

Understanding the functional effects of DNA sequence variants is of critical importance for studies of basic biology, evolution, and medical genetics, but measuring these effects in a high-throughput manner is a major challenge. One promising avenue is precise editing with the CRISPR/Cas9 system, which allows generation of DNA double-strand breaks (DSBs) at genomic sites matching the targeting sequence of a guide RNA (gRNA). Recent studies have used CRISPR libraries to generate many frameshift mutations genome-wide through faulty repair of CRISPR-directed breaks by nonhomologous end-joining (NHEJ)1. Here, we developed a CRISPR library-based approach for highly efficient and precise genome-wide variant engineering. We used our method to examine the functional consequences of premature termination codons (PTCs) at different locations within all annotated essential genes in yeast. We found that most PTCs were highly deleterious unless they occurred close to the 3’ end of the gene and did not affect an annotated protein domain. Surprisingly, we discovered that some putatively essential genes are dispensable, while others have large dispensable regions. This approach can be used to profile the effects of large classes of variants in a high-throughput manner.
Precise gene editing by CRISPR/Cas9 requires providing a DNA template to be used for homology-directed repair (HDR)2, in the process incorporating desired sequence variants encoded on the template into the genomic locus. Generating many uniquely edited cells in parallel thus requires each cell to receive the correct gRNA-repair template pair. We devised an approach that accomplishes such pairing by encoding gRNA targeting sequences and their corresponding repair templates in cis on oligonucleotides generated in bulk with high-throughput synthesis. These oligonucleotide libraries are then used to generate pools of plasmids pairing the two components for delivery into yeast cells (Supplementary Figure 1). We used this approach to understand the consequences of one important class of genetic variants: premature termination codons (PTCs).
PTCs interrupt the open reading frames (ORFs) of protein-coding genes. Such mutations are generally expected to have strong deleterious effects, either by abrogating or changing the functions of the encoded proteins or by causing mRNA degradation through the nonsense-mediated decay (NMD) surveillance pathway. More than 10% of annotated pathogenic human variants are PTCs3,4. Nonetheless, our understanding of the detrimental effects of PTCs is incomplete, particularly when they occur near the 3’ ends of genes. Such mutations may not shorten the encoded proteins sufficiently to affect their function, and often escape NMD.
We first tested gene editing that employs a plasmid-encoded paired gRNA and repair template (figure 1a) by targeting eight specific PTCs to the S. cerevisiae genome. S. cerevisiae has a naturally high propensity to repair DSBs through HDR5, which we enhanced by using a haploid yeast strain in which NHEJ is abolished by a deletion of the NEJ1 gene6 (nej1Δ; Supplementary Table 1). For each targeted mutation, we sequenced the corresponding genomic locus in thousands of transformed yeast cells. In all eight cases, the desired mutation was present in >95% of sequencing reads, demonstrating the high efficiency of this strategy (Table 1). We also tested editing in wildtype diploid yeast, where NHEJ is inactive6, and observed high efficiency at most sites (Supplementary Table 2). None of the sites showed a high rate of indel formation in either the nej1Δ or diploid strains, consistent with NHEJ being inactive.
Figure 1:
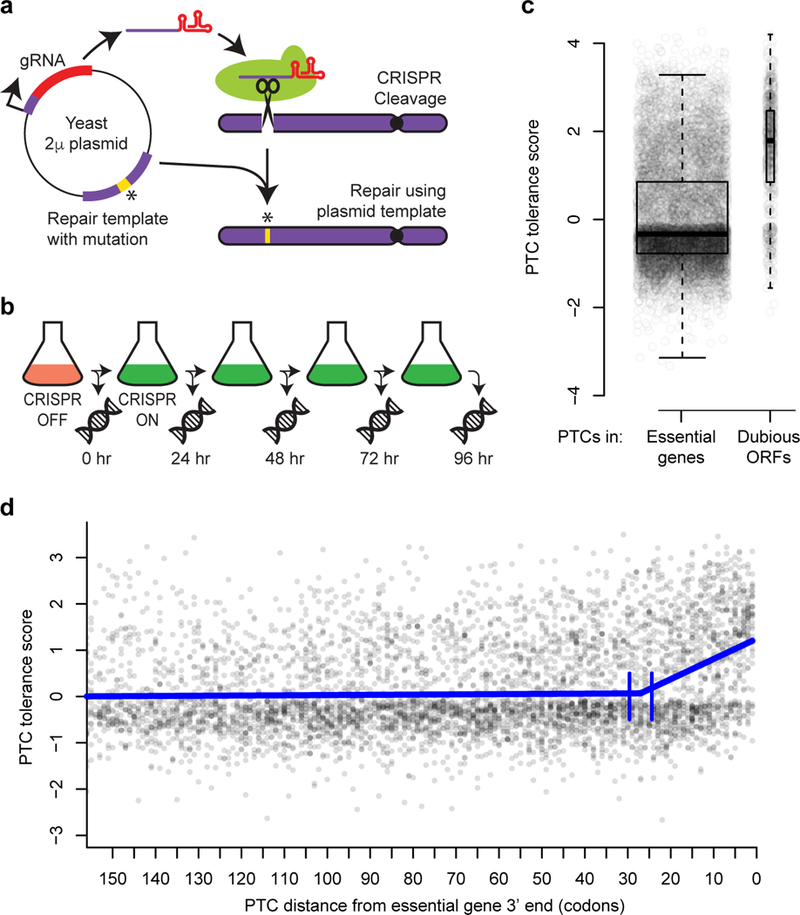
Measuring the effects of engineered PTCs in essential genes. a, Schematic of pairing of CRISPR gRNA and repair template on plasmids. b, Experimental design. Following Cas9 induction, DNA was extracted every 24 hours. At each time point, edit-directing plasmids were quantified by sequencing. c, Tolerance scores for n = 8,353 PTCs targeting essential genes and n = 694 PTCs targeting dubious ORFs are shown, with overlaid boxplots. The centerline of each box corresponds to the data’s median value; the top and bottom of the box span from the first quartile to the third quartile of the data; and the whiskers reach to either the data’s most extreme values or 1.5 times the interquartile range. P < 2 × 10−16, two-sided Wilcoxon rank sum test. d, Scatterplot of PTC tolerance scores versus distance in codons from the 3’ ends of essential genes. The thick blue line shows a segmented regression fit. Vertical blue lines indicate the 95% confidence interval for the boundary between the segments. The segmented regression was fit on PTC tolerance scores for n = 7,583 PTCs that were within 500 codons of the 3’ end of a gene.
Table 1:
Assessing the efficiency of edit-directing plasmids. Outcomes of directed mutations at eight loci in nej1Δ cells, as determined by classifying paired-end Illumina reads of PCRs of genomic DNA at each locus.
| Expected edit | Unedited | Mismatch | Indel | |
|---|---|---|---|---|
| ho-G582Stop | 98.51% | 0.08% | 1.40% | 0.00% |
| his2-E308Stop | 99.83% | 0.07% | 0.10% | 0.00% |
| mnd1-V219Stop | 99.35% | 0.53% | 0.12% | 0.00% |
| spo11-F381Stop | 95.56% | 4.24% | 0.19% | 0.00% |
| spo13-P252Stop | 99.67% | 0.20% | 0.13% | 0.00% |
| ste3-P469Stop | 99.75% | 0.12% | 0.13% | 0.00% |
| can1-G121Stop | 99.81% | 0.06% | 0.13% | 0.00% |
| can1-G70Stop | 99.80% | 0.03% | 0.17% | 0.00% |
We next scaled up the approach by using large-scale oligonucleotide synthesis to generate a pool of over 10,000 distinct paired gRNA-repair template plasmids (Supplementary figure 1). These plasmids targeted PTCs to different sites in 1034 yeast genes considered essential for viability7,8. Each gene was targeted at 10 sites, chosen with a preference for sites closer to the 3’ end (Supplementary Figure 2). Targeted PTCs were represented by multiple independent barcoded plasmids. We transformed haploid nej1Δ yeast in bulk with plasmid pools in two independent replicate experiments. After inducing Cas9 expression, we collected millions of surviving transformed cells every 24 hours for four days (figure 1b). PTCs that disrupt the function of genes essential for viability are expected to drop out of the pool over time, while those that do not are expected to persist.
We determined the abundance of each barcoded edit-directing plasmid at each time point by bulk sequencing, and computed a “PTC tolerance score” based on the persistence of the barcoded plasmids over the duration of the time-course experiment (Materials and Methods). PTC tolerance scores from the replicate experiments were correlated at r = 0.6 (p < 2 × 10−16) (Supplementary figure 3). As controls, we used a set of 90 “dubious ORFs,” which were originally annotated as genes but later reclassified due to lack of conservation and ascribable function9. As expected, PTCs in essential genes were much less tolerated than those in dubious ORFs (Wilcoxon rank sum test, P < 2 × 10−16) (figure 1c). As a further control, 71 sites in essential genes were targeted with two plasmids that had the same gRNA but different repair templates, only one of which introduced a PTC. Plasmids that introduced a PTC were significantly less tolerated (Supplementary Figure 3) (paired t-test t = 6.5, P = 8 × 10−9), showing that the observed phenotypic effects are predominantly due to specific introduction of the desired mutations, rather than repair-template-independent Cas9 activities.
One possibility for the observed PTC intolerance is that most truncations of essential genes fatally disrupt the function of the encoded proteins. Another possibility is that NMD removes most transcripts carrying PTCs, which is fatal in the case of PTCs in essential genes. We tested these alternatives by introducing PTCs in a strain that is NMD-deficient10. PTCs in this strain were similarly deleterious (Supplementary Figure 4) (χ2 = 1.66, P = 0.20) (Supplementary Table 3), suggesting that protein truncation, rather than degradation of transcripts via NMD, explains the observed PTC intolerance.
Although most PTCs in annotated essential genes were highly deleterious, some appeared to be tolerated. We examined the relationship between tolerance scores and locations of PTCs. PTCs were generally deleterious when located more than 27 codons away from the gene end (figure 1d). Within the 27 terminal codons, the tolerance scores rose toward the 3’ end. PTCs were also more tolerated if they did not interrupt or remove an annotated protein domain11 (χ2 = 317.2, P = 5.86 × 10−71) (Supplementary figure 5, Supplementary Table 3). PTCs that disrupted protein domains tended to be deleterious even when they fell close to gene ends. Evolutionary conservation of the truncated region among related yeast species12 also had an effect on PTC tolerance (χ2 = 49.8, P = 1.66 × 10−12) (Supplementary figure 5).
We built a model to more precisely delineate dispensable 3’ ends of essential genes. While our experiment is not designed to comprehensively rule out the existence of small dispensable C-termini, it is interesting to note that 517 genes did not appear to tolerate any tested PTCs, in some cases even very close to their ends (Supplementary figure 6, Supplementary Note 1). In contrast to these highly PTC-intolerant genes, 101 genes tolerated five or more PTCs, suggesting that these genes have large dispensable C-termini (Supplementary Figure 6). We computed the overall tolerance of PTCs for each gene and observed considerable variation among genes (figure 2a). A gene ontology enrichment analysis13 showed that genes encoding proteins with catalytic activity were significantly less PTC-tolerant than other genes (Kolmogorov-Smirnov test, Bonferroni corrected P = 0.0024) (Supplementary Table 4, Supplementary Figure 7), while genes with functions relating to mRNA splicing and processing were significantly more PTC-tolerant (Kolmogorov-Smirnov test, Bonferroni corrected P = 0.0017).
Figure 2:
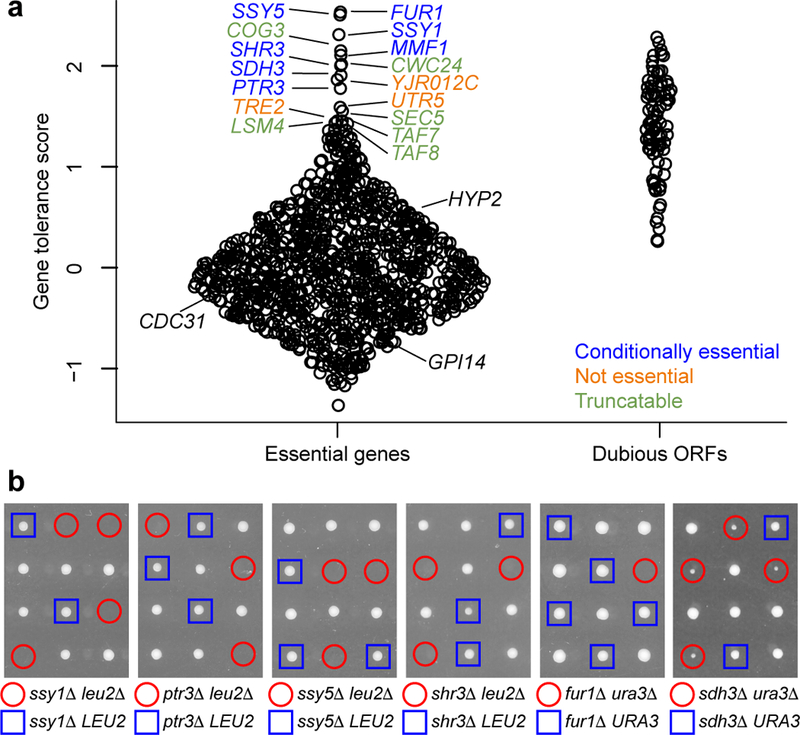
PTC tolerance of genes. a, Gene tolerance scores for essential genes and dubious ORFs, shown as a violin plot that displays the individual data points. b, Analysis of conditionally essential genes in yeast tetrads. Each vertical set of four colonies corresponds to the four haploid meiotic products from a diploid yeast strain. Each diploid was heterozygous for a deletion mutation of interest and for an interacting mutation. Haploid colonies carrying the deletion of interest are highlighted in red or blue based on their genotype at the interacting locus. Absence of a visible colony (first five panels) indicates a lethal interaction; small colonies (last panel) indicate an interaction causing poor growth. n = 10 tetrads were examined for the ssy1Δ, ptr3Δ, ssy5Δ, and fur1Δ interactions; n = 6 tetrads were examined for the shr3Δ interaction.
To better understand why some genes annotated as essential could tolerate many PTCs, we closely examined the 16 most PTC-tolerant genes (figure 2a). We found that three of these genes had been misannotated as essential because their deletion disrupts the function of a nearby essential gene, illustrating the value of PTC introduction for characterization of gene essentiality (Supplementary Figure 8, figure 2a, Supplementary Note 1). PTC-tolerant genes also included SSY1, PTR3, and SSY5, which encode the three members of the SPS (Ssy1-Ptr3-Ssy5) plasma membrane amino acid sensor system14, as well as SHR3, required for SPS cell-surface localization15. Defects in SPS function compromise leucine uptake, and the strain originally used to determine which genes are essential is deficient in leucine biosynthesis and thus requires leucine uptake, which explains the lethality of SPS mutations in this strain16,17. We confirmed that deletions of these genes were viable in yeast that could synthesize leucine, but lethal in yeast that could not (figure 2b). Similarly, the PTC-tolerant gene FUR1 is required for the utilization of exogenous uracil18, and uracil biosynthesis is also disrupted in the strain used to annotate essential genes. We confirmed that FUR1 is only essential in yeast which cannot synthesize uracil (figure 2b), consistent with previous synthetic lethality results19. Unexpectedly, we also observed poor growth of yeast with deletions of both URA3 and the PTC-tolerant gene SDH3 (figure 2b), a member of the mitochondrial inner membrane protein translocase complex20, which suggests that proper uracil utilization may involve an unknown mitochondrial function. These examples illustrate that genes not universally essential for yeast viability can appear essential in a specific genetic background. Another PTC-tolerant gene, MMF1, is viable in our growth conditions, but not in those used to define the set of essential genes (Supplementary Figure 9, Supplementary Note 1), providing an example of environment-dependent essentiality.
Six PTC-tolerant essential genes encode proteins with large dispensable C-terminal regions. One striking case is CWC24, a highly conserved member of the spliceosome. Cwc24 has a CCCH-type Zinc finger domain (Znf) and a RING-type Znf domain. Analysis of the effect of PTCs in CWC24 suggested the RING finger domain was dispensable while the CCCH Znf was essential, which we confirmed by engineering CWC24 truncations (figure 3a; see also Wu et al., 201621). It is interesting to note that a PTC after the RING finger domain of the essential22 human homolog of CWC24, RNF113A, is also viable31. Four other PTC-tolerant genes, TAF7, TAF8, COG3, and LSM4, have been reported to tolerate large truncations24–27. We verified that SEC5, a 971-amino acid member of the essential exocyst complex28, tolerates truncation of at least 615 amino acids (figure 3b). Our observation that 101 genes tolerated five or more PTCs suggests that many additional genes have dispensable C-terminal regions.
Figure 3:
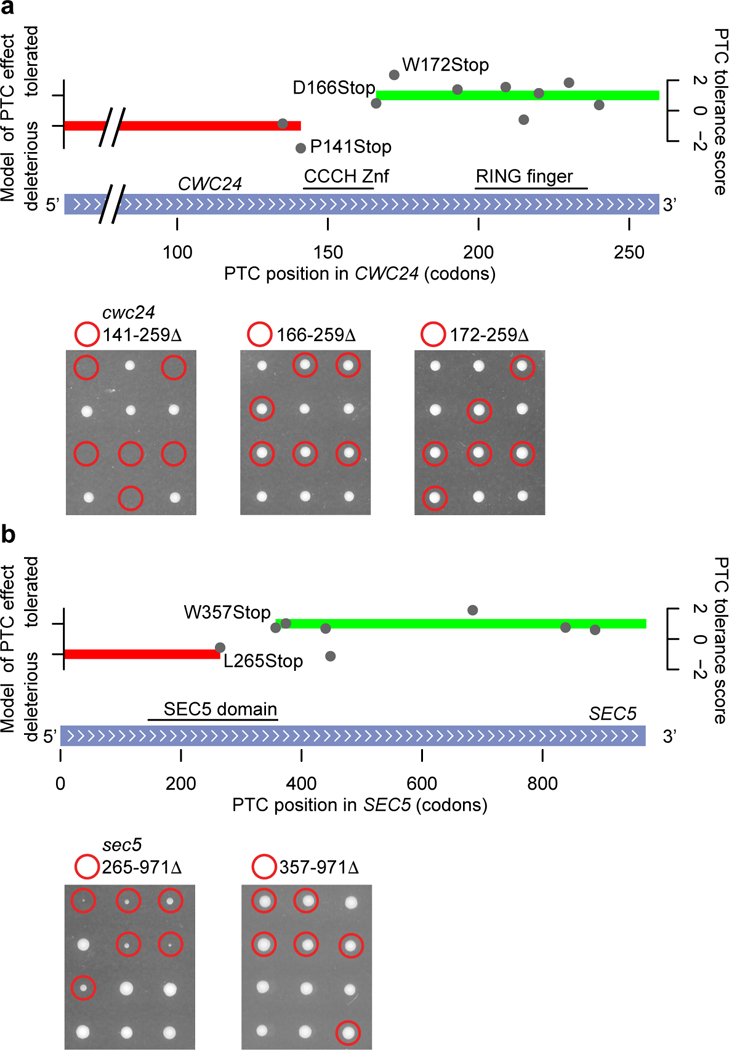
Selected truncatable essential genes. a, Tolerance scores for 10 PTCs in CWC24 are shown by gray circles; red and green bars indicate HMM calls of ‘deleterious’ and ‘tolerated’, respectively (top). The RING finger and CCCH Znf domains of Cwc24 are highlighted. Analysis of deleterious and tolerated truncations of CWC24 in yeast tetrads, displayed as in figure 2 (bottom). Deletions of the last 88 and 94 codons of CWC24 are tolerated (middle and right panels), while deletion of the last 119 codons is not (left panel). n = 10 tetrads were examined for each tested deletion. b, Tolerance scores for eight PTCs in SEC5 are shown by gray circles; red and green bars indicate HMM calls of ‘deleterious’ and ‘tolerated’, respectively (top). The Pfam-annotated “SEC5 domain” is highlighted. Analysis of deleterious and tolerated truncations of SEC5 in yeast tetrads (bottom). Deletion of the last 615 codons of SEC5 is tolerated (right panel), while deletion of the last 707 codons is not (left panel). n = 8 tetrads were examined for the deletion of 615 codons, and n = 12 tetrads were examined for the deletion of 707 codons.
Our results improve the annotation of essential genes in the well-studied yeast genome. We discovered several cases of genes that appeared to be essential as a consequence of the specific strain and growth conditions originally used to test viability of gene deletions. These results were consistent with those recently reported based on transposon mutagenesis29 (Supplementary Note 2; Supplementary Figure 10). A deletion screen in a different yeast isolate also revealed examples of conditionally essential genes30. Applying our approach and related methods in a diverse set of isolates and growth conditions will further refine the core set of essential yeast genes.
PTCs are prioritized in studies of human genetic variants because of the high likelihood that they abolish gene function. Our results suggest that PTCs are most likely to be deleterious when they disrupt annotated protein domains or truncate more than 27 amino acids, and these criteria may improve filtering of candidate causal variants. We observed that NMD did not make a strong contribution to PTC tolerance. This result is consistent with recent findings that NMD in yeast acts most strongly on transcripts with PTCs toward their 5’ ends31. PTCs near the ends of human genes are also likely to escape NMD according to the 50-base-pair rule32 (Supplementary figure 11), and our criteria may be especially useful for predicting their effects.
In our study we carried out a pooled screen of the functional effects of approximately 10,000 directed mutations in eukaryotic cells. A similar method was recently reported in bacteria33. Our method has higher editing efficiency, which enables screens that do not rely on positive selection of mutants, as demonstrated here in the PTC depletion experiments. Further, our strategy of using barcodes provides higher power to detect the effects of introduced edits by generating multiple independent observations for each targeted edit. We also demonstrate that our method can efficiently generates homozygous targeted mutations in diploids. These features of the method enable many applications in yeast, including targeted genome-wide mutagenesis screens, deep mutational scanning of specific genes, and assessment of phenotypic effects of natural variants. Notably, the method can be used in strains other than the S288c reference strain, where tools such as deletion libraries are largely unavailable.
Multiplex CRISPR-based editing has been reported at single loci in human cells34. This method differs from ours in that it uses a single gRNA in combination with a library of repair templates to generate many distinct edits in a small genomic region. The method also requires selective enrichment of edited DNA as a consequence of the low usage of HDR in DSB repair in human cells. This is a general limitation that applies to all uses of editing in mammalian cells, including potential extensions of our method. Another challenge to extending our method from yeast to mammalian cells is the need for longer homology regions in the repair templates. Improving DNA synthesis and delivery and enhancing the efficiency of HDR in mammalian cells are active areas of research35–37, and we anticipate that advances on these fronts will facilitate the development of a mammalian version of our system.
The approach we describe can be extended to assess the functional effects of any desired nucleotide variants in a highly parallel manner. The ability to profile the impact of broad classes of alleles, including missense and regulatory variants, will enable a more fine-grained understanding of the relationship between genotypes and phenotypes.
Methods
Strain and plasmids used
Strains used in this study are listed in Supplementary Table 5; plasmids used in this study are listed in Supplementary Table 6; and oligonucleotides used in cloning are listed in Supplementary Table 7.
Assessing the efficiency of edit-directing plasmids
We designed nine edit-directing plasmids to assess the efficiency of the method. The targeted edits were chosen such that we expected minimal fitness effects (Table 1); each targeted a PTC to a gene that was not expected to be important for vegetative yeast growth. The sites were distributed across six chromosomes. We included two gRNAs targeting CAN1 that have previously been characterized 2, including one (can1-G121Stop) that targeted a site reported to have lower-efficiency Cas9 targeting 38. To generate edit-directing plasmids, DNA fragments carrying the desired gRNA as well as the appropriate repair template were synthesized as gBlocks (Integrated DNA Technologies, Coralville, IA), which were then cloned into pLK78 (p426 SNR52p-gRNA.CAN1.Y-Sup4t, a plasmid for expressing gRNAs under an SNR52 promoter, kindly provided by George Church2) by Gibson assembly 39.
YLK3221 (nej1Δ with pLK77) was transformed with edit-directing plasmids by standard lithium acetate transformation 40. YLK3229 (nej1Δ nmd2Δ with pLK77; see Supplementary Table 8), YLK2525 (NEJ1 NMD2) with pLK77 (see Supplementary Table 1), and YLK3257 with pLK77 (see Supplementary Table 2) were also transformed with select edit-directing plasmids to test the effects of NMD2, NEJ1, and diploidy, on editing efficiency, respectively. pLK77 encodes Streptococcus pyogenes Cas9 under the Gal1L galactose-inducible promoter, and was kindly provided by George Church2. Edit-directing plasmids were transformed into yeast on glucose plates to repress Cas9 expression. Single colonies were picked and grown in glucose overnight, after which approximately 1,000 cells of each culture were plated on galactose plates to induce Cas9 expression. Colonies were allowed to grow for approximately 72 hours, after which cells were washed off the plates. Genomic DNA was extracted from harvested cells with a DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany), with a yeast-optimized protocol using Zymolyase (AMSBIO, Abingdon, United Kingdom). For each targeted site, genomic DNA was amplified with Pfu Ultra II (Agilent Technologies, Santa Clara, CA). To specifically amplify genomic DNA rather than the edit-directing plasmid repair template DNA, primers were chosen to anneal outside the regions of homology in the repair templates.
The primer pairs were designed to create PCR products that matched the product of the Illumina Nextera transposon reaction (Illumina, San Diego, CA). Indexed sequencing libraries were generated from these PCR products by proceeding with the amplification and indexing as described in the Nextera DNA Library Prep Protocol Guide. Libraries were pooled before sequencing on an Illumina MiSeq Sequencer with 300 bp paired-end reads. Our PCR products were designed to generate paired-end reads with extensive regions of overlap; these overlaps were stitched together with PEAR, version 0.9.6, (with parameters -v10 -m 400 –q 20) 41 to reduce errors introduced by mistaken sequencing calls. Reads were trimmed to remove adaptor sequences using trimmomatic, version 0.32 42 and matched to the expected set of targeted sequences in the SacCer3 reference assembly using bwa mem, version 0.7.12–5 43. Reads were assigned to expected regions with bwa mem and then realigned using a Smith-Waterman local alignment as implemented in the R package Biostrings, version 2.46. As parameters for the realignment we used +1 for match, −3 for mismatch, 5 for gap-opening and 2 for gap-extension. Custom R code was used to call variants and count the number of reads containing the set of expected edits. For nej1Δ, nej1Δ nmd2Δ, and NEJ1, we examined a 50-nucleotide (nt) window centered on the Cas9 protospacer-adjacent motif (PAM) site, and classified reads as (1) perfectly matching the unmutated sequence, (2) showing the desired mutation, (3) showing undesired mutations in the absence of the desired mutation, or (4) showing undesired insertion/deletion mutations. For the diploid sequencing, we classified the reads as (1) perfectly matching the unmutated targeted bases, (2) perfectly matching the targeted edits, (3) having at least one targeted base matching neither the unmutated bases or targeted edits, with a quality score (Q-score) greater than 25 (reads with low quality called mutations in the targeted sites were filtered), or (4) having an undesired insertion/deletion mutation in a 50-nt window centered on the PAM site.
Design of oligonucleotide library coupling targeting gRNAs to PTC-containing repair templates
We devised a strategy to edit any PAM site (sequence NGG) in a coding sequence such that the PAM site is disrupted and an in-frame TGA stop codon is introduced (Supplementary Table 9). The edits are designed to ensure that the targeted PAMs are changed to a sequence other than NGG or NAG, which can support a low level of continued Cas9 targeting44. We located all PAM sites in coding regions of annotated yeast ORFs. We designed all 124,087 potential repair templates that would disrupt the PAM site through the introduction of a TGA stop codon for the 1,034 annotated essential genes and 90 dubious ORFs. All 90 are adjacent to or overlapping a tested essential gene; 59 of these were originally annotated as essential, likely due to their deletion disrupting the adjacent or overlapping essential gene 7–9. For each gene, we randomly selected 10 PTCs from the available set. In the random sampling procedure, PTCs were weighted by 1/n, where n is the distance in codons from the 3’ end of the gene, to enrich for targeted sites near the 3’ ends of genes. Sampling was performed without replacement. As some dubious ORFs overlapped essential genes, in some cases the same gRNA was used to create a chosen PTC in a dubious ORF and in its overlapping essential gene. These were examined in Supplementary figure 3.
For each selected site, we designed a 181-nucleotide DNA oligonucleotide (Supplementary Table 7, oligonucleotide named OLS Library). Each oligonucleotide was designed as in Supplementary Figure 1; in brief, the sequences were flanked with 15 constant nucleotides on either end for PCR amplification, and contained a paired gRNA targeting sequence and 101-bp repair template sequence, along with appropriate cloning sites to generate edit-directing plasmids. These cloning sites were: a BstEII cloning site between the 5’ amplification sequence and the gRNA targeting sequence, and SphI and MluI cloning sites between the gRNA targeting sequence and the repair template sequence. An eight-nucleotide constant sequence (CGATCGAT) separated the SphI and MluI sites. We modified the SNR52 promoter and gRNA structural sequence to contain the BstEII and SphI sites, respectively (Supplementary Figure 12). We note that the plasmids used in the pilot experiments assessing the efficiency of edit-directing plasmids (see above; Table 1 and Supplementary Table 10) also had these modifications. Prior to oligonucleotide library synthesis, we filtered out oligonucleotides that contained additional EagI, MluI, SphI, or BstEII sites, as such oligonucleotides could create nonfunctional edit-directing plasmids. This filtering gave us 10,971 total sites to target.
Generation of barcoded edit-directing plasmid pools
The single-stranded oligonucleotides were synthesized by the Oligo Library Synthesis (OLS) platform (Agilent Technologies, Santa Clara, CA) in either the Watson or Crick orientation in order to minimize each oligonucleotide’s frequency of adenine bases. From the oligonucleotide pool generated by OLS, we generated edit-directing plasmid pools via a ligation-mediated cloning scheme described below (graphically summarized in Supplementary Figure 1). Oligonucleotides were amplified on an AriaMx real-time PCR system (Agilent Technologies, Santa Clara, CA), using the KAPA Library Amplification kit (Kapa Biosystems, Wilmington, MA). The amplification primers were designed to introduce an EagI cut site into the 3’ end of the amplification product (Supplementary Table 7, primers named OLS Library Amplification F and R). Reactions were stopped during linear amplification, and the amplified library was then purified with the QIAquick PCR purification kit (Qiagen, Hilden, Germany), digested with BstEII-HF and EagI-HF (New England Biolab, Ipswich, MA), and purified again.
The amplified library was cloned into pLK88, a version of pLK78 modified to include the BstEII and SphI sites. pLK88 was isolated with a QIAGEN Plasmid Plus Maxiprep kit (Qiagen, Hilden, Germany) from 200 milliliters of Escherichia coli culture. 20 micrograms of plasmid was then digested by BstEII-HF and EagI-HF, treated with Shrimp Alkaline Phosphatase (New England Biolabs, Ipswich, MA), and purified with the Qiagen PCR purification kit. We tested two ligation reactions: 1 microgram of digested vector was ligated with either 100 nanograms or 800 nanograms of the digested insert, with 4 microliters of T4 DNA Ligase M0202M (New England Biolabs, Ipswich, MA), in an 800 or 200 microliter reaction, respectively, at room temperature for 10 minutes. Concurrently, we ran negative control ligations lacking the insert DNA. Ligation reactions were stopped on ice. To test ligation efficiency, 0.5 microliter of each ligation was transformed into OneShot chemically competent E. coli (Thermo Fisher Scientific, Waltham, MA). Both ligations were successful, so we proceeded to pool the ligations and concentrated them with the DNA Clean and Concentrator 25 kit (Zymo Research, Irvine, CA), eluting in 25 microliters. Then, we transformed 10 microliters of concentrated ligation product into 10 reactions of Supreme DUO electro-competent E. coli cells (Lucigen, Middleton, WI) with a Bio-Rad Micropulser (Bio-Rad Laboratories, Inc., Hercules, CA) and 0.1 cm E. coli Pulser cuvettes (Bio-Rad Laboratories, Inc., Herculues, CA). After one hour of rescue growth, cells were transferred to 200 milliliters of LB medium with 100 μg/mL Ampicillin (Sigma-Aldrich Corporation, St. Louis, MO), and grown overnight. From serial dilutions plated after the transformation, we estimated that approximately 700,000 E. coli cells were transformed. Plasmids were maxiprepped from 150 milliliters of culture.
Next, we cloned in the remaining gRNA structural region and terminator between the gRNA targeting sequence and repair template of the first-step cloning product, while adding a 12-nt barcode adjacent to the repair template (Supplementary Figure 1). The cloning insert also included a Kan-resistance gene to facilitate enrichment for the correct cloning product. The insert sequence was Pfu Ultra II PCR-amplified from pLK89 (the amplification primers used are named Insert Amplification in Supplementary Table 7). The barcode was introduced during the insert PCR amplification by use of mixed bases in the synthesis of the reverse primer. Two separate possible barcode sequence classes were used, to generate two edit-directing plasmid pools distinguishable by their barcodes. One pool had barcodes of the form NNNNNNNNSWWS, while the other had barcodes of the form NNNNNNNNWSSW, where S can be either a cytosine or guanine base, W can be either an adenine or thymine base, while N can be any of the four bases. 10 micrograms of each PCR product was digested with MluI-HF and SphI-HF and gel extracted with the QIAquick Gel Extraction kit (Qiagen, Hilden, Germany).
20 micrograms of the first-step cloning product was digested with MluI-HF and SphI-HF (New England Biolabs, Ipswich, MA), treated with Shrimp Alkaline Phosphatase, and purified with the QIAquick PCR purification kit. 2 micrograms of the purified product was ligated with 0.7 micrograms of PCR-amplified insert (described above) to give the final barcoded edit-directing plasmid pool. Ligations were done in 800 microliter volumes with 8 microliters of T4 DNA ligase, at room temperature for 15 minutes. E. coli was then transformed with the ligation product. E. coli cells were grown in LB medium containing both 100 μg/mL Ampicillin and 50 μg/mL Kanamycin (Thermo Fisher Scientific, Waltham, MA). Plasmid DNA was extracted as above. We estimated that approximately 1.5E6 E. coli cells were transformed with each barcoded pool.
PTC induction in yeast pools
Strains YLK3221 (nej1Δ) and YLK3229 (nej1Δ nmd2Δ) were each separately transformed with both the WSSW and SWWS plasmid pools. For these large-scale transformations, cells were grown in yeast extract peptone dextrose medium (YPD) to an OD600 of approximately 0.5, then approximately 1E9 cells went into a transformation reaction with 10 micrograms of either plasmid pool. Each transformation was plated to four 15-cm YNB + glucose + CSM – Ura – Leu (Sunrise Science, San Diego, CA) plates. We estimated that 100,000 yeast cells were successfully transformed for each of the four combinations of strain and plasmid pool.
After growth of transformed colonies, cells were collected from all plates by washing with phosphate-buffered saline (PBS). We took approximately 3E8 cells of each pool, and washed twice more with PBS. Then, we combined nej1Δ cells carrying SWWS plasmids together with nej1Δ nmd2Δ cells carrying WSSW plasmids, and vice versa. In these combined pools, the SWWS/WSSW barcode difference marks the strain background genotype. This allowed us to test the effects of PTCs in nej1Δ and nej1Δ nmd2Δ cells in the same flask. Approximately 1E8 cells from each combined culture were frozen for a pre-Cas9-induction time-point, then approximately 2E8 cells of each combined culture were transferred to 300 milliliters of YNB + galactose + CSM – Ura – Leu to induce Cas9 (shaking, at 30C). Then, every 24 hours for the next 96 hours, we froze pellets of approximately 2E8 cells, while transferring 0.5 milliliters of culture (approximately 2.2E6 – 7E6 cells) to 300 milliliters of medium for continuing growth. During this time, the cultures never left the exponential growth phase, as OD600 was always less than 0.75.
PTC repair template and barcode sequencing
DNA, including edit-directing plasmid DNA, was extracted from the harvested frozen cell pellets with the DNeasy Blood and Tissue kit. We designed PCR primers to amplify both the barcode and repair template sequences from the plasmids for Illumina sequencing (the amplification primers used are named Repair Template Amplification for Illumina in Supplementary Table 7). The PCRs were performed using the KAPA Library Amplification kit, and then sequencing libraries were generated with a unique index for each culture at each time-point. Libraries were pooled and sequenced on four lanes of an Illumina HiSeq 2500 sequencing system with 150 bp paired-end reads. Reads were demultiplexed using custom R code that tolerates up to one-edit distance error in each of the indices. Invariant sequences at the ends of the library were trimmed using cutadapt, version 1.15 45 (with parameters –n 3 –m 40 –g leftseq –b rightseq). Observed reads were matched to the set of 10,971 synthesized repair template sequences using bwa mem.
Processing of plasmid barcode and repair template sequences
Each indexed sequencing library contained reads from both the nej1Δ and nej1Δ nmd2Δ strain backgrounds, as a feature of the construction of PTC-induction time-course strategy (see above). For any given library, the 12-bp internal barcode distinguishes whether the PTCs were generated in the nej1Δ or nej1Δ nmd2Δ strain background, which we used to further demultiplex the reads to their corresponding strains. Reads with barcodes that did not perfectly match the expectation for either of the barcode pools (SWWS or WSSW, as described above) were discarded.
We tracked each observed barcode for each PTC across the timepoints. For each PTC, all observed corresponding barcode sequences were collected across the multiple time points and across the nej1Δ and nej1Δ nmd2Δ strain backgrounds. Sequencing and PCR errors can falsely generate novel barcodes; we used the following approach to collapse similar barcodes together. A matrix of pairwise Levenshtein edit distances was calculated between all pairs of unique barcode sequences observed for each PTC. Barcodes were hierarchically clustered by edit distance and grouped together if they had an edit distance less than 3. For each unique barcode group per PTC, the most commonly observed repair template sequence was used for all downstream analyses. Repair template sequences were locally realigned as described in ‘Assessing the efficiency of edit-directing plasmids’ above, and differences relative to the expected sequence were cataloged for the filtering below. Hereafter, we use the term “barcode” to refer to a unique combination of barcode, repair template, and strain background.
Filtering of repair templates with errors
We removed any barcode with fewer than 20 read counts at the initial time-point from all downstream analyses. Errors introduced during oligonucleotide synthesis in the repair template sequence generate edit-directing plasmids that can direct additional unwanted edits. Barcoded PTCs were discarded from the analysis if their corresponding repair template sequence contained differences in the expected engineered TGA stop codon or in the 20-nt sequence immediately upstream of the engineered TGA. We included barcoded PTCs with up to two mismatched bases and two inserted or deleted bases in the region more than 20 nucleotides upstream of the engineered TGA, and up to four mismatched bases and four inserted or deleted bases downstream of the engineered TGA. Clipping of up to 4 nucleotides at either end of the repair template was also allowed. 9,047 and 9,041 total targeted sites were observed with at least one barcode before galactose induction in the nej1Δ and nej1Δ nmd2Δ backgrounds, respectively (9,990 sites in total). Observed sites were represented by an average of 4.7 and 4.6 barcodes in the nej1Δ and nej1Δ nmd2Δ backgrounds, respectively (Supplementary figure 13).
Calculating PTC tolerance scores and gene tolerance scores
To obtain slope (theta) and intercept (u) estimates for each tracked barcoded plasmid, we fit a generalized linear model using the glm function in R:
| (1) |
where we normalize for the differing read depths for the distinct time points by including total_counts, the vector of total observed read counts across all barcodes for each time-point. The observed distribution of slopes (thetas) was distinctly bimodal (Supplementary Figure 14). We interpreted this bimodality as representing persisting and depleted barcoded plasmids and classified barcoded plasmids as “persisting” or “depleted” according to whether they had a theta estimate above or below −0.025; this value was chosen by visual inspection to best separate the two modes.
To calculate the ‘PTC tolerance’ score and the ‘gene tolerance’ score, we fit logistic mixed effect models on these binarized barcoded plasmid persistences using the lme4 R package 46 and the glmer function. This analysis method was chosen because it handles the nested structure of the experiment, with varying numbers of barcodes per targeted PTC, and multiple PTCs per gene47. The dependent variable was the classification of each barcoded plasmid as persisting or depleted, as determined above. We included the following fixed effects to control for technical factors that could affect CRISPR function: [1] an indicator variable for potential off-target gRNA hits as determined by BLAST searching, [2] an indicator variable for having TTTT in the gRNA targeting sequence (potential terminator for PolIII 48), [3] a gRNA efficiency score as calculated with the CRISPR Efficiency Predictor tool from the Drosophila RNAi Screening Center, [4] the total number of barcodes observed for each PTC, [5] the u term as estimated in Eq(1), representing an estimate of the number of observations of each barcode at the initial timepoint, [6] the GC content of the gRNA sequence, and [7] the number of bases edited to generate the PTC. These terms were included to control for potential confounding technical effects on PTC tolerance.
As PTCs behaved similarly in the nej1Δ or nej1Δ nmd2Δ strain background (Supplementary figure 4b), for subsequent analyses and figures we used both datasets in the models and included a fixed effect [8] for whether the barcoded PTC was observed in the nej1Δ or nej1Δ nmd2Δ strain background (figures 2 and 3, Supplementary figures 3, 5, 6, and 7). Thus, we note that certain figures do not include a strain background effect: figure 1 and Supplementary figure 4b (bottom) show results from the nej1Δ strain background only, while Supplementary figure 4a and 4b (top) show results from the nej1Δ nmd2Δ strain background only. The models included one or two random effects: one for the specific PTC directed by each barcoded plasmid, and optionally one for the gene each barcoded plasmid targeted. For analyses comparing essential gene PTCs to non-PTCs or dubious ORF PTCs we used a model that did not include the effect of gene (figures 1c and 3, Supplementary figures 3, 4a, and 6); all other analyses are from models including both random effects. The analyses of the trend of PTC tolerance as a function of distance from the end of the gene did not include dubious ORF PTCs. The PTC tolerance scores and gene tolerance scores were calculated using the ranef function, and are the best linear unbiased predictors (BLUPs) for the PTC and gene, respectively. Each score is the log odds ratio of (persistence/depletion) conditional on the other terms in the model, either at the level of a particular tested PTC, or at the level of an individual gene. All PTC tolerance scores and gene tolerance scores are given in Supplementary Tables 11 and 12, respectively. We further note that tolerance scores in figure 1d, and Supplementary figures 4b and 5 were calculated using only PTCs targeting essential genes.
To determine the correlations for both PTC tolerance and gene PTC tolerance scores between replicate experiments, the model described above was fit separately on SWWS and WSSW barcoded plasmids from the nej1Δ background (Supplementary figure 3). This analysis indicated that the scores from the replicate flasks were well correlated, and we therefore combined the replicates into one dataset for all subsequent analyses in order to increase statistical power.
Gene ontology enrichment analysis was performed with TopGO, version 2.30.0, and is presented in Supplementary Table 449.
Extended model with additional features
To determine how features of the barcoded PTCs and their targeted genes affected PTC tolerance, we extended the mixed effects model analysis. We restricted the analysis to essential genes only. In addition to the eight fixed effects and two random effects listed above, we modeled the following fixed effect terms (Supplementary Table 3): [9] whether the PTC disrupted an annotated Pfam domain, by occurring either within or upstream of it 11, [10] the average conservation across the amino acid sequence of the targeted gene, where conservation was calculated using a five-species amino acid alignment 12 and the conserv() function in the bio3d 50 R package, version 2.3–3, [11] the average conservation as calculated above, but limited to the sequence from the targeted PTC to the C-terminal end of the protein, [12] gene evolvability categorizations as determined by Liu et al. 51, [13] the distance in amino acids of the PTC from the C-terminal end of the protein, [14] whether the PTC disrupted a low complexity region 52, [15] whether any allele of the gene has ever been annotated as viable on the Saccharomyces Genome Database, and [16] the overall length of the gene targeted.
Coefficients were obtained as above from the glmer function (Supplementary Table 3). Type III analysis-of-variance tables were computed for the fixed effect terms in the model with the Anova() function in the car R package, version 2.1–6 53. Likelihood ratio chi-square values and p-values for the fixed-effect terms in the model were also computed using this function. Tjur’s D was used to calculate a pseudo R2 statistic for overall model fit (Tjur’s D = 0.39)54.
Segmented regression
We fit two-segment segmented regressions for PTC tolerance scores given the distance of a PTC, in codons, from the 3’ end of a targeted gene and obtained 95% confidence intervals for the breakpoint, using functions provided in the R package segmented, version 0.5–3.0 55.
Hidden Markov model of dispensable 3’ ends of genes
We built a hidden Markov model (HMM) for each gene to more precisely delineate dispensable 3’ ends of essential genes. We ordered PTCs from the 3’ to 5’ end of the gene. For each introduced PTC in a gene, the binary hidden states represented that PTC being deleterious or tolerated. The observations were the binarized barcoded plasmid persistences of each PTC, as described above in the section ‘Calculating PTC tolerance scores and gene tolerance scores.’ We assumed that all PTCs 5’ of a deleterious PTC would also be deleterious. This was represented in the model by setting the transition probabilities (moving from the 3’ to 5’ end of the gene) from “deleterious” to “deleterious” at 1 and from “deleterious” to “tolerated” at 0. The transition probabilities from “tolerated” to “deleterious” and from “tolerated” to “tolerated” were set at 0.5. We also set the prior probabilities of the 3’-most PTC being deleterious or tolerated at 0.5. The depletion or persistence of an individual barcoded plasmid may not always faithfully represent whether the underlying targeted PTC is deleterious or tolerated. For instance, some gRNAs may target with lower efficiency, which could make a deleterious PTC appear tolerated. Conversely, some tolerated PTCs may appear deleterious if the corresponding edit-directing plasmids drop out of the pool due to off-target cutting or stochastic fluctuations in frequency. To take such errors into account, we set the emission probabilities of a tolerated PTC generating an observation of a barcoded plasmid as “persisting” at 0.5676 and as “depleted” at 0.4324. These parameters were estimated as the fractions of barcoded plasmids targeting dubious ORFs that were classified as “persisting” and “depleted.” Similarly, we set the emission probabilities of a deleterious PTC generating an observation of a barcoded plasmid as “depleted” at 0.8056 and as “persisting” at 0.1944. These parameters were estimated as the fraction of barcoded plasmids at the 5’-most position of essential genes that were classified as “depleted” and “persisting.” We used the Viterbi algorithm to identify the most likely hidden state for each PTC.
Testing individual genes
Essential genes predicted to be particularly PTC-tolerant or PTC-intolerant were verified by direct modification of yeast genes, either through ORF deletion or ORF truncation via partial deletion. The partial deletions differed from standard yeast gene deletions in that the deleted 3’ end of the gene was replaced by a cassette encoding a stop codon, a synthetic short terminator sequence (TATATAACTGTCTAGAAATAAATTTTTTCAAA) 56, and the selectable KanMX marker. All modifications were made as heterozygous alleles in diploid yeast strains, which were then sporulated in liquid medium. In brief, cells were grown to log phase in rich medium (YPD or YP-galactose), to which YP with 2% potassium acetate was added for overnight growth, and then were transferred to H2O with 2% potassium acetate and minimal supplementation of nutrients if required by strain auxotrophies. Once asci were observed under a light microscope (typically 3–7 days), asci were digested with Zymolyase and tetrads were dissected with a Singer MSM 400 System dissection microscope (Singer Instruments, Somerset, United Kingdom). Unless otherwise mentioned, tetrad dissections were done on YPD plates and photographed after two days of growth. Dissections done on “defined medium” plates used YNB + CSM. Genotypes of resulting colonies were determined by replica plating.
URLs
The CRISPR efficiency Predictor tool was accessed from http://www.flyrnai.org/evaluateCrispr/. The Saccharomyces Genome Database was accessed at http://yeastgenome.org. Code and data has been deposited at https://github.com/joshsbloom/coubledCRISPR_essentialStops. Illumina read sequences have been deposited at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA421550. The plasmids used for cloning the edit-directing plasmid pool can be obtained from https://www.addgene.org/Leonid_Kruglyak.
Data availability
Code and data can be found on github (see URLs). Illumina read sequences are available from the Sequence Read Archive under project accession PRJNA421550. Plasmids are available from Addgene (see URLs).
Supplementary Material
Acknowledgements
We thank Kruglyak laboratory members, F. Albert, M.P. Hughes, and J. Rine for helpful discussion, R. Cheung and E. Pham for technical assistance, and G. Church for plasmids. Funding was provided by the Howard Hughes Medical Institute and NIH grants R01 GM102308 (L.K.) and F32 GM116318 (M.J.S.).
Footnotes
Competing Financial Interests Statement
The authors declare no competing financial interests.
Author contributions
Experiments were designed by M.J.S, J.S.B., J.J.S., S.K., and L.K. Experiments were performed by M.J.S. and L.D. Data were analyzed by M.J.S., J.S.B., and L.K. The manuscript was written by M.J.S., J.S.B., and L.K., and incorporates comments by all other authors.
References
- 1.Shalem O, Sanjana NE & Zhang F High-throughput functional genomics using CRISPR–Cas9. Nat. Rev. Genet. 16, 299–311 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dicarlo JE et al. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 41, 4336–4343 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Landrum MJ et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–D868 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stenson PD et al. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 133, 1–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teo S-H & Jackson SP Identification of Saccharomyces cerevisiae DNA ligase IV: involvement in DNA double-strand break repair. EMBO J. 16, 4788–4795 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valencia M et al. NEJ1 controls non-homologous end joining in Saccharomyces cerevisiae. Nature 414, 666–669 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Giaever G et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Kastenmayer JP et al. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Res. 365–373 (2006). doi: 10.1101/gr.4355406.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fisk DG et al. Saccharomyces cerevisiae S288C genome annotation: a working hypothesis. Yeast 23, 857–865 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He F & Jacobson A Identification of a novel component of the nonsense-mediated mRNA decay pathway by use of an interacting protein screen. Genes Dev. 9, 437–54 (1995). [DOI] [PubMed] [Google Scholar]
- 11.Finn RD et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scannell D et al. The Awesome Power of Yeast Evolutionary Genetics: New Genome Sequences and Strain Resources for the Saccharomyces sensu stricto Genus. G3 Genes, Genomes, Genet. 1, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Consortium TGO Gene Ontology Consortium: going forward. Nucleic Acids Res. 43, D1049–D1056 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forsberg H & Ljungdahl PO Genetic and Biochemical Analysis of the Yeast Plasma Membrane Ssy1p-Ptr3p-Ssy5p Sensor of Extracellular Amino Acids. Mol. Cell. Biol. 21, 814–826 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klasson H, Fink GR & Ljungdahl PO Ssy1p and Ptr3p Are Plasma Membrane Components of a Yeast System That Senses Extracellular Amino Acids. Mol Cell Biol 19, 5405–5416 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forsberg H, Hammar M, Andréasson C, Molinér A & Ljungdahl PO Suppressors of ssy1 and ptr3 null mutations define novel amino acid sensor-independent genes in Saccharomyces cerevisiae. Genetics 158, 973–988 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ljungdahl PO, Gimeno CJ, Styles CA & Fink GR SHR3: a novel component of the secretory pathway specifically required for localization of amino acid permeases in yeast. Cell 71, 463–78 (1992). [DOI] [PubMed] [Google Scholar]
- 18.Kern L, de Montigny J, Jund R & Lacroute F The FUR1 gene of Saccharomyces cerevisiae: cloning, structure and expression of wild-type and mutant alleles. Gene 88, 149–157 (1990). [DOI] [PubMed] [Google Scholar]
- 19.Koren A, Ben-Aroya S, Steinlauf R & Kupiec M Pitfalls of the synthetic lethality screen in Saccharomyces cerevisiae: an improved design. Curr. Genet. 43, 62–69 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Gebert N et al. Dual function of Sdh3 in the respiratory chain and TIM22 protein translocase of the mitochondrial inner membrane. Mol. Cell 44, 811–818 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Wu N-Y, Chung C-S & Cheng S-C The Role of Cwc24 in the First Catalytic Step of Splicing and Fidelity of 5’ Splice Site Selection. Mol. Cell. Biol. (2016). doi: 10.1128/MCB.00580-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang T et al. Identification and characterization of essential genes in the human genome. Science 350, 1–10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corbett MA et al. A novel X-linked trichothiodystrophy associated with a nonsense mutation in RNF113A. J. Med. Genet. 52, 269–274 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Matangkasombut O, Buratowski RM, Swilling NW & Buratowski S Bromodomain factor 1 corresponds to a missing piece of yeast TFIID. Genes Dev. 14, 951–62 (2000). [PMC free article] [PubMed] [Google Scholar]
- 25.Volanakis A et al. Spliceosome-mediated decay (SMD) regulates expression of nonintronic genes in budding yeast. Genes Dev. 27, 2025–2038 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spelbrink RG & Nothwehr SF The yeast GRD20 gene is required for protein sorting in the trans-Golgi network/endosomal system and for polarization of the actin cytoskeleton. Mol. Biol. Cell 10, 4263–81 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Decker CJ, Teixeira D & Parker R Edc3p and a glutamine/asparagine-rich domain of Lsm4p function in processing body assembly in Saccharomyces cerevisiae. J. Cell Biol. 179, 437–449 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.TerBush DR, Maurice T, Roth D & Novick P The Exocyst is a multiprotein complex required for exocytosis in Saccharomyces cerevisiae. EMBO J. 15, 6483–94 (1996). [PMC free article] [PubMed] [Google Scholar]
- 29.Michel AH et al. Functional mapping of yeast genomes by saturated transposition. Elife 6, e23570 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dowell RD et al. Genotype to phenotype: a complex problem. Science 80309 (2010). doi: 10.1126/science.1189015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Decourty L et al. Long Open Reading Frame Transcripts Escape Nonsense-Mediated mRNA Decay in Yeast. Cell Rep. 6, 593–598 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Nagy E & Maquat LE A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem. Sci. 23, 198–9 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Garst AD et al. Genome-wide mapping of mutations at single-nucleotide resolution for protein, metabolic and genome engineering. Nat. Biotechnol. 1–12 (2016). doi: 10.1038/nbt.3718 [DOI] [PubMed] [Google Scholar]
- 34.Findlay GM, Boyle EA, Hause RJ, Klein JC & Shendure J Saturation editing of genomic regions by multiplex homology-directed repair. Nature 513, 120–123 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chu VT et al. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 33, (2015). [DOI] [PubMed] [Google Scholar]
- 36.Lin S, Staahl BT, Alla RK & Doudna JA Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 3, e04766 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song J et al. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat. Commun. 7, 1–7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References
- 38.Bao Z et al. Homology-Integrated CRISPR-Cas (HI-CRISPR) System for One-Step Multigene Disruption in Saccharomyces cerevisiae. ACS Synth. Biol. (2014). doi: 10.1021/sb500255k [DOI] [PubMed] [Google Scholar]
- 39.Gibson DG et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–5 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Becker DM & Lundblad V in Current protocols in molecular biology (ed. Ausubel FM) Unit13.7 (2001). doi: 10.1002/0471142727.mb1307s27 [DOI] [PubMed] [Google Scholar]
- 41.Zhang J, Kobert K, Flouri T & Stamatakis A PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bolger AM, Lohse M & Usadel B Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang W, Bikard D, Cox D, Zhang F & Marraffini LA RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 31, 233–239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10 (2011). [Google Scholar]
- 46.Bates D, Mächler M, Bolker B & Walker S Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 67, 1–48 (2015). [Google Scholar]
- 47.Searle SR, Casella G & McCulloch CE Variance components. (Wiley, 2006). [Google Scholar]
- 48.Nielsen S, Yuzenkova Y & Zenkin N Mechanism of Eukaryotic RNA Polymerase III Transcription Termination. Science 340, 1577–1580 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alexa A & Rahnenfuhrer J topGO: Enrichment Analysis for Gene Ontology. (2016). [Google Scholar]
- 50.Grant BJ, Rodrigues APC, ElSawy KM, McCammon JA & Caves LSD Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics 22, 2695–2696 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Liu G et al. Gene Essentiality Is a Quantitative Property Linked to Cellular Evolvability. Cell 163, 1–12 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Wootton JC & Federhen S Analysis of compositionally biased regions in sequence databases. Methods Enzymol. 266, 554–71 (1996). [DOI] [PubMed] [Google Scholar]
- 53.Fox J & Weisberg S An {R} Companion to Applied Regression. (Sage, 2011). at <http://socserv.socsci.mcmaster.ca/jfox/Books/Companion> [Google Scholar]
- 54.Tjur T Coefficients of Determination in Logistic Regression Models—A New Proposal: The Coefficient of Discrimination. Am. Stat. 63, 366–372 (2009). [Google Scholar]
- 55.Muggeo VM R. segmented: an R Package to Fit Regression Models with Broken-Line Relationships. R News 8, 20–25 (2008). [Google Scholar]
- 56.Curran KA et al. Short Synthetic Terminators for Improved Heterologous Gene Expression in Yeast. ACS Synth. Biol. 4, 824–832 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Albert FW, Muzzey D, Weissman JS & Kruglyak L Genetic Influences on Translation in Yeast. PLoS Genet. 10, e1004692 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rhee HS & Pugh BF Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature 483, 295–301 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Code and data can be found on github (see URLs). Illumina read sequences are available from the Sequence Read Archive under project accession PRJNA421550. Plasmids are available from Addgene (see URLs).