

Abstract
Coordination of redox-active ligands to metals is a compelling strategy for making reduced complexes more accessible. In this work, we explore the use of redox-active formazanate ligands in low-coordinate iron chemistry. Reduction of an iron(II) precursor occurs at milder potentials than analogous non-redox-active β-diketiminate complexes, and the reduced three-coordinate formazanate-iron compound is characterized in detail. Structural, spectroscopic and computational analysis show that the formazanate ligand undergoes reversible ligand-centered reduction to form a formazanate radical dianion in the reduced species. The less negative reduction potential of the reduced low-coordinate iron formazanate complex leads to distinctive reactivity with formation of a new N-I bond that is not seen with the β-diketiminate analogue. Thus, the storage of an electron on the supporting ligand changes the redox potential and enhances certain reactivity.
Keywords: Redox-active ligand, low-coordinate, iron, ligand-centered-radical, formazanate
FULL PAPER

Low-coordinate iron formazanate complexes undergo reversible ligand-centered reduction, which is possible at milder potentials than related iron complexes with non-redox-active β-diketiminate ligands. The ligand-centered radical avoids iron(I), yet retains the capability to abstract halide atoms
Introduction
β-Diketiminate ligands have found extensive use as auxiliary ligands to stabilize unusual metal complexes throughout the periodic table.1 In particular, highly reduced low-coordinate base metal complexes featuring β-diketiminate ligands are able to activate a number of challenging chemical bonds.2 Reductive processes in β-diketiminate complexes are normally metal-centered due to the very negative potentials required for ligand-centered reduction.3 However, the negative potential required for metal-centered reduction (placing electrons in the LUMO at the left of Figure 1) requires the use of harsh reductants such as Na or KC8, which are often incompatible with exogenous substrates for catalytic transformations. Moreover, catalytic turnover is often not feasible because the reduced states are too high in energy.
Figure 1.
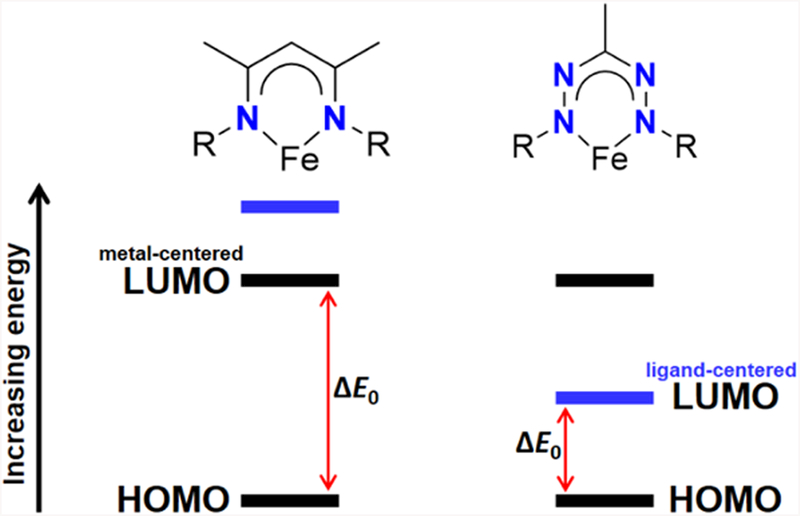
Conceptual representation of how the redox-active formazanate ligand can provide access to a more accessible reduced state.
The ability of redox-active ligands to reversibly accept and donate electrons offers an appealing way to generate more accessible reduced states: as shown on the right of Figure 1, the ligand orbital can be lower in energy, moderating the redox potential. Accessible ligand-centered redox processes can expand upon the intrinsic reactivity of metal centers.4 Redox-active ligands have enabled multi-electron processes in systems where the transition metal changes oxidation states by less than two electrons, thereby facilitating elementary reactions such as oxidative addition and reductive elimination.5 Alternatively, redox-active ligands can enable single-electron transformations without changing metal oxidation state.6 A recent computational investigation by Hu and Chen7 demonstrated the utility of multiconfigurational calculations for characterizing the redox-active bisiminopyridine ligands, which enable facile reductive elimination in iron-catalyzed [2+2] cycloadditions developed by Chirik and coworkers.8 Given the wide use of β-diketiminates, there is motivation for exploring a redox-active ligand that is structurally similar, but can readily accept electrons, enabling one-electron reduction at less negative potentials.
Recent studies have shown that β-diketiminates can undergo ligand-centered oxidation in homoleptic complexes of zinc,9 cobalt,10 and nickel,11,12 but ligand-centered reductions on transition metals have not been reported. Here, we use a formazanate as a redox-active ligand that is structurally similar to β-diketiminates, but which can accept electrons. By placing an unoccupied ligand orbital low in energy, they may make the complex easier to reduce (Figure 1, right). Otten has shown that formazanates undergo facile ligand-centered reduction when bound to boron and to zinc.13,14 Although spectroscopically observed,15 no formazanate complex of any redox-active transition metal that shows ligand-centered reduction has yet been isolated. Moreover, a recently reported homoleptic formazanate iron(II) complex adopted a low-spin iron(I) configuration upon one-electron reduction rather than generating a ligand-centered radical,16 highlighting the need for further study.
Herein, we describe the synthesis, characterization and reactivity of some new low-coordinate iron formazanate complexes and their one-electron reduction products. Spectroscopic and computational data are used to characterize the frontier orbitals. These studies reveal that the iron-formazanate complexes undergo ligand-centered reduction at relatively mild potentials, and demonstrate an isolable example of a redox-active metal with a formazanate radical dianion. In order to illustrate the new opportunities coming from redox activity at the formazanate ligand, we demonstrate how the formazanate complexes activate chemical bonds like low-coordinate β-diketiminate iron complexes, but in contrast are solely mediated by ligand-centered redox changes. In addition, we show that the less electron donating formazanate ligand affects the stability of higher metal oxidation states through an unusual N-I elimination reaction.
Results and Discussion
Synthesis and characterization.
The dropwise addition of a suspension of the known17 formazan 1 to a stirred solution of Fe[N(SiMe3)2]2 in pentane gives the three-coordinate formazanate complex 2 in near quantitative yield (Scheme 1). The 1H NMR spectrum of complex 2 in C6D6 at 298 K shows the expected number of paramagnetically shifted resonances between +82 and −40 ppm, which are tentatively assigned based on their relative integration and distance from the metal center (Figure S1). The presence of a single equivalent of THF in the synthesis of complex 2 results in the exclusive formation of the four-coordinate complex 3 (Scheme 1). Hence, care must be taken to avoid the presence of THF in the solvent and starting materials.18 We observed that the resonance for the SiMe3 protons shifts >40 ppm upfield upon the addition of one equiv of THF to complex 2. Due to fast exchange on the NMR time scale and an equilibrium constant > 5000 M−1, the chemical shift can be used as an indication of the THF content of the sample (Figure S27).
Scheme 1.
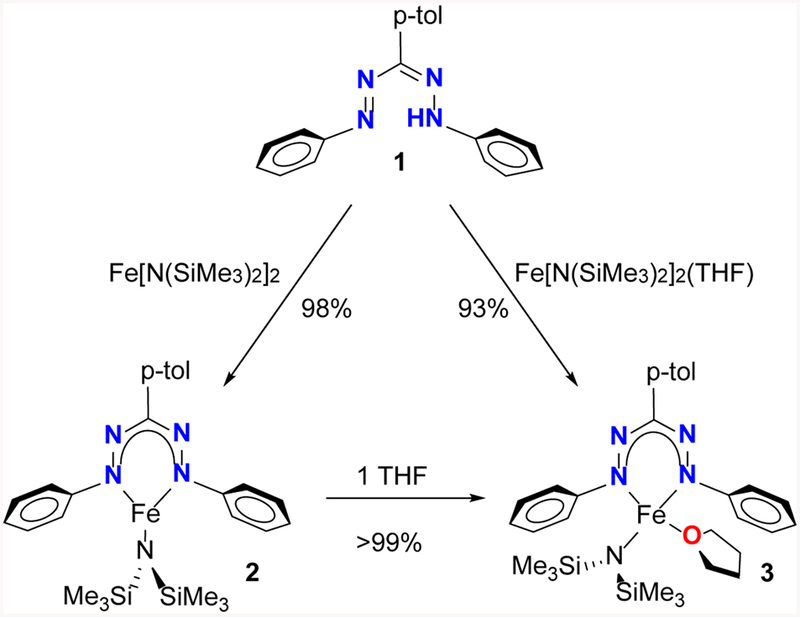
Synthesis of complexes 2 and 3.
Cooling a solution of 2 or 3 in pentane yielded crystals suitable for single crystal X-ray diffraction. In 2, two crystallographically independent molecules were found in the asymmetric unit, which share a π–π interaction between the tolyl groups in the formazanate ligands (Figure S35). Each molecule displays a planar, three-coordinate geometry at iron where the sum of the three angles is 359.8 ± 0.5 ° (Figure 2, left). A notable feature in the solid state structure of 2 is the small dihedral angle between the formazanate and N-phenyl groups (1.6 – 28.3°), which is very different from analogous three-coordinate Fe complexes containing a N(SiMe3) and bulky β-diketiminate ligands (65 – 90°).19 We attribute this difference to the lack of sterically demanding ortho-substituents on the N-phenyl groups in complex 2. The Fe–N bond lengths to the formazanates (1.95 – 1.97 Å) are shorter than those in analogous three-coordinate Fe complexes containing a N(SiMe3) and bulky β-diketiminate ligands (1.99 – 2.03 Å).20 The N–N bond lengths (1.319(5) – 1.327(5) Å) are slightly longer than commonly observed for formazanate complexes (1.30−1.31 Å)14e,15, which was also observed for the reported homoleptic iron formazanate complex.17 A notable difference is the significantly longer Fe–N bonds (1.955(4) – 1.966(4) Å) in 2 when compared with the homoleptic bis(formazanate)iron(II) species (1.817(2) − 1.833(2) Å).17 The solid state structure of complex 3 (Figure 2, right) shows that the iron center adopts a distorted trigonal pyramidal geometry (τ4 = 0.70 and 0.72)20 for both molecules in the asymmetric unit. As expected, coordination of an additional ligand causes all Fe–N bond lengths in 3 to be elongated (1.979(2) – 2.000(2) Å) compared to 2. Interestingly, the bulky amide forces the bound THF into a geometry wherein the C atoms lie over the formazanate ligand.
Figure 2.
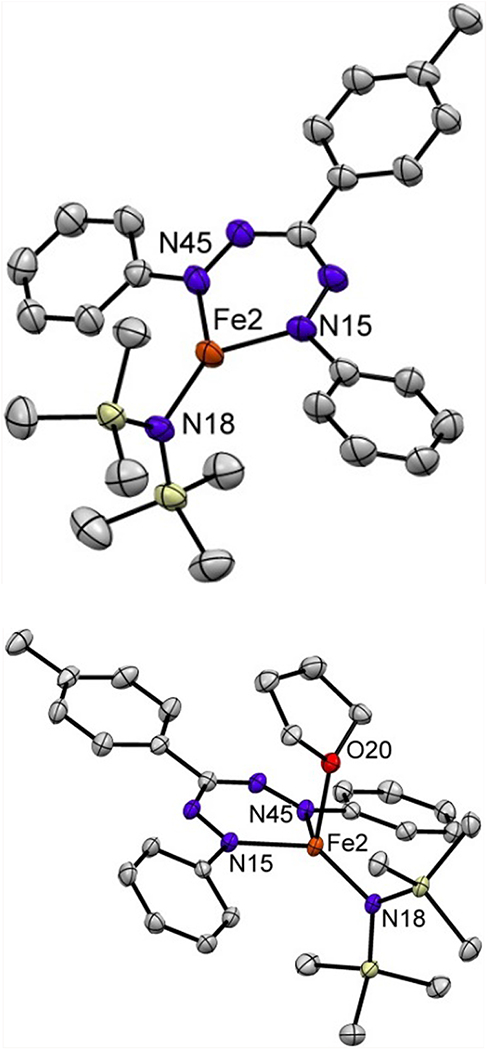
Displacement ellipsoid plots (50% probability) of complex 2 and 3. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°) for 2: Fe2–N18 1.903(3); Fe2–N15 1.964(4); Fe2–N45 1.966(4); N18–Fe2–N15 132.5(2); N18–Fe2–N45 135.2(2); N15–Fe2–N45 91.7(1). For 3: Fe2–N18 1.937(2); Fe2–N15 1.979(2); Fe2–N45 1.984(2); Fe2–O20 2.145(1); N18–Fe2–N15 130.40(7); N18–Fe2–N45 127.99(6); N15–Fe2–N45 91.59(6).
Cyclic voltammetry of complex 3 in THF revealed two reversible one-electron reduction events at E½red1 = −1.65 V and E½red2 = −2.47 V vs Fc+/Fc (Figure S29). The first reduction occurs at a potential ~0.8 V more positive than the corresponding three coordinate β-diketiminate complexes (E½red = −2.4 to −2.6 V vs Fc+/Fc).21 The nature of this reduction will be discussed below. The addition of CoCp*2 to a solution of 2 or 3 in hexanes gave the one-electron reduced species 4, which precipitated as a green solid (Scheme 2). The 1H NMR spectrum of 4 in THF-d8 at 298 K shows the expected number of paramagnetically shifted resonances between +105 and −25 ppm consistent with a single C2v symmetric species (Figure S9). Intraligand bond lengths in redox-active ligands can often be used to determine the locus of a redox event.4–7,15,22 Unfortunately, we were unable to grow crystals of complex 4 that were suitable for X-ray structure determination. Although complex 4 is stable in the solid state for months, it decomposes in THF solution at room temperature within hours to form a mixture of species, concomitant with a color change from dark green to brown. X-ray diffraction of crystals obtained from this mixture revealed a complex wherein the formazanate has undergone N–N bond scission (Figure S37). However, we were unable to isolate sufficient amounts of this unusual product for full characterization, and thus we sought a more stable analogue. Since the C-H bonds in CoCp*2 and [CoCp*2]+ are susceptible to deprotonation or C-H activation,23 and because of potential stabilizing effects of alkali cations,24 we used an equivalent of elemental Na to reduce complex 3 in THF, which resulted in a green solution similar in color to 4. Addition of 2 equiv of 12-crown-4 and cooling the THF mixture gave 5 as a green crystalline solid in 67% yield (Scheme 2). The 1H NMR (Figure S15) and zero-field Mӧssbauer spectra (vide infra) of 5 are nearly identical to those for complex 4. However, unlike 4, complex 5 is stable in solution for days under inert atmosphere. Single crystals suitable for X-ray structure determination were obtained by cooling a THF solution of 5. The solid state structure reveals that the Na is sandwiched between two crown ethers, and the resulting cation is separated from the anionic formazanate complex (Figure 3). Although the anion in 5 is isostructural to neutral complex 2, there are some remarkable differences in the bond lengths and angles. The iron amide bond (Fe1–N14) is longer in complex 5 by ca. 0.05 Å, suggestive of a more electron-rich Fe center. The N–N bonds within the formazanate are elongated (1.377(6) and 1.345(6) Å) and the Fe–N (formazanate) bonds (1.951(5) and 1.958(5) Å) are shortened compared to 2 (Figure 2), which is consistent with either formazanate-centered reduction or significant π-backdonation into the formazanate ligand.
Scheme 2.
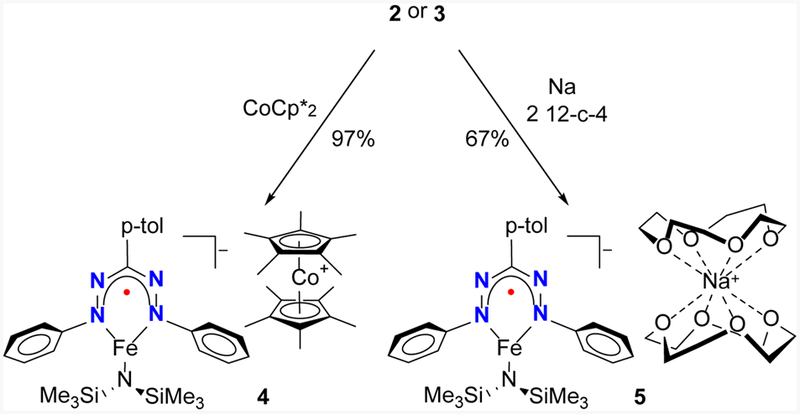
Synthesis of complexes 4 and 5.
Figure 3.

Displacement ellipsoid plot (50% probability) of complex 5. Hydrogen atoms and a THF molecule have been omitted. Selected bond lengths (Å) and angles (°): Fe1–N11 1.935(5); Fe1–N14 1.951(5); Fe1–N41 1.958(5); N11–N21 1.377(6); N31–N41 1.345(6); N11–Fe1–N14 134.9(2); N11–Fe1–N41 96.5(2); N14–Fe1–N41 128.6(2).
Magnetism and EPR spectroscopy.
The temperature dependence of the magnetic susceptibility of solid samples of complexes 2–5 was studied in the range 2–290 K using a SQUID magnetometer, and at room temperature in solution using the Evans method. The room temperature magnetic moments of complex 2 and 3 in C6D6 solution were 4.8 ± 0.1 μB and 4.7 ± 0.1 μB, respectively, in agreement with a high-spin iron(II) configuration for both complexes (S = 2, spin-only value μs.o. = 4.9 μB). In the solid state, three-coordinate 2 and four-coordinate 3 have nearly identical, temperature independent magnetic moments of 5.1 μB over the range 30 – 290 K (Figures S4 and S8), which is near the solution values and consistent with a high-spin iron(II) (S = 2) ion in each complex. Below 30 K, the μeff for 2 decreases under the influence of zero-field splitting. The sign and magnitude of the zero-field parameters were determined from simulations of μeff(T) in combination with isofield magnetization measurements at 1, 4, and 7 T (Figure S4 inset). Global fitting of the magnetic data yielded D = −3.8 cm–1 and rhombicity E/D = 0.19. The temperature profile is different for 3, where there is a slight rise in μeff below 30 K, presumably due to intermolecular interactions. The best fit was obtained for D = 3.1 cm–1 and E/D = 0.30, where the sign of D is opposite to that for three-coordinate 2, though the sign loses meaning so close to full rhombicity. The similarity in the magnitude of these spin-Hamiltonian parameters for 2 and 3 reveal only a very minor change to the electronic structure upon THF coordination.
Room temperature magnetic moments of 3.8 ± 0.1 μB and 3.9 ± 0.1 μB were determined from THF solutions of 4 and 5, respectively, suggesting S = 3/2 ground states for both compounds (μs.o. = 3.9 μB). Both 4 and 5 exhibited temperature independent effective magnetic moments of 4.0 μB over the range 50 – 290 K (Figures S14 and S18), consistent with these values. Much like 2, 4 shows a low-temperature downturn in μeff due to zero-field splitting but the more gentle decrease is modelled with a larger D = −16.8 cm–1 and large rhombicity E/D = 0.29. For 5, on the other hand, the downturn in μeff is more abrupt, suggesting intermolecular interactions like those observed in 3. Nevertheless an excellent fit was obtained from modelling the combined μeff(T) and isofield magnetization plots (including an intermolecular interaction approximation), which gave D = −10.1 cm–1 and E/D = 0.26. These solid state measurements were augmented with an EPR analysis of the odd-electron complexes 4 and 5. Frozen solution spectra recorded in THF at 10 K highlighted the aforementioned solution instability of 4. EPR spectra of 4 (Figure S12) show a rhombic S = 3/2 signal, and a strong rhombic S = 1/2 signal that originates from (formazanate)2FeI,17 which is one of the decomposition product of 4 (see SI for more details). The S = 3/2 signal has peaks at resonant field positions consistent with an S = 3/2 species having g = (2.17, 2.24, 1.87), D = −5.7 cm–1, and E/D = 0.19 (Figure S12). The greater solution stability displayed by 5 yielded an EPR spectrum (Figure 4) wholly consisting of an S = 3/2 signal with geff = (2.68, 2.03, 2.34), D = 16 cm–1 and E/D = 0.33.
Figure 4.

X-band EPR spectrum of 5 in THF at 10 K. Experimental data are represented by the black line; simulation is depicted by the red trace. Asterisk denotes trace impurity contributing <0.5% to the total signal. Experimental conditions: frequency 9.3701 GHz; power 0.2 mW; modulation 2.0 mT.
Minor deviations in g and E/D may be due to structural differences of solid and solution samples (D was taken from magnetic data because the second Kramers doublet could not be determined due to relaxation issues). The increased quality of the S = 3/2 signal in the sample of 5 is consistent with the greater solution stability of this compound. Our assignment of 5 is bolstered by the agreement of g values and zfs parameters between EPR and magnetometry.
Mӧssbauer spectroscopy.
The zero-field Mӧssbauer spectra of solid samples of 2 and 3 at 80 K (Figure 5) each showed a quadrupole doublet, with 2 at δ = 0.63 mm s–1 and |ΔEQ| = 2.48 mm s–1, and 3 at δ = 0.71 mm s–1 and |ΔEQ| = 1.46 mm s–1. These parameters are within the range previously observed for three-25 and four-coordinate26 high-spin iron(II) diketiminate complexes. The smaller quadrupole splitting and larger isomer shift of 3 in comparison to 2 are consistent with the increase in coordination number.27 The zero-field Mӧssbauer spectrum of a solid sample of 4 at 80 K shows an asymmetric doublet with δ = 0.48 mm s–1 and |ΔEQ| = 1.25 mm s–1 (Figure 5, bottom), which shows no significant changes upon warming to 173 K. The zero-field Mӧssbauer spectrum of a solid sample of 5 at 80 K is nearly identical to that of 4, with δ = 0.49 mm s–1 and |ΔEQ| = 1.30 mm s–1 (Figure S16). The asymmetric broadening of the doublets is common for Kramers systems as a result of intermediate rates of spin relaxation.28 Interestingly, the change in isomer shift between 2/3 and 4/5 is comparable, but opposite in direction from the reduction of the homoleptic iron formazanate complex reported by Otten and co-workers,17 which undergoes metal-centered reduction.
Figure 5.

Zero-field Mössbauer spectrum of a solid samples of complex 2 (top), 3 (middle), 4 (bottom) at 80 K. The black circles are experimental data with the fit represented by the black line. The thin grey trace is the residual of the fit.
Though it is tempting to use the shift in Mössbauer parameters to assign the metal oxidation state, previous results advise caution. In earlier work we described a di(μ-hydrido)diiron(II) complex29 with a high-spin electronic configuration (S = 2) supported by bulky β-diketiminate ligand that gave a mononuclear three-coordinate iron(I) hydride complex with an S = 3/2 electronic configuration.30 The change in observed isomer shift in the zero-field Mӧssbauer spectra of solid samples of the di(μ-hydrido)diiron(II) complex (δ = 0.59 mm s–1) and mononuclear three-coordinate iron(I) hydride (δ = 0.40 mm s–1) at 80 K is comparable to the decrease in isomer shift between 2/3 and 4/5. On the other hand, we have also observed similar changes in isomer shift from changes in ligand oxidation state. Namely, oxidation of a four-coordinate high-spin iron(II) complex (S = 2) with a dianionic redox-active tetrazene ligand (δ = 0.81 mm s–1) gives a complex where a tetrazene radical anion (S = 1/2) is strongly antiferromagnetically coupled to a high-spin iron(II) center (S = 2) to give an overall S = 3/2 electronic configuration (δ = 0.69 mm s–1).31 Consequently, the observed changes in isomer shift between 2/3 and 4/5 could be attributed to either ligand- or metal-centered reduction.
UV-Vis and X-ray absorption spectroscopy, and computations.
The iron(II) complexes 2 and 3 in hexane solution each show an intense absorption in the visible region at 524 nm (19 × 103 cm−1), which we assign as the formazanate π−π* transition by analogy to intense bands at similar energies uncoordinated formazans.32 The absorption in these compounds is red-shifted by ca. 30 nm (~1 × 103 cm−1) relative to the protonated formazanate ligand.14e In contrast, the electronic spectrum of 5 recorded at in THF at ambient temperature (Figure 6) exhibits prominent absorption bands at 414 and 696 nm (ε = 25 × 103 and 10 × 103 M−1 cm−1). Absorption bands with comparable intensity and position have been observed in reduced triaryl-formazanates bound to redox-inert atoms (carbon:33 λ = 398 and 721 nm, ε = 7.5 × 103 and 3.5 × 103 M−1 cm−1; boron:14a λ = 454 and 716 nm, ε = 15 × 103 and 7.5 × 103M−1 cm−1; zinc:14a λ = 462 and 755 nm, ε = 20 × 103 and 10 × 103 M−1 cm−1), and are diagnostic of a formazanate radical dianion. Together with the Mӧssbauer and EPR spectrum and magnetism of 5, the absorption spectrum supports an electronic configuration where a ligand-centered radical (SL = 1/2) is strongly antiferromagnetically coupled to a high-spin iron(II) center (SFe = 2) to give an overall Stotal = 3/2 ground state.
Figure 6.
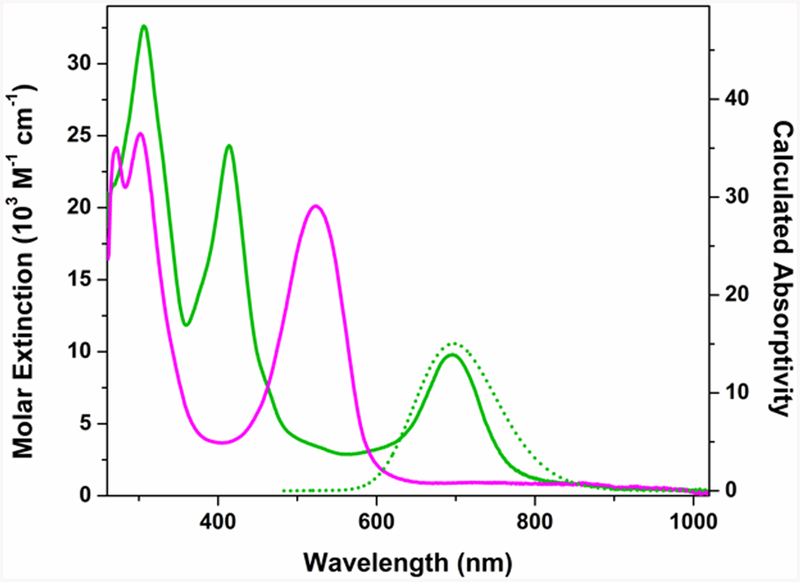
Experimental (solid) and calculated (dotted, SORCI) absorption spectra for complex 3 (pink) and 5 (green) in THF solution.
The Fe K-edge X-ray absorption spectra of 2 – 4 are overlaid in Figure 7. They are characterized by electric dipole forbidden but quadrupole-allowed 1s → 3d pre-edge transitions that appear at the base of the dipole-allowed 1s → np rising edge that dominates a K-edge spectrum. Since the ligand field typically increases with increasing oxidation state for a given ligand set, the pre-edge energy often provides a useful marker for oxidation state. The centers of mass for pre-edge peaks in spectra obtained for 2 – 4 are identical within experimental error at 7112.2 ± 0.1 eV. As a ~1 eV increase in energy is expected per unit of oxidation, the similar edge positions indicate that the physical Fe oxidation state is the same for the Fe ion in neutral 2 and 3, and the monoanion in 4. Moreover, it is noteworthy that the pre-edge peak is of the same intensity for each complex. Electric quadrupole allowed 1s → 3d pre-edge transitions gain intensity through 4p mixing into the 3d orbitals and therefore are sensitive to the symmetry of the complex.34 Here, the uniform peak height is consistent with the dominant trigonal symmetry in this series where the inclusion of a THF ligand in 3 has little effect on its electronic structure (vide supra). In neutral 2 and 3, there is a shoulder to higher energy at ~7113.2 eV. In contrast, the shoulder in 4 lies to lower energy at 7111.5 eV, and is tentatively assigned as transition to an orbital with significant ligand character.35
Figure 7.

Overlay of the normalized Fe K-edge XAS spectra of 2 (red), 3 (blue), and 4 (green). Inset shows an expansion of the pre-edge region with a comparison of the experimental spectra above the corresponding calculated spectra obtained from B3LYP TD-DFT calculations. Calculated intensity in arbitrary units.
Although the rising K-edge is frequently used as an additional metric of physical oxidation state in first-row metals, the rising-edge energies are also influenced by coordination geometry and ligand identity.36 In the present case, similar edge positions are displayed by three-coordinate 2 and 4 of 7117.0 and 7117.4 eV, respectively, while the edge is shifted by 0.7 eV to higher energy at 7118.1 eV for four-coordinate 3. Thus, these do not change the conclusion from the pre-edge features, that all the complexes have the same oxidation state at iron.
Density functional theory calculations were carried out to facilitate spectroscopic and electronic structure interpretations. In particular, time-dependent DFT (TDDFT) calculations are known to reproduce metal K-edge XAS with remarkable fidelity after accounting for inaccurate modeling of the 1s core potential with either a scalar or linear energy shift.37 Time-dependent (TD) DFT calculations (B3LYP hybrid density functional and CP(PPP) basis set on Fe with ZORA-def2-TZVP(-f) on all remaining atoms) were performed on the crystallographic coordinates of each complex. For 2 and 3, these reproduce the pre-edge region with good fidelity (Fig 7 inset), but the TD-DFT calculation for 4 produces a pre-edge with an energy maximum that is underestimated by 1 eV. Inspection of the frontier orbitals calculated for 4 using this level of theory revealed a straightforward unrestricted Kohn-Sham solution consistent with (S = 3/2) iron(I). Given the consistency of the spectroscopic data described above with an iron(II) configuration, we evaluated a broken-symmetry (BS) option where the four electrons from high-spin iron(II) are allowed to participate in a magnetic interaction with an S = 1/2 formazanate radical anion (BS 4,1). While this approach reproduces the Fe K pre-edge with remarkable fidelity, the BS calculation converges to a ferromagnetic S = 5/2 solution, which conflicts with the magnetic measurements. Thus, the single-reference DFT methods fail to acceptably reproduce physical/spectroscopic properties of 4.
We hypothesized that the ground states of the complex ions in 4 and 5 are multiconfigurational, explaining the poor performance of DFT. Thus, we carried out a multireference configuration interaction (MRCI) calculation using the spectroscopy oriented configuration interaction (SORCI) method.38 We employed an active space of 11 electrons and 9 orbitals [CAS(11,9)] chosen to include five Fe 3d orbitals and four electrons from the formazanate π system. Five quartet, doublet, and sextet states were calculated. The chosen active space gave ≥ 90% reference weights for all states. The resulting state energies are given in Table 1. SORCI appropriately predicts a ground-state quartet, with the lowest spin-forbidden excited state–a sextet–occurring at ca. 4300 cm–1 to higher energy. The UV-vis absorption spectrum calculated by SORCI reproduces the intense absorption at 14,000 cm–1.
Table 1.
SORCI-calculated [CAS(11,9)] states for the Fe complex anion in 4.
| State | Spin multiplicity | Energy (cm−1) |
|---|---|---|
| 0 | 4 | 0 |
| 1 | 4 | 823 |
| 2 | 6 | 3953 |
| 3 | 4 | 4297.7 |
| 4 | 6 | 4402.3 |
| 5 | 4 | 5689.2 |
| 6 | 6 | 5792.2 |
| 7 | 6 | 8840 |
| 8 | 2 | 12537.3 |
| 9 | 2 | 14095 |
| 10 | 4 | 14254 |
| 11 | 2 | 17470 |
| 12 | 2 | 18342.5 |
| 13 | 6 | 19032.2 |
| 14 | 2 | 28867 |
Inspection of the quartet ground state exposes substantial multiconfigurational character. Five configurations have greater than 5% participation in the ground state, with the two leading configurations contributing ca. 25% each. Details of these two principal leading configurations are shown in Figure 8. Evaluating the composition of each configuration reveals that the ground state is best described as a nearly equal mix of configurations falling into two electronic structure categories. In one category, high-spin iron(II) participates in antiferromagnetic coupling with the unpaired electron residing in an a2-symmetry formazanate π* MO. The second category represents high-spin iron(I) without ligand participation.
Figure 8.

Calculated SORCI averaged atomic natural orbitals (AANOs) comprising the CAS(11,9) reference as well as the two leading configurations comprising the ground-state quartet of the complex ion in 4. AANOs comprising principally Fe 3d character (the Fe ligand field) are boxed, and the antiferromagnetic (AF) interaction between Fe 3dxy and the a2 formazanate (Fz) π* is indicated with a red bracket. Orbitals are plotted at an isovalue of 0.03 au.
In further agreement with our assignment, the homoleptic formazanate iron complex reported by Otten and co-workers,17 which is proposed to undergo metal-centered reduction, lacks the characteristic absorption bands for ligand-centered reduction that 5 displays at 414 and 696 nm. Overall, the electronic absorption spectra show that the one-electron reduction of the formazanate complexes involves substantial ligand participation.
Reactivity of the reduced complexes.
No differences are found in the 1H NMR spectra of complexes 4 and 5, except for resonances attributable to the countercation. Yet, complex 4 decomposes within hours in solution whereas no signs of decomposition were observed for solutions of 5 over the course of several days (vide supra). To investigate whether this was from reaction between the anion and CoCp*2 or from sodium stabilization that slows decomposition, NaBArF4 was added to a solution of complex 4 and [CoCp*2]PF6 was added to a solution of 5. In both cases decomposition was observed to the same mixture of unidentified species, showing that the decomposition of the one-electron reduced complex is brought about by the presence of the CoCp*2 cation. Though we have not been able to determine the fate of the CoCp*2 cation, it is possible that decomposition occurs through a pathway involving C-H activation through deprotonation of one of the Cp* ligands.24
Reduced low-coordinate iron complexes often abstract halide atoms from sp2 and sp3 carbon atoms. Considering the significantly less negative reduction potential of 5, we wanted to evaluate its competence in the activation of C-X (X = halogen bonds). Addition of alkyl and aryl iodides to solutions of 5 results in rapid formation of iron(II) iodide-amide complex 6, which can also be prepared by reaction of 5 with 0.5 equiv of I2 or by addition of 1 equiv NaI to solutions of 2 or 3 (Scheme 3). Addition of trityl chloride to a solution of 5 showed formation of triphenylmethyl radical (observed as Gomberg’s dimer)39 and the chloride analogue of 6.
Scheme 3.

Reactivity of complex 5 and independent synthesis of reaction products.
Accessing the ferric oxidation state was possible by a reaction of 2 or 3 with 0.5 equiv of I2 giving complex 7, which has a high-spin iron(III) (S = 5/2) configuration (see Supporting Information). Accordingly, we expected that treatment of 5 with one equiv of I2, or 6 with 0.5 equiv of I2 in THF, would also give 7. However, these reactions result in the formation of complex 8 that still has a high-spin iron(II) (S = 2) center in 82% isolated yield. The only additional reaction product in the oxidations of 5 and 6 is NaI. To test our hypothesis that complex 7 is formed in these reactions but undergoes a decomposition pathway involving NaI we modified the reaction conditions to enable monitoring by 1H NMR spectroscopy. Indeed, the addition of NaI to a THF-d8 solution of complex 7 gave 8 in 65% spectroscopic yield together with 6. Similarly, the addition of I2 to a THF-d8 solution of complex 5 gave 8 in 70 % spectroscopic yield together with 6. In addition, formation of varying amounts of I-N(SiMe3)2 was observed in both reactions, and its presence was confirmed by spiking the mixture with an independently synthesized sample of I-N(SiMe3)2.40
In the reaction of 7 with NaI an intermediate with very broad resonances is observed, and the apparent concentration is more pronounced at higher NaI concentration. We propose that this intermediate is a five-coordinate Fe(III) species that is formed from 7 and NaI. This five-coordinate species either reductively eliminates I-N(SiMe3)2 in analogy to the reaction known for a Ni(IV) complex,41 or loses •N(SiMe3)242 as observed for lanthanide complexes.43 In the latter scenario the formation of I-N(SiMe3)2 can be explained by abstraction of an I atom from Fe by •N(SiMe3)2.
In contrast, the β-diketiminate analogue of complex 7 (see Supplementary Information) does not react with stoichiometric or excess NaI. This difference suggests that the formazanate ligand is beneficial for N-I elimination. Both of the potential pathways, reductive elimination and Fe-N homolysis, are thermodynamically more favorable when the lower oxidation state is favored, indicating that the substitution of diketiminate for formazanate successfully modulates the electron density of the iron center in a way that enables new reactivity.
Conclusions
This manuscript has described the synthesis of low-coordinate iron formazanate complexes that can reversibly undergo reduction at a less negative potential compared to analogous iron β-diketiminate species. Distinguishing the high-spin iron(I) description from the antiferromagnetically coupled iron(II)-ligand radical description is challenging, and the distinctive absorption spectra support the latter model. Moreover, multiconfigurational calculations support the presence of a substantial character of iron(II) with a formazanate radical dianion. Thus, in contrast with the earlier reported bis(formazanate) complexes that have predominant iron(I) character,17 these highly electron-rich amido complexes have electron density on the supporting ligand. To our knowledge, 5 is the first well-characterized complex of a redox-active metal with a formazanate radical dianion. Despite the less negative reduction potential, 5 activates carbon halogen bonds mimicking the reactivity of low valent β-diketiminate iron complexes. In an important difference, the less-electron donating formazanate does not stabilize the higher ferric oxidation state, and therefore a different reaction pathway is followed, leading to N-I elimination.
Supplementary Material
Acknowledgements
This work was supported by The Netherlands Organization for Scientific Research (Rubicon Postdoctoral Fellowship 680-50-1517 to D.L.J.B.) and the National Institutes of Health (Grant GM-065313 to P.L.H.). K.M. L. thanks the A. P. Sloan Foundation and the National Science Foundation (CHE-1454455) for funding. P.L.H. thanks the Alexander von Humboldt Foundation for support. We thank Prof. Gary Brudvig for access to an EPR spectrometer.
Footnotes
Conflict of interest
The authors declare no competing financial interest.
References
- 1.a) Bourget-Merle L; Lappert MF; Severn JR Chem. Rev 2002, 102, 3031–3066; [DOI] [PubMed] [Google Scholar]; b) Kundu S; Stieber SCE; Ferrier MG; Kozimor SA; Bertke JA; Warren TH Angew. Chem. Int. Ed, 2016, 55, 10321–10325. [DOI] [PubMed] [Google Scholar]
- 2.Selected examples MacLeod KC; Lewis RA; DeRosha DE; Mercado BQ; Holland PL Angew. Chem. Int. Ed 2017, 56, 1069–1072;Pfirrmann S; Limberg C; Herwig C; Stӧsser R; Ziemer B Angew. Chem. Int. Ed 2009, 48, 3357–3361;Dugan TR; Bill E; MacLeod KC; Christian GJ; Cowley RE; Brennessel WW; Ye S; Neese F; Holland PL J. Am. Chem. Soc 2012, 134, 20352–20364;Horn B; Limberg C; Herwig C; Braun B Chem. Commun 2013, 49, 10923;Dai X; Kapoor P; Warren TH J. Am. Chem. Soc 2004, 126, 4798–4799.
- 3.For exceptions see: Eisenstein O; Hitchcock PB; Khvostov AV; Lappert MF; Maron L; Perrin L; Protchenko AV J. Am. Chem. Soc, 2003, 125, 10790–10791.Avent AG; Hitchcock PB; Khvostov AV; Lappert MF; Protchenko AV Dalton Trans, 2004, 33, 2272–2280.
- 4.a) Broere DLJ; Plessius R; van der Vlugt JI Chem. Soc. Rev 2015. 44, 6886–6915; [DOI] [PubMed] [Google Scholar]; b) Luca OR; Crabtree RH Chem. Soc. Rev 2013, 42, 1440–1459; [DOI] [PubMed] [Google Scholar]; c) Praneeth VKK; Ringenberg MR; Ward TR Angew. Chem. Int. Ed. 2012, 51, 10228–10234; [DOI] [PubMed] [Google Scholar]; d) van der Vlugt JI Eur. J. Inorg. Chem. 2012, 363–375; [Google Scholar]; e) Lyaskovskyy V; de Bruin B; ACS Catal. 2012, 2, 270–279; [Google Scholar]; f) Kaim W Inorg. Chem. 2011, 50, 9752–9765; [DOI] [PubMed] [Google Scholar]; g) Chirik PJ; Wieghardt K Science 2010, 327, 794–795. [DOI] [PubMed] [Google Scholar]
- 5.a) Johnson SA; Higgins RF; Abu-Omar MM; Shores MP; Bart SC Organometallics 2017, 36, 3491–3497; [Google Scholar]; b) Wong JL; Hernández Sánchez RH; Glancy Logan J; Zarkesh RA; Ziller JW; Heyduk AF Chem. Sci. 2013, 4, 1906–1910; [Google Scholar]; c) Kraft SJ; Fanwick PE; Bart SC J. Am. Chem. Soc. 2012, 134, 6160–6168; [DOI] [PubMed] [Google Scholar]; d) Haneline MR; Heyduk AF J. Am. Chem. Soc. 2006, 128, 8410–8411. [DOI] [PubMed] [Google Scholar]
- 6.a) Jacquet J, Cheaib K, Ren Y, Vezin H, Orio M, Blanchard S, Fensterbank L, M. Desage-El Murr Chem. Eur. J. 2017, 23,15030–15034; [DOI] [PubMed] [Google Scholar]; b) Fujita D, Sugimoto H, Shiota Y, Morimoto Y, Yoshizawa K, Itoh S, Chem. Commun. 2017, 53, 4849–4852; [DOI] [PubMed] [Google Scholar]; c) Jacquet J, Blanchard S, Derat E, Desage-El Murr M, Fensterbank L, Chem. Sci. 2016, 7, 2030–2036; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Broere DLJ, Modder DK, Blokker E, Siegler MA, van der Vlugt JI, Angew. Chem. Int. Ed. 2016, 55, 2406–2410; [DOI] [PubMed] [Google Scholar]; e) Broere DLJ, de Bruin B, Reek JNH, Lutz M, Dechert S, van der Vlugt JI, J. Am. Chem. Soc. 2014, 136, 11574–11577: [DOI] [PubMed] [Google Scholar]
- 7.Hu L; Chen HJ Am. Chem. Soc. 2017, 139, 15564–15567. [DOI] [PubMed] [Google Scholar]
- 8.a) Hoyt JM; Schmidt VA; Tondreau AM; Chirik PJ Science 2015, 349, 960–963; [DOI] [PubMed] [Google Scholar]; b) Bouwkamp MW; Bowman AC; Lobkovsky E; Chirik PJ J. Am. Chem. Soc. 2006, 128, 13340–13341; [DOI] [PubMed] [Google Scholar]; c) Hoyt JM; Sylvester KT; Semproni SP; Chirik PJ J. Am. Chem. Soc. 2013, 135, 4862–4877; [DOI] [PubMed] [Google Scholar]; d) Russell SK; Lobkovsky E; Chirik PJ J. Am. Chem. Soc. 2011, 133, 8858–8861. [DOI] [PubMed] [Google Scholar]
- 9.Khusniyarov MM; Bill E; Weyhermüller T; Bothe E; Wieghardt K Angew. Chem., Int. Ed. 2011, 50, 1652–1655. [DOI] [PubMed] [Google Scholar]
- 10.Marshak MP; Chambers MB; Nocera DG Inorg. Chem. 2012, 51, 11190–11197. [DOI] [PubMed] [Google Scholar]
- 11.Takaichi J; Morimoto Y; Ohkubo K; Shimokawa C; Hojo T; Mori S; Asahara H; Sugimoto H; Fujieda N; Nishiwaki N; Fukuzumi S; Itoh S Inorg. Chem, 2014, 53, 6159–6169. [DOI] [PubMed] [Google Scholar]
- 12.For a review on the non-innocence of β-diketiminate ligands see: Camp C; Arnold J; Dalton Trans 2016, 45, 14462–14498.
- 13.a) Chang M-C; Otten E Chem. Commun. 2014, 50, 7431–7433; [DOI] [PubMed] [Google Scholar]; b) Chang M-C; Chantzis A; Jacquemin D; Otten E Dalton Trans. 2016, 45, 9477–9484; [DOI] [PubMed] [Google Scholar]; c) Barbon SM; Price JT; Reinkeluers PA; Gilroy JB Inorg. Chem. 2014, 53, 10585–10593; [DOI] [PubMed] [Google Scholar]; d) Mondol R; Snoeken DA; Chang M-C; Otten E Chem. Commun. 2017, 53, 513–516; [DOI] [PubMed] [Google Scholar]; e) Gilroy JB; Ferguson MJ; McDonald R; Patrick BO; Hicks RG Chem. Commun 2007, 126–128. [DOI] [PubMed] [Google Scholar]
- 14.a) Chang M-C; Dann T; Day DP; Lutz M; Wildgoose GG; Otten E Angew. Chem. Int. Ed. 2014, 53, 4118–4122; [DOI] [PubMed] [Google Scholar]; b) Chang M-C Roewen P; Travieso-Puente R; Lutz M; Otten E; Inorg. Chem. 2015, 54, 379–388. [DOI] [PubMed] [Google Scholar]
- 15.a) Mandal A; Schwederski B; Fiedler J; Kaim W; Lahiri GK Inorg. Chem. 2015, 54, 8126–8135; [DOI] [PubMed] [Google Scholar]; b) Kabir E; Wu C-H; Wu JI; Teets TS Inorg. Chem. 2016, 55, 956–963; [DOI] [PubMed] [Google Scholar]; c) Gilroy JB; Patrick BO; McDonald R; Hicks RG Inorg. Chem. 2008, 47, 1287–1294; [DOI] [PubMed] [Google Scholar]; d) Hong S; Hill LMR; Gupta AK; Naab BD; Gilroy JB; Hicks RG; Cramer CJ; Tolman WB Inorg. Chem. 2009, 48, 4514–4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Travieso-Puente R; Broekman JOP; Chang M-C; Demeshko S; Meyer F; Otten E; J. Am. Chem. Soc. 2016, 138, 5503–5506. [DOI] [PubMed] [Google Scholar]
- 17.Gilroy JB; Ferguson MJ; McDonald R; Patrick BO Hicks RG; Chem. Commun. 2007,126–128. [DOI] [PubMed] [Google Scholar]
- 18.Broere DLJ; Čorić I; Brosnahan A; Holland PLH Inorg. Chem. 2017, 56, 3140–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Panda A; Stender M; Wright RJ; Olmstead MM; Klavins P; Power PP Inorg. Chem. 2002, 41, 3909–3916. [DOI] [PubMed] [Google Scholar]
- 20.Yang L; Powell DR; Houser RP Dalton Trans. 2007, 955–964. [DOI] [PubMed] [Google Scholar]
- 21.Chiang KP; Barrett PM; Ding F; Smith JM; Kingsley S; Brennessel WW; Clark MM; Lachicotte RJ; Holland PL Inorg. Chem. 2009, 48, 5106–5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a) Brown SN Inorg. Chem. 2012, 51, 1251–1260; [DOI] [PubMed] [Google Scholar]; b) Broere DLJ; Plessius R; Tory J; Demeshko S; de Bruin B; Siegler MA; Hartl F; van der Vlugt JI Chem. Eur. J. 2016, 22, 13965–13975; [DOI] [PubMed] [Google Scholar]; c) Broere DLJ; van Leest NP; Siegler MA; van der Vlugt JI Inorg. Chem. 2016, 55, 8603–8611; [DOI] [PubMed] [Google Scholar]; d) Sproules S; Wieghardt K. Coord. Chem.Rev. 2010, 254, 1358–1382. [Google Scholar]
- 23.MacLeod KC; McWilliams SF; Mercado BQ; Holland PL Chem. Sci. 2016, 7, 5736–5746; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ohki Y; Murata A; Imada M; Tatsumi K Inorg. Chem. 2009, 48, 4271–4273. [DOI] [PubMed] [Google Scholar]
- 24.a) MacLeod KC; Menges FS; McWilliams SF; Craig SM; Mercado BQ; Johnson MA; Holland PL J. Am. Chem. Soc 2016, 138, 11185–11191; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) McWilliams SF; Rodgers KR; Lukat-Rodgers G; Mercado BQ; Grubel K; Holland PL Inorg. Chem 2016, 55, 2960–2968; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Grubel K; Brennessel WW; Mercado BQ; Holland PL J. Am. Chem. Soc. 2014, 136, 16807–16816; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Connor GP; Holland PL Catalysis Today 2017, 286, 21–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McWilliams SF; Brennan-Wydra E; MacLeod KC Holland PL ACS Omega 2017, 2, 2594–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.a) Arnet NA; Dugan TR; Menges FS; Mercado BQ; Brennessel WW; Bill E; Johnson MA; Holland PL J. Am. Chem. Soc. 2015, 137, 13220–13223; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lewis RA; MacLeod KC; Mercado BQ; Holland PL Chem. Commun. 2014, 50, 11114–11117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.a) Kleinlein C; Bendelsmith AJ; Zheng S-L; Betley TA Angew.Chem. Int.Ed 2017, 56, 12197–12201; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dunsford JJ; Evans DJ; Pugh T; Sha SN; Chilton NF; Ingleson MJ Organometallics 2016, 35, 1098–1106; [Google Scholar]; c) Evans DJ; Hughes DL; Silver J Inorg. Chem. 1997, 36, 747–748. [Google Scholar]
- 28.Gülich P; Bill E; Trautwein AX Mӧssbauer spectroscopy and transition metal chemistry, Springer, Heidelberg, 2011. [Google Scholar]
- 29.Initial synthesis: Smith JM; Lachiocotte RJ; Holland PL J. Am. Chem. Soc. 2003i, 125, 15752–15753;; Mӧssbauer spectrum and alternative synthesis: Dugan TR; Holland PL J. Organomet. Chem. 2009, 694, 2825–2830.
- 30.Chiang KP; Scarborough CC; Horitani M; Lees NS; Ding K; Dugan TR; Brennessel WW; Bill E; Hoffman BM; Holland PL Angew. Chem. Int. Ed. 2012, 51, 3658–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cowley RE; Bill E; Neese F; Brenessel WW; Holland PL Inorg. Chem. 2009, 48, 4828–4836. [DOI] [PubMed] [Google Scholar]
- 32.a) Tezcan H; Can S; Tezcan R Dyes Pigm. 2002, 52, 121; [Google Scholar]; b) Ibrahim YA; Abbas AA; Elwaby AHMJ; Heterocycl. Chem. 2004, 41, 135. [Google Scholar]
- 33.Gilroy JB; McKinnon SDJ; Koivisto BD; Hicks RG Org. Lett. 2007, 9, 4837–4840. [DOI] [PubMed] [Google Scholar]
- 34.Shulman RG; Yafet Y; Eisenberger P; Blumberg WE Proc. Natl. Acad. Sci. U.S.A. 1976, 73, 1384–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Banerjee P; Sproules S; Weyhermüller T; DeBeer George S; Wieghardt K Inorg. Chem, 2009, 48, 5829–5847. [DOI] [PubMed] [Google Scholar]
- 36.a) MacMillan SN; Lancaster KM ACS Catal. 2017, 7, 1776–1791; [Google Scholar]; b) Kau LS; Spira-Solomon DJ; Penner-Hahn JE; Hodgson KO; Solomon EI J. Am. Chem. Soc. 1987, 109, 6433–6442. [Google Scholar]
- 37.DeBeer George S; Petrenko T; Neese FJ Phys. Chem. A 2008, 112, 12936–12943. [DOI] [PubMed] [Google Scholar]
- 38.Neese FJ Chem. Phys. 2003, 119, 9428. [Google Scholar]
- 39.Lankamp H; Nauta WT MacLean C Tet. Lett. 1968, 9, 249–254. [Google Scholar]
- 40.Bailey RE; West RJ Organomet. Chem. 1965, 4, 430–439. [Google Scholar]
- 41.Laarhoven JJ; Mulder PJ Phys. Chem. B 1997, 101, 73–77. [Google Scholar]
- 42.Cook MD; Roberts BP; Singh KJ Chem. Soc, Perkin Trans 2, 1983, 635–643. [Google Scholar]
- 43.a) Lewis AJ; Carroll PJ; Schelter EJ J. Am. Chem. Soc 2013, 135, 511–518. [DOI] [PubMed] [Google Scholar]; (c) Mullane KC; Lewis AJ; Yin H; Carroll PJ; Schelter EJ Inorg. Chem 2014, 53, 9129–9139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.