

Abstract
It was previously shown that the estrogen-receptor negative breast cancer cell line MBA-MD-231 expresses high levels of A2B adenosine receptors as the sole adenosine receptor subtype. These receptors couple to both, stimulation of adenylyl cyclase and a Ca2+ signal. In order to establish a potential role of A2B adenosine receptors in tumor growth and development MAPK signaling was investigated in these breast cancer cells. Although it is known that A2B adenosine receptors may stimulate MAPK it was found that in MBA-MD-231 cells ERK1/2 phosphorylation is reduced upon agonist-stimulation of A2B adenosine receptors. This reduction is also triggered by forskolin, but abolished by the PKA inhibitor H89, suggesting an important role for the cAMP-PKA pathway. Likewise, a role for intracellular Ca2+ was established as the Ca2+ chelator 1,2-bis-(o-aminophenoxy)-ethane-N,N,N’,N’-tetraacetic acid, tetraacetoxymethyl ester (BAPTA-AM) abolished the reduction of ERK1/2 phosphorylation triggered by A2B stimulation. It was shown that various pathways downstream from A2B adenosine receptors resulted in a stimulation of MAPK phosphatase-1 (MKP-1) which dephosphorylates phospho ERK1/2, and thus plays a critical role in the regulation of the phosphorylation state of ERK1/2. The reduction of ERK1/2 phosphorylation mediated by A2B adenosine receptors might provide an interesting approach for adjuvant treatment leading to reduced growth of certain tumors expressing the A2B subtype.
Introduction
Adenosine receptors (ARs) comprise a family of four G protein-coupled receptors which are identified as A1, A2A, A2B, and A3 subtypes [1, 2]. The canonical signal of the A1 and A3 subtypes is inhibition of adenylyl cyclase whereas the A2 subtypes mediate a stimulation of cAMP production. Adenosine affects the function of virtually every organ due to abundant expression of one or several AR subtypes [1–3]. Consequently, adenosine receptors are considered as interesting drug targets for the treatment of numerous conditions including cardiovascular, inflammatory and neurological diseases [3, 4]. Increasing evidence suggests that adenosine may also be involved in the regulation of cellular growth control and proliferation via all four receptor subtypes and, therefore, these may serve as therapeutic targets to combat tumor development and growth [5–8].
We have previously characterized the expression of adenosine receptors in the estrogen receptor-negative breast cancer cell line MDA-MB-231 [9]. These cells express A2B adenosine receptors as the sole subtype at a very high density of > 1 pmol/mg membrane protein [9]. In an attempt to characterize a potential pathophysiological role of this highly expressed AR subtype we characterized MAPK signaling in these cells and discovered that stimulation of A2BARs causes a reduction of ERK1/2 phosphorylation. Stimulation of A2BARs might therefore help to control growth and proliferation of these and other cancer cells expressing A2BARs. The goal of this study was to understand the signaling pathway(s) leading to the inhibition of MAPK signaling as shown by a reduction of ERK1/2 phosphorylation as a consequence of stimulation of A2BARs.
The observed reduction of ERK1/2 phosphorylation might be the result of inhibition of kinases upstream from ERK1/2. One well-known GPCR-Gs-meditated signalling pathway that can lead to ERK1/2 inhibition and anti-proliferative effects in cancer cells is for example the cAMP-PKA-mediated Raf1 inhibition via PKA-phosphorylation at serine 259 [10–12]. Alternatively, the stimulation of a phosphatase could be responsible for the inhibitory input on MAPK signaling in MDA-MB-231 breast cancer cells [13, 14]. We found that several A2BAR mediated signaling pathways control MKP-1 activity which plays an important role in the regulation of ERK1/2 phosphorylation and, thus, may control growth and proliferation of these cells.
Materials and methods
Materials
The breast cancer cell line MDA-MB-231 was provided by the Institut für Zellbiologie (Tumorforschung), Universitätsklinik Essen, Germany. Cell culture media and fetal calf serum were purchased from PanSystems, Aidenbach, Germany. Penicillin (100 U/ml), streptomycin (100 μg/ml), L-glutamine and G-418 were from Gibco-Life Technologies, Eggenstein, Germany. N-[2-[[3-(4-Bromophenyl)-2-propenyl]amino]ethyl]-5-isoquinolinesulfonamide (H-89) and BAPTA-AM were purchased from Calbiochem, Bad Soden, Germany. All inhibitors were used at concentrations at least 10 fold higher than reported IC50 values. The adenosine receptor ligands 9-ethyl-8-furyl adenine (EFA, antagonist) and HEMADO (agonist) were provided by Prof. Rosaria Volpini, School of Pharmacy, Medicinal Chemistry Unit, University of Camerino. Other adenosine receptor agonists and antagonists were from Sigma/RBI, Taufkirchen, Germany and cAMP derivatives from BioLog Life Science Institute, Bremen, Germany. Sources for antibodies were as follows: anti-phospho-ERK1/2 and anti-ERK1/2, New England Biolabs, Frankfurt, Germany; anti MKP-1 and anti-GAPDH, Santa Cruz, Heidelberg, Germany; anti-β-tubulin, Sigma, Taufkirchen, Germany; secondary HRP-conjugated goat anti-rabbit antibodies, Dianova, Hamburg, Germany. Other materials were obtained from sources as described earlier [9, 15].
Cell culture
MDA-MB-231 breast cancer cells were grown adherently and maintained at 37°C in DMEM containing 10% fetal calf serum, penicillin (100 U/ml), streptomycin (100 μg/ml), L-glutamine (2 mM) in 5% CO2/95% air. Cells were split 2 or 3 times weekly at a ratio between 1:4 to 1:8 [9]. Fetal calf serum was omitted on the day of the experiment.
Immunoblot analysis
Prior to an experiment, confluent MDA-MB-231 cells were washed with PBS, then trypsinized and seeded at 50,000 cells/well in precoated six well plates in medium containing 10% fetal calf serum for 24 h and then treated with the indicated inhibitors for the respective time periods. When inhibitors were used, they were added 30 min prior to an agonist. Agonist was added and incubated for 30 min. After washing them with ice-cold PBS cells were lysed in electrophoresis sample buffer for 45 min on a shaker at room temperature followed by sonification. Proteins were separated by SDS polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. For the immunoblot analysis, the membranes were blocked with 5% dry milk powder on a shaker at room temperature and incubated overnight at 4°C with the appropriate primary antibody. Equal loading was confirmed with GAPDH (see S1 Fig) or β-tubulin antibody. After incubation with horseradish peroxidase–coupled secondary antibody, antigen-antibody complexes were visualized by the ECL method and quantified by densitometry with ImageJ (for details see [16]).
Data analysis
All experiments were repeated 4–10 times. Statistical difference between two groups of data was tested by the paired Student’s t test and the difference among three or more groups was assessed by a one-way Anova followed by a Bonferroni post-test. These analyses were performed with the statistical software package GraphPad Prism 6.0 (GraphPad Software Inc., La Jolla, CA, USA) and a 0.05 significance level was assumed to indicate statistical significance. Results are expressed as treated/untreated ratio (mean ± S.E.M).
Results
We have previously shown that MDA-MB 231 breast cancer cells express very high levels of A2B receptors as the sole adenosine receptor subtype [9]. In addition to the canonical stimulation of adenylyl cyclase an A2B-mediated Ca2+ signal was observed in these cells. In this study, we investigated a potential activation of MAP kinase signaling via A2B receptors as a signaling pathway known to be activated by this receptor subtype [17]. As shown in Fig 1, MDA-MB 231 cells exhibited a considerable basal ERK1/2 phosphorylation which was reduced by the nonselective adenosine receptor agonist adenosine-5’-N-ethyluronamide (NECA) in a concentration and time dependent manner (Fig 1A and 1B). In contrast, no such reduction was observed with the subtype selective agonists CCPA (A1) and HEMADO (A3) (Fig 1C). The AR agonist 2-[p-(2-carboxyethyl)phenylethylamino]adenosine-5’-N-ethyluronamide (CGS 21680) that does not stimulate the A2B subtype, did also not show any effect (Fig 1C). These data support the notion that the observed reduction of ERK1/2 phosphorylation was mediated by the A2B adenosine receptor.
Fig 1. Reduction of ERK1/2 phosphorylation by the adenosine receptor agonist NECA.

(A) NECA reduced the ERK1/2 phosphorylation in a concentration-dependent manner (EC50 5.85 nM, 95% confidence limit 2.91–11.8). (B) The reduction was time-dependent with a maximum after 30 min. (C) Only NECA as a nonselective agonist showed this effect while activation of A1 with CCPA, A2A (also A1 and A3 to a certain degree) with CGS 21680, and A3 with HEMADO showed no effect. Panel A and the Western blots in B and C show representative experiments, the columns in B and C show data from n = 7 and 5 independent experiments, respectively (* p < 0.001, significantly different from control).
This suggestion was further confirmed by the pharmacological profile of adenosine receptor antagonists. The NECA-induced reduction of ERK1/2 phosphorylation was antagonized by the nonselective adenosine receptor antagonist EFA (Fig 2A) and the A1/A2B antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) (Fig 2B) with an IC50 indicative of an A2B receptor-mediated effect. As shown in Fig 2C and 2D, both the A2A selective antagonist 2-(2-furanyl)-7-(2-phenylethyl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine (SCH 58261) and the A3 selective antagonist N-[9-chloro-2-(2-furanyl)[1,2,4]-triazolo[1,5-c]quinazolin-5-yl]benzene acetamide (MRS 1220) had no effect corroborating the A2B-mediated effect on ERK1/2 phosphorylation.
Fig 2. NECA-induced reduction of ERK1/2 phosphorylation and effect of adenosine receptor antagonists.

The nonselective antagonist EFA (10 μM) blocked the NECA (100 nM) effect (A) and so did DPCPX at 10 μM, a concentration high enough to block A2BAR (B). In contrast, the A2A selective antagonist SCH 58261 (C) and the A3 selective antagonist MRS 1220 (D), both at 10 μM, had no effect. Western blots show a representative experiment, the columns represent mean values of n = 8 (A), 6 (B), 4 (C), and 5 (D) independent experiments (* p < 0.001, significantly different from control).
Fig 3A shows that reduction of ERK1/2 phosphorylation was also achieved with forskolin suggesting the involvement of cAMP in the effect. This was further confirmed using the membrane-permeable cAMP analogue adenosine-3’,5’-cyclic monophosphate, acetoxymethyl ester (cAMP-AM, ‘caged cAMP’) which releases cAMP after activation by esterases inside the cell (Fig 3B). Also, inhibition of phosphodiesterase-4 with 4-(3-butoxy-4-methoxyphenyl)methyl-2-imidazolidone (Ro 20–1724) resulted in a reduction of ERK1/2 phosphorylation comparable to the NECA-effect (Fig 3C) further supporting the assumption that cAMP is mediating the effect. The subsequent activation of PKA seems to be necessary as inhibition of PKA with H89 blocked the NECA-induced effect on ERK1/2 phosphorylation (Fig 4).
Fig 3. Reduction of ERK1/2 phosphorylation by cAMP.

The effect of 100 nM NECA on ERK1/2 phosphorylation was mimicked by compounds increasing intracellular cAMP, like 1 μM forskolin (A), 100 μM cAMP-AM (“caged cAMP”, B), or the PDE inhibitor Ro20-1724 (C) at 100 μM. Western blots show a representative experiment, the columns represent mean values from n = 6 (A), 4 (B), and 5 (C) independent experiments (* p < 0.001, significantly different from control).
Fig 4. Effect of the PKA inhibitor H-89 on NECA-mediated reduction of ERK1/2 phosphorylation.
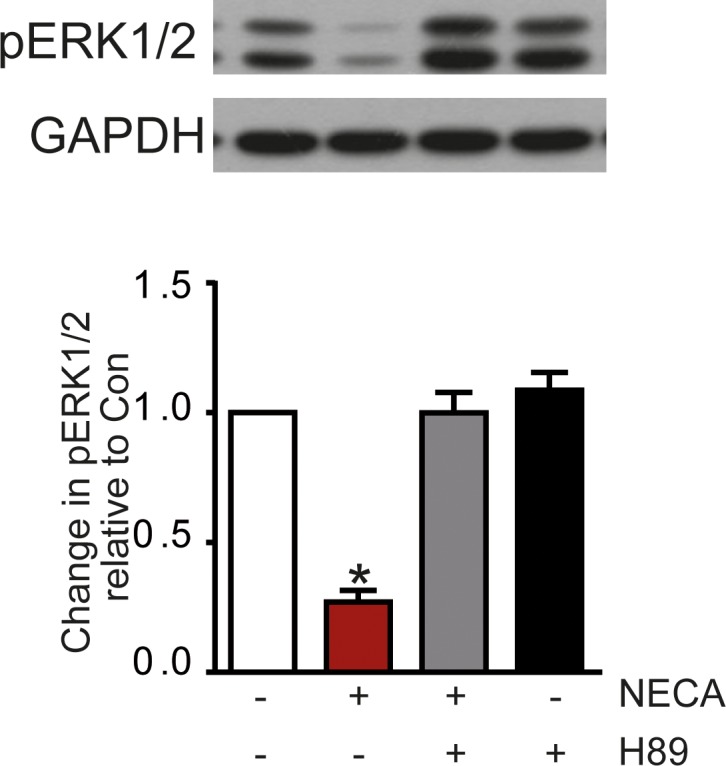
Preincubation (30 min) of MDA-MB-231 cells with 10 μM of the PKA inhibitors H-89 prevented the NECA effect on ERK1/2 phosphorylation. The Western blot shows a representative experiment, the columns represent mean values from n = 7 independent experiments (* p < 0.001, significantly different from control).
We have previously shown that stimulation of the A2B receptor in MDA-MB-231 cells triggers a Ca2+-response in addition to the activation of adenylyl cyclase (Panjehpour et al., 2005). Therefore, we investigated whether Ca2+ signaling might be involved in the NECA-induced reduction of ERK1/2 phosphorylation. As shown in Fig 5A the chelator BAPTA (applied as the cell-permeable precursor BAPTA-AM) completely abolished the NECA effect. Consequently, triggering a Ca2+-signal with the P2Y2/4 agonist UTP (Fig 5B) resulted in a time-dependent reduction of ERK1/2 phosphorylation (Fig 5C).
Fig 5. Role of Ca2+ for reduction of ERK1/2 phosphorylation.

The NECA-induced reduction of ERK1/2 phosphorylation was blocked by the Ca2+-chelator BAPTA intracellularly released from 30 μM of the cell-penetrating derivative BAPTA-AM (A). Consequently, an intracellular increase of Ca2+ triggered by 100 μM UTP (B) resulted in a reduction of ERK1/2 phosphorylation similar to increasing levels of cAMP (C). Western blots and the Ca2+ trace in B show a representative experiment, the columns represent mean values from n = 7 (A) and 8 (C) independent experiments (* p < 0.001, significantly different from control).
Inhibition of protein synthesis with cycloheximide abolished the NECA effect on ERK1/2 phosphorylation (Fig 6A). We identified MKP-1, a MAPK phosphatase, as a candidate whose expression might be involved in reduction of ERK1/2 phosphorylation by A2B receptor stimulation with NECA. Fig 6B shows the time-dependent increase in MKP-1 expression upon stimulation with NECA. This effect was blocked by the addition of cycloheximide (Fig 6C) confirming a role of MKP-1 in the regulation of the phosphorylation state of ERK1/2.
Fig 6. Effect of cycloheximide on ERK-1/2 phosphorylation and MKP-1 expression.

The inhibition of ERK1/2 phosphorylation by NECA was blocked by 10 μg/ml of cycloheximide (CHX) suggesting that protein synthesis is mandatory for the NECA effect (A). Stimulation of A2B receptors in MDA-MB-231 with NECA cells causes and increase in MKP-1 expression (B), treatment with 1 μM forskolin shows the same effect (C). The increase caused by NECA is blocked by cycloheximide (D). Western blots show a representative experiment, the columns represent mean values from n = 7 (A), 4 (B), 7 (C), and 6 (D) independent experiments (* p < 0.001, significantly different from control).
Regulation of MKP-1 activity was further established as a relevant mechanism contributing to the NECA-mediated reduction of ERK1/2 phosphorylation. The stimulation of adenylyl cyclase with forskolin resulted in an increased expression of MKP-1 similar to that seen after stimulation of the A2B receptor with NECA (Fig 6D). The Ca2+-chelator BAPTA inhibited the NECA-effect on MKP-1 expression (Fig 7), suggesting that a Ca2+-signal may upregulate the level of MKP-1.
Fig 7. Ca2+ effect on MKP-1 expression.
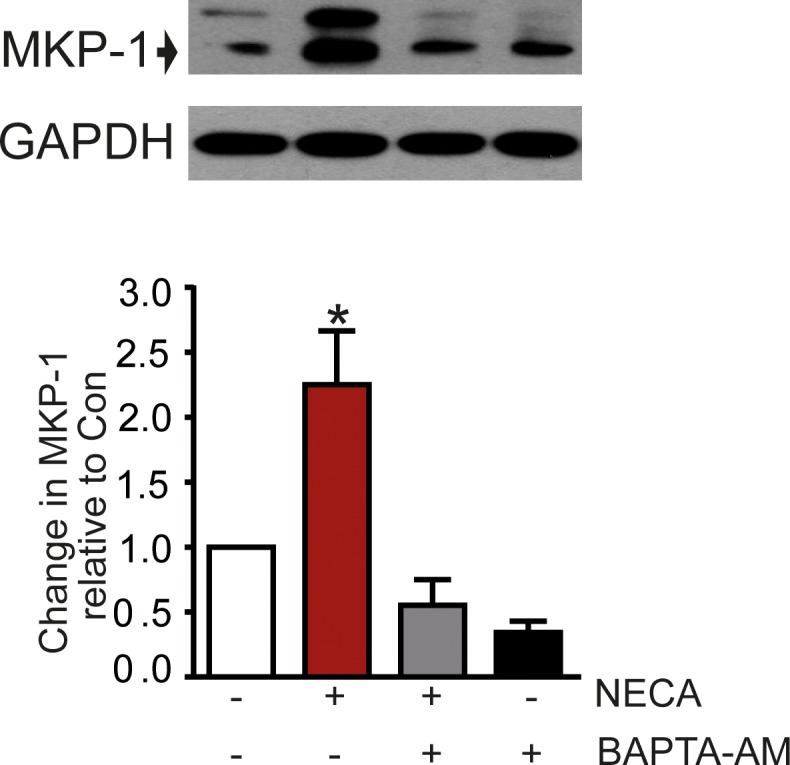
The NECA-induced increase of MKP-1 expression was blocked by the Ca2+-chelator BAPTA (addition of 30 μM BAPTA-AM). The Western blot shows a representative experiment, the columns represent mean values from n = 4 independent experiments (* p < 0.001, significantly different from control).
Although Ca2+ is necessary for the NECA-mediated reduction of ERK1/2 phosphorylation it is not sufficient as is shown by the effect of UTP which elicited a Ca2+-signal via P2Y2/4 receptors (Fig 5B), but diminished ERK1/2 phosphorylation consistently to a lesser extent compared to NECA (Fig 5A and 5C). Given that the A2B adenosine receptor triggers both an increase in cAMP and a Ca2+-signal it might be possible that these dual signals act in concert on the phosphorylation state of ERK1/2. Such a synergism was observed when combining UTP with a subthreshold concentration of forskolin (30 nM) with no effect on ERK1/2 phosphorylation on its own. In such an experiment forskolin increased the effect of UTP (Fig 8) supporting a role of synergism between signaling pathways regulating ERK1/2 phosphorylation.
Fig 8. Reduction of ERK1/2 phosphorylation by UTP compared to NECA.
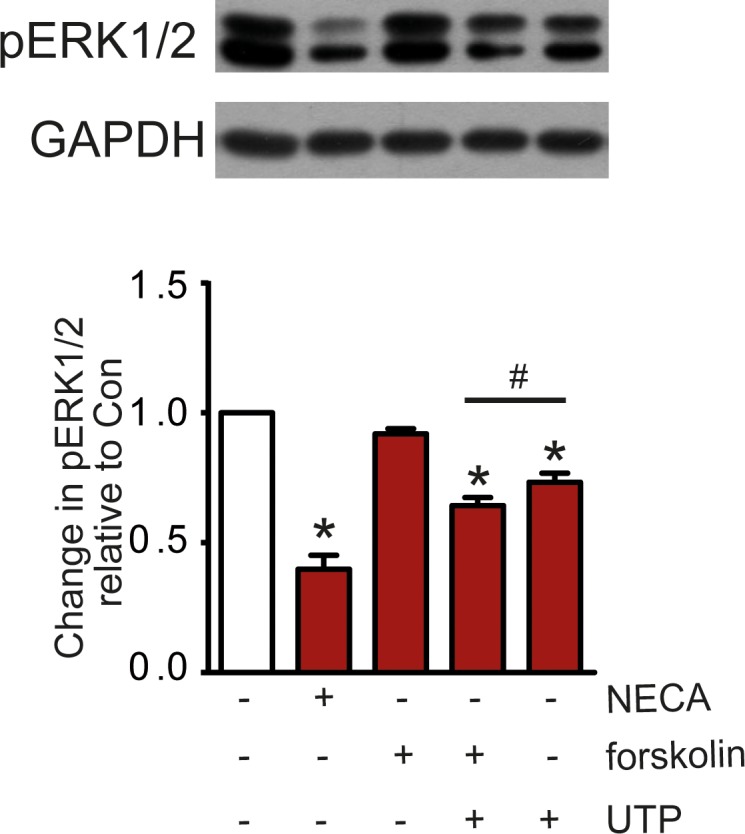
The effect of 100 μM UTP was consistently lower than of 100 nM NECA. Addition of 30 nM forskolin to 100 μM UTP increased the UTP effect although forskolin at this concentration had no effect on its own. The Western blot shows a representative experiment, the columns represent mean values from from n = 7 independent experiments (* p < 0.001, significantly different from control; # p < 0.05, significantly different from each other).
An additional contribution to a reduced phosphorylation of ERK1/2 by A2BAR stimulation was observed by another pathway leading to an A2B receptor-dependent dephosphorylation of c-Raf. Stimulation of the A2B receptor with NECA caused a time-dependent reduction of c-Raf phosphorylation on S338 which is a key activation site (Fig 9A). The same effect was observed after forskolin treatment suggesting a cAMP-dependent process (Fig 9B).
Fig 9. Phosphorylation of S338 of c-Raf is reduced by NECA and forskolin.

A time-dependent inhibition of phosphorylation of c-Raf at S338 is observed by stimulation of the A2B receptor with NECA (A). A similar effect was seen with forskolin (B). Western blots show a representative experiment, the columns represent mean values from n = 5 (A) and 10 (B) independent experiments (* p < 0.001, significantly different from control).
As a consequence of reduced ERK1/2 phosphorylation one would expect an effect on cell proliferation. S2 Fig confirms that stimulation of A2BARs may reduce cell growth as shown by a reduced [3H]thymidine incorporation.
Discussion
A plethora of data suggests that adenosine receptors play a role in the control of growth and proliferation of tumor cells [5, 6, 8]. In particular A3 adenosine receptors have been identified as important regulators of tumor progression [18–21]. Recently, we have shown that the estrogen receptor-negative cell line MDA-MB-231 expressed very high levels of A2B adenosine receptors mediating both a stimulation of adenylyl cyclase and an activation of PLC with a consecutive Ca2+ signal [9]. Data from other cells show that the A2B subtype may also stimulate the MAPK pathway [17] and thereby increase proliferation.
Attempts to identify a potential A2B-mediated stimulatory MAPK signal in MDA-MB-231 cells failed, probably due to high basal ERK1/2 phosphorylation in these cells. The surprising observation was that stimulation of A2B adenosine receptors with the agonist NECA caused a reduction of the marked constitutive ERK1/2 phosphorylation. The effect occurred with the appropriate pharmacology that would be expected for an A2BAR-mediated response. Although reports exist of a receptor-mediated inhibition of MAPK signaling [22–24] the common observation is that G protein-coupled receptors mediate an activation of this pathway. The study presented here was initiated to understand the signaling cascade leading from A2B activation to the reduction of ERK1/2 phosphorylation.
Our data reveal that multiple pathways are involved in this pathway as both cAMP as well as a Ca2+ signal is sufficient for a reduction of ERK1/2 phosphorylation to occur. The finding that protein synthesis is required for this inhibitory effect led to the hypothesis that a MAPK phosphatase (MKP) might be involved as the activity of these enzymes is subject to regulation by their expression level [25]. It was indeed observed that both signals triggered by stimulation of A2BAR with NECA (cAMP and Ca2+ signal) increased MKP-1 expression. It was previously established that intracellular Ca2+ is involved in the regulation of MKP-1 expression [26].
The reduction of ERK1/2 phosphorylation triggered by stimulation of an AR does not seem to be strictly dependent on A2BARs. We could show that stimulation of the Gs-coupled β adrenergic receptors with 100 nM isoproterenol caused in addition to the canonical activation of adenylyl cyclase a Ca2+ signal, in analogy to what was observed for A2BARs (S3 Fig). Also in line with the effect observed for the A2BAR, stimulation of β adrenergic receptors resulted in a reduction of ERK1/2 phosphorylation (S3 Fig). It was recently reported that β2 receptors in HEK293 cells also trigger a cAMP-independent intracellular Ca2+ mobilization similar to the observation in MDA-MB-231 cell [27]. This result suggests that not just stimulation of A2BARs may help to control tumor growth, the same effect might be exploitable in cancer cells expressing other Gs-coupled receptors which in addition to an increase in cAMP mediate a concomitant Ca2+ signal.
It was striking that a Ca2+ signal as elicited by UTP via P2Y2/4 receptors caused a smaller reduction of ERK1/2 phosphorylation than stimulation of A2BARs. This might be explained by the lack of a stimulatory effect of P2Y2/4 receptors on adenylyl cyclase as opposed to A2BAR signaling. Indeed, application of 30 nM forskolin, a concentration that had no effect on ERK1/2 phosphorylation of its own, along with UTP reduced ERK1/2 phosphorylation to the same degree as NECA, acting through A2BARs. This result demonstrated that the cAMP signal and the Ca2+ signal synergistically reduced ERK1/2 phosphorylation. In addition, we could show that an A2B receptor-mediated dephosphorylation of c-Raf at S338 also contributed directly to a reduction of ERK1/2 phosphorylation. A similar effect involving c-Raf was already observed in the past [28].
In summary, our data establish that activation of the A2BAR in MBA-MD-231 cells leads to an ERK1/2 dephosphorylation which is known to reduce cell proliferation as a result of inhibition of MAPK signaling. The A2BAR (and possibly other Gs-coupled receptors) may be an attractive target for adjuvant treatment of tumors expressing this adenosine receptor subtype. It was found that MKP-1 played an important role for the reduction of ERK1/2 phosphorylation seen in the breast cancer cell line MBA-MD-231 after stimulation of A2BARs. Several signaling pathways including an increase of cAMP or intracellular Ca2+ lead to an increased expression of MKP-1, thus, resulting in a reduction of ERK1/2 phosphorylation (Fig 10).
Fig 10. Overview of A2B signaling pathways.

The scheme summarizes signaling pathways contributing to a reduction of ERK1/2 phosphorylation through stimulation of A2B adenosine receptors in MDA-MB-231 breast cancer cells. p-Raf denotes c-Raf phosphorylated at S338.
Supporting information
Unphosphorylated ERK1/2 could not successfully be used as a loading control as the intensity of the staining with both ERK1/2 and pERK1/2 antibodies prevented sufficient stripping of the labeled bands before labeling the overlapping second set of bands. It is shown that GAPDH and ERK1/2 staining (same gels) result in identical documentation of equal loading of the gels. pERK1/2 labeling is shown with the same samples on a separate gel for comparison. The columns show the means of n = 5 experiments with SEM; * p < 0.001.
(PDF)
[3H]Thymidine incorporation was determined as described earlier (S1 Text). It is shown that cell proliferation is inhibited to about 80% after 12 and 24h of incubation with NECA. Data show mean values with SEM of n = 8 experiments performed in triplicates. The NECA values are significantly different from controls with p = 0.0011 and 0.0004 for the 12 and 24h time points, respectively (paired Student’s t-test).
(PDF)
(PDF)
(DOCX)
Acknowledgments
The expert technical assistance of Ms. Sonja Kachler is gratefully acknowledged. Some preliminary experiments for this study were contributed by Mojtaba Panjehpour, Rahul Yadav, and Daniela Bieber.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
The authors received no specific funding for this work.
References
- 1.Fredholm BB, IJzerman AP, Jacobson KA, Klotz K-N, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001,53: 1–26. [PMC free article] [PubMed] [Google Scholar]
- 2.Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller C. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors.-.an update. Pharmacol Rev. 2011,63: 1–34. 10.1124/pr.110.003285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gessi S, Merighi S, Varani K, Borea PA. Adenosine receptors in health and disease. Adv Pharmacol. 2011,61: 41–75. 10.1016/B978-0-12-385526-8.00002-3 [DOI] [PubMed] [Google Scholar]
- 4.Fredholm BB. Adenosine receptors as drug targets. Exp Cell Res. 2010,316: 1284–1288. 10.1016/j.yexcr.2010.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merighi S, Mirandola P, Varani K, Gessi S, Leung E, Baraldi PG, et al. A glance at adenosine receptors: novel target for antitumor therapy. Pharmacol Ther. 2003,100: 31–48. [DOI] [PubMed] [Google Scholar]
- 6.Fishman P, Bar-Yehuda S, Synowitz M, Powell JD, Klotz K-N, Gessi S, et al. Adenosine receptors and cancer. Handb Exp Pharmacol. 2009,193: 399–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gessi S, Merighi S, Sacchetto V, Simioni C, Borea PA. Adenosine receptors and cancer. Biochim Biophys Acta. 2011,1808:1400–1412. 10.1016/j.bbamem.2010.09.020 [DOI] [PubMed] [Google Scholar]
- 8.Antonioli L, Blandizzi C, Pacher P, Haskó G. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer. 2013,13: 842–857 10.1038/nrc3613 [DOI] [PubMed] [Google Scholar]
- 9.Panjehpour M, Castro M, Klotz K-N. The human breast cell line MDA-MB-231 expresses endogenous A2B adenosine receptors mediating a Ca2+ signal. Br J Pharmacol., 2005155: 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhillon AS, Pollock C, Steen H, Shaw PE, Mischak H, Kolch W. Cyclic AMP-dependent kinase regulates Raf-1 kinase mainly by phosphorylation of serine 259. Mol Cell Biol. 2002,22: 3237–3246. 10.1128/MCB.22.10.3237-3246.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naviglio S, Caraglia M, Abbruzzese A, Chiosi E, Di Gesto D, Marra M, et al. Protein kinase A as a biological target in cancer therapy. Expert Opin Ther Targets. 2009,13: 83–92. 10.1517/14728220802602349 [DOI] [PubMed] [Google Scholar]
- 12.Naviglio S, Di Gesto D, Romano M, Sorrentino A, Illiano F, Sorvillo L, et al. Leptin enhances growth inhibition by cAMP elevating agents through apoptosis of MDA-MB-231 breast cancer cells. Cancer Biol Ther. 2009,8: 1183–1190. [DOI] [PubMed] [Google Scholar]
- 13.Boutros T, Chevet E, Metrakos P. Mitogen-Activated Protein (MAP) Kinase/MAP Kinase Phosphatase regulation: Roles in cell growth, death, and cancer. Pharmacol Rev. 2008,60: 261–310. 10.1124/pr.107.00106 [DOI] [PubMed] [Google Scholar]
- 14.Tuglu MM, Bostanabad SY, Ozyon G, Dalkiliç B, Gurdal H. The role of dual‑specificity phosphatase 1 and protein phosphatase 1 in β2‑adrenergic receptor‑mediated inhibition of extracellular signal regulated kinase 1/2 in triple negative breast cancer cell lines. Mol Med Rep. 2018,17: 2033–2043. 10.3892/mmr.2017.8092 [DOI] [PubMed] [Google Scholar]
- 15.Klotz K- N, Hessling J, Hegler J, Owman C, Kull B, Fredholm BB, et al. Comparative pharmacology of human adenosine receptor subtypes–characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedeberg’s Arch Pharmacol. 1998,357: 1–9. [DOI] [PubMed] [Google Scholar]
- 16.Ruppert C, Deiss K, Herrmann S, Vidal M, Oezkur M, Gorski A, et al. Interference with ERKThr188 phosphorylation impairs pathological but not physiological cardiac hypertrophy. Proc Natl Acad Sci USA. 2013,110: 7440–7445. 10.1073/pnas.1221999110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schulte G, Fredholm BB. Signalling from adenosine receptors to mitogen-activated protein kinases. Cell Signal. 2003,15: 813–827. [DOI] [PubMed] [Google Scholar]
- 18.Ohana G, Bar-Yehuda S, Barer F, Fishman P. Differential effect of adenosine on tumor and normal cell growth: Focus on the A3 adenosine receptor. J Cell Physiol. 2001,186: 19–23. [DOI] [PubMed] [Google Scholar]
- 19.Merighi S, Mirandola P, Milani D, Varani K, Gessi S, Klotz K-N, et al. Adenosine receptors as mediators of both cell proliferation and cell death of cultured human melanoma cells. J Invest Dermatol. 2002,119: 923–933. 10.1046/j.1523-1747.2002.00111.x [DOI] [PubMed] [Google Scholar]
- 20.Madi L, Bar-Yehuda S, Barer F, Ardon E, Ochaion A, Fishman P. A3 adenosine receptor activation in melanoma cells. Association between receptor fate and tumor growth inhibition. J Biol Chem. 2003,278: 42121–42130. 10.1074/jbc.M301243200 [DOI] [PubMed] [Google Scholar]
- 21.Madi L, Ochaion A, Rath-Wolfson L, Bar-Yehuda S, Erlanger A, Ohana G, et al. The A3 adenosine receptor is highly expressed in tumor versus normal cells: Potential target for tumor growth inhibition. Clin Cancer Res. 2004,10: 4472–4479. 10.1158/1078-0432.CCR-03-0651 [DOI] [PubMed] [Google Scholar]
- 22.Haneda M, Araki S-i, Sugimoto T, Togawa M, Koya D, Kikkawa R. Differential inhibition of mesangial MAP kinase cascade by cyclic nucleotides. Kidney Int. 1996,50: 384–391. [DOI] [PubMed] [Google Scholar]
- 23.Pandey KN, Nguyen HT, Li M, Boyle JW. Natriuretic peptide receptor-A negatively regulates mitogen-activated protein kinase and proliferation of mesangial cells: Role of cGMP-dependent protein kinase. Biochem Biophys Res Commun. 2000,271: 374–379. 10.1006/bbrc.2000.2627 [DOI] [PubMed] [Google Scholar]
- 24.Li R-c, Cindrova-Davies T, Skepper JN, Sellers LA. Prostacycline induces apoptosis of vascular smooth muscle cells by a cAMP-mediated inhibition of extracellular signal-regulated kinase activity and can counteract the mitogenic activity of endothelin-1 or basic fibroblast growth factor. Circ Res, 2004,94: 759–767. 10.1161/01.RES.0000121568.40692.97 [DOI] [PubMed] [Google Scholar]
- 25.Brion L, Maloberti PM, Gomez NV, Poderoso C, Gorostizaga B, Sequeiros Garcia MM, et al. MAPK phosphatase-1 (MKP-1) expression is up-regulated by hCG/cAMP and modulates steroidogenesis in MA-10 Leydig cells. Endocrinology. 2011,152: 2665–2677.3445 10.1210/en.2011-0021 [DOI] [PubMed] [Google Scholar]
- 26.Scimeca J-C, Servant MJ, Dyer J-O, Meloche S. Essential role of calcium in the regulation of MAP kinase phosphatase-1 expression. Oncogene. 1997,15: 717–725. 10.1038/sj.onc.1201231 [DOI] [PubMed] [Google Scholar]
- 27.Galaz-Montoya M, Wright SJ, Rodriguez GJ, Lichtarge O, Wensel TG. β2-Adrenergic receptor activation mobilizes intracellular calcium via a non-canonical cAMP-independent signaling pathway. J Biol Chem. 2017,292: 9967–9974. 10.1074/jbc.M117.787119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cook SJ, McCormick F. Inhibition by cAMP of Ras-dependent activation of Raf. Science. 1993,262: 1069–1072. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Unphosphorylated ERK1/2 could not successfully be used as a loading control as the intensity of the staining with both ERK1/2 and pERK1/2 antibodies prevented sufficient stripping of the labeled bands before labeling the overlapping second set of bands. It is shown that GAPDH and ERK1/2 staining (same gels) result in identical documentation of equal loading of the gels. pERK1/2 labeling is shown with the same samples on a separate gel for comparison. The columns show the means of n = 5 experiments with SEM; * p < 0.001.
(PDF)
[3H]Thymidine incorporation was determined as described earlier (S1 Text). It is shown that cell proliferation is inhibited to about 80% after 12 and 24h of incubation with NECA. Data show mean values with SEM of n = 8 experiments performed in triplicates. The NECA values are significantly different from controls with p = 0.0011 and 0.0004 for the 12 and 24h time points, respectively (paired Student’s t-test).
(PDF)
(PDF)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.