3-mer of 1. Within a single aggregate, all of the diagonal striations appeared to be parallel, indicating that chains in the aggregate all had the same helical handedness. Such homochirality is required for the close packing of helices, to allow their screw threads to interdigitate.
We thus infer these crystalline aggregates to consist of individual chains of 1 that are packed into the grooves of neighboring helices, affording a close-packed structure (Figure 8d). This stepwise assembly bears a resemblance to the hierarchical assembly of helically organized amyloid peptides into macroscopic fibrils.98,99
The spontaneous formation of ordered aggregates of a single helical handedness is relevant for the incorporation of these conjugated, double-helical metallopolymers into devices that exploit their conductive or emissive properties. These applications are currently under investigation, in addition to exploring means of isolating bulk quantities of enantiopure polymer.
Conclusions
The strength of chiral induction in these helical metallopolymers is dependent upon the choice of chiral induction mechanism, in addition to the length and regiochemistry of the polymer. Three distinct methods were demonstrated to provide control over polymer handedness: (1) chiral monomer, (2) chiral solvent, and (3) chiral initiator. Each method provides distinct advantages, and each is associated with a characteristic strength of polymer helicity induction.
With the design rules in hand to control the triad of length, regiochemistry, and stereochemistry, and an understanding of the length scale over which the excited states are delocalized and the polymers assemble, future work will focus upon rationally exploring this new class of materials and their ordered aggregates that were observed by TEM, for applications in polarized light emission and charge transport in a helical environment.
Acknowledgments
This work was funded by the UK Engineering and Physical Sciences Research Council (EPSRC EP/P027067/1 and EP/M01083x/1) and the European Research Council (ERC 695009). We are grateful to David Jefferson for assistance with TEM and to Haim Weissman and Boris Rybtchinski for informative discussions regarding the TEM images.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b06195.
Synthetic details and characterization data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Naaman R.; Waldeck D. H. J. Phys. Chem. Lett. 2012, 3, 2178. 10.1021/jz300793y. [DOI] [PubMed] [Google Scholar]
- Michaeli K.; Kantor-uriel N.; Naaman R.; Waldeck D. H. Chem. Soc. Rev. 2016, 45, 6478. 10.1039/C6CS00369A. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Correa R.; Smilgies D.; Campbell A. J.; Fuchter M. J. Adv. Mater. 2013, 25, 2624. 10.1002/adma.201204961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Correa R.; Fuchter M. J.; Campbell A. J. Nat. Photonics 2013, 7, 634. 10.1038/nphoton.2013.176. [DOI] [Google Scholar]
- Mtangi W.; Tassinari F.; Vankayala K.; Vargas Jentzsch A.; Adelizzi B.; Palmans A. R. A.; Fontanesi C.; Meijer E. W.; Naaman R. J. Am. Chem. Soc. 2017, 139, 2794. 10.1021/jacs.6b12971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nuzzo D.; Kulkarni C.; Zhao B.; Smolinsky E.; Tassinari F.; Meskers S. C. J.; Naaman R.; Meijer E. W.; Friend R. H. ACS Nano 2017, 11, 12713. 10.1021/acsnano.7b07390. [DOI] [PubMed] [Google Scholar]
- Sato I.; Yamashima R.; Kadowaki K.; Yamamoto J.; Shibata T.; Soai K. Angew. Chem., Int. Ed. 2001, 40, 1096.. [DOI] [PubMed] [Google Scholar]
- Chow W. Y.; Bihan D.; Forman C. J.; Slatter D. A.; Reid D. G.; Wales D. J.; Farndale R. W.; Duer M. J. Sci. Rep. 2015, 5, 1. 10.1038/srep12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck K.; Brodsky B. J. Struct. Biol. 1998, 29, 17. 10.1006/jsbi.1998.3965. [DOI] [PubMed] [Google Scholar]
- Aida T.; Meijer E. W.; Stupp S. I. Science 2012, 335, 813. 10.1126/science.1205962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali D.; Bycroft M.; Fersht A. R. Nature 1988, 335, 740. 10.1038/335740a0. [DOI] [PubMed] [Google Scholar]
- Doyle D. A.; Cabral M.; Pfuetzner R. A.; Kuo A.; Gulbis J. M.; Cohen S. L.; Chait B. T.; Mackinnon R. Science 1998, 280, 69. 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Sahoo J.; Arunachalam R.; Subramanian P. S.; Suresh E.; Valkonen A.; Rissanen K.; Albrecht M. Angew. Chem., Int. Ed. 2016, 55, 9625. 10.1002/anie.201604093. [DOI] [PubMed] [Google Scholar]
- Khan M. K.; Hamad W. Y.; Maclachlan M. J. Adv. Mater. 2014, 26, 2323. 10.1002/adma.201304966. [DOI] [PubMed] [Google Scholar]
- Wang P.-X.; Hamad W. Y.; MacLachlan M. J. Nat. Commun. 2016, 7, 11515. 10.1038/ncomms11515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashima E.; Maeda K. Acc. Chem. Res. 2008, 41, 1166. 10.1021/ar800091w. [DOI] [PubMed] [Google Scholar]
- Kumar M.; Ing N. L.; Narang V.; Wijerathne N. K.; Hochbaum A. I.; Ulijn R. V. Nat. Chem. 2018, 10, 696. 10.1038/s41557-018-0047-2. [DOI] [PubMed] [Google Scholar]
- Brandt J. R.; Wang X.; Yang Y.; Campbell A. J.; Fuchter M. J. J. Am. Chem. Soc. 2016, 138, 9743. 10.1021/jacs.6b02463. [DOI] [PubMed] [Google Scholar]
- Schulte T. R.; Holstein J. J.; Krause L.; Michel R.; Stalke D.; Sakuda E.; Umakoshi K.; Longhi G.; Abbate S.; Clever G. H. J. Am. Chem. Soc. 2017, 139, 6863. 10.1021/jacs.7b03963. [DOI] [PubMed] [Google Scholar]
- Harano K.; Kawai T.; Nishihara H. J. Am. Chem. Soc. 2017, 139, 16024. 10.1021/jacs.7b07077. [DOI] [PubMed] [Google Scholar]
- Ikai T.; Wada Y.; Awata S.; Yun C.; Maeda K.; Mizuno M.; Swager T. M. Org. Biomol. Chem. 2017, 15, 8440. 10.1039/C7OB02046E. [DOI] [PubMed] [Google Scholar]
- Albrecht M. Angew. Chem., Int. Ed. 2005, 44, 6448. 10.1002/anie.200501472. [DOI] [PubMed] [Google Scholar]
- Lohr A.; Würthner F. Angew. Chem. 2008, 120, 1252. 10.1002/ange.200704550. [DOI] [Google Scholar]
- Greenfield J. L.; Rizzuto F. J.; Goldberga I.; Nitschke J. R. Angew. Chem., Int. Ed. 2017, 56, 7541. 10.1002/anie.201702320. [DOI] [PubMed] [Google Scholar]
- Arias S.; Núñez-Martínez M.; Quiñoá E.; Riguera R.; Freire F. Polym. Chem. 2017, 8, 3740. 10.1039/C7PY00561J. [DOI] [PubMed] [Google Scholar]
- Yashima E.; Maeda K.; Iida H.; Furusho Y.; Nagai K. Chem. Rev. 2009, 109, 6102. 10.1021/cr900162q. [DOI] [PubMed] [Google Scholar]
- Arias S.; Rodriguez R.; Quiñoá E.; Riguera R.; Freire F. J. Am. Chem. Soc. 2018, 140, 667. 10.1021/jacs.7b09965. [DOI] [PubMed] [Google Scholar]
- Yashima E.; Maeda K.; Nishimura T. Chem. - Eur. J. 2004, 10, 42. 10.1002/chem.200305295. [DOI] [PubMed] [Google Scholar]
- Freire F.; Quiñoá E.; Riguera R. Chem. Rev. 2016, 116, 1242. 10.1021/acs.chemrev.5b00280. [DOI] [PubMed] [Google Scholar]
- Nakano T.; Okamoto Y. Chem. Rev. 2001, 101, 4013. 10.1021/cr0000978. [DOI] [PubMed] [Google Scholar]
- Salerno F.; Berrocal J. A.; Haedler A.; Zinna F.; Meijer E. W.; Di Bari L. J. Mater. Chem. C 2017, 5, 3609. 10.1039/C7TC00281E. [DOI] [Google Scholar]
- Yang Y.; Rice B.; Shi X.; Brandt J. R.; Correa Da Costa R.; Hedley G. J.; Smilgies D. M.; Frost J. M.; Samuel I. D. W.; Otero-De-La-Roza A.; Johnson E. R.; Jelfs K. E.; Nelson J.; Campbell A. J.; Fuchter M. J. ACS Nano 2017, 11, 8329. 10.1021/acsnano.7b03540. [DOI] [PubMed] [Google Scholar]
- Palmans A. R. A.; Meijer E. W. Angew. Chem., Int. Ed. 2007, 46, 8948. 10.1002/anie.200701285. [DOI] [PubMed] [Google Scholar]
- Rodríguez R.; Sagués F.; Quiñoá E.; Riguera R.; Freire F. Nanoscale 2016, 8, 3362. 10.1039/C5NR07990J. [DOI] [PubMed] [Google Scholar]
- Ikeda M.; Tanaka Y.; Hasegawa T.; Furusho Y.; Yashima E. J. Am. Chem. Soc. 2006, 128, 6806. 10.1021/ja0619096. [DOI] [PubMed] [Google Scholar]
- Maeda K.; Yashima E. Top. Curr. Chem. 2017, 375, 1. 10.1007/s41061-017-0161-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto A.; Ide T.; Kaimori Y.; Fujiwara S.; Soai K. Chem. Lett. 2015, 44, 688. 10.1246/cl.150052. [DOI] [Google Scholar]
- Dawson S. J.; Mészáros Á.; Petho L. Eur. J. Org. Chem. 2014, 2014, 4265. 10.1002/ejoc.201402247. [DOI] [Google Scholar]
- Zhang W.; Jin W.; Fukushima T.; Mori T.; Aida T. J. Am. Chem. Soc. 2015, 137, 13792. 10.1021/jacs.5b09878. [DOI] [PubMed] [Google Scholar]
- Smulders M. M. J.; Schenning A. P. H. J.; Meijer E. W. J. Am. Chem. Soc. 2008, 130, 606. 10.1021/ja075987k. [DOI] [PubMed] [Google Scholar]
- Kang J.; Miyajima D.; Mori T.; Inoue Y.; Itoh Y.; Aida T. Science 2012, 347, 646. 10.1126/science.aaa4249. [DOI] [PubMed] [Google Scholar]
- Ghosh S.; Li X.; Stepanenko V.; Würthner F. Chem. - Eur. J. 2008, 14, 11343. 10.1002/chem.200801454. [DOI] [PubMed] [Google Scholar]
- Qiu H.; Gilroy J. B.; Manners I. Chem. Commun. 2013, 49, 42. 10.1039/C2CC37026C. [DOI] [PubMed] [Google Scholar]
- Dolain C.; Jiang H.; Leger J.; Guionneau P.; Huc I. J. Am. Chem. Soc. 2005, 127, 12943. 10.1021/ja0527828. [DOI] [PubMed] [Google Scholar]
- Guichard G.; Huc I. Chem. Commun. 2011, 47, 5933. 10.1039/c1cc11137j. [DOI] [PubMed] [Google Scholar]
- Huc I. Eur. J. Org. Chem. 2004, 2004, 17. 10.1002/ejoc.200300495. [DOI] [Google Scholar]
- Clayden J.; Castellanos A.; Solà J.; Morris G. A. Angew. Chem. 2009, 121, 6076. 10.1002/ange.200901892. [DOI] [PubMed] [Google Scholar]
- Byrne L.; Solà J.; Boddaert T.; Marcelli T.; Adams R. W.; Morris G. A.; Clayden J. Angew. Chem. 2014, 126, 155. 10.1002/ange.201308264. [DOI] [PubMed] [Google Scholar]
- Brown R. A.; Marcelli T.; De Poli M.; Solà J.; Clayden J. Angew. Chem., Int. Ed. 2012, 51, 1395. 10.1002/anie.201107583. [DOI] [PubMed] [Google Scholar]
- Sato K.; Itoh Y.; Aida T. Chem. Sci. 2014, 5, 136. 10.1039/C3SC52449C. [DOI] [Google Scholar]
- Castilla A. M.; Miller M. A.; Nitschke J. R.; Smulders M. M. J. Angew. Chem., Int. Ed. 2016, 55, 10616. 10.1002/anie.201602968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram J.; Weissman H.; Rybtchinski B. J. Phys. Chem. B 2014, 118, 12068. 10.1021/jp507945t. [DOI] [PubMed] [Google Scholar]
- Zhao D.; Moore J. S. J. Am. Chem. Soc. 2002, 124, 9996. 10.1021/ja026957e. [DOI] [PubMed] [Google Scholar]
- De Greef T. F. A.; Smulders M. M. J.; Wolffs M.; Schenning A. P. H. J.; Sijbesma R. P.; Meijer E. W. Chem. Rev. 2009, 109, 5687. 10.1021/cr900181u. [DOI] [PubMed] [Google Scholar]
- Piguet C. Chem. Commun. 2010, 46, 6209. 10.1039/c0cc00811g. [DOI] [PubMed] [Google Scholar]
- Jha S. K.; Cheon K.; Green M. M.; Selinger J. V. J. Am. Chem. Soc. 1999, 121, 1665. 10.1021/ja983202s. [DOI] [Google Scholar]
- Moad G.; Rizzardo B. E.; Thang S. H. Aust. J. Chem. 2009, 62, 1402. 10.1071/CH09311. [DOI] [Google Scholar]
- Kamigaito M.; Ando T.; Sawamoto M. Chem. Rev. 2001, 101, 3689. 10.1021/cr9901182. [DOI] [PubMed] [Google Scholar]
- Matyjaszewski K.; Xia J. Chem. Rev. 2001, 101 (9), 2921. 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]
- Smulders M. M. J.; Nieuwenhuizen M. M. L.; de Greef T. F. A.; Van der Schoot P.; Schenning A. P. H. J.; Meijer E. W. Chem. - Eur. J. 2010, 16, 362. 10.1002/chem.200902415. [DOI] [PubMed] [Google Scholar]
- Ogi S.; Stepanenko V.; Sugiyasu K.; Takeuchi M.; Würthner F. J. Am. Chem. Soc. 2015, 137 (9), 3300. 10.1021/ja511952c. [DOI] [PubMed] [Google Scholar]
- Zhao D.; Moore J. S. Org. Biomol. Chem. 2003, 1, 3471. 10.1039/B308788C. [DOI] [PubMed] [Google Scholar]
- Van der Zwaag D.; Pieters P. A.; Korevaar P. A.; Markvoort A. J.; Spiering A. J. H.; de Greef T. F. A.; Meijer E. W. J. Am. Chem. Soc. 2015, 137, 12677. 10.1021/jacs.5b08138. [DOI] [PubMed] [Google Scholar]
- Tidhar Y.; Weissman H.; Wolf S. G.; Gulino A.; Rybtchinski B. Chem. - Eur. J. 2011, 17, 6068. 10.1002/chem.201003419. [DOI] [PubMed] [Google Scholar]
- Korevaar P. A.; George S. J.; Markvoort A. J.; Smulders M. M. J.; Hilbers P. A. J.; Schenning A. P. H. J.; De Greef T. F. A.; Meijer E. W. Nature 2012, 481, 492. 10.1038/nature10720. [DOI] [PubMed] [Google Scholar]
- Mattia E.; Otto S. Nat. Nanotechnol. 2015, 10, 111. 10.1038/nnano.2014.337. [DOI] [PubMed] [Google Scholar]
- Campbell E.; Nitschke J. R. Synlett 2008, 20, 3077. 10.1055/s-0028-1087361. [DOI] [Google Scholar]
- Whittell G. R.; Hager M. D.; Schubert U. S.; Manners I. Nat. Mater. 2011, 10 (3), 176. 10.1038/nmat2966. [DOI] [PubMed] [Google Scholar]
- Way A. E.; Korpusik A. B.; Dorsey T. B.; Buerkle L. E.; Von Recum H. A.; Rowan S. J. Macromolecules 2014, 47, 1810. 10.1021/ma402618z. [DOI] [Google Scholar]
- Korevaar P. A.; Grenier C.; Meijer E. W. J. Polym. Sci., Part A: Polym. Chem. 2015, 53 (2), 385. 10.1002/pola.27446. [DOI] [Google Scholar]
- Das A.; Vantomme G.; Markvoort A. J.; Ten Eikelder H. M. M.; Garcia-Iglesias M.; Palmans A. R. A.; Meijer E. W. J. Am. Chem. Soc. 2017, 139 (20), 7036. 10.1021/jacs.7b02835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkata Rao K.; Miyajima D.; Nihonyanagi A.; Aida T. Nat. Chem. 2017, 9 (11), 1133. 10.1038/nchem.2812. [DOI] [PubMed] [Google Scholar]
- Ogi S.; Stepanenko V.; Sugiyasu K.; Takeuchi M. J. Am. Chem. Soc. 2015, 137, 3300. 10.1021/ja511952c. [DOI] [PubMed] [Google Scholar]
- Dobrawa R.; Lysetska M.; Ballester P.; Grüne M.; Würthner F. Macromolecules 2005, 38 (4), 1315. 10.1021/ma047737j. [DOI] [Google Scholar]
- Mastalerz M.; Rivera H. J. E.; Oppel I. M.; Dyker G. CrystEngComm 2011, 13, 3979. 10.1039/c1ce05234a. [DOI] [Google Scholar]
- Ellis W. W.; Schmitz M.; Arif A. A.; Stang P. J. Inorg. Chem. 2000, 39, 2547. 10.1021/ic991315m. [DOI] [PubMed] [Google Scholar]
- Garner M. H.; Hoffmann R.; Rettrup S.; Solomon G. C. ACS Cent. Sci. 2018, 4, 688. 10.1021/acscentsci.8b00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutin M.; Cramer C. J.; Gagliardi L.; Rehaman A.; Shahi M.; Cerny R.; Nitschke J. R. J. Am. Chem. Soc. 2007, 129, 8774. 10.1021/ja070320j. [DOI] [PubMed] [Google Scholar]
- Amendola V.; Fabbrizzi L.; Mangano C.; Pallavicini P.; Roboli E.; Zema M. Inorg. Chem. 2000, 39, 5803. 10.1021/ic0005700. [DOI] [PubMed] [Google Scholar]
- Johnson G. F.; Graves D. J. Biochemistry 1966, 5, 2906. 10.1021/bi00873a019. [DOI] [PubMed] [Google Scholar]
- Wakabayashi M.; Yokojima S.; Fukaminato T.; Shiino K.; Irie M.; Nakamura S. J. Phys. Chem. A 2014, 118, 5046. 10.1021/jp409559t. [DOI] [PubMed] [Google Scholar]
- Salvadori P.; Bertucci C.; Rosini C.; Cnr C.; Stereordinate M. Chirality 1991, 3, 376. 10.1002/chir.530030424. [DOI] [Google Scholar]
- Rajca A.; Pink M.; Xiao S.; Miyasaka M.; Rajca S.; Das K.; Plessel K. J. Org. Chem. 2009, 74, 7504. 10.1021/jo901769c. [DOI] [PubMed] [Google Scholar]
- Damle P.; Ghosh A.; Datta S. Phys. Rev. B: Condens. Matter Mater. Phys. 2001, 64, 201403. 10.1103/PhysRevB.64.201403. [DOI] [Google Scholar]
- Stepanenko V.; Li X.; Gershberg J.; Würthner F. Chem. - Eur. J. 2013, 19, 4176. 10.1002/chem.201204146. [DOI] [PubMed] [Google Scholar]
- Pereira C. S. M.; Silva V. M. T. M.; Rodrigues E. Green Chem. 2011, 13, 2658. 10.1039/c1gc15523g. [DOI] [Google Scholar]
- Aparicio S.; Alcalde R. Green Chem. 2009, 11, 65. 10.1039/B811909K. [DOI] [Google Scholar]
- Smulders M. M. J.; Riddell I. A.; Browne C.; Nitschke J. R. Chem. Soc. Rev. 2013, 42, 1728. 10.1039/C2CS35254K. [DOI] [PubMed] [Google Scholar]
- Maurizot V.; Dolain C.; Huc I. Eur. J. Org. Chem. 2005, 2005, 1293. 10.1002/ejoc.200400641. [DOI] [Google Scholar]
- Wilson A. J.; Masuda M.; Sijbesma R. P.; Meijer E. W. Angew. Chem., Int. Ed. 2005, 44, 2275. 10.1002/anie.200462347. [DOI] [PubMed] [Google Scholar]
- George S. J.; Tomovic Z.; Smulders M. M. J.; Greef T. F. A. De; Lecl P. E. L. G.; Meijer E. W.; Schenning A. P. H. J. Angew. Chem., Int. Ed. 2007, 46, 8206. 10.1002/anie.200702730. [DOI] [PubMed] [Google Scholar]
- In Comprehensive Chiroptical Spectroscopy; Berova N., Polavarapu P. L., Nakanishi K., Woody R. W., Eds.; John Wiley & Sons, 2012; Vol. 2. [Google Scholar]
- Kumar J.; Nakashima T.; Kawai T. J. Phys. Chem. Lett. 2015, 6, 3445. 10.1021/acs.jpclett.5b01452. [DOI] [PubMed] [Google Scholar]
- Chen S. H.; Katsis D.; Schmid A. W.; Mastrangelo J. C.; Tsutsui T.; Blanton T. N. Nature 1999, 397, 506. 10.1038/17343. [DOI] [Google Scholar]
- Weight M.; Kline B. R. J.; Mcgehee M. D.; Kadnikova E. N.; Liu J.; Frøchet J. M. J. Adv. Mater. 2003, 15 (18), 1519. 10.1002/adma.200305275. [DOI] [Google Scholar]
- Tassinari F.; Mathew S. P.; Fontanesi C.; Schenetti L.; Naaman R. Langmuir 2014, 30, 4838. 10.1021/la500657e. [DOI] [PubMed] [Google Scholar]
- Hoeben F. J. M.; Jonkheijm P.; Meijer E. W.; Schenning A. P. H. J. Chem. Rev. 2005, 105, 1491. 10.1021/cr030070z. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Wang T.; Shen Z.; Liu M. Adv. Mater. 2016, 28, 1044. 10.1002/adma.201502590. [DOI] [PubMed] [Google Scholar]
- Lee C. C.; Grenier C.; Meijer E. W.; Schenning A. P. H. J. Chem. Soc. Rev. 2009, 38 (3), 671. 10.1039/B800407M. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.