

Abstract
Stereoselective formation of glycosidic bonds remains one of the most challenging topics in carbohydrate chemistry. The predominant method for stereoselective construction of 1,2-trans-glycosidic bonds is through the neighboring group participation effect (NGPE), which proved to be less successful in synthesizing Galβ(1→3)GalNAc disaccharide. The steric effect that overshadows NGPE and the impacts of substituents at the 3-O- and 2-N-positions of donors and acceptors, respectively, on this synthesis were systematically examined to lead to some practical guidelines for choosing protecting groups towards the successful synthesis of Galβ(1→3)GalNAc and similar disaccharides.
Keywords: neighboring group participation effect, steric effect, galactose, galactosamine, β-glycosylation
Graphical Abstract
The result is dependent on the properties of R, R1 and R2.
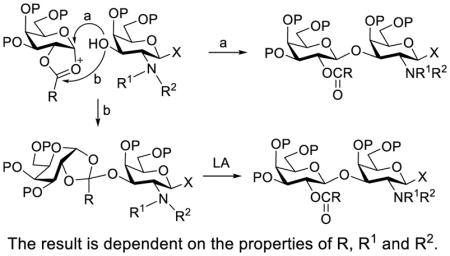
Introduction
After over a century of research, stereoselective formation of glycosidic bonds still remains one of the most challenging topics in organic and carbohydrate chemistry. In this context, many new glycosylation strategies, such as in situ anomerization,1 intramolecular aglycone delivery,2 stereo-controlled or conformation-directed glycosylation,3,4 etc., have been developed to achieve desired stereochemical outcomes. Among all existing strategies, stereoselective glycosylation based on the neighboring group participation effect (NGPE), involving mainly the protecting group at the 2-O-position of glycosyl donors (Scheme 1), is considered the most reliable strategy in constructing 1,2-trans-glycosidic bonds. Over the years, this strategy has been progressively improved. For example, reaction conditions have been extensively studied and perfected, whereas the pool of available participating groups has been expanded from ester to ether,5 amine,6 and heterocycle.7 Consequently, NGPE-based glycosylation has become the ubiquitous method for constructing 1,2-trans-glycosidic bonds in complex carbohydrate synthesis.
Scheme 1.

NGPE-directed 1,2-trans-glycosylation
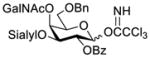
However, occasionally the NGPE can be outweighed by other factors to give rise to unexpected glycosylation results. For instance, in the course of globo H synthesis, it was observed that constructing the β-linked disaccharide, Galβ(1→3)GalNAc, based on the NGPE was troublesome.8,9 When a D-galactosamine derivative such as 2a having the 2-N-position protected with a phthalyl (Phth) group (Scheme 2) was employed as the glycosyl acceptor, very low yields of the desired β-isomer were obtained along with substantial amounts of the α-isomer, even when a benzoyl group (Bz) with great potential of NGPE was utilized to protect the 2-O-position of the glycosyl donor 1a. Therefore, in spite of some obvious advantages of the Phth group as a protecting group for amines, such as its excellent NGPE ability and the good stability of the resulting phthalimide, Phth-protected galactosamine derivatives have been rarely used as the glycosyl acceptor in the synthesis of Galβ(1→3)GalNAc. Even if the Phth group was used in the glycosyl acceptor synthesis, it was usually switched to a 2,2,2-trichloroethoxycarbonyl (Troc) or an azido group just before the glycosylation.9,10 These findings call for a better mechanistic understanding of the NGPE to exploit its full potential in this synthesis.
Scheme 2.

Attempted assembly of the Galβ(1→3)GalNAc motif in globo H synthesis
In the meantime, Galβ(1→3)GalNAc is an important structural motif widely present in glycosphingolipids and many tumor-associated carbohydrate antigens (TACAs), such as GM1, TF, and globo series antigens. Because of its universal overexpression on cancer cells, a screening method, known as the Shams’ test, to diagnose colorectal cancer has been developed based on the enzymatic detection of this structural motif.11 Therefore, an efficient and reliable method to construct the Galβ(1→3)GalNAc motif should be very useful in the chemical synthesis of TACAs and related glycans. The current work aimed to address this frequently overlooked but nonetheless important problem by detailed analysis of the influence of various substituents in the glycosyl donor and glycosyl acceptor on the stereochemical outcome of 3-O-galactosylation of galactosamine.
Results and Discussion
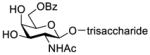
As reported in the literature,10,12–25 a number of TACAs and glycoconjugates containing the Galβ(1→3)GalNAc motif have been synthesized. Among these syntheses, the widely adopted strategy was based on the β-galactosylation of preassembled oligosaccharides or glycoconjugates that had a galactosamine residue at the non-reducing end (Table 1).10,16–24 In this case, the protecting group at the galactosamine 2-N-position could be and indeed was usually switched to the small and desired acetyl group before galactosylation (entries 1–9, Table 1), and most glycosylation reactions using 2-O-acyl protected galactosyl donors afforded good yields and stereoselectivity. However, if the galactosamine 2-N-position was protected with a Troc or a trichloroacetyl (TCA) group, it was not required to be switched to an acetyl group for the successful assembly of the Galβ(1→3)GalNAc motif (entries 10 and 11, Table 1).
Table 1.
Construction of the Galβ(1→3)GalNAc motif using GalNAc in oligosaccharides or glycoconjugates as the galactosyl acceptor
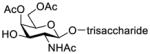
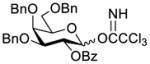
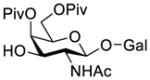
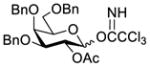
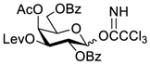
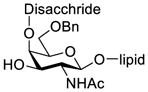
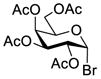
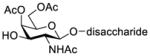
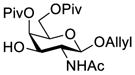
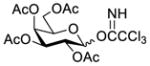
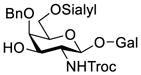
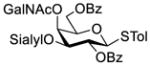
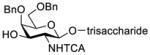
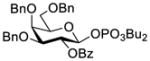
Another strategy that has been adopted for the construction of glycans containing the Galβ(1→3)GalNAc motif was to assemble this disaccharide unit first (Table 2), which was then used as a glycosyl donor for the next glycosylation reaction.13,25–27 In this case, the galactosamine 2-N-position was usually protected with a Troc or TCA group (entries 1–4, Table 2), which was demonstrated to possess the participating ability and facilitate subsequently NGPE-assisted glycosylation.13,25,27,28 In addition, the 2-azido derivatives of galactosamine were also used as glycosyl acceptors for β-galactosylation to successfully fashion the Galβ(1→3)GalNAc motif (entry 5, Table 2), with the cost of further manipulation to convert the azido protection to the desired acetamino group.9
Table 2.
Construction of the Galβ(1→3)GalNAc motif using GalNTroc, GalNTCA, and 2-azido derivatives of galactosamine as glycosyl acceptors
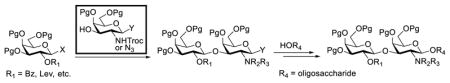
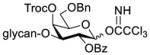
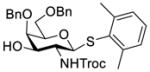
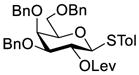
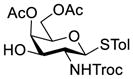
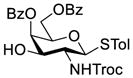
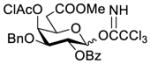
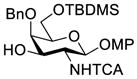
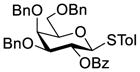
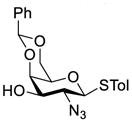
Clearly, galactosamine derivatives with the 2-N-position protected with Ac, Troc, and TCA group or as an azide could be successfully 3-O-β-galactosylated in good yields and stereoselectivity. In contrast, when the galactosamine 2-N-position was protected with a Phth group, the galactosylation reaction failed to give the desired β-anomer, even when an acyl group was present at the galactosyl donor 2-O-position (Scheme 2). These results may suggest the great influence of the protecting group at the galactosamine 2-N-position on its 3-O-galactosylation. The major and shared difference of the Phth group from Ac, Troc, TCA and azido groups is the steric bulk. Based on these discoveries, we suspected that during the galactosylation of galactosamine, the steric effect might surpass NGPE to affect the stereochemical outcome. To probe this hypothesis and find a solution to the problem, we designed and performed the following systematic studies.
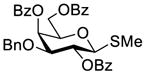
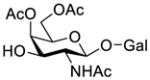
First, we probed the reaction between glycosyl donor 1a and glycosyl acceptor 2b29 with the Phth group in 2a substituted for the Troc group, which should be less sterically demanding but as effective as Phth in directing stereoselective glycosylation.26 Under the preactivation condition used in Scheme 2, the reaction between 1a and 2b went smoothly to give β-linked disaccharide 3a (JH1,2 = 7.9 Hz at δ = 4.86 ppm) as the only isomer in a very good yield (87%). This experiment clearly indicated that the bulky Phth group in 2a was probably the major reason to cause failed glycosylation in Scheme 2.
Since steric interaction should involve two parties, next, we furthered our investigation to examine the influence of substituents in the glycosyl donor on this glycosylation reaction. Accordingly, we probed the reaction of 2a with thioglycoside 1b (Scheme 4), which had an acetyl group, instead of a Bz group, at the 2-O-position under the same condition and expected to obtain even better results than Scheme 3 due to reduced steric interactions. To our surprise, however, this reaction produced only a trace amount of desired disaccharide 3b observed by mass spectrometry. TLC monitoring and analysis of the reaction proved that the activation of glycosyl donor 1b, just like the activation of 1a, was successful and complete, but the activated donor and acceptor did not react at −78 °C and no reaction was observed even after the mixture was allowed to warm to room temperature. The reaction became very complex after overnight stirring at room temperature, whereas the acceptor was recovered.
Scheme 4.

Galactosylation using 2-O-Ac-protected donors under preactivation condition
Scheme 3.
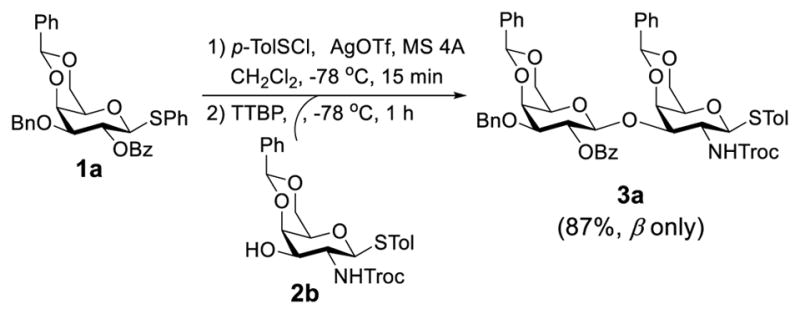
Galactosylation of less sterically demanding 2b under preactivation condition
To find out how the benzylidene ring in the glycosyl donor might have affected the reaction,30 we synthesized tetraacetylated thioglycoside 1c and studied its reaction with 2b under the same preactivation condition. To our surprise again, this reaction was rather complex with orthoester 4 (Scheme 4), instead of the desired disaccharide, isolated as a major product in a low yield. The structure of 4 was characterized by NMR spectrometry. For example, the chemical shift of proton signal of the orthoester methyl group was 1.67 ppm opposed to that of the acetyl methyl groups (>2.0 ppm), and the chemical shift of the orthoester C-1 signal was 121.3 ppm as compared to 170 ppm for carbonyl signals of the three acetyl groups. Furthermore, the NOESY spectrum of 4 also showed strong NOE correlations between the orthoester methyl protons and H-3 and H-3′ protons.
It is well known that when 2-O-acylated sugars are used as donors, some glycosylation reactions, especially those of sterically demanding or relatively unreactive substrates, go through the mechanism involving orthoester intermediates that can rearrange to afford the desired glycosides under acidic conditions.31–33 Alternatively, orthoester may be obtained as a common byproduct of glycosylation.34 The isolation of orthoester 4 from the reaction mixture of 1c and 2b suggested that it was probably quite difficult for the acceptor 2b to attack the anomeric carbon in the activated intermediate derived from 1c but to attack the C-1 position of the participating 2-O-acetyl group instead. On the other hand, orthoester 4 could not rearrange to form the corresponding glycoside under the neutral or mildly basic condition, namely, in the presence of 2,4,6-tri-tert-butylpyrimidine (TTBP) employed to neutralize the acid formed from the glycosylation reaction. This rationale can also be used to explain the low reactivity between 1b and 2a which in fact was even more sterically demanding than 2b. We further hypothesized that the reaction of 1b and 2a might have formed the corresponding orthoester as well, but it was presumably unstable because of increased steric interactions and ring strains to give rise to complex results.
To gain more insights into this glycosylation reaction, we converted thioglycoside 2a into methyl glycoside 2c and investigated its coupling with 1b and 1c (Scheme 5), which allowed for glycosylation reactions under normal conditions, namely, in the presence of Lewis acids. Both reactions using N-iodosuccinimide (NIS) and silver triflate (AgOTf) as the promoters, which were also reported to promote orthoester rearrangement to generate glycosides,35 proceeded smoothly to give the desired disaccharides 3c (JH1,2 = 8.3 Hz at δ = 4.57 ppm) and 3d (JH1,2 = 8.1 Hz at δ = 4.57 ppm) in good yields and stereoselectivity (β only). These results supported a hypothesis that in addition to steric factors, the failure of reactions in Scheme 4 to produce the desired disaccharides was probably because of the reaction condition used, which could not promote orthoester rearrangement.
Scheme 5.

2-O-Ac-directed galactosylation under acidic condition
To further confirm the above rationale, we studied the glycosylation reactions of 2c with more sterically demanding donors 1a and 1c under the promotion of NIS and AgOTf (Scheme 6), which turned out to be very sluggish and gave only low yields of the desired disaccharides 3c (25%, β major) and 3f (20%, α:β = 1:1). A comparison of the results of Schemes 5 and 6 clearly indicated that steric interactions were indeed the major factor to cause the failure of 3-O-galactosylation of galactosamine when larger protecting groups were present at the C-2-positions of both the donor and the acceptor.
Scheme 6.

2-O-Bz-directed galactosylation under acidic conditions
The above discoveries together with literature results led us to conclude that when the Gal 2-O-position and galactosamine 2-N-position were both protected with large groups, such as the Bz and Phth groups respectively, severe steric interactions between the donor and the acceptor as shown in Figure 1A prohibited effective glycosylation, to result in poor reaction yields. The relatively poor stereoselectivity might result from a SN2 type of glycosylation reaction, namely attack at the anomeric carbon of the oxonium intermediate by less sterically demanding nucleophiles such as triflate to form reactive glycosyl triflate followed by nucleophilic displacement of the triflate with galactosamine acceptor. Such reaction mechanism was reported previously.36,37 This was applicable to glycosylation reactions carried out under both preactivation conditions (Scheme 2) and conventional conditions (Scheme 6). When the 2-N-Phth group in galactosamine acceptors was replaced with a smaller protecting group, such as Ac (Table 1) and Troc (Table 2 and Scheme 3), which would reduce steric hindrance, the reactions went smoothly under both conditions to afford the desired β-linked oligosaccharides in very good yields and stereoselectivity. The result of Scheme 3 further suggested that the glycosylation reaction might go through a mechanism of direct acceptor attack at the anomeric carbon of the oxonium cation intermediate of the activated donor with NGPE involvement (Figure 1B) to give high stereoselectivity, because the reaction was performed under mildly basic conditions that did not allow for orthoester rearrangement. This should be reasonable since a large group linked to the carbonyl carbon of the oxonium cation would make acceptor attack at the carbonyl carbon unfavorable. On the other hand, when the donor had an acetyl group at the 2-O-position, its reaction with acceptors having the large Phth group at the 2-N-position might go through the orthoester intermediate (Figure 1C), because nucleophilic attach at the less sterically hindered carbonyl carbon would become more favorable. This was supported by the isolation of a substantial amount of orthoester 4 and the complex results from the reactions in Scheme 4 under the preactivation condition that prohibited orthoester rearrangement to form glycosides. However, when glycosylation reactions were performed under acidic conditions that could promote orthoester rearrangement, they afforded the desired glycosides in good yields and excellent stereoselectivity (Scheme 5). Finally, when the protecting groups at both the Gal 2-O-position and the galactosamine 2-N-position were small, literature results (Tables 1 and 2) showed that the reactions gave the desired glycosides in relatively good yields and stereoselectivity, presumably through the mechanism outlined in Figure 1B.
Figure 1.

Steric interactions during the synthesis of Galβ(1→3)GalN motif
In summary, we have systematically studied and analyzed the steric factors that might affect 3-O-β-glycosylation of galactosamine with galactose donors having acyl protecting groups at the 2-O-position, of which the NGPE failed in several cases to work or to assist stereoselective 1,2-trans-glycosylation. The failure of NGPE in directing stereoselective glycosylation was usually ascribed to solvent effects9 or conformational influences.8 Our results demonstrated that during 3-O-galactosylation of galactosamine steric interactions between the glycosyl donor and acceptor were also vital. Consequently, 2-O-benzylated galactosyl donors could not be used to react with galactosamine acceptors having 2-N-Phth protection effectively to give the desired 1,2-trans-glycosylation products, because the steric factors might have outweighed the NGPE. Replacing the 2-N-Phth or 2-O-Bz group or both with less steric demanding protecting groups could alter the reaction pathways and put the NGPE back into action. These results may also be used to interpret the “matched and mismatched glycosylation” theory.38
It is also worth noting that 3-O-β-galactosylation of galactosamine derivatives could proceed smoothly for a variety of donors and acceptors under acidic conditions except for the 2-O-benzoylated donor and 2-N,N-phthalylated acceptor pair as shown in Scheme 6. Therefore, under these glycosylation conditions, a variety of combinations of protecting groups for the glycosyl donor and acceptor can be selected. However, under neutral and basic glycosylation conditions, relatively large acyl groups should be used to protect the donor 2-O-position in order to suppress orthoester formation. This has put limitations on our selection of the proper protections for the acceptor 2-N-position, that is, only small protecting groups should be used. Consequently, this work helped us develop some useful principles that can not only provide general guidelines for our future design of successful synthetic schemes for the Galβ(1→3)GalNAc motif but also help the synthesis of other similarly β-1,3-linked disaccharides.
Experimental Section
General Procedures
Chemicals and materials were purchased from commercial sources and were used as received without further purification unless otherwise noted. Molecular sieves 4Å (MS 4Å) were flame-dried under high vacuum and used immediately after cooling to rt under a N2 atmosphere. Analytical TLC was carried out on silica gel 60Å F254 plates with detection by a UV detector and/or by charring with 10% (v/v) H2SO4 in EtOH. Flash column chromatography was performed on silica gel 60 (230–400 Mesh). NMR spectra were acquired on a 400, 500 or 600 MHz machine with chemical shifts reported in ppm (δ) and referenced with CHCl3 (1H NMR δ 7.26 ppm) or CDCl3 (13C NMR δ 77.0 ppm). Peak and coupling constant assignments are based on 1H NMR, 1H–1H COSY, 1H–13C HMQC and 1H–13C HMBC experiments.
p-Tolyl (2-O-benzoyl-3-O-benzyl-4,6-O-benzylidene-β-D-galactopyranosyl)-(1→3)- 4,6-O-benzylidene-2-(2,2,2-tri-chloroethoxycarbonylamido)-2-deoxy-1-thio-β-D-galactopyranoside (3a)
Galactosyl donor 1a (55 mg, 0.1mmol) and freshly activated MS 4Å (1.0 g) in CH2Cl2 (3 mL) was stirred at rt for 1 h. The mixture was cooled to −78 °C before extra dry silver triflate (77 mg, 0.3 mmol, 3.0 eq.) in acetonitrile (0.3 mL) was added to the reaction mixture, which was followed by the addition of p-TolSCl (16 μL, 0.1 mmol) 15 min later. After complete activation of galactosyl donor 1a was confirmed by TLC, the CH2Cl2 solution (1 mL) of galactosamine acceptor 2b (50 mg, 0.09 mmol) and TTBP (25 mg, 0.1 mmol) were added. The reaction was kept at −78 °C until its completion (in about 1 h), which was indicated by the disappearance of 2b, and the reaction mixture was filtered through a Celite pad. The solid was thoroughly washed with DCM and the filtrates were combined and washed with saturated aq. solution of NaHCO3. The organic layer was then dried over Na2SO4 and the solvent was removed by vacuum followed by purification of the residue with silica gel column chromatography (hexane/EtOAc = 1/1) to afford 3a as a white solid (86 mg, 87% yield). 1H NMR (500 MHz, CDCl3): δ 8.03 (d, 2H, J = 7.8 Hz, ArH), 7.59-7.54 (m, 3H), 7.45-7.35 (m, 7H, ArH), 7.22-7.16 (m, 10H, ArH), 6.97 (d, 2H, J = 7.8 Hz, ArH), 5.60 (dd, 1H, J = 9.9, 8.5 Hz, H-2), 5.53 (s, 1H, PhCH), 5.37 (s, 1H, PhCH), 5.27 (d, 1H, J = 7.7 Hz, NH), 4.88 (d, 1H, J = 10.7 Hz, H-1′), 4.86 (d, 1H, J = 7.9 Hz, H-1), 4.82 (d, 1H, J = 12.3 Hz, CCl3CH2), 4.67 (d, 1H, J = 13.5 Hz, PhCH2), 4.57 (d, 1H, J = 13.5 Hz, PhCH2), 4.45 (d, 1H, J = 3.2 Hz, H-4′), 4.40-4.27 (m, 4H. CCl3CH2, H-6a, H-3′, H-6a′), 4.23 (d, 1H, J = 2.9 Hz, H-4), 4.08 (d, 1H, J = 12.4Hz, H-6b), 3.98 (d, 1H, J = 13.0 Hz, H-6b′), 3.64 (dd, 2H, J = 10.4, 3.8 Hz, H-3, H-2′), 3.46 (s, 1H, H-5), 3.39 (m, 1H, H-5′), 2.29 (s, 3H, PhCH3); 13C NMR (125 MHz, CDCl3): δ 165.2, 154.0, 137.9, 137.8, 137.5, 133.6, 133.2, 129.9, 129.8, 129.6, 129.0, 128.5, 128.4, 128.1, 128.0, 127.7, 126.5, 101.0, 100.9, 100.0, 96.0, 85.4, 77.2, 77.19, 75.1, 74.1, 72.7, 71.1, 70.3, 70.0, 69.3, 69.2, 66.9, 51.5, 21.1; HR ESI-TOF-MS: [M + Na]+ C50H48Cl3NO12SNa+ m/z calcd 1014.1855, found 1014.1898.
p-Tolyl 3-O-[(3,4,6-tri-O-acetyl-β-D-galactopyranos-1,2-O-orthoacet)-yl]-4,6-O-benzylidene-2-(2,2,2-tri-chloroethoxycarbonylamido)-2-deoxy-1-thio-β-D-galactopyranoside (4)
Compound 4 was obtained as a white solid (yield 30%) from the reaction between 1c and 2b under the same conditions described above for the synthesis of 3a. 1H NMR (500 MHz, CDCl3): δ 7.54 (d, 2H, J = 8.2 Hz, ArH), 7.47-7.45 (m, 2H, ArH), 7.38-7.36 (m, 3H, ArH), 7.03 (d, 2H, J = 8.0 Hz, ArH), 5.72 (d, 1H, J = 4.9 Hz, H-1), 5.56 (s, 1H, PhCH), 5.39 (m, 1H, J = 3.4, 2.0 Hz, H-4), 5.37 (d, 1H, J = 10.0 Hz, H-1′), 5.19 (d, 1H, J = 7.2 Hz, NH), 5.00 (dd, 1H, J = 7.0, 3.5 Hz, H-3), 4.78 (d, 1H, J = 12.2 Hz, CCl3CH2), 4.68 (d, 1H, J = 12.2 Hz, CCl3CH2), 4.46 (dd, 1H, J = 10.3, 3.1 Hz, H-3′), 4.39 (dd, 1H, J = 12.2, 2.0 Hz, H-6a′), 4.37 (d, 1H, J = 3.1 Hz, H-4′), 4.26-4.23 (m, 2H. H-2, H-5), 4.16 (dd, 1H, J = 11.4, 6.5 Hz, H-6a), 4.07 (dd, 1H, J = 11.7, 6.5 Hz, H-6b), 4.05 (dd, 1H, J = 12.5, 1.2 Hz, H-6b′), 3.58 (s, 1H, H-5′), 3.39 (m, 1H, H-2), 2.34 (s, 3H, PhCH3), 2.06 (s, 3H, COCH3), 2.04 (s, 3H, COCH3), 2.03 (s, 3H, COCH3), 1.67 (s, 3H, CH3, orthoester); 13C NMR (125 MHz, CDCl3): δ170.4, 170.0, 169.8, 153.7, 138.4, 137.9, 134.2, 129.8, 129.1, 126.5, 121.3, 100.9,97.7, 95.3, 83.5, 75.1, 74.5, 71.1, 70.5, 69.9, 69.4, 69.2, 69.1, 65.7, 61.2, 51.3, 23.5, 21.2, 20.7, 20.68, 20.5; HR MALDI-TOF-MS: [M + Na]+ C37H42Cl3NO15SNa+ m/z calcd 900.1233, found 900.1273.
Methyl (2-O-acetyl-3-O-benzyl-4,6-O-benzylidene-β-D-galactopyranosyl)-(1→3)-4,6-O-benzylidene-2-deoxy-2-phthalimido)-β-D-galactopyranoside (3c)
A mixture of galactosyl donor 1b (68 mg, 0.12 mmol), galactosamine acceptor 2c (41 mg, 0.1 mmol) and freshly activated MS 4Å (1.0 g) in CH2Cl2 (3 mL) was stirred at rt for 1 h. The mixture was cooled to −78 °C before NIS (30 mg, 0.13 mmol) and dry AgOTf (8 mg, 0.03 mmol) were added to the reaction mixture. The reaction was allowed to warm to −30 °C and stirred for 1 h, when TLC indicated the completion of reaction. The reaction was quenched with Et3N, followed by filtration through a pad of Celite. The solid was thoroughly washed with DCM and the filtrates were combined and washed with saturated aq. solution of Na2SO3, water, and brine. The organic layer was dried over Na2SO4 and the solvent was removed under vacuum followed by purification of the residue with silica gel column chromatography (hexane/EtOAc = 1/1) to afford the 3c as a white solid (70 mg, 88% yield). 1H NMR (500 MHz, CDCl3): δ 7.86-7.83 (m, 2H), 7.75-7.73 (m, 2H, ArH), 7.61-7.59 (m, 2H, ArH), 7.53-7.51 (m, 2H, ArH), 7.35-7.33 (m, 7H, ArH), 7.26-7.19 (m, 4H, ArH), 5.61 (s, 1H, PhCH), 5.40 (s, 1H, PhCH), 5.22 (dd, 1H, J = 10.0, 8.1 Hz, H-2), 5.14 (d, 1H, J = 8.5 Hz, H-1′), 4.82 (dd, 1H, J = 11.2, 3.6 Hz, H-3′), 4.73 (dd, 1H, J = 11.2, 8.5 Hz, H-2′), 4.57 (d, 1H, J = 8.3 Hz, H-1), 4.56 (d, 1H, J = 12.6 Hz, PhCH2), 4.49 (d, 1H, J = 12.6 Hz, PhCH2), 4.48 (d, 1H, J = 3.4 Hz, H-4′), 4.37 (dd, 1H, J = 12.2, 1.2, Hz, H-6a′), 4.11 (dd, 1H, J = 12.2, 1.2, Hz, H-6b′), 4.06 (d, 1H, J = 3.3 Hz, H-4), 3.99 (d, 1H, J = 11.4 Hz, H-6a), 3.91 (d, 1H, J = 11.4 Hz, H-6b), 3.59 (s, 1H, H-5′), 3.44 (s, 3H, OMe), 3.25 (s, 1H, H-5), 1.58 (s, 3H, OAc); 13C NMR (125 MHz, CDCl3): δ 170.5, 169.5, 128.3, 128.2, 128.1, 127.4, 126.6, 126.5, 101.3, 100.9, 100.5, 99.2, 77.5, 75.6, 73.0, 72.97, 70.9, 70.3, 69.3, 69.2, 69.1, 68.9, 66.9, 66.8, 56.1, 51.7, 29.7, 25.6, 20.3; HR ESI-TOF-MS: [M + Na]+ C44H43NO13Na+ m/z calcd 816.2627, found 816.2655.
Methyl (2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-(1→3)-4,6-O-benzylidene-2-deoxy-2-phthalimido-β-D-galactopyranoside (3d)
Compound 3d was obtained as a white solid (65 mg, 85%) from the reaction between 1d and 2c under the same conditions described above for the synthesis of 3c. 1H NMR (500 MHz, CDCl3): δ 7.88-7.86 (m, 2H,, ArH), 7.76-7.74 (m, 2H, ArH), 7.62-7.60 (m, 2H, ArH), 7.42-7.36 (m, 3H,, ArH), 5.61 (s, 1H, PhCH), 5.30 (dd, 1H, J = 3.5, 1.2 Hz, H-4), 5.10 (dd, 1H, J = 10.5, 7.8 Hz, H-2), 5.06 (d, 1H, J = 8.0 Hz, H-1′), 4.83 (dd, 1H, J = 10.4, 3.4 Hz, H-3), 4.76-4.74 (m, 2H,, H-3′, H-2′), 4.57 (d, 1H, J = 8.1 Hz, H-1), 4.39 (ddd, 2H, J = 6.0, 4.9, 1.3 Hz, H-4′, H-6a′), 4.13 (dd, 1H, J = 12.3, 1.5 Hz, H-6b′), 4.10-4.05 (m, 2H, H-6a, H-6b, ), 3.82 (dt, 1H, J = 6.5, 1.1 Hz, H-5), 3.60 (s, 1H, H-5′), 3.44 (s, 3H, OCH3), 2.13 (s, 3H, COCH3), 2.04 (s, 3H, COCH3), 1.88 (s, 3H, COCH3), 1.42 (s, 3H, COCH3); 13C NMR (150 MHz, CDCl3): δ 170.5, 170.4, 170.3, 169.3, 168.9, 167.4, 137.9, 134.4, 134.3, 131.9, 131.8, 129.1, 128.3, 126.6, 123.9, 123.5, 101.6, 101.1, 99.3, 75.8, 75.0, 71.1, 70.9, 69.4, 68.7, 67.0, 66.9, 61.4, 56.3, 51.6, 32.1, 29.8, 29.5, 22.8, 20.88, 20.86, 20.7, 20.6, 19.9; HR HR ESI-TOF-MS: [M + Na]+ C36H39NO16Na+ m/z calcd 764.2161, found 764.2194.
Methyl (2-O-benzoyl-3-O-benzyl-4,6-O-benzylidene-β-D-galactopyranosyl)-(1→3)-4,6-O-benzylidene-2-deoxy-2-phthalimido-α,β-D-galactopyranoside (3e)
A mixture of galactosyl donor 1a (68 mg, 0.12 mmol), galactosamine acceptor 2c (41 mg, 0.1 mmol) and freshly activated MS 4Å (1.0 g) in CH2Cl2 (3 mL) was stirred at rt for 1 h. The mixture was cooled to −78 °C before NIS (30 mg, 0.13 mmol) and dry AgOTf (8 mg, 0.03 mmol) were added. The mixture was allowed to warm to −30 °C and stirred for 1 h, whereas TLC showed incomplete reaction. Thus, the mixture was warmed to rt and stirred at rt overnight. TLC showed that the acceptor disappeared but no further progress of the reaction was observed. The reaction was quenched with Et3N followed by filtration through a pad of Celite. The solid was thoroughly washed with DCM and the filtrates were combined and washed with saturated aq. solution of Na2SO3, water, and brine. The organic layer was dried over Na2SO4 and the solvent was removed under vacuum, which was followed by purification of the residue with silica gel column chromatography (hexanes/EtOAc = 1/1) to 3e as a white solid (22 mg, 25% yield) along with recovered acceptor 2c (about 50%). Purified major isomer 3e-β: 1H NMR (500 MHz, CDCl3): δ 7.93-7.86 (m, 4H), 7.78-7.76 (m, 2H, ArH), 7.50-7.39 (m, 5H, ArH), 7.33-7.16 (m, 13H, ArH), 5.50 (d, 1H, J = 4.0 Hz, H-1), 5.37 (dd, 1H, J = 10.6, 4.0 Hz, H-2), 5.26 (s, 1H, PhCH), 5.09 (d, 1H, J = 8.2 Hz, H-1′), 5.01 (s, 1H, PhCH), 4.72-4.63 (m, 3H, H-2′, H-3′, PhCH2), 4.58 (d, 1H, J = 12.2 Hz, PhCH2), 4.25 (dd, 1H, J = 12.3, 0.7, Hz, H-6a′), 4.03 (dd, 1H, J = 10.5, 3.4, Hz, H-3), 3.98 (m, 2H, H-4, H-4′), 3.82 (dd, 1H, J = 12.2, 1.6 Hz, H-6b′), 3.47 (s, 1H, H-5′), 3.41 (s, 3H, OMe), 3.36 (dd, 1H, J = 12.5, 1.3 Hz, H-6a), 3.28 (s, 1H, H-5), 2.98 (dd, 1H, J = 12.2, 1.7 Hz, H-6b); 13C NMR (125 MHz, CDCl3): δ 168.5, 167.9, 166.0, 137.6, 137.4, 134.5, 134.3, 133.2, 131.8, 131.6, 129.7, 129.6, 128.9, 128.6, 128.4, 128.3, 128.1, 128.0, 127.6, 127.5, 126.2, 126.1, 123.5, 123.4, 100.8, 100.3, 99.1, 98.7, 76.4, 74.0, 73.6, 73.4, 72.1, 70.8, 69.0, 68.4, 66.4, 63.4, 56.4, 52.3, 36.6, 29.7; HR ESI-TOF-MS: [M + Na]+ C49H45NO13Na+ m/z calcd 878.2783, found 878.2804.
Methyl (2,3,4,6-tetra-O-benzoyl-α-D-galactopyranosyl)-(1→3)-4,6-O-benzylidene-2-deoxy-2-phthalimido-α,β-D-galactopyranoside (3f)
Compound 3f was obtained as a white solid (85% yield for both isomers) from the reaction between 1c and 2c under the same conditions described above for the synthesis of 3e, and the two anomers were separated by silica gel column chromatography (hexane/EtOAc = 1/1). 3f-α: 1H NMR (600 MHz, CDCl3): δ 7.95-7.92 (m, 4H, ArH), 7.88-7.87 (m, 1H, ArH), 7.82-7.80 (m, 3H, ArH), 7.44-7.37 (m, 7H, ArH), 7.31-7.26 (m, 2H, ArH), 7.20-7.11 (m, 7H, ArH), 5.79 (d, 1H, J = 2.2 Hz, H-4), 5.74 (dd, 1H, J = 10.6, 3.3 Hz, H-3), 5.72 (d, 1H, J = 3.9 Hz, H-1), 5.50 (dd, 1H, J = 11.0, 3.9 Hz, H-2), 5.17 (s, 1H, PhCH), 5.14 (d, 1H, J = 7.6 Hz, H-2′), 4.85-4.82 (m, 2H, H-1′, H-3′), 4.28 (d, 1H, J = 12.3 Hz, H-6a′), 4.20-4.18 (m, 2H, H-5, H-4′), 4.02 (dd, 1H, J = 10.5, 8.1 Hz, H-6a), 3.93-3.90 (m, 2H,, H-6b, H-6b′), 3.43 (s, 3H, OCH3), 3.40 (s, 1H, H-5′); 13C NMR (150 MHz, CDCl3): δ 169.0, 167.5, 166.3, 165.4, 165.3, 165.0, 137.1, 134.4, 134.3, 133.5, 133.4, 133.2, 133.0, 131.5, 131.4, 129.8, 129.77, 129.74, 129.6, 129.4, 129.1, 129.0, 128.6, 128.5, 128.46, 128.4, 128.35, 128.3, 128.2, 127.9, 125.9, 124.0, 123.1, 100.2, 99.1, 95.3, 73.8, 72.1, 69.2, 69.0, 68.3, 67.8, 67.4, 66.4, 61.3, 60.4, 56.3, 51.8, 29.7; HR ESI-TOF-MS: [M + Na]+ C56H47NO16Na+ m/z calcd 1012.2787, found 1012.2790. 3f-β: 1H NMR (600 MHz, CDCl3): δ 7.95-7.93 (m, 2H, ArH), 7.89-7.87 (m, 1H, ArH), 7.82-7.78 (m, 4H, ArH), 7.66-7.65 (m, 1H, ArH), 7.62-7.53 (m, 7H, ArH), 7.40-7.30 (m, 12H, ArH), 7.13-7.10 (m, 2H, ArH), 6.00 (d, 1H, J = 5.2 Hz, H-1), 5.63-5.62 (m, 1H), 5.51 (s, 1H, PhCH), 5.31 (dd, 1H, J = 5.9, 4.2 Hz), 5.07 (d, 1H, J = 8.5 Hz, H-1′), 4.58 (dd, 1H, J = 11.0, 8.8 Hz), 4.52 (dd, 1H, J = 114, 6.8 Hz), 4.39-4.36 (m, 2H), 4.34-4.32 (m, 2H), 4.26 (dd, 1H, J = 10.5, 5.6 Hz), 4.16 (d, 1H, J = 3.8 Hz), 4.04 (dd, 1H, J = 12.2, 1.5 Hz), 3.44 (s, 1H), 3.39 (s, 3H, OCH3); 13C NMR (150 MHz, CDCl3): δ 168.2, 167.4, 165.9, 165.0, 164.9, 137.7, 135.5, 134.0, 133.8, 133.5, 133.3, 133.2, 131.9, 131.8, 129.8, 129.75, 129.72, 129.4, 129.0, 128.8, 128.4, 128.35, 128.3, 128.2, 126.4, 126.38, 125.8, 123.6, 122.9, 119.9, 101.3, 98.9, 98.3, 74.5, 72.7, 69.4, 69.2, 69.17, 68.6, 66.6, 66.0, 62.2, 56.2, 51.7, 29.7; HR ESI-TOF-MS: [M + Na]+ C56H47NO16Na+ m/z calcd 1012.2787, found 1012.2811.
Supplementary Material
Acknowledgments
We thank NIH/NCI for the support (grant R01CA095142) of this research work.
References
- 1.Lemieux RU, Hendriks KB, Stick RV, James K. J Am Chem Soc. 1975;97:4056–4062. [Google Scholar]
- 2.Jung KH, Mueller M, Schmidt RR. Chem Rev. 2000;100:4423–4442. doi: 10.1021/cr990307k. [DOI] [PubMed] [Google Scholar]
- 3.Crich D, Sun S. J Am Chem Soc. 1997;119:11217–11223. [Google Scholar]
- 4.Zhu X, Kawatkar S, Rao Y, Boons GJ. J Am Chem Soc. 2006;128:11948–11957. doi: 10.1021/ja0629817. [DOI] [PubMed] [Google Scholar]
- 5.Chao C-S, Lin C-Y, Mulani S, Hung W-C, Mong K-kT. Chem Eur J. 2011;17:12193–12202. doi: 10.1002/chem.201100732. [DOI] [PubMed] [Google Scholar]
- 6.Jiao H, Hindsgaul O. Angew Chem Int Ed. 1999;38:346–348. doi: 10.1002/(SICI)1521-3773(19990201)38:3<346::AID-ANIE346>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 7.Smoot JT, Pornsuriyasak P, Demchenko AV. Angew Chem Int Ed. 2005;44:7123–7126. doi: 10.1002/anie.200502694. [DOI] [PubMed] [Google Scholar]
- 8.Mandal SS, Liao G, Guo Z. RSC Adv. 2015;5:23311–23319. doi: 10.1039/C5RA00759C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Z, Zhou L, El-Boubbou K, Ye XS, Huang X. J Org Chem. 2007;72:6409–6420. doi: 10.1021/jo070585g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chuang HY, Ren CT, Chao CA, Wu CY, Shivatare SS, Cheng TJR, Wong CH. J Am Chem Soc. 2013;135:11140–11150. doi: 10.1021/ja403609x. [DOI] [PubMed] [Google Scholar]
- 11.Sakamoto K, Nagamachi Y, Sugawara I. Gan No Rinsho. 1990;36:865–870. [PubMed] [Google Scholar]
- 12.Amer H, Hofinger A, Kosma P. Carbohydr Res. 2003;338:35–45. doi: 10.1016/s0008-6215(02)00355-5. [DOI] [PubMed] [Google Scholar]
- 13.Karst N, Jacquinet JC. Eur J Org Chem. 2002:815–825. [Google Scholar]
- 14.Komba S, Kitaoka M, Kasumi T. Eur J Org Chem. 2005:5313–5329. [Google Scholar]
- 15.Arosio D, Vrasidas I, Valentini P, Liskamp RMJ, Pieters RJ, Bernardi A. Org Biomol Chem. 2004;2:2113–2124. doi: 10.1039/b405344c. [DOI] [PubMed] [Google Scholar]
- 16.Bosse F, Marcaurelle LA, Seeberger PH. J Org Chem. 2002;67:6659–6670. doi: 10.1021/jo025834+. [DOI] [PubMed] [Google Scholar]
- 17.Fujikawa K, Nakashima S, Konishi M, Fuse T, Komura N, Ando T, Ando H, Yuki N, Ishida H, Kiso M. Chem Eur J. 2011;17:5641–5651. doi: 10.1002/chem.201003357. [DOI] [PubMed] [Google Scholar]
- 18.Guillemineau M, Auzanneau FI. Carbohydr Res. 2012;357:132–138. doi: 10.1016/j.carres.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 19.Hirose H, Tamai H, Gao C, Imamura A, Ando H, Ishida H, Feizi T, Kiso M. Org Biomol Chem. 2015;13:11105–11117. doi: 10.1039/c5ob01744k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Komori T, Imamura A, Ando H, Ishida H, Kiso M. Carbohydr Res. 2009;344:1453–1463. doi: 10.1016/j.carres.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 21.Konishi M, Imamura A, Fujikawa K, Ando H, Ishida H, Kiso M. Molecules. 2013;18:15153–15181. doi: 10.3390/molecules181215153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lay L, Panza L, Poletti L, Prosperi D, Canevari S, Perico ME. Eur J Org Chem. 2001:4331–4336. [Google Scholar]
- 23.Zhang P, Wang K, Zhang J, Li C, Guan H. Eur J Org Chem. 2015;2015:570–583. [Google Scholar]
- 24.Sawada N, Ito M, Ishida H, Kiso M. Tetrahedron Lett. 2001;42:1745–1747. [Google Scholar]
- 25.Burkhart F, Zhang Z, Wacowich-Sgarbi S, Wong CH. Angew Chem Int Ed. 2001;40:1274–1277. doi: 10.1002/1521-3773(20010401)40:7<1274::aid-anie1274>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Z, Ollmann IR, Ye XS, Wischnat R, Baasov T, Wong CH. J Am Chem Soc. 1999;121:734–753. [Google Scholar]
- 27.Mong TKK, Lee HK, Duron SG, Wong CH. Proc Natl Acad Sci USA. 2003;100:797–802. doi: 10.1073/pnas.0337590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanaka H, Nishiura Y, Takahashi T. J Am Chem Soc. 2008;130:17244–17245. doi: 10.1021/ja807482t. [DOI] [PubMed] [Google Scholar]
- 29.Sun B, Jiang H. Tetrahedron Lett. 2012;53:5711–5715. [Google Scholar]
- 30.Crich D, Li L. J Org Chem. 2007;72:1681–1690. doi: 10.1021/jo062294y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gass J, Strobl M, Loibner A, Kosma P, Zaehringer U. Carbohydr Res. 1993;244:69–84. doi: 10.1016/0008-6215(93)80005-y. [DOI] [PubMed] [Google Scholar]
- 32.Sznaidman ML, Johnson SC, Crasto C, Hecht SM. J Org Chem. 1995;60:3942–3943. [Google Scholar]
- 33.Wang W, Kong F. J Org Chem. 1998;63:5744–5745. doi: 10.1021/jo981135e. [DOI] [PubMed] [Google Scholar]
- 34.Seeberger PH, Eckhardt M, Gutteridge CE, Danishefsky SJ. J Am Chem Soc. 1997;119:10064–10072. [Google Scholar]
- 35.Forman A, Auzanneau FI. Carbohydr Res. 2016;425:10–21. doi: 10.1016/j.carres.2016.02.012. [DOI] [PubMed] [Google Scholar]
- 36.Crich D, Chandrasekera NS. Angew Chem, Int Ed. 2004;43:5386–5389. doi: 10.1002/anie.200453688. [DOI] [PubMed] [Google Scholar]
- 37.Crich D. Acc Chem Res. 2010;43:1144–1153. doi: 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]
- 38.Spijker NM, Van Boeckel CAA. Angew Chem Int Ed Engl. 1991;30:180–183. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.