

Abstract
Objective
Raynaud’s Phenomenon is common in rheumatic diseases with anti-ribonucleoprotein antibodies (RNP+), but is not itself known to be caused by autoimmunity. This study assessed for autoantibodies that could mediate this process.
Methods
Antibodies from patient sera and from murine models of anti-RNP autoimmunity were screened for the ability to induce Raynaud’s-like tissue ischemia and endothelial apoptosis in murine models and in vitro systems.
Results
RNP+ human sera from Raynaud’s patients and murine sera from RNP+ B cell adoptive transfer recipients induced Raynaud’s-like tissue ischemia and endothelial apoptosis. Proteomic analysis identified Cytokeratin 10 (K10) as a candidate Raynaud’s autoantigen. Monoclonal anti-K10 antibodies reproduced patterns of ischemic tissue loss and endothelial apoptosis; K10 knockout mice or serum depletion of anti-K10 activity were protective. Cold exposure enhanced K10 expression and in vivo tissue loss.
Conclusions
Anti-K10 antibodies are sufficient to mediate Raynaud’s-like ischemia in murine models, and are implicated in Raynaud’s pathogenesis in patients with anti-RNP autoimmunity.
Raynaud’s Phenomenon (Raynaud’s, RP), cold-induced peripheral vasospasm, exists in two forms. “Benign” Raynaud’s occurs in 10% of healthy young women and is associated with minimal morbidity (1). The other form, autoimmunity-associated Raynaud’s, is common in Systemic Sclerosis (scleroderma) and related autoimmune rheumatic diseases in which anti-RNP antibodies are present (2). This second form can be associated with significant morbidity, including gangrene and tissue loss of fingers and toes (3). Current therapy for autoimmunity-associated Raynaud’s uses vasodilator drugs to reduce local manifestations of ischemia (4), but does not address the underlying pathogenesis of the process. Studies of Raynaud’s pathogenesis have identified abnormalities in vascular tone and response to neuroendocrine stimuli (5), but have struggled to connect Raynaud’s to autoimmunity.
Endothelial apoptosis has been regarded as a central event in scleroderma pathogenesis, with the potential to drive both vasospastic and fibrotic disease manifestations (6). Sera from scleroderma patients have previously been observed to induce apoptosis of cultured endothelial cells (7,8). A spontaneous avian model of Raynaud’s has been described in which increased apoptosis of endothelial cells in the area of vasospasm can be observed, and in which sera from affected birds also induces endothelial apoptosis (9,10). A pathway whereby scleroderma antisera could induce apoptosis of endothelial progenitor cells has been identified, in which serum-induced inhibition of Akt signaling leads to upregulation of Bim expression and hence apoptosis, but the target antigen/receptor has not been defined (11). This report addresses the specificity of antisera that mediate endothelial apoptosis, and connects this process to novel in vivo animal models.
Ear and tail vessels in mice have thermoregulatory function similar to finger and toe vessels in humans, respond similarly to human digital arteries when exposed to vasoconstrictors implicated in episodes of Raynaud’s (12), and would be the presumed targets of Raynaud’s in mice. (In contrast, murine digits have not been observed to share the thermoregulatory function seen in human digits.) We have previously developed an induced murine model of anti-ribonucleoprotein (RNP) autoimmunity with lung and renal manifestations consistent with human Mixed Connective Tissue Disease (MCTD) (13,14). However, this murine model does not develop Raynaud’s manifestations, a finding present in over 90% of human MCTD patients (15).
Case reports of improving Raynaud’s after anti-B cell therapy in anti-RNP autoimmunity have been published (16,17). Supporting a link between humoral autoimmunity and Raynaud’s, some anti-RNP antibodies have been shown to bind endothelium (18). We therefore hypothesized that a previously uncharacterized set of autoantibodies that induces endothelial apoptosis could be pathogenic for Raynaud’s and that patients with Raynaud’s develop high titers of these antibodies. Although we have previously reported immunologically distinct anti-RNP responses in patients with Raynaud’s (2), a specific target antigen that is expressed on endothelium, that induces endothelial apoptosis when bound by a cognate antibody, and that can induce Raynaud’s -like ischemia of thermoregulatory tissues has not previously been described.
This report presents murine models of Raynaud’s-like ischemic lesions that can be induced by B cell transfer, murine serum transfer, transfer of human Raynaud’s patient serum, or transfer of monoclonal antibodies to the novel autoantigen Cytokeratin 10 (K10). It shows that anti-K10 antibodies can be found in Raynaud’s patient sera, that anti-K10 antibodies can induce endothelial apoptosis in vitro, and that anti-K10-mediated apoptosis and tissue loss are prevented in K10-knockout mice. We also show that Bim-knockout mice are resistant to antibody-induced tissue ischemia. Collectively, these results establish novel murine models of Raynaud’s, demonstrate that Raynaud’s can be an autoimmune process mediated through anti-intermediate filament antibodies, and indicate that the Cytokeratin 10 antibody/antigen system (and its downstream signaling pathway) may be a relevant target for novel diagnostic and therapeutic approaches to Raynaud’s.
Materials and Methods
Experimental Design
The primary study design was of controlled laboratory experiments. Pre-defined outcomes included the frequency and extent to local tissue ischemia of the ears and tails of study mice, and the extent of apoptosis induction as measured by caspase 3/7 activation using a commercial colorimetric indicator. Power analyses were performed prior to each experiment based on preliminary data. Studies were powered to a Beta of at least 0.8 for Alpha of 0.05 or less. Observations of tissue ischemia were made by trained study personnel blinded to the treatment arm(s) of the study mice. Studies with human sera were performed using a sample of clinically characterized RNP+ patients recruited for study at our center for whom antisera were available, and a cohort of healthy human subjects (19).
Study Approval
All human samples and data were collected and used following IRB-approved protocols. Patients and normal controls were recruited, and written informed consent for study participation was provided by study subjects, following IRB-approved protocols. All animal studies were performed according to Institutional Animal Care and Use Committee (IACUC)-approved protocols in Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC International)-approved facilities, in adherence to the NIH Guide for the Care and Use of Laboratory Animals.
Human and Animal Studies
Patients seen at our center between 2006 and 2015 were characterized as RNP+ if they had a positive clinical laboratory anti-RNP test, and/or anti-RNP antibodies were identified in our lab by ELISA or immunoblot (19,20). Subject data were obtained by structured interview, physical exam, laboratory screening and review of medical records (15). Raynaud’s Phenomenon was designated if patients gave a history of cold-induced color changes in the digits following the validated picture-question format of Maricq and colleagues (21). Sera obtained from study subjects at the time of a research encounter was kept in frozen aliquots at −80°C.
C57BL/6 mice were from Jackson Labs except as indicated below. HLA-DR4tg mice on a B6 background, as previously described, were from Taconic (13). To induce anti-RNP autoimmunity, female 8–10 week old mice were immunized subcutaneously, as previously described (13,14), with 50 micrograms each of 70k (as an MBP fusion protein, 70k-FP) and U1-RNA together in 50 microliters of sterile PBS. Successful immunizations were confirmed by 70k ELISA as previously described (19,20). K10 knockout mice and control BALB/C mice (the background strain in which the K10 knockout was produced) were obtained from Thomas M. Magin (22), confirmed to be genetically homozygous for the K10 knockout genotype by sequencing of tail clip samples (Transnetyx, Cordova TN), and established to have undetectable endothelial levels of K10 protein by immunohistochemistry and immunoblotting. Bim knockout mice were generated by crossing Bimfl/fl mice on a C57/B6 background with a ROSA26-cre mouse resulting in a germline deletion of the floxed allele of Bim (23,24). Mice were crossed until homozygous deleted mice were obtained. Inbred littermate B6 mice were used as controls.
In vivo cold exposure experiments were performed on mice under isoflurane inhaled anesthesia, following IACUC-approved protocols. Specific body parts (one ear) were exposed to a 4°C thermal pad for one minute, then returned to usual body temperature warming pad conditions.
Cell culture
All cell culture experiments were performed in accordance with institutional biosafety committee-approved protocols. HUVEC cells (Lonza, Allendale, NJ) were maintained in EGM Complete medium (Lonza, Cat# 3024A). Short-term murine endothelial progenitor cell cultures were developed from bone marrow of B6, BALB/C, or K10 knockout mice, following the protocol of Chunming Dong (25). Studies were performed on HUVEC and endothelial progenitor cells at 8 or fewer passages.
Cold exposure studies were performed in vitro by placing the cells in medium cooled to 4°C for the indicated time points (typically 1 minute for cold pre-exposure) before replacing the medium with 37°C medium.
Bacterial transfection and culture to express and purify K10 proteins was performed as previously described (20,26). Plasmids expressing K10-MBP and hisK10 were obtained respectively from Thomas M. Magin and Jose L. Jorcano. Protein was purified from sonicated cell lysate by column chromatography (amylose or nickel columns), and following gel purification, was validated by MSMS and by immunoblot.
B Cell and antibody adoptive transfer studies
Single cell suspensions of spleens from immunized and control mice were prepared as previously described (14,26). B cells were purified by AutoMACS B cell separation kit and found to have consistently over 90% purity for B cell markers by FACS. Adoptive transfers were performed with 5 × 105 cells in 400 microliters of sterile PBS, injected into the tail vein of 10–12 week old syngeneic naïve mice with a sterile 26g needle, as previously described with T cell and dendritic cell transfer studies (14,26). Serum (up to 50 microliters) and commercial antibody (up to 50 micrograms in 50 microliters) transfer studies were likewise performed by a single tail vein injection into 10–12 week old recipient mice. Recipient mice were followed for two weeks prior to sacrifice. At the time of sacrifice, serum was collected and tissues were processed for histological evaluation (14,26). Anti-K10 monoclonal antibodies included Clone LH2, a murine IgG1 monoclonal raised against human K10 with reported reactivity against both human and mouse cytokeratin 10 (LifeSpan BioSciences, Seattle); Clone VIK-10, a murine monoclonal IgG1 with anti-K10 specificity raised against a human epidermal cytoskeletal extract (Thermo Fisher Scientific, Waltham MA); and clone AE20, a murine monoclonal IgG specific for human K10 (Genway Biotech, San Diego). Isotype controls included Clone AE1, a mouse monoclonal IgG1 raised against human epidermis with reported activity against both human and murine acidic cytokeratins prominently including cytokeratin 9 (Genway Biotech, San Diego). Conjugations of antibodies to fluorescent markers were performed by the commercial suppliers. For indirect immunofluorescence with human sera, FITC-conjugated goat anti-human IgG was used at a 1:500 dilution. Anti-K10 ELISAs were performed similarly to our anti-70k ELISAs. For the IgG ELISAs, K10-MBP was used as to coat plate wells and purified MBP was used to coat wells in parallel to define the background staining. For the IgM ELISAs, hexahistidine-tagged K10 was used as to coat wells in the same buffer and at the same concentration, and compared to wells incubated in blocking buffer to define the background staining. Goat anti-human IgM-HRP (Southern Biotech, Birmingham, AL) was used at 1:5000 as the second antibody for the IgM ELISA studies.
Immunoprecipitation and protein identification
The reverse immunophenotyping approach of Ascherman was used to identify the 56kD band induced in endothelial cells and recognized in endothelial cell lysates by RP sera but not control sera (27). Briefly, serum samples from representative anti-RNP positive patients with RP versus without RP were bound to protein A–Sepharose beads. HUVEC cells were lysed in sample buffer and used as the antigen sources. Lysates were either used unmodified or pre-cleared with the RP+ or RP- anti-RNP sera. Antibody-conjugated beads and lysates were allowed to co-incubate overnight at 4° C, then were spun and washed in immunoprecipitation buffer, suspended in sample buffer, electrophoresed on a standard-size 10% polyacrylamide gel, and imaged by Coomassie staining. The candidate 56kD band preferentially immunoprecipitated by sera from patients with RP was excised, digested with trypsin in situ, eluted, and subjected to tandem matrix-assisted laser desorption ionization–time-of-flight mass spectrometry for sequence analysis of derivative peptides. Use of the Sequest search engine and Proteome Discoverer software generated probability scores for the identity of electrophoretically separated parent proteins.
Histopathology
Conventional sections from murine tissue samples were fixed in UMFix and stained with H&E as previously described (13,14). In vivo TUNEL staining was performed on mice labelled in vivo with the ApopTag Plus Peroxidase Kit (EMD Millipore, Billerica MA) and sacrificed 32 hours after exposure to 50 microliters of a human RNP+ RP+ serum or normal control human serum. Caspase 3/7 staining was performed in vitro with CellEvent Caspase 3/7 Green Detection Reagent (Thermo Fisher Scientific, Waltham MA) following the manufacturer’s instructions. For the Caspase assays, fluorescence images of intact unpermeabilized cells were captured using an Olympus (1×71) Inverted Fluorescence Microscope.
Statistical Analysis
Groups were compared using Fisher’s Exact test for categorical data, and T tests for quantitative data using Prism 5.0 and Graphpad QuickCalcs (Graphpad Software, San Diego). Odds Ratios were calculated with MedCalc (Medcalc Software, Belgium).
Results
Murine models of anti-RNP thermoregulatory tissue ischemia
We previously have observed distinct clinical phenotypes associated with adoptive transfer of specific subsets of spleen cells (T cells or dendritic cell subsets) from RNP-immunized mice into syngeneic controls (14). We performed similar studies to assess whether splenic B cell adoptive transfers would likewise induce a particular phenotype. We found that adoptive transfers of splenic B cells from RNP immunized HLA-DR4 transgenic B6 mice, but not from naïve syngeneic B cell donors, induced ear and tail ischemia and tissue loss within two weeks of cell transfer in mice housed under standard conditions (Figure 1), with ischemic lesions evident in 14/17 (82%) recipients of RNP+ B Cell donors versus 0/5 recipients of naïve splenic B cells (0%, Fisher’s Exact p = 0.002). Out of over 500 HLA-DR4 transgenic B6 mice directly immunized to develop anti-RNP antibodies in our concurrent and prior studies, no other cases of spontaneous ear or tail gangrene and tissue loss had occurred.
Figure 1.
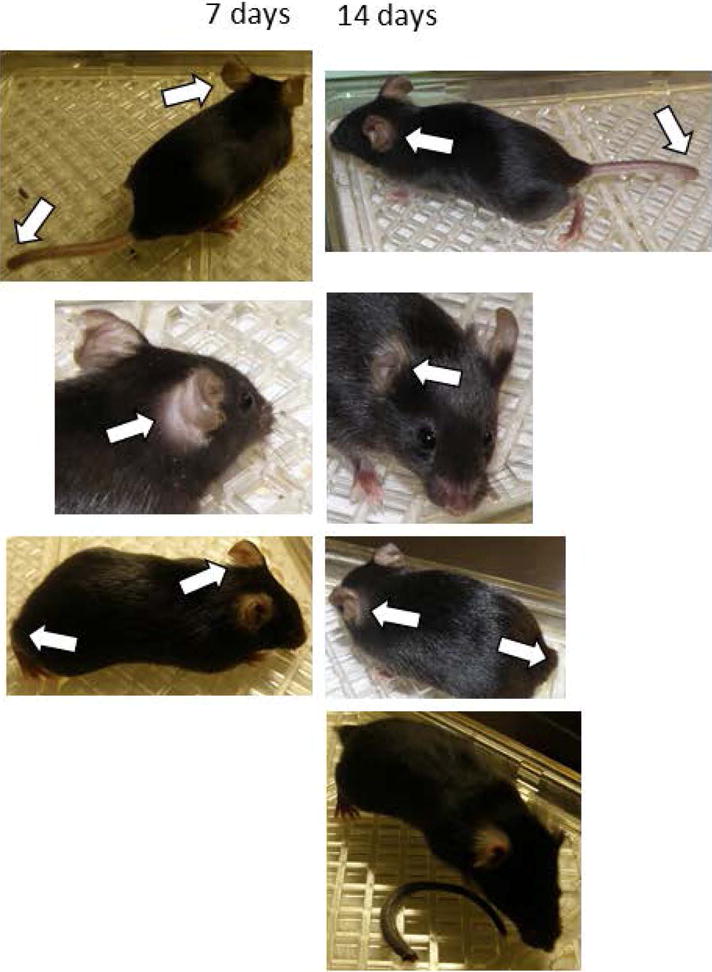
B cell transfer-induced murine model of ischemic Raynaud’s. Progressive ear and tail loss are noted in syngeneic B cell recipients from 70k-immunized HLA-DR4tg B6 mice. In the top three rows, the same mice are shown 7 and 14 days after cell transfer; arrows indicate areas of tissue loss. At the bottom, an additional recipient mouse that lost its tail 14 days after transfer is shown along with the ischemic tail.
Sera from the RNP+ B cell recipients that developed ear and tail ischemia also induced ear and tail ischemia within two weeks of IV infusion into naïve syngeneic mice (housed under standard conditions), in a dose-dependent manner (Figure 2A). Similar ischemic lesions were not observed with serum transfer from either naïve mice (not shown) or from RNP-immunized mice that served as B cell donors.
Figure 2.
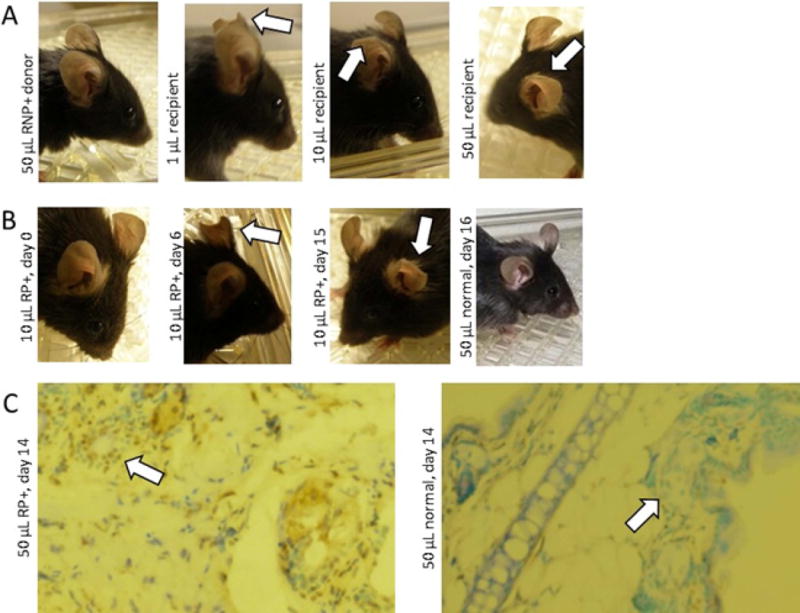
Serum transfer of ischemia phenotype. A. Murine serum transfer from ischemic B cell recipients as in Figure 1 induces dose-dependent tissue loss by 14 days (arrows), not seen with B cell donor sera. B. Human Raynaud’s patient serum transfer (10 microliters) following the same protocol as in A induces progressive ear tissue loss (arrows) from Day 0 to Day 6 to Day 15 in the same mouse, not seen with healthy control human serum. C. Raynaud’s serum transfer induces vascular apoptosis. Mice received human Raynaud’s or healthy control sera. Two weeks later, ear tissue (10×) showed increased TUNEL staining (brown) in the Raynaud’s serum recipient compared to the normal control, prominently near the lumen of blood vessels (arrows).
We performed similar serum transfers into naïve mice of the same strain using human serum from an MCTD patient with anti-RNP autoimmunity and a history of active Raynaud’s. The patient’s serum likewise induced progressive ear ischemia and tissue loss within 2 weeks (Figure 2B). No effects were noted with healthy human control serum transfer. Histological analysis of Raynaud’s serum recipients two weeks after transfer revealed no significant internal organ derangements, and no evidence of vasculitis or thrombosis (Supplementary Data, Figure S1). Increased apoptosis by TUNEL staining was observed in the ear and tail vasculature of mice receiving ischemia-inducing sera (Figure 2C).
Identification of K10 as target autoantigen and association of anti-K10 antibodies with Raynaud’s-like ischemia
Ischemia-inducing human anti-RNP+ serum from a Raynaud’s patient but not control human anti-RNP+ serum from a patient without Raynaud’s showed anti-endothelial cell reactivity by indirect immunofluorescence (Figure 3A). Studies performed with RNP+ sera from patients with Raynaud’s Phenomenon showed similar results regardless of the clinical diagnosis (primarily SLE and MCTD) of the patients (Supplementary Data, Tables S1 and S2). Immunoprecipitation of HUVEC cells with the human MCTD patient serum (from the patient with Raynaud’s as in Figure 2) that induced murine ischemia identified a 56kD band not present in RNP+ serum from a patient without Raynaud’s that did not induce ischemia in mice after serum transfer (Figure 3B). Mass spectroscopy analyses identified the immunoprecipitated 56kD band as Cytokeratin 10 (K10).
Figure 3.
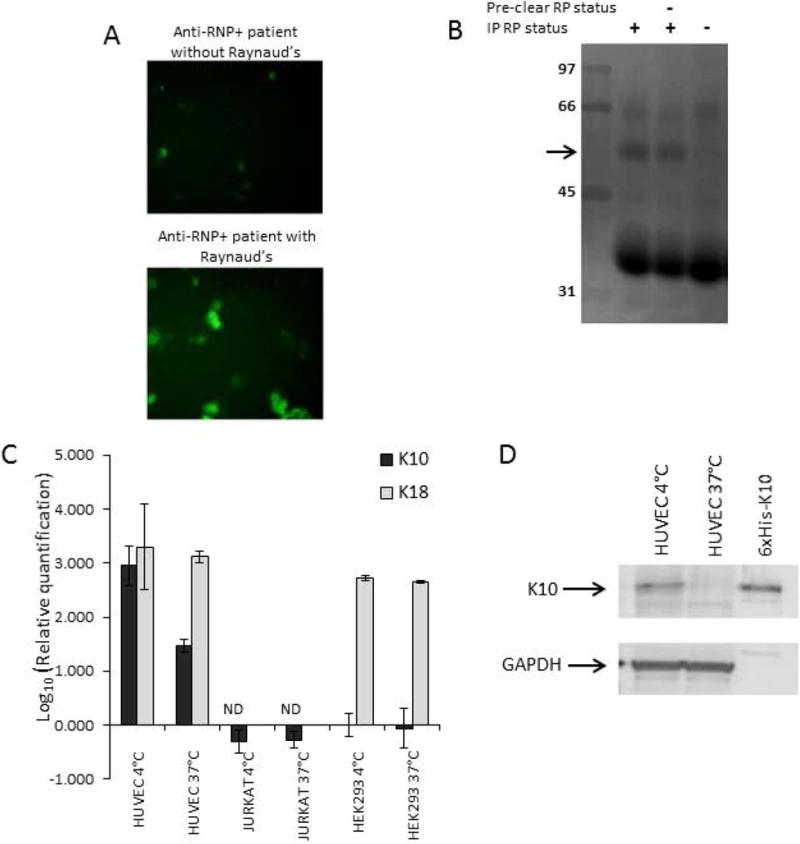
K10 as a cold-inducible target of Raynaud’s-associated antibodies. Cultured cells were kept at 37°C in complete medium (see Methods). Where indicated for cold exposure, cells were exposed to 4°C medium for 1 minute 24 hours before cell harvesting, then returned to 37°C. A. Anti-endothelial activity by immunofluorescence of HUVEC (see Methods) with an RNP+ serum from a Raynaud’s patient. B. Reverse immunophenotyping immunoprecipitation (see Methods) of HUVEC lysates with an anti-RNP+ human Raynaud’s Phenomenon serum identified a ~56kD band (arrow) that was not pre-cleared by an RNP+ serum from a patient without Raynaud’s Phenomenon (RP-), for MSMS analysis. C. Expression and cold-induced upregulation of mRNA levels by real time PCR were quantitated for K10 and control cytokeratin 18 (K18), normalized to expression of 18S ribosomal RNA. The endothelial cell line HUVEC expressed much higher levels of K10 message than epithelial HEK293 cells or lymphoid Jurkat cells at 37°C, and further dramatically upregulated K10 but not K18 expression levels after cold exposure. D. Expression and upregulation of K10 protein in HUVEC. While low levels of K10 protein were present in HUVEC cells at 37°C, the quantity of protein dramatically increased in cold-exposed cells (compared to GAPDH loading control).
Though prior reports of K10 expression have been primarily in skin cells, we found that endothelial cells do express K10 mRNA and protein (Figure 3C & 3D). In contrast to other endothelial cytokeratins, endothelial K10 expression levels were substantially upregulated at the mRNA and protein levels by a brief exposure of endothelial cell cultures to cold temperature. Similar changes in expression were also evident by immunofluorescence with directly FITC-conjugated anti-K10 (Figure S2).
To test whether anti-K10 antibodies were sufficient to induce ischemic lesions, we performed IV treatment of B6 HLA-DR4tg study mice with commercially available anti-K10 monoclonal antibodies. Anti-K10 monoclonal antibodies with established reactivity with murine K10 (both the LH2 and VIK10 antibodies) induced ischemic ear lesions similar to those seen with the murine and human sera (Figure 4).
Figure 4.

Anti-K10 monoclonal antibodies induce ischemic tissue loss. Female 10 week old C57BL/6 mice received 50 μg of anti-mouse anti-K10 monoclonal antibody by IV tail vein infusion (“Anti-K10 Monoclonal”, n = 5), or an equal amount of normal mouse immunoglobulin (“Control Serum”, n = 5). Representative images are shown at Days 0 and 15 for each of the 10 mice. None of the Control Serum mice had any clinical manifestations (0/5). In contrast, 5/5 Anti-Mouse K10 recipients (100%, Fisher’s Exact p = 0.008) developed cyanosis and ischemia of the ears, with progressive tissue loss (bilateral, though most prominent at arrows). Results are representative of 3 separate experiments.
Pursuing the hypothesis that anti-K10 antibodies in Raynaud’s sera induce endothelial cell apoptosis, we assessed in vitro endothelial caspase 3/7 activation after 24 hour serum incubations using anti-RNP+ patient sera and healthy control human sera. The presence of anti-K10 IgG antibodies by ELISA in RNP+ patients was correlated with induction of high caspase signaling, both in patients with clinical Raynaud’s and in patients in whom clinical RP had not been appreciated, i. compared to RNP+ patients without anti-K10 antibodies (either with or without Raynaud’s) and ii. compared to control sera from healthy subjects (Figure 5A). All 11 of the RNP+/K10+ sera tested showed high levels of in vitro endothelial cell caspase activation (defined as 2 standard deviations above the mean caspase activation by control sera), as did 3/6 of the sera in the RNP+/K10-/RP+ group (Fisher’s Exact p = 0.029 versus the anti-K10+ sera), 0/6 of the sera in the RNP+/K10-/RP- group (Fisher’s Exact p < 0.0001 versus the RNP+/K10+ sera), and 0/11 of the K10- healthy control sera (Fisher’s Exact p < 0.0001 versus the RNP+/K10+ sera and p = 0.036 versus the RNP+/K10-/RP+ group). Differences in caspase activation were reflected in differences in cell death by morphological criteria in HUVEC cells (Figure S3).
Figure 5.
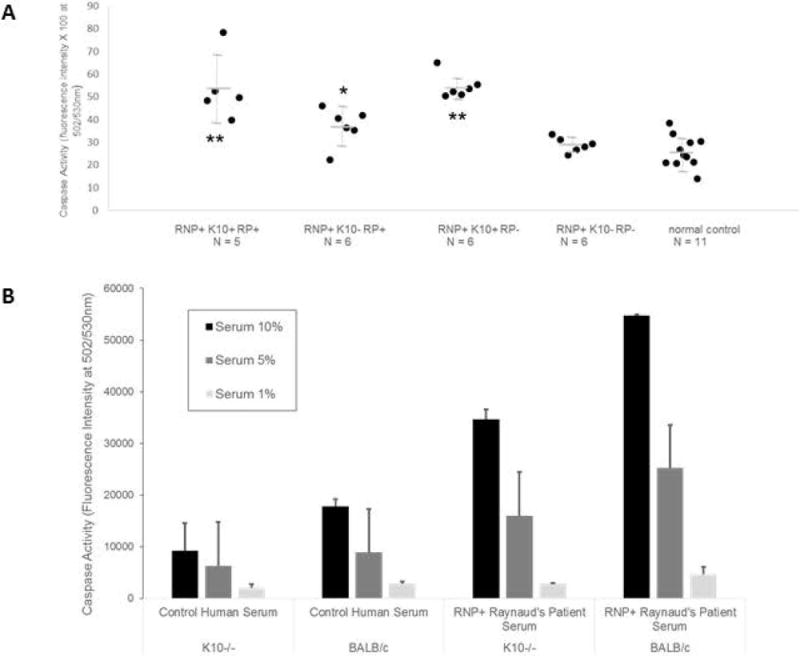
K10-dependent endothelial apoptosis. A. HUVEC were exposed for 24 hours to 10% dilutions of sera from healthy subjects (Ctrl) or RNP+ patients with or without Raynaud’s (RP+ or -) and anti-K10 IgG antibodies (ELISA > 2 S.D. above healthy subject mean to be anti-K10+), loaded with CellEvent Caspase 3/7 Green Detection Reagent (10μM/ml) and assayed for green absorbance in 24 hours in duplicate wells. Anti-K10+ sera induced high levels of caspase activity (an indicator of apoptosis) from patients with or without RP. ** t test p < 0.041 versus K10-RP+ sera, p < 0.003 versus K10-RP- sera, and p <= 0.0001 versus Ctrl K10- sera; * t test p < 0.048 versus K10-RP- sera and p < 0.009 versus Ctrl K10- sera. Results were representative of 3 separate experiments. B. Endothelial cell cultures concurrently generated from BALB/c, and K10-knockout BALB/c mice (see Methods) were exposed to dilutions of K10- control serum or K10+ RNP+ RP+ serum for 24 hours, and assayed for caspase activity as above (representative of 2 separate experiments). The Raynaud’s serum induced increased dose-dependent caspase activity in both K10-intact and K10−/− cells compared to control serum, but much more so in K10-intact cells.
Over a larger number of well-characterized RNP+ patients at our center, ELISA screening showed that anti-K10 antibodies (either IgG or IgM) were detectable in 48% (56/116) of patients (Supplementary data Table S1). The clinical diagnoses for the patients in this cohort included SLE (77, 66%), MCTD (34, 29%), myositis (2, 2%), scleroderma (1, <1%), and autoimmune hepatitis (1, <1%) (Supplementary data Table S2).
To further investigate the role of K10 in humorally-mediated induction of endothelial cell apoptosis, we tested endothelial cell cultures from K10 knockout mice versus their background strain (BALB/c) for responsiveness to dilutions of human serum at 37°C (Figure 5B). As expected, we found that caspase 3/7 activation was high in K10-intact endothelium when exposed to increasing concentrations of RNP+ serum from a patient with Raynaud’s Phenomenon whose serum had been shown to have high titer anti-K10 IgG and IgM antibodies by ELISA and to induce clinical ear and tail ischemia when transferred into study mice. Much less caspase activation was observed with increasing concentrations of a normal control human serum. Caspase activation induced by human sera was substantially attenuated in endothelial cells from K10 knockout mice compared to K10-intact endothelial cells.
Despite exposure to the same doses of anti-K10+ RP serum used in B6/HLA-DR4tg mice, germline K10-expressing BALB/C mice developed ischemic lesions less often than B6/HLA-DR4tg mice, with 0/5 mice showing ear lesions other than subtle ruffling of the ear edge and only one case of unequivocal tail ischemia out of 5 treated mice (Supplementary data Figure S4). To increase the expression of disease in the BALB/C mice, we used targeted cold exposure with the intention of upregulating local K10 expression (as in Figure 3). When we pre-treated one ear of BALB/C mice with 1 minute of 4°C cold exposure 48 hours and again 24 hours before infusing the mice with anti-K10+ patient serum, we could identify definite ear tissue loss—only on the cold-pre-treated side—in 4/5 mice (Fisher’s Exact p = 0.047) (Figure 6A). Age and sex-matched K10-knockout BALB/c mice were concurrently treated with cold exposure prior to administration of the same dose of anti-K10+ patient serum. The K10 knockout mice showed no ear or tail ischemic tissue loss. Indeed, when we measured the ear lengths of the germline BALB/C versus K10 knockout mice starting at 10 weeks of age, we observed that the K10 knockout mice showed interval increases of their ear lengths that were unaffected by the presence or absence of cold exposure or anti-K10 antibodies, whereas the BALB/C mice showed unchanged ear size with anti-K10 antibodies in the absence of cold exposure and clear tissue loss with the combination of cold pre-exposure and anti-K10 antibodies.
Figure 6.
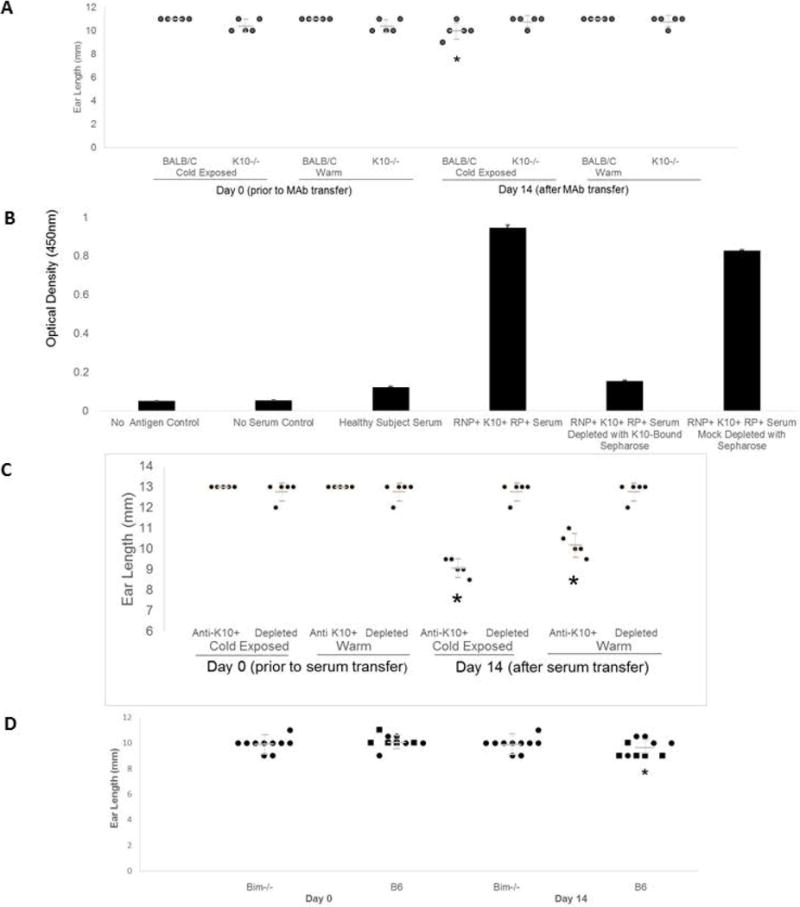
K10−/−, Anti-K10 depletion, or Bim−/− prevent ischemia. 24h before antibody injections, one ear of each mouse was exposed to 4°C for one minute. Ear lengths were measured at baseline and Day 14. A. Anti-K10 MAb (50 μg) injected via tail vein (n = 5). In K10−/−, no ear loss was observed, regardless of cold exposure. BALB/C mice had ear loss after cold exposure in 4/5 mice (*change in ear length versus K10−/− mice T Test p = 0.008). B. Anti-K10+ human Raynaud’s serum depletion by incubation with his6-K10-loaded nickel-Sepharose beads yielded control serum levels of anti-K10 ELISA reactivity. Nickel-Sepharose mock depletion had minimal effect. C. In B6 mice, ear tissue was lost in 5/5 recipients of mock-depleted serum, each worse on the cold-exposed side, but in 0/5 recipients of anti-K10 depleted serum (*Fisher’s Exact p = 0.008). D. Bim−/− and germline control B6 mice (n = 10) received 50 μl anti-K10+ human serum; only cold-exposed ears shown. Squares: ear loss observed; Circles: no ear loss. Bim−/− mice were protected from ear loss versus Bim-intact mice (*Fisher’s Exact p = 0.033).
To further establish that anti-K10 antibodies mediated the ischemia-inducing effects of human Raynaud’s sera on study mice, we depleted anti-K10 from a patient serum (from an anti-RNP+ anti-K10+ Raynaud’s patient) using Sepharose beads bound to his6-K10, and transferred depleted or mock-depleted serum to BALB/C and K10−/− study mice as above (Figure 6B & 6C). Depletion of anti-K10 prevented the development of ear ischemic lesions in study mice, whether or not the ears received cold pre-exposure (though cold pre-exposure exacerbated tissue loss with mock-depleted serum).
Collectively, these observations provide further evidence of K10’s role in mediating cold-associated ischemic tissue loss induced by Raynaud’s patient sera.
Relevance of Bim to Raynaud’s Serum-Induced Ischemia
A Raynaud’s-associated pathway of serum-induced endothelial progenitor cell apoptosis mediated through Akt inhibition and subsequent upregulation of Bim has previously been identified by Zhu and colleagues (11). We therefore assessed whether Bim knockout mice would be protected from anti-K10 induced tissue loss (Figure 6D). We observed that Bim knockout mice were protected from anti-K10-induced tissue ischemia changes (after cold pre-exposure) compared to the background Bim-intact B6 strain.
Discussion
This report links Raynaud’s Phenomenon with an autoimmune response to a specific autoantigen, Cytokeratin 10. It demonstrates that direct serum transfer of anti-K10 antibodies is sufficient to induce RP-like ischemia in mice, shows that K10-knockout mice are protected from the development of ischemic tissue loss, and demonstrates in anti-K10 depletion studies that other antibodies providing redundant ischemia-inducing activity need not be present in anti-RNP sera.
Previous studies of Raynaud’s-associated serum-induced endothelial progenitor cell apoptosis have implicated Akt inactivation-induced Bim upregulation in the pathogenic pathway (11). In keratinocytes, K10 overexpression has been shown to lead to K10 aggregates that sequester and inactivate Akt (28). The indications in our studies that higher titers of anti-K10 antibodies may be more strongly linked to Raynaud’s manifestations supports the idea that anti-K10 antibodies may be inducing the formation of similar K10 aggregates that mediate endothelial apoptosis. Our findings of the Bim-dependence of serum-induced tissue ischemia in our murine model (Figure 6) are consistent with this mechanism of action.
Our observations that cold exposure enhances endothelial K10 expression (Figure 5) may help explain the predilection of Raynaud’s Phenomenon for cold-exposed areas, and may also help account for the reported seasonal variation in rates of RP flares. The mechanism for RP induction (and cold sensing) that we propose is dependent on anti-K10 antibodies and K10-expressing endothelial cells, without a requirement for neurological signals. However, this hypothesis does not exclude roles that neurologic signals can have in Raynaud’s Phenomenon. Rather, it identifies new pathways to explore to understand the mechanisms of neurological effects on RP. For example, nerve signals could potentially alter the set point for endothelial apoptotic induction (for example, by changing expression levels of K10, Bim or of other modulators of endothelial apoptosis). Alternatively, nerve signals could influence manifestations of Raynaud’s Phenomenon downstream of the induction of endothelial apoptosis (such as by influencing the extent/intensity of local vasospasm induced by an area of endothelial apoptosis).
Limitations of this work include disease-restricted (anti-RNP autoimmunity) assessment of the relationship between anti-K10 antibodies and Raynaud’s Phenomenon. Further study of patients with other forms of Raynaud’s-associated autoimmunity (such as subtypes of scleroderma with different clinical and autoantibody profiles) is needed to assess how general the association of anti-K10 with autoimmune RP may be.
Our results show that anti-K10 antibodies (even ones demonstrated to induce endothelial cell death) can be observed in RNP+ patients who either do or do not have clinical Raynaud’s Phenomenon. As shown in Table S1, anti-K10 positivity is barely seen in the majority of Raynaud’s patients in our cohort, and is seen in almost as large a percentage of patients without Raynaud’s as in those with Raynaud’s. While it is possible that some patients in whom anti-K10 antibodies were present at high titers but Raynaud’s had not been appreciated clinically may develop Raynaud’s in the future, our animal studies suggest that high titer anti-K10 antibodies are not the only factor that determines whether or not clinical Raynaud’s will emerge. While anti-K10 antibodies often led to profound tissue loss in B6 mice in our studies, in germline BALB/C mice high levels of anti-K10 antibodies typically led to only subtle (if at all noticeable) clinical changes, unless the endothelial apoptosis pathway activated by anti-K10 antibodies was primed (such as by prior cold induction of endothelial K10 expression). BALB/C mice express lower levels of K10 than B6 mice; sufficient levels of K10 expression are likely required for clinical expression of anti-K10-induced Raynaud’s. Our K10 depletion studies in vitro suggest that anti-RNP+ sera even in the absence of anti-K10/K10 interactions can induce at least some level of endothelial apoptosis, and may therefore potentiate the effects of anti-K10. Antibodies to such non-K10 specificities could also be associated with anti-K10 negative cases of Raynaud’s Phenomenon. In our experiments (Figure 5), it was not unusual for sera from RNP+ patients with Raynaud’s Phenomenon but with negative ELISA assays for anti-K10 to still show caspase-activating effects. Further studies will be needed to assess if anti-K10+ Raynaud’s patients have a worse prognosis regarding ischemic complications than anti-K10- Raynaud’s patients.
The rate of anti-K10 positivity observed in this anti-RNP+ cohort is similar to the percentage of MCTD patients reported by Szodoray and colleagues that develop an aggressive vasculopathic phenotype (29). Since the in vitro experiments in this study were performed in the absence of IL-6 and neutrophils, future studies will be needed to determine if the IL-6-associated apoptosis-inducing effects of scleroderma patient sera previously identified by Barnes, et al. may also interact with the K10-mediated endothelial apoptosis pathway (30). Future studies are warranted to explore whether endothelial expression levels of K10, Bim, and other components of this apoptotic pathway are linked with risk of Raynaud’s Phenomenon (and other forms of vasculopathy) in human cohorts, and whether modulation of the K10-induced apoptotic pathway may have diagnostic, prognostic, or therapeutic relevance in RP-associated autoimmune diseases.
Overall, our findings suggest a direct pathogenic link for the first time between humoral autoimmunity and Raynaud’s Phenomenon. They suggest a new series of potential therapeutic approaches to be tested in Raynaud’s Phenomenon that focus on the K10/Bim-mediated endothelial apoptosis pathway. The use of Raynaud’s Phenomenon animal models may expedite this discovery process and facilitate identification of therapeutic candidates for human trials. We have thus far only used our animal models in short-term studies to assess the effects of autoantibodies on the induction of tissue ischemic injury. These models, applied more chronically, could provide insight regarding the extent to which humorally-induced endotheliopathy might be a sufficient stimulus to reconstitute additional elements of scleroderma pathology.
Supplementary Material
Acknowledgments
We thank Judith Pignac-Kobinger, Lisa Harlow, and Ying Wang for technical assistance.
Grant Support/Funding: This work was supported by the United States Department of Veterans Affairs (ELG: Merit Review Grant); the National Institute of Arthritis and Musculoskeletal and Skin Diseases (awards AR48805 to ELG and AR071369 to DPA); the Scleroderma Foundation (ELG); the Community Foundation of Broward; the Audrey Love Foundation; and a gift from Mr. and Mrs. Terry Taylor.
Footnotes
The authors do not have any financial interests that could be perceived as being a conflict of interest. No patents pertaining to the results presented in the paper have been awarded or filed.
References
- 1.Voulgari PV, Alamanos Y, Papazisi D, Christou K, Papanikolaou C, Drosos AA. Prevalence of Raynaud’s phenomenon in a healthy Greek population. Ann Rheum Dis. 2000;59:206–10. doi: 10.1136/ard.59.3.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greidinger EL, Casciola-Rosen L, Morris SM, Hoffman RW, Rosen A. Autoantibody recognition of distinctly modified forms of the UI-70-kd antigen is associated with different clinical disease manifestations. Arthritis Rheum. 2000;43:881–888. doi: 10.1002/1529-0131(200004)43:4<881::AID-ANR20>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 3.Amanzi L, Braschi F, Fiori G, Galluccio F, Miniati I, Guiducci S, et al. Digital ulcers in scleroderma: staging, characteristics and sub-setting through observation of 1614 digital lesions. Rheumatology (Oxford) 2010;49:1374–82. doi: 10.1093/rheumatology/keq097. [DOI] [PubMed] [Google Scholar]
- 4.Chung L, Shapiro L, Fiorentino D, Baron M, Shanahan J, Sule S, et al. MQX-503, a novel formulation of nitroglycerin, improves the severity of Raynaud’s phenomenon: a randomized, controlled trial. Arthritis Rheum. 2009;60:870–7. doi: 10.1002/art.24351. [DOI] [PubMed] [Google Scholar]
- 5.Cooke JP, Marshall JM. Mechanisms of Raynaud’s disease. Vasc Med. 2005;10:293–307. doi: 10.1191/1358863x05vm639ra. [DOI] [PubMed] [Google Scholar]
- 6.Kahaleh B. The microvascular endothelium in scleroderma. Rheumatology (Oxford) 2008 Oct;47(Suppl 5):v14–5. doi: 10.1093/rheumatology/ken279. [DOI] [PubMed] [Google Scholar]
- 7.Bordron A, Dueymes M, Levy Y, Jamin C, Leroy JP, Piette JC, et al. The binding of some human antiendothelial cell antibodies induces endothelial cell apoptosis. J Clin Invest. 1998;101:2029–35. doi: 10.1172/JCI2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sgonc R, Gruschwitz MS, Boeck G, Sepp N, Gruber J, Wick G. Endothelial cell apoptosis in systemic sclerosis is induced by antibody-dependent cell-mediated cytotoxicity via CD95. Arthritis Rheum. 2000;43:2550–62. doi: 10.1002/1529-0131(200011)43:11<2550::AID-ANR24>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 9.Sgonc R, Gruschwitz MS, Dietrich H, Recheis H, Gershwin ME, Wick G. Endothelial cell apoptosis is a primary pathogenetic event underlying skin lesions in avian and human scleroderma. J Clin Invest. 1996;98:785–92. doi: 10.1172/JCI118851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Worda M, Sgonc R, Dietrich H, Niederegger H, Sundick RS, Gershwin ME, et al. In vivo analysis of the apoptosis-inducing effect of anti-endothelial cell antibodies in systemic sclerosis by the chorionallantoic membrane assay. Arthritis Rheum. 2003;48:2605–14. doi: 10.1002/art.11179. [DOI] [PubMed] [Google Scholar]
- 11.Zhu S, Evans S, Yan B, Povsic TJ, Tapson V, Goldschmidt-Clermont PJ, et al. Transcriptional regulation of Bim by FOXO3a and Akt mediates scleroderma serum-induced apoptosis in endothelial progenitor cells. Circulation. 2008;118:2156–65. doi: 10.1161/CIRCULATIONAHA.108.787200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eid AH, Maiti K, Mitra S, Chotani MA, Flavahan S, Bailey SR, et al. Estrogen increases smooth muscle expression of alpha2C-adrenoceptors and cold-induced constriction of cutaneous arteries. Am J Physiol Heart Circ Physiol. 2007;293(3):H1955–61. doi: 10.1152/ajpheart.00306.2007. [DOI] [PubMed] [Google Scholar]
- 13.Greidinger EL, Zang Y, Jaimes K, Hogenmiller S, Nassiri M, Bejarano P, et al. A Murine Model of Mixed Connective Tissue Disease (MCTD) Induced with U1-snRNP Autoantigen. Arthritis Rheum. 2006;54:661–9. doi: 10.1002/art.21566. [DOI] [PubMed] [Google Scholar]
- 14.Greidinger EL, Zang Y, Fernandez I, Berho M, Nassiri M, Martinez L, et al. Tissue targeting of anti-RNP autoimmunity: Effects of T cells and myeloid dendritic cells in a murine model. Arthritis Rheum. 2009;60:534–542. doi: 10.1002/art.24256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maldonado M, Perez M, Pignac-Kobinger J, Triana E, Tozman E, Greidinger EL, et al. Clinical and Immunologic Manifestations of Mixed Connective Tissue Disease (MCTD) in Miami Population Compared to Midwestern Caucasian MCTD Population. J Rheumatol. 2008;35:429–37. [PMC free article] [PubMed] [Google Scholar]
- 16.Haroon M, O’Gradaigh D, Foley-Nolan D. A case of Raynaud’s phenomenon in mixed connective tissue disease responding to rituximab therapy. Rheumatology (Oxford) 2007;46:718–9. doi: 10.1093/rheumatology/kem003. [DOI] [PubMed] [Google Scholar]
- 17.Rudolph SE, Kouba M, Hrdlicka P. Severe corticoid-refractory autoimmune thrombocytopenia associated with mixed connective tissue disease (Sharp’s syndrome). Treatment with rituximab. Dtsch Med Wochenschr. 2009;134:1734–8. doi: 10.1055/s-0029-1234008. [DOI] [PubMed] [Google Scholar]
- 18.Okawa-Takatsuji M, Aotsuka S, Uwatoko S, Takaono M, Iwasaki K, Kinoshita M, et al. Endothelial cell-binding activity of anti-U1-ribonucleoprotein antibodies in patients with connective tissue diseases. Clin Exp Immunol. 2001;126:345–54. doi: 10.1046/j.1365-2249.2001.01669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carpintero MF, Martinez L, Fernandez I, Romero AC, Mejia C, Zang YJ, et al. Diagnosis and risk stratification in patients with anti-RNP autoimmunity. Lupus. 2015;24:1057–66. doi: 10.1177/0961203315575586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greidinger EL, Foecking MF, Magee J, Wilson L, Ranatunga S, Ortmann RA, et al. A major B cell epitope present on the apoptotic but not the intact form of the U1-70-kDa ribonucleoprotein autoantigen. J Immunol. 2004;172:709–16. doi: 10.4049/jimmunol.172.1.709. [DOI] [PubMed] [Google Scholar]
- 21.Maricq HR, Weinrich MC. Diagnosis of Raynaud’s phenomenon assisted by color charts. J Rheumatol. 1988;15(3):454–9. [PubMed] [Google Scholar]
- 22.Reichelt J, Furstenberger G, Magin TM. Loss of keratin 10 leads to mitogen-activated protein kinase (MAPK) activation, increased keratinocyte turnover, and decreased tumor formation in mice. J Invest Dermatol. 2004;123:973–81. doi: 10.1111/j.0022-202X.2004.23426.x. [DOI] [PubMed] [Google Scholar]
- 23.Takeuchi O, Fisher J, Suh H, Harada H, Malynn BA, Korsmeyer SJ. Essential role of BAX, BAK in B cell homeostasis and prevention of autoimmune disease. Proc Natl Acad Sci U S A. 2005;102:11272–11277. doi: 10.1073/pnas.0504783102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nature Genetics. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 25.Zhu S, Liu X, Li Y, Goldschmidt-Clermont PJ, Dong C. Aging in the atherosclerosis milieu may accelerate the consumption of bone marrow endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2007;27:113–9. doi: 10.1161/01.ATV.0000252035.12881.d0. [DOI] [PubMed] [Google Scholar]
- 26.Zang Y, Martinez L, Fernandez I, Pignac-Kobinger J, Greidinger EL. Conservation of pathogenic TCR homology across class II restrictions in anti-ribonucleoprotein autoimmunity: extended efficacy of T cell vaccine therapy. J Immunol. 2014;192(9):4093–102. doi: 10.4049/jimmunol.1203197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harlow L, Rosas IO, Gochuico BR, Mikuls TR, Dellaripa PF, Oddis CV, et al. Identification of citrullinated hsp90 isoforms as novel autoantigens in rheumatoid arthritis-associated interstitial lung disease. Arthritis Rheum. 2013;65:869–79. doi: 10.1002/art.37881. [DOI] [PubMed] [Google Scholar]
- 28.Paramio JM, Segrelles C, Ruiz S, Jorcano JL. Inhibition of protein kinase B (PKB) and PKCzeta mediates keratin K10-induced cell cycle arrest. Mol Cell Biol. 2001;21:7449–59. doi: 10.1128/MCB.21.21.7449-7459.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szodoray P, Hajas A, Kardos L, Dezso B, Soos G, Zold E, et al. Distinct phenotypes in mixed connective tissue disease: subgroups and survival. Lupus. 2012;21:1412–22. doi: 10.1177/0961203312456751. [DOI] [PubMed] [Google Scholar]
- 30.Barnes TC, Spiller DG, Anderson ME, Edwards SW, Moots RJ. Endothelial activation and apoptosis mediated by neutrophil-dependent interleukin 6 trans-signalling: a novel target for systemic sclerosis? Ann Rheum Dis. 2011;70:366–72. doi: 10.1136/ard.2010.133587. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.