

Abstract
Background
Autosomal-dominant polycystic kidney disease (ADPKD) is the leading inherited renal disease worldwide. The proproliferative function of macrophages is associated with late-stage cyst enlargement in mice with PKD; however, the way in which macrophages act on cyst-lining epithelial cells (CLECs) has not been well elucidated.
Methods
We generated a rapid-onset PKD mouse model by inactivating Pkd1 on postnatal day 10 (P10) and compared cell proliferation and differential gene expression in kidney tissues of the PKD mice and wild-type (WT) littermates.
Results
The cystic phenotype was dominant from P18. A distinct peak in cell proliferation in polycystic kidneys during P22–P30 was closely related to late-stage cyst growth. Comparisons of gene expression profiles in kidney tissues at P22 and P30 in PKD and WT mice revealed that arginine metabolism was significantly activated; 204 differentially expressed genes (DEGs), including Arg1, an arginine metabolism–associated gene, were identified in late-stage polycystic kidneys. The Arg1-encoded protein, arginase-1 (ARG1), was predominantly expressed in macrophages in a time-dependent manner. Multiple-stage macrophage depletion verified that macrophages expressing high ARG1 levels accounted for late-stage cyst enlargement, and inhibiting ARG1 activity significantly retarded cyst growth and effectively lowered the proliferative indices in polycystic kidneys. In vitro experiments revealed that macrophages stimulated CLEC proliferation, and that L–lactic acid, primarily generated by CLECs, significantly upregulated ARG1 expression and increased polyamine synthesis in macrophages.
Conclusions
Interactions between macrophages and CLECs promote cyst growth. ARG1 is a key molecule involved in this process and is a potential therapeutic target to help delay ADPKD progression.
Keywords: polycystic kidney disease, metabolism, arginine, proliferation, macrophage
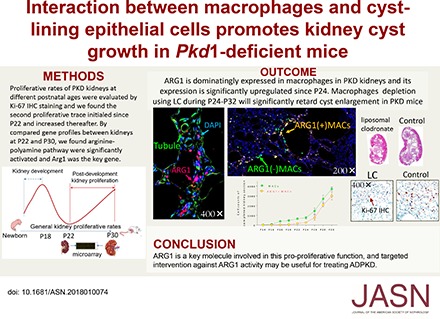
Autosomal-dominant polycystic kidney disease (ADPKD) is the leading cause of inherited kidney disease.1,2 The pathogens that cause ADPKD are complicated and related to intricate interactions among many cell types within polycystic kidney tissue. The conditional Pkd1 knockout mouse is an ideal model that mimics the course of ADPKD in humans with PKD1 gene deficiency. By comparing the gene expression profiles in middle- and late-stage kidneys from rapid-onset polycystic kidney disease (PKD) mice, we discovered that many metabolic elements participated in late-stage cyst growth and that arginine metabolism played an important role. Arginase-1 (ARG1) not only plays a key role in arginine metabolism, but also acts as a classic functional marker for macrophages. Macrophages infiltrate cysts in polycystic kidneys of late-stage patients and PKD animal models.3 Although the proproliferative function of macrophages in connection with cyst-lining epithelial cells (CLECs) has been explored in a few studies, the specific mechanism remains unclear.4–6 Macrophages are not homogenous, and they are generally categorized into two distinct phenotypes: classically activated (M1) or alternatively activated (M2).7 ARG1 is a classic M2-like molecular marker for macrophages and abundant M2-type macrophages are detected in polycystic kidney tissue.6 This histopathologic feature hints that ARG1 might play an important role in promoting late-stage cyst enlargement, which Leonhard et al.8 called the “snowball of cyst growth.” Thus, we hypothesized that there might be a correlation between the function of ARG1 in macrophages and the “snowball phenomenon” in polycystic kidneys.
Methods
Animal Experiments
Establishment of Rapid and Chronic-Onset PKD Mice
We crossed C57/BL6 Pkd1cond/cond mice with C57/BL6 tamoxifen-Cre (B6. Cg-Tg [Cre/Esr1]) 5Amc/J mice (stock 004682; Jackson Laboratories) to produce the mice used in this study. To generate the rapid-onset PKD mouse model, we induced Cre recombinase activity in mice at postnatal day 10 (P10) by intraperitoneal (i.p.) injection with tamoxifen (10 mg/kg) in coin oil (Sigma, St. Louis, MO).3 To generate the chronic-onset PKD mouse model, we induced Cre recombinase activity in mice at P30 by i.p. injection with tamoxifen (total dose: 300 and 100 mg/kg on three sequential days) in coin oil.9 All mice were euthanized by isoflurane treatment, and kidneys were processed for DNA and RNA extraction and histologic examination (fixed in 4% paraformaldehyde buffered solution, pH 7.3–7.4). A sample of the kidney specimens was flash frozen in liquid nitrogen and immediately stored at −80°C for further investigation. All experiments were performed using protocols approved by the Second Military Medical University Animal Care and Use Committee (SMMU-ACUC-20150430).
Macrophage Depletion in PKD Mice
A total of 37 rapid-onset PKD mice were randomized into four groups: stage 1‒liposomal clodronate (LC), which received 8-day (P16‒P23) LC treatment via i.p. injection (F70101C-N, Clophosome; 1 µl/g, daily) followed by 8-day (P24‒P32) liposomal vehicle (LV) treatment via i.p. injection (F70101C-NC, Clophosome; 1 µl/g, daily); stage 2‒LC, which received 8-day LV treatment (P16‒P23) followed by 8-day LC treatment (P24‒P32); stage 1+2‒LC, which received 16-day LC treatment; and stage 1+2‒LV, which received 16-day LV treatment.
Arginase Inhibition in PKD Mice
ARG1 was inhibited by once-daily i.p. injection of Nω-hydroxyl-L-arginine (nor-NOHA; Bachem, Bubendorf, Switzerland) after microcyst formation in the kidneys (from P14 in rapid-onset PKD mice and P180 for chronic-onset PKD mice). The doses of nor-NOHA used were 10 and 20 mg/kg, after reference to previous studies.10–13 Control PKD mice were treated with normal saline (1.5 µl/g, daily) via i.p. injection. The body weights of all mice were recorded daily.
Measurement of Renal Function
Plasma levels of creatinine were measured using a mouse creatinine assay kit (#80350; Crystal Chem, IL). Plasma levels of BUN were measured using a BUN colorimetric detection kit (#K024-H5; Arbor Assays, Ann Arbor, MI).
Cystic Index Calculation
Kidney sections (2-µm thick) were stained with hematoxylin and eosin using routine procedures. Cyst formation was quantified from sagittal sections of whole kidneys. All specimens were scanned using Aperio XT, Leica Aperio (Leica Biosystems), and the raw data were read using Imagescope (version 12.3). The cystic indices (CIs) of whole kidney sections were calculated. Briefly, high-resolution whole kidney images were divided (along the longitudinal axis) into 20–30 small regions. Cystic areas in each split image and the whole kidney area were measured using Image Pro Plus (version 6.0) and the cystic areas were summed using Microsoft Excel 2010 as follows:
 |
CIs were calculated from 5–6 kidney sections at each postnatal age and the mean CI was determined. The change in CI (ΔCI) was calculated using the formula:
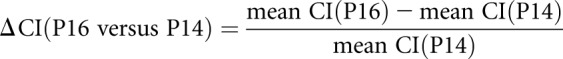 |
Identification of Cell Proliferation in Kidney Sections
Ki-67 immunohistochemical (IHC) staining was performed on paraffin sections (2-µm thick) of formalin-fixed tissue. All kidney sections were first incubated in 3% hydrogen peroxide (H2O2) followed by 4% serum from the host animal in which the relevant secondary antibody was generated. Then sections were incubated overnight at 4°C with primary antibodies against Ki-67 (#sc7844, 1:100; Santa Cruz Biotechnology; #ab15580, 1:100; Abcam). Signals were detected using Dako EnVision Detection Systems Peroxidase/DAB kit, Rabbit/Mouse (#K5007), and VECTASTAIN Elite ABC-HRP kits (peroxidase, goat IgG, #PK-6105, CA). Micrographs were randomly generated with a light microscope at ×400 magnification. The numbers of positive and total nuclei were counted using Image Pro Plus (version 6.0). The ratio of Ki-67–positive nuclei to the total number of nuclei was calculated as a percentage using Microsoft Excel (version 2010). Images were captured from 5–6 kidney specimens at each postnatal age and 12‒15 images were taken from each specimen (covering the cortex, cortico-medullary junction, and medulla).
Identification of Apoptotic Cells in Polycystic Kidneys
Apoptotic cells in renal sections were labeled using terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling (TUNEL) according to the manufacturer’s protocol (in situ Cell Death Detection Kit, Fluorescein [FITC-conjugated, green], #11684–795–910; Roche, Auckland, New Zealand). 4,6-Diamino-2-phenyl indole (DAPI, #P-36931; Thermo Fisher Scientific, blue) was applied to illuminate the nuclei. The apoptotic index was calculated as TUNEL-labeled nuclei/Total nuclei×100 (%).
RNA Isolation and Microarray Analyses
The Agilent Sure Print G3 Mouse GE Microarray (8×60K, ID: 028005) was used. Total RNA was quantified with a Nano Drop ND-2000 (Thermo Scientific), and RNA integrity was assessed with an Agilent Bioanalyzer 2100 (Agilent Technologies). Sample labeling, microarray hybridization, and washing were performed according to the manufacturer’s instructions. Briefly, total RNA was transcribed to double-strand cDNA, synthesized into cRNA, and labeled with cyanine-3-CTP; the labeled cRNA was hybridized onto the microarray. After three washes, the arrays were scanned with the Agilent Scanner G2505C. Feature Extraction (version 10.7.1.1; Agilent Technologies) was used to analyze array images to obtain raw data. Gene Spring (version 13.1; Agilent Technologies) was used to complete basic analyses of the raw data. The raw data were first normalized with the quantile algorithm. Probes with at least 100% of the values in any one of the conditions with flags in “detected” were chosen for further data analyses. Then we identified differentially expressed genes (DEGs) through fold changes. The threshold set for up- and downregulated genes was a fold change ≥2.0. Afterward, gene ontology and KEGG analyses were used to determine the roles of the differentially expressed mRNA.
Real-Time PCR
Total RNA was extracted using an RNase mini-kit (Invitrogen, Carlsbad, CA) and reverse-transcribed. The primer sequences were as follows: Arg1, primer sense (F), 5′-CTC CAA GCC AAA GTC CTT AGA G-3′ and primer anti-sense (R), 5′-GGA GCT GTC ATT AGG GAC ATC-3′; Nos2, primer F, 5′-GTT CTC AGC CCA ACA ATA CAA GA-3′ and primer R, 5′-GTG GAC GGG TCG ATG TCA C-3′; Mrc1, primer F, 5′-AAG GCA AGG ATG GAT ACT G-3′ and primer R, 5′-GTG AAG GTG GAT AGA GTG G-3′; Odc1, primer F, 5′-TTG CCC AGA GTC TGT TGG TG-3′ and primer R, 5′- GGG TTG AGG GAA CAG TCA GG-3′; Smox, primer F, 5′-AGC TCC AAG ACA GCG CAT AG-3′ and primer R, 5′-GGT CCA GGG ATT GTT CTG GG-3′; and Gapdh, primer F, 5′-AGA ACA TCA TCC CTG CAT CC-3′ and primer R, 5′-ATA CCA GGA AAT GAG CTT GAC-3′. Real-time PCR was performed using SYBR Green PCR Master Mix (Toyobo, Osaka, Japan) and the Rotor-Gene 3000A real-time PCR system (Corbett, Sydney, Australia) according to the manufacturers’ instructions. In brief, the PCR amplification reaction mixture (20 μl) contained 2 μl cDNA, 0.4 μl F primer, 0.4 μl R primer, and 10 μl SYBR Green I. After initial denaturation at 95°C for 1 minute, the reaction was cycled 45 times. Each cycle consisted of denaturation at 95°C for 15 seconds and primer annealing and extension at 60°C for 31 seconds. Results are shown as the relative expression of Arg1, Nos2, Mrc1, Odc1, and Smox normalized to the expression of Gapdh. Real-time PCR was performed in triplicate for each experiment, and the average values were measured. Each experiment was repeated three times. Using the gene-specific efficiencies, mRNA relative expression folds were calculated as 2−ΔΔ circle threshold.14
Western Blotting Analyses
Snap-frozen kidney tissue was homogenized in freshly prepared tissue protein extraction reagent (Pierce Bioscience, Rockford, IL). Homogenates were centrifuged and the supernatants were stored at −80°C. Lysates in SDS sample buffer were boiled for 5 minutes at 95°C, and equal amounts of protein were resolved by SDS-PAGE before transfer to a polyvinylidene difluoride membrane. The membrane was probed with primary antibody overnight at 4°C, followed by incubation with the secondary antibody for 2 hours at room temperature. Proteins of interest were visualized by enhanced chemiluminescence. The average densitometry values of three independent experiments were measured. The primary antibodies were as follows: anti-ARG1 (#ab91279, 1:1000; Abcam), anti-ODC1 (#ab193338, 1:2000; Abcam), anti-SMOX (#ab213631, 1:2000; Abcam), anti-P-ERK 1/2 (#4370, 1:1500; CST), anti-ERK 1/2 (#4695, 1:1000; CST), and anti-GAPDH (#ab8245, 1:3000; Abcam).
Laser-Scanning Confocal Imaging
For multiple fluorescence–IHC staining, deparaffinized kidney sections were incubated in 4% FBS in PBS for 30 minutes followed by coincubation with FITC-conjugated anti-F4/80 (#11–4801–82, 1:50; eBioscience, green), anti-NOS2 (#ab15323, 1:50; Abcam), and anti-ARG1 (#ab60176, 1:50; Abcam) antibodies overnight at 4°C. Then, secondary antibodies were applied according to the origin of the primary antibodies: Alexa Fluor 647-conjugated donkey anti-rabbit IgG (#ab150075, red), Alexa Fluor 647-conjugated donkey anti-goat IgG (#ab150131, red), Alexa Fluor 488-conjugated goat anti-rabbit IgG (#ab150077, green), Alexa Fluor 488-conjugated donkey anti-goat IgG (#ab150129, green), and Alexa Fluor 405-conjugated donkey anti-rabbit IgG (#ab175651, blue). Anti-wheat germ agglutinin with autofluorescence (FITC-conjugated WGA, #ab20528; Abcam, green) was used to illuminate the tubular structures. Samples were mounted in Prolong Gold Antifade with DAPI (blue) and visualized using a Leica TCS SP5 confocal scanning laser microscope (Leica Microsystems, IL) and LAS AF Lite (version 2.6). Red, green, and blue images were merged using Image Pro Plus (version 6.0). Field images were taken with the ×400‒600 objective.
Images were acquired for positive whole-cell quantification using the automated image acquisition mode of Panoramic P250 (3DHISTECH, Budapest, Hungary). High-resolution images of whole sagittal sections were divided into 40–50 parts, and cells in each part were counted with Image Pro Plus (version 6.0). Microsoft Excel 2010 was used to determine the total count.
L-Lactate Assays
Kidney tissue (50 mg) was homogenized in 1 ml ice-cold 10% TCA on ice. The same kidney tissue/TCA ratio was used for all samples. Samples were centrifuged for 5 minutes at maximum speed. A 0.5-ml aliquot of the supernatant was transferred to a new microtube. Saturated ether (0.5 ml) was added to the supernatant, and the samples were vortexed for 20 seconds. The last two steps were repeated three times. Samples were left open in the fume hood for 30 minutes, and 50 µl each sample was mixed with 50 μl polyethylene glycol solution. The solution was mixed vigorously, incubated on ice for 30 minutes, and centrifuged at 13,000 rpm for 5 minutes at 4°C. The supernatant was transferred to a fresh tube and diluted five-fold with ice-cold dH2O before the assay. Then, an L-lactate (L-LA) assay kit (Cat#: A-108S and A-108L; Department of Biochemistry, University at Buffalo, Buffalo, NY) was used to measure the levels of L-LA in the kidney samples.
Nitric Oxide Assays
Nitric oxide (NO) levels in all samples were assayed using a Quanti Chrom Nitric Oxide Assay Kit (D2NO-100; BioAssay Systems, Hayward, CA). Tissues were homogenized in PBS and centrifuged at 10,000 rpm at 4°C; the supernatant was used for NO assays. All samples were deproteinated before assay. The standards (100, 60, 30, and 0 μM) were prepared using Premix and H2O. Before the reaction, working reagent for all samples and standards was first prepared as follows: 100 µl Reagent A+4 µl Reagent B+100 µl Reagent C for each sample. Then, 200 µl mixed working reagent was added to each sample and incubated for 10 minutes at 60°C. A total of 250 µl each sample and standard was transferred to 96-well plates and the OD at 540 nm was read. The OD was plotted against the standard concentration and the slope was calculated. Finally, NO concentrations were calculated as (OD [sample]−OD [blank])/Slope.
Targeted Metabolomics Analyses of L-Arginine, L-Ornithine, and Polyamine
An aliquot of each individual kidney sample or collected cells was precisely weighed and transferred to a microfuge tube. After adding 1 ml extraction solution (acetonitrile/methanol/water=2:2:1), the samples were vortexed for 30 seconds, homogenized at 45 Hz for 4 minutes, and sonicated for 5 minutes in an ice-water bath. The homogenization and sonication steps were repeated three times, followed by incubation at −20°C for 1 hour and centrifugation at 12,000 rpm and 4°C for 15 minutes. An 80-µl aliquot of the clear supernatant was transferred to an autosampler vial for liquid chromatography tandem mass spectrometry analyses. Stock solutions were prepared individually by dissolving or diluting each standard substance to a final concentration of 10 mmol/L. All standards were purchased from Sigma-Aldrich: putrescine (51799), spermine (S4264), spermidine (S0266), mixed amino acid standards (A9906), and 4-aminobutyraldehyde (A44150). A series of calibration standard solutions was prepared by stepwise dilution of this mixed standard solution with extraction solution. UHPLC separation was performed using an Agilent 1290 Infinity I Series UHPLC system (Agilent Technologies) equipped with a Water Acquity UPLC BEH Amide column (2.1×100 mm, 1.7 μm). An Agilent 6460 triple quadrupole mass spectrometer equipped with an AJS-ESI interface was used for assay development. The MRM parameters were optimized for each of the targeted analytes using flow injection analyses by injecting the standard solutions of the individual analytes into the API source of the mass spectrometer. At least two MRM transitions (i.e., the Q1/Q3 pairs) per analyte were obtained, and the two most sensitive transitions were used in the MRM scan mode to optimize the collision energy for each Q1/Q3 pair. Between the two MRM transitions per analyte, the Q1/Q3 pairs that showed the highest sensitivity and selectivity were used as the MRM transitions for quantitative monitoring. The additional transitions acted as qualifiers for verifying the identity of the target analytes. Agilent Mass Hunter Work Station (B.08.00) was used for MRM data acquisition and processing. The working standard solution was diluted in a stepwise manner with the extraction solution, with double dilution factors. Standard solutions were subjected to UHPLC-MRM-MS analyses. Signal-to-noise ratios were used to determine LLODs and LLOQs. LLODs and LLOQs were defined as the analyte concentrations that led to peaks with signal-to-noise ratios of 5 and 20, respectively.
In Vitro Experiments
Mouse Kidney–Derived Macrophage Cultures
The method used has previously been described in human and swine macrophage isolations and cultures.15,16 Briefly, after anesthesia and euthanasia, the kidneys were dissected from P22, P26, and P30 PKD mice. The kidney capsule was carefully removed and the kidneys were cut into small pieces (1‒2 mm3). Next, the minced pieces were digested by incubation with dissociation medium (PBS solution containing 100 μg/ml collagenase/dispase [#10269638001; Roche] and 10% FBS [Gibco, CA]; the medium was sterilized on a 0.22-µm filter before application) for 1 hour at 37°C in a gently shaking water bath or rotator. Then the kidney tissue was poured onto a 100-µm cell strainer in PBS using a scraper while the liquid was collected into a 50-ml tube underneath. Next, the tissue was rinsed twice using 2‒3 ml cold PBS containing 5 mM EDTA (#798681; Sigma) and then gently pushed through the strainer using the rubber end of a syringe plunger. Then the plunger and mesh screen were rinsed three times with PBS-EDTA. After the tube was left on ice for 4 minutes until the particulate matter settled, the supernatant was transferred to a new tube, centrifuged for 10 minutes at 1500 rpm, and the sediment washed with DMEM/F12 medium supplemented with 10% FBS. Then the cells were resuspended in macrophage growth medium composed of DMEM/F12 medium containing 10% FBS, 100 μM β-mercaptoethanol (#97622; Sigma), 5 μg/ml insulin, streptomycin-penicillin (1:1000), and 10 μg/ml Fungin (#ant-fn-2; InvivoGen, CA). Next, the cells were split into Thermo Scientific Nunc EasY Flasks (75 cm2) and cultured at 37°C in an atmosphere of 95% air/5% CO2. The culture media were replaced every 3 days. After 7 days of culture, a mixed monolayer cell sheet was observed and macrophage-like cells began to proliferate on the cell sheets. Because macrophages can adhere firmly to substrata when cultured on plastic flasks in the presence of serum, the adherent cells were detached using PBS supplemented with 5 mM EDTA. Cells (0.5‒1.0×105 cells/flask) were harvested every 3–4 days.
Isolating Mouse Bone Marrow–Derived Macrophages
Bone marrow–derived macrophages (BMDMs) were isolated from P22, P26, and P30 wild-type (WT) mice. The method used has been reported previously.4,5 Briefly, a 23-gauge needle was inserted into bone marrow cavities and bone marrow cells were flushed out into a 50-ml tube; the process was done three times to release as many cells as possible. Next, cells were spun down at 1500 rpm for 5 minutes at room temperature, the supernatant was discarded, and the cells were resuspended in 5 ml Roswell Park Memorial Institute (RPMI)–1640 medium (Gibco) using a syringe fitted with a blunt-ended 18-gauge needle. Next, the cells were plated on 100-mm plates in 10 ml RPMI-1640 medium supplemented with 20 ng/ml colony stimulating factor 1 (#SRP3221; Sigma). Cells were maintained in this media for 1 week at 37°C with 95% air/5% CO2 with minimal media changes to prevent contaminating cell growth. On day 7, the cells were washed with PBS, detached by incubating with PBS-EDTA for 10 minutes, and centrifuged at 1500 rpm for 5 minutes; the sediment was collected. Finally, the macrophages were cultivated in warm RPMI-1640 medium.
Transwell Coculture of Human CLECs and RAW264.7 Macrophages
Human immortalized CLECs (OX-161) and renal tubular epithelial cells (RTECs) (UCL-93) were kindly provided by Professor A.C. Ong (University of Sheffield, Sheffield, UK) and cultured as described previously.17 RAW264.7 cells and CLECs were cocultured in a transmembrane system (Transwell; Corning, Lowell, MA, USA) with 0.4-μm pores. In shared coculture media, CLECs were cultured in the lower well and RAW264.7 cells were cultured in the upper well. The ratio of cell seeding of RAW264.7 cells to CLECs was 1:5.
Urea Detection
Urea levels were assessed using a Megazyme L-arginine/urea/ammonia rapid assay kit (K-LARGE; Megazyme International Ireland, Wicklow, Ireland) according to the manufacturer’s instructions.
Cell Cycle Analyses
After washing with PBS, CLECs were suspended in 70% ice-cold ethanol and kept overnight at 4°C. Then they were rehydrated in PBS at a density of 1×106 cells/ml. After RNase digestion, the CLECs were stained with 50 μg/ml propidium iodide. Flow cytometry analyses were performed on the basis of red emissions at 630 nm and the data were analyzed using a Beckman Coulter CyAn ADP analyzer (Sheffield, UK).
5-Ethynyl-2’-Deoxyuridine Assay for Cell Proliferation
The proliferation of CLECs was evaluated using Molecular Probes Click-iT EdU Imaging Kit (Invitrogen, Carlsbad, CA). First, the cells were plated on coverslips overnight and 2×5-ethynyl-2’-deoxyuridine (EdU) working solution was used to dilute the media containing cells. Then the cells were incubated for 4 hours followed by immediate fixation and permeabilization. The Click-iT reaction cocktail (0.5 ml) was added to each well with a coverslip and incubated for 30 minutes in the dark. DNA was stained with Hoechst 33342 and the samples were imaged via Olympus BX-51 fluorescence microscopy. Each well was washed twice with 1 ml PBS between the two steps.
Statistical Analyses
All results are presented as mean±SD. Variables were compared using t tests and Mann–Whitney U tests as appropriate. Data analyses were performed using SPSS (version 16.0), and P<0.05 was considered to indicate significance.
Results
Cell Proliferation in Kidney Tissues of PKD Mice
The Pkd1 gene was inactivated by tamoxifen at P10 to generate a rapid-onset conditional knockout mouse model (PKD mice).3 IHC staining for Ki-67 was used to evaluate the proliferative indices of polycystic and WT kidneys at different postnatal ages (Figure 1A). Most Ki-67–stained nuclei were located in the cyst-lining epithelial tissue and remaining tubules; only a few were located in mesenchyme cells (Figure 1B). In rapid-onset polycystic kidneys, the distribution of proliferating epithelial cells was homogeneous and widely covered the renal cortex, medulla, and junctional distinct (Figure 1C). There were two marked peaks of proliferation over the course of the disease. The first peak occurred during the developmental period from P14 to P18; however, the second peak, which occurred from P22 to P30, occurred only in polycystic kidneys. The proliferative indices of polycystic kidneys broke through the physiologic breaking point during the kidney development and further increased from P22 onward (Figure 1D). In addition, the two-kidney weight/body weight (2KW/BW) ratios began to increase at P22 in a time-dependent manner (Figure 1E).
Figure 1.

Cell proliferation in the kidney tissues of mice with PKD. (A) Polycystic kidney and age-matched WT kidney sections were stained with Ki-67 (red arrows, ×400). (B) Multichannel immunofluorescence colocalization of Ki-67 (red), fluorescein wheat germ agglutinin (WGA; green, to illuminate the tubular structures), and DAPI (blue) in P30 polycystic kidney sections, ×400. (C) The distribution of proliferative cells in polycystic kidney tissues at P30, as shown by Ki-67 IHC staining, ×600. (D and E) Comparison of the (D) proliferative indices (percentage of positive nuclei stained by Ki-67) and (E) 2KW/BW ratios between PKD mice and WT littermates; *P<0.05. (F) Whole kidney section scans after hematoxylin and eosin staining. Microcysts formed at P14. (G) CIs (red trace, shown on left y axis) and the change in CI (ΔCI, blue trace, shown on right y axis) of polycystic kidneys at different postnatal ages. Data are presented as means±SDs of five or six mice at different postnatal ages. C-M, cortico-medullary; vs, versus.
Whole kidney sections stained with hematoxylin and eosin were scanned and CIs were calculated (Figure 1F). CIs increased with postnatal age and the ΔCI exhibited two increasing phases (Figure 1G). In parallel with cyst enlargement, renal functions deteriorated continuously with postnatal age in PKD mice (Supplemental Figure 1). TUNEL staining revealed that the apoptotic cell count was persistently low in polycystic kidneys during P22‒P30 (Supplemental Figure 2).
Gene Expression Profiles in Kidney Tissues from PKD Mice
Gene expression profiles were compared between polycystic kidney tissues at P22 and P30 to discover which elements contributed to “snowball” cyst enlargement (Figure 2A). Compared with P22 WT littermates, the complement and coagulation cascades were the most activated pathways in age-matched polycystic kidney tissues (Figure 2B), whereas metabolic pathways were the most activated pathways in P30 polycystic kidney tissues (Figure 2C). Of all metabolism-associated pathways, the arginine-polyamine pathway was the most significantly activated (Figure 2D). Three enzymes, ARG-1, ornithine decarboxylase-1 (ODC-1), and spermine oxidase (SMOX), regulate this pathway.18 As shown in Supplemental Figure 3A, Arg1 and Smox, but not Odc1, were significantly upregulated in polycystic kidneys at P30 compared with at P22 and compared with WT kidneys at P30 and P22. The cellular localization and expression of the three enzymes in polycystic kidneys are shown in Supplemental Figure 3, B‒F and Table 1. Compared with WT littermates, significantly higher levels of L-arginine, L-ornithine, and spermidine were detected in the cystic kidneys of PKD mice at P30 compared with P22 counterparts. Interestingly, spermine was not detected in any polycystic or WT kidney specimens (Supplemental Figure 3G). Thus, the expression of ARG1 and SMOX were significantly upregulated and more polyamine metabolites were produced in polycystic kidneys at P30 compared with age-matched WT littermates and polycystic counterparts at P22 (Figure 2E).
Figure 2.

Arginine-polyamine pathway is activated in late-stage polycystic kidneys. (A) Kidney specimens from P22 and P30 PKD mice and age-matched WT littermates were collected and microarrays were used to compare DEGs and to analyze significantly activated signaling pathways (Kyoto Encyclopedia of Genes and Genomes [KEGG] pathway analyses). P22 and P30 were the starting point and peak, respectively, of the second proliferative index of polycystic kidneys. (B) KEGG signaling pathway analyses of kidney tissues from P22 PKD mice versus WT littermates. (C) KEGG signaling pathway analyses of kidney tissues from P30 PKD mice versus WT littermates. (D) Many metabolic pathways were significantly activated in kidney tissues from P30 PKD mice versus WT littermates; arginine metabolism was the leading activated pathway. (E) Substrate, enzymes, and metabolites of the arginine-polyamine pathway. Red up arrow, upregulation; green down arrow, downregulation; black up arrow, significantly increased metabolite production; solid line, activated pathway; dashed line, inactivated pathway. (F) Multi-pool comparison of kidney DEGs: Pool-1, P30 versus P22 PKD mice (yellow circle); Pool-2, P22 PKD mice versus WT littermates (blue circle); Pool-3, P30 PKD mice versus WT littermates (green circle); Pool-4, P30 versus P22 WT mice (red circle). In total, 204 DEGs were expressed specifically in P30 polycystic kidneys. Arg1 was included in the 204 DEGs. vs, versus.
Table 1.
Location and expression of ARG1, ODC1, and SMOX in polycystic kidneys
| Location/Expression | Key Enzymes | ||
|---|---|---|---|
| ARG1 | ODC1 | SMOX | |
| Cellar location | |||
| CLECs | √ | ||
| RTECs | √ | ||
| Mesenchymal cells | √ | √ | √ |
| Subcellular localization | |||
| Cytoplasm | √ | √ | |
| Nuclei | √ | ||
| mRNA expression in kidneys | |||
| P22 PKs versus P22 WTKs | ↑ | ||
| P30 PKs versus P30 WTKs | ↑ | ↑ | |
| P30 PKs versus P22 PKs | ↑ | ↑ | |
√, position where the enzymes were located; ↑, upregulation; PKs, polycystic kidneys; WTKs, WT kidneys.
DEGs were compared between polycystic kidney tissues at P30 and P22 (Pool-1), polycystic and WT kidneys at P22 (Pool-2), polycystic and WT kidneys at P30 (Pool-3), and WT kidneys at P30 and P22 (Pool-4). Ultimately, 204 DEGs were identified in the overlapping regions of Pool-1, Pool-2, and Pool-3, and the DEGs from Pool-4 were excluded (Figure 2F). Arg1 is an exclusive arginine metabolism gene that was also included in the candidate DEGs (Supplemental Figure 4).
Localization and Expression of ARG1 in Kidney Tissues of PKD Mice
The expression of ARG1, Arg1 encoding protein, was investigated in infiltrating macrophages of polycystic kidneys. ARG1(+) and total macrophages in whole kidney sections over the disease course are summarized in Figure 3A. F4/80 is a specific marker of mouse monocytes/macrophages.19 Infiltration of F4/80-stained macrophages was more easily observed in cystic kidneys at P18, but ARG1 expression lagged approximately 6‒8 days behind that of F4/80. Approximately 50% of ARG1(+) macrophages costained with Ki-67 in P22 polycystic kidneys but the population presented continuous decrease onward (Supplemental Figure 5). The mRNA expression of kidney-derived macrophages isolated from polycystic kidneys at P22, P26, and P30 and BMDMs isolated from age-matched WT littermates was investigated further. The expression of M2-like markers Arg1 and Mrc1 was upregulated in kidney-derived macrophages at all postnatal ages compared with BMDMs, and in polycystic kidneys in a time-dependent manner (P30>P26>P22, P<0.05). The expression of gene encoding NO synthase 2 (Nos2) exhibited the opposite (Figure 3B). As shown in Figure 3C, ARG1 was mostly located in interstitial tissues rather than tubular structures. Multichannel-immunofluorescent cell localization revealed that macrophages were the principal source of ARG1 in polycystic kidneys and that, interestingly, ARG1 exhibited time-dependent upregulation in macrophages (Figure 3D). However, the expression of NOS2 in macrophages had the opposite trend (Figure 3E). The populations of NOS2(+)/ARG1(−), NOS2(−)/ARG1(+), and NOS2(−)/ARG1(−) macrophages are compared among different postnatal ages in Supplemental Figure 6.
Figure 3.

Localization and expression of ARG1 and NOS2 in kidney tissues from PKD mice. (A) Total macrophages (labeled by F4/80, green) and ARG1-positive macrophages (bilabeled by F4/80 and ARG1, yellow) in whole polycystic kidney sections were counted, ×200. (B) mRNA expression of Arg1, Mrc1, and Nos2 in kidney-derived macrophages (KDMs) isolated from cystic kidney specimens of P22, P26, and P30 PKD mice versus BMDMs that were isolated from age-matched WT littermates; *P<0.05 versus age-matched polycystic kidneys. (C) Trichannel laser scanning confocal microscopy for WGA (green, to illuminate the tubular structure), ARG1 (red), and DAPI (blue) in polycystic kidney sections at P30, ×400. (D and E) Trichannel laser scanning confocal microscopy for F4/80 (green), ARG1/NOS (red), and DAPI (blue) in polycystic kidney sections (P22–P30); the ratios of NOS2- and ARG1-stained macrophages in polycystic kidney tissues were also compared. *P<0.05 versus P22 cystic kidneys; #P<0.05 versus P24 cystic kidneys; ψP<0.05 versus P26 cystic kidneys; ϕP<0.05 versus P28 cystic kidneys. Data are presented as means±SDs with 10‒12 images taken from 5–6 mice at different postnatal ages.
Role of Multiple-Range Macrophage Depletion in Postponing Cyst Growth
During P16‒P23, NOS2(+)/ARG1(−) and NOS2(−)/ARG1(−) macrophages played a dominant role in the population. From P24, macrophages began to upregulate ARG1 and the NOS2(−)/ARG1(+) macrophage count increased continuously thereafter (Figure 4A). On the basis of these findings, multistage macrophage depletion was performed to explore the effects of macrophages expressing ARG1 on cyst growth (Figure 4B). Macrophages were depleted with LC via i.p. injection (1 μl/g, daily) from P16 to P23 (stage 1); stage 2 was defined as the period from P24 to P32. Age-matched PKD mice received equal volumes of LV (1 μl/g, daily, i.p.). Unfortunately, LC intervention failed to significantly improve the general survival rates of PKD mice compared with those receiving LV (P=0.08, Mantel–Cox test, Figure 4C). LC can be swallowed by macrophages, resulting in cell death within 24 hours. At P32, LV-treated PKD mice exhibited numerous infiltrating macrophages in the kidneys, whereas few were detected in their LC-treated counterparts (Figure 4D). Macrophage depletion during stage 2 or stage 1+2 postponed cyst growth, lowered 2KW/BW ratios, and improved CIs, whereas stage 1 macrophage depletion failed to slow cyst growth compared with LV-treated PKD mice (Figure 4, E‒G). Moreover, reduced cyst growth was combined with valid renal protection (Figure 4H) and inhibited L-LA production in LC-treated polycystic kidneys (Figure 4I). Ki-67 staining revealed significantly lower proliferative indices in stage 2 and stage 1+2 LC-treated polycystic kidneys compared with their stage 1 LC- or LV-treated counterparts, although no significant differences were found between the latter two groups (Figure 4J).
Figure 4.

Role of multiple-range macrophage depletion in postponing cyst growth in PKD mice. (A) The transition of macrophage populations over the disease course in rapid-onset polycystic kidneys: green cells, NOS2(−)/ARG1(−) macrophages; blue cells, NOS2(+)/ARG1(−) macrophages; and red cells, NOS2(−)/ARG1(+) macrophages. (B) LC-mediated multistage macrophage depletion was performed in PKD mice to investigate the proproliferative function of macrophages expressing ARG1. Stage 1, P16‒P23; stage 2, P24‒P32. (C) The general survival rates were compared among PKD mice receiving stage 1 LC, stage 2 LC, stage 1+2 LC, and stage 1+2 LV treatment. (D) Compared with controls treated with LV, macrophages (green) were thoroughly eliminated from the kidneys, ×200. Nuclei were illuminated by DAPI (blue). (E and F) Kidney CIs showed a significant decrease in stage 2/1+2 LC-treated PKD mice compared with LV-treated and stage 1 LC-treated PKD mice, paralleling (G) notably lower 2KW/BW ratios, (H) improved renal function, and (I) significantly inhibited L–lactic acid production in kidney tissues. *P<0.05 versus stage 1 LC-treated PKD mice; #P<0.05 versus stage 2 LC-treated PKD mice; ψP<0.05 versus stage 1+2 LC-treated PKD mice; ϕP<0.05 versus LV-treated PKD mice. (J) Cell proliferation was significantly reduced in the kidneys of stage 2/1+2 LC-treated mice compared with LV-treated and stage 1 LC-treated PKD mice, ×400. *P<0.05 versus stage 1 LC-treated PKD mice; #P<0.05 versus stage 2 LC-treated PKD mice; ψP<0.05 versus stage 1+2 LC-treated PKD mice; ϕP<0.05 versus LV-treated PKD mice. Data are presented as means±SDs, with 10‒12 images taken from 4–9 mice undergoing the different treatments.
Effects of ARG1 Inhibition on Cyst Enlargement
The balance of ARG1 and NOS2 activities in arginine metabolism closely relates to cellular functions and behaviors,12,20–22 and Nor-NOHA was applied to inhibit ARG1 activity in polycystic kidneys (Figure 5A). In general, short-term nor-NOHA treatment failed to improve survival rates in PKD mice (Figure 5B). However, nor-NOHA treatment protected renal function and lowered 2KW/BW ratios, slowed cyst growth, and decreased proliferative indices (Figure 5, C–G). Reduced cyst enlargement was paralleled by significantly decreased L-LA production in polycystic kidney tissues (Figure 5H). Nor-NOHA treatment (20 mg/kg) not only significantly downregulated the expression of ARG1 in kidney-derived macrophages (Figure 5I, also see Supplemental Figure 7, A and B), but also remarkably inhibited the generation of polyamine metabolites in kidney tissues compared with normal saline–treated controls (also see Supplemental Figure 7C). To investigate the efficiency of long-term ARG1 inhibition in ADPKD, we generated a chronic-onset PKD model. Compared with short-term treatment, nor-NOHA intervention for 4 months was superior at slowing cyst growth, protecting renal functions, and improving survival rates in chronic-onset PKD mice. The data are detailed in the supplemental material (Supplemental Material, Supplemental Figure 8).
Figure 5.

Role of inhibiting ARG1 in delaying cyst growth in PKD mice. (A) Twenty-eight PKD mice were randomized into two groups: one group (n=14) received 18-day nor-NOHA treatment (seven mice received nor-NOHA at a dosage of 10 mg/kg daily and seven mice received nor-NOHA at a dosage of 20 mg/kg daily, i.p.) and the other group (n=14) received normal saline (NS, 1.5 μl/g, daily, i.p.). The intervention was initiated at P14 and all mice were euthanized at P32. (B) Eighteen-day nor-NOHA treatment failed to significantly improve the general survival rates in PKD mice (P=0.14, Mantel–Cox test). (C‒E) Treatment with any dose of nor-NOHA significantly improved the renal function of PKD mice ((C), creatinine [Cr]; (D), BUN) and (E) lowered their 2KW/BW ratios. *P<0.05 versus 10 mg/kg nor-NOHA–treated PKD mice; #P<0.05 versus 20 mg/kg nor-NOHA–treated PKD mice; ψP<0.05 versus NS-treated PKD mice. (F) Treatment with nor-NOHA moderately but significantly improved CIs. *P<0.05 versus 10 mg/kg nor-NOHA–treated PKD mice; #P<0.05 versus 20 mg/kg nor-NOHA–treated PKD mice. (G) Nor-NOHA inhibited cell proliferation in a dose-dependent manner; Ki-67 IHC staining, ×400. (H) Treatment with nor-NOHA significantly alleviated the production of L–lactic acid in kidney tissues. *P<0.05 versus 10 mg/kg nor-NOHA–treated PKD mice; #P<0.05 versus 20 mg/kg nor-NOHA–treated PKD mice; ψP<0.05 versus NS-treated PKD mice. (I) Any dose of nor-NOHA remarkably downregulated the expression of ARG1 in polycystic kidneys in a dose-dependent manner. *P<0.05 versus 20 mg/kg nor-NOHA–treated PKD mice; #P<0.05 versus 10 mg/kg nor-NOHA–treated PKD mice; ψP<0.05 versus NS-treated PKD mice. Data are presented as means±SDs, with 4‒6 mice treated with different interventions.
Interactions between Macrophages and CLECs
L-LA levels significantly increased with increasing postnatal age in polycystic kidney tissues (Figure 6A). In addition, CLECs secreted L-LA in a time-dependent manner. CLECs were incubated with RAW264.7 cells in a Transwell system (TS). The concentrations of L-LA were significantly higher in DMEM from the TS compared with CLECs at 72 and 96 hours; CLECs produced significantly more L-LA than RTECs at 96 hours. The production of L-LA in RAW264.7 cells was extremely low compared with CLECs and RTECs (Figure 6B); therefore, the L-LA detected in the DMEM of TS was predominantly derived from CLECs rather than RAW264.7 cells.
Figure 6.

Interaction between macrophages and CLECs. (A) L–lactic acid (L-LA) accumulated in cystic kidneys. *P<0.05 versus P14–P16 PKD mice; #P<0.05 versus P18–P20 PKD mice; ψP<0.05 versus P22–P24 PKD mice; ϕP<0.05 versus P26–P28 PKD mice. (B) RAW264.7 cells were incubated with human CLECs to generate a TS. Dynamic comparison of L-LA concentrations between DMEMCLECs, DMEMRTECs, DMEMRAW264.7 cells, and DMEMTS. *P<0.05 versus 24-hour incubation; #P<0.05 versus 48-hour incubation; ψP<0.05 versus 72-hour incubation. (C) ARG1 was expressed in the cytoplasm of RAW264.7 cells and significantly upregulated under L-LA stimulation. Nuclei were stained with DAPI (blue), ×1600. (D and E) Comparison of the (C) mRNA and (D) protein expression of Arg1 in RAW264.7 cells incubated in DMEM containing different treatments. *P<0.05 versus control; #P<0.05 versus 15 mM L-LA; ψP<0.05 versus 18 mM L-LA; ϕP<0.05 versus TS; ∆P<0.05 versus TS+18 mM L-LA. (F) The arginine-polyamine metabolic pathway consists of two parts: arginine cycle (blue circle) and polyamine synthesis (red line). (G and H) L-arginine, urea, L-ornithine, and putrescine levels in RAW264.7 cells incubated in DMEM with different treatments. *P<0.05 versus control; #P<0.05 versus 18 mM L-LA; ψP<0.05 versus TS; ϕP<0.05 versus TS+18 mM L-LA; ΔP<0.05 versus TS+nor-NOHA. (I) EdU (red) nuclei staining was used to compare the proliferative rates of CLECs under different incubations, ×200. Nuclei were stained with DAPI (blue). *P<0.05 versus control; #P<0.05 versus 18 mM L-LA; ψP<0.05 versus TS; ϕP<0.05 versus TS+18 mM L-LA. Data are presented as means±SDs, with 4‒5 cell specimens for each different treatment.
L-LA can regulate the M2-like differentiation of macrophages in vitro.23,24 In addition, L-LA induced RAW264.7 cells to express Arg1 in a dose-dependent manner. Arg1 expression was significantly higher in RAW264.7 cells incubated in TS than in cells stimulated with L-LA alone. Adding L-LA to DMEM in TS promoted Arg1 expression in RAW264.7 cells to a significantly greater extent than the TS system or L-LA stimulation alone (Figure 6, C–E). As shown in Figure 6F, ornithine and putrescine are key intermediates of arginine-polyamine metabolism. Arg1 upregulation promoted the generation of urea, ornithine, and putrescine in RAW264.7 cells, paralleling the absorption of arginine (Figure 6, G and H). Thus, nor-NOHA treatment markedly hindered the absorption of arginine, decreased the biosynthesis of urea and polyamine in RAW264.7 cells in TS, and indirectly inhibited the expression of Arg1 in RAW264.7 cells. EdU can be incorporated into DNA when CLECs proliferate. Compared with negative control, L-LA treatment did not have an obvious effect on CLEC proliferation; however, it significantly promoted CLEC proliferation in TS. Adding nor-NOHA to DMEM markedly counteracted the L-LA–dependent stimulation of CLECs in TS (Figure 6I). In addition, ARG1 directly stimulated CLEC proliferation via an arginine metabolism–independent way. These data are detailed in the supplemental material (Supplemental Material, Supplemental Figure 9).
Discussion
Pkd1 inactivation in mouse models at different times results in completely disparate pathogenic courses,25 and in our study proliferative indices underwent a two-peak course in rapid-onset polycystic kidney tissues. The first peak was also observed in age-matched WT mice, albeit with a lower value and earlier physiologic breaking point. This suggests that the first proliferative index peak is driven by the combined effects of physiologic proliferation and Pkd1 deficiency. The second peak began at P22. Because it occurred after kidney development, there was no influence of other physiologically expressed genes; therefore, it was directly caused by Pkd1 gene deficiency. We assumed that it might be closely related to ARG1-expressing macrophages that infiltrated polycystic kidneys during the same period.
Macrophages have been detected in the kidney tissues of individuals with ADPKD and in related animal models.1–3 In this study, two phenotypes of macrophages were detected in the polycystic kidney tissues of rapid-onset mice. NOS2 is an M1-like marker for macrophages. The contribution of NOS2 and its metabolite NO to the development of ADPKD is related to endothelial function and oxidative stress rather than macrophage functions.26,27 We discovered a macrophage population transition from NOS2-like priority to ARG1-like priority in polycystic kidney tissues. The phenomenon was also detected during AKI,1,28 and AKI was considered a third hit to promote cystogenesis in adult PKD mice.29 Thus, we suggest that NOS2(+) macrophages likely contributed to initial cystogenesis in PKD mice.
M2-like macrophages have been identified in the kidneys of patients with ADPKD and mouse models.5,6,30 The M2-like phenotype can mediate early repair and regenerative processes, including stimulating epithelial cell proliferation after AKI.28 Our work enriches understanding of the proproliferative function of macrophages in cyst enlargement and, more importantly, focuses this function on the protein ARG1, which is highly expressed in M2-like macrophages. The proproliferative function of arginine has previously been demonstrated; for example, arginine deprivation significantly downregulates mTOR protein in tumors and intestinal cells.31,32 It also induces autophagy and negatively regulates cell growth.33 A recent study showed that ARG1 directly promotes epidermal cell proliferation.34 Krischel et al.35 suggested that it might rely on the biphasic regulation of NO.
Clodronate liposomes are widely used in animal experiments to investigate the function of macrophages,36,37 and we applied multiple-range macrophage depletion to successfully inhibit cyst enlargement in PKD mice. Although it is impractical to deplete macrophages in humans to postpone cyst growth, selectively weakening macrophage activation might be feasible to treat the disease. Nor-NOHA is a homolog of a natural form of the compound that emerges as an intermediate during NOS-catalyzed NO synthesis.38 It can disrupt the products of polyamine, which consist of putrescine, spermidine, and spermine. Putrescine can penetrate the cytomembrane and stimulate cell proliferation.39 The transformation of spermine to spermidine can generate high levels of H2O2.40 DNA damage induced by high levels of H2O2 silences many tumor suppressor genes, resulting in uncontrolled cell growth.41,42 Inhibition of spermine synthase and activation of SMOX both drive the spermine-to-spermidine transformation,43 which may explain why spermine was nearly undetectable in polycystic kidney tissues. Nor-NOHA has been widely administered to improve endothelial function in patients with diabetic nephrology.44,45 Thus, our findings provide a novel intervention for ADPKD.
Similar to neoplastic diseases, the development of ADPKD is closely related to the specific microenvironment. In a polycystic kidney microenvironment (PKM), CLECs are able to reprogram their metabolism according to their needs and can also reprogram the metabolism of surrounding cells to respond to their demands and fuel their proliferation.46 As one of most important immunocytes, macrophages are also notably influenced by multimetabolites from CLECs in PKMs; among them, glycometabolism is a hotspot issue.47,48 A high glycolysis rate enables M1-type macrophages to survive in anoxic microenvironments.49,50 By contrast, M2-type macrophages have an intact Krebs cycle and preferentially exploit oxidative phosphorylation and mitochondrial fatty acid oxidation51–53; highly active oxidative glucose metabolism provides the required energy to promote effector cell proliferation.50,54 However, because hypoxia is a key attribute of polycystic kidney tissues,55 an interesting question is why macrophages present as M2-type rather than M1-type in late-stage PKM. One contribution of our work is that it demonstrates that L-LA plays a crucial role in macrophage polarization and reveals that Pkd1 gene deficiency drives CLECs to overproduce L-LA in polycystic kidney tissues. In turn, L-LA induces macrophages to promote CLEC proliferation. Thus, a vicious circle is formed in the PKM, which drives the development of ADPKD (Figure 7).
Figure 7.

Vicious circle between CELCs and macrophages. (A) CELCs secrete L-LA to promote the differentiation of macrophages. (B) L-LA induced macrophages to upregulate ARG1 and activate the arginine-polyamine metabolic pathway. (C) Polyamine stimulates CLEC proliferation. (D) ARG1 stimulates CLEC proliferation (requires further verification). (E) Proliferative CLECs produce more L-LA in the microenvironment. (F) Nor-NOHA can disrupt the synthesis of polyamine. MACs, macrophages; L-LA, L–lactic acid.
There were some limitations to this study. First, Pkd1 gene inactivation was systemic in PKD mice but the effects of Pkd1 gene deficiency on macrophage function were not assessed. Second, the definitive mechanism through which polyamine affects cell proliferation has not been completely clarified. Third, the concrete mechanism underlying how L-LA affects macrophage polarization has not been elucidated. Overall, our findings indicate that ARG1 is a novel potential therapeutic target for ADPKD, and deepen understanding of the interactions between macrophages and CLECs in the PKM.
Disclosures
None.
Supplementary Material
Acknowledgments
This work was supported by the following grants: (1) the National Natural Science Foundation of China (81700579, 81670612), (2) the Three-Year Project of Action for Shanghai Public Health System (GWIV‒18), (3) the National Key Research and Development Program of China (2016YFC0901502), and (4) Shanghai Top Priority Key Clinical Disciplines Construction Project (2017ZZ02009).
C.M. and Y.Y. designed the study; Y.Y., M.C., J.Z., J.L., S.S., L.F., J.C., and M.Y. carried out experiments; Y.Y., M.C., and J.Z. analyzed the data; Y.Y. made the figures; Y.Y., M.C., and C.M. drafted and revised the paper; all authors approved the final version of the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2018010074/-/DCSupplemental.
References
- 1.Prasad S, McDaid JP, Tam FW, Haylor JL, Ong AC: Pkd2 dosage influences cellular repair responses following ischemia-reperfusion injury. Am J Pathol 175: 1493–1503, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zeier M, Fehrenbach P, Geberth S, Möhring K, Waldherr R, Ritz E: Renal histology in polycystic kidney disease with incipient and advanced renal failure. Kidney Int 42: 1259–1265, 1992 [DOI] [PubMed] [Google Scholar]
- 3.Su Z, Wang X, Gao X, Liu Y, Pan C, Hu H, et al.: Excessive activation of the alternative complement pathway in autosomal dominant polycystic kidney disease. J Intern Med 276: 470–485, 2014 [DOI] [PubMed] [Google Scholar]
- 4.Karihaloo A, Koraishy F, Huen SC, Lee Y, Merrick D, Caplan MJ, et al.: Macrophages promote cyst growth in polycystic kidney disease. J Am Soc Nephrol 22: 1809–1814, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peda JD, Salah SM, Wallace DP, Fields PE, Grantham CJ, Fields TA, et al.: Autocrine IL-10 activation of the STAT3 pathway is required for pathological macrophage differentiation in polycystic kidney disease. Dis Model Mech 9: 1051–1061, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swenson-Fields KI, Vivian CJ, Salah SM, Peda JD, Davis BM, van Rooijen N, et al.: Macrophages promote polycystic kidney disease progression. Kidney Int 83: 855–864, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parisi L, Gini E, Baci D, Tremolati M, Fanuli M, Bassani B, et al.: Macrophage polarization in chronic inflammatory diseases: Killers or builders? J Immunol Res 2018: 8917804, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leonhard WN, Zandbergen M, Veraar K, van den Berg S, van der Weerd L, Breuning M, et al.: Scattered deletion of PKD1 in kidneys causes a cystic snowball effect and recapitulates polycystic kidney disease. J Am Soc Nephrol 26: 1322–1333, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rogers KA, Moreno SE, Smith LA, Husson H, Bukanov NO, Ledbetter SR, et al.: Differences in the timing and magnitude of Pkd1 gene deletion determine the severity of polycystic kidney disease in an orthologous mouse model of ADPKD. Physiol Rep 4, pii: e12846, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akazawa Y, Kubo M, Zhang R, Matsumoto K, Yan F, Setiawan H, et al.: Inhibition of arginase ameliorates experimental ulcerative colitis in mice. Free Radic Res 47: 137–145, 2013 [DOI] [PubMed] [Google Scholar]
- 11.Hernandez LF, Buchwald P, Abdulreda MH: Effect of Arginase-1 inhibition on the incidence of autoimmune diabetes in NOD mice. Curr Res Diabetes Obes J 5, pii: 555661, 2018 [PMC free article] [PubMed] [Google Scholar]
- 12.Lasch M, Caballero-Martinez A, Troidl K, Schloegl I, Lautz T, Deindl E: Arginase inhibition attenuates arteriogenesis and interferes with M2 macrophage accumulation. Lab Invest 96: 830–838, 2016 [DOI] [PubMed] [Google Scholar]
- 13.Moura VB, Silva MM, Batista LF, Gomes CM, Leenen PJ, Lino RS Jr, et al.: Arginase activity is associated with fibrosis in experimental infection with Taenia crassiceps, but does not play a major role in resistance to infection. Exp Parasitol 135: 599–605, 2013 [DOI] [PubMed] [Google Scholar]
- 14.Pfaffl MW: A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: e45, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takenouchi T, Suzuki S, Shinkai H, Tsukimoto M, Sato M, Uenishi H, et al.: Extracellular ATP does not induce P2X7 receptor-dependent responses in cultured renal- and liver-derived swine macrophages. Results Immunol 4: 62–67, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rustenhoven J, Park TI, Schweder P, Scotter J, Correia J, Smith AM, et al.: Isolation of highly enriched primary human microglia for functional studies. Sci Rep 6: 19371, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu M, Chen M, Jing Y, Gu J, Mei S, Yao Q, et al.: The C-terminal tail of polycystin-1 regulates complement factor B expression by signal transducer and activator of transcription 1. Am J Physiol Renal Physiol 310: F1284–F1294, 2016 [DOI] [PubMed] [Google Scholar]
- 18.Hesterberg RS, Cleveland JL, Epling-Burnette PK: Role of polyamines in immune cell functions. Med Sci (Basel) 6, pii: E22, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belliere J, Casemayou A, Ducasse L, Zakaroff-Girard A, Martins F, Iacovoni JS, et al.: Specific macrophage subtypes influence the progression of rhabdomyolysis-induced kidney injury. J Am Soc Nephrol 26: 1363–1377, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olivon VC, Fraga-Silva RA, Segers D, Demougeot C, de Oliveira AM, Savergnini SS, et al.: Arginase inhibition prevents the low shear stress-induced development of vulnerable atherosclerotic plaques in ApoE-/- mice. Atherosclerosis 227: 236–243, 2013 [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Wehling-Henricks M, Samengo G, Tidball JG: Increases of M2a macrophages and fibrosis in aging muscle are influenced by bone marrow aging and negatively regulated by muscle-derived nitric oxide. Aging Cell 14: 678–688, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhatta A, Yao L, Xu Z, Toque HA, Chen J, Atawia RT, et al.: Obesity-induced vascular dysfunction and arterial stiffening requires endothelial cell arginase 1. Cardiovasc Res 113: 1664–1676, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohashi T, Aoki M, Tomita H, Akazawa T, Sato K, Kuze B, et al.: M2-like macrophage polarization in high lactic acid-producing head and neck cancer. Cancer Sci 108: 1128–1134, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al.: Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513: 559–563, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG: A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med 13: 1490–1495, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klawitter J, Reed-Gitomer BY, McFann K, Pennington A, Klawitter J, Abebe KZ, et al.: Endothelial dysfunction and oxidative stress in polycystic kidney disease. Am J Physiol Renal Physiol 307: F1198–F1206, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eroglu E, Kocyigit I: Induction of nitric oxide release with nebivolol may improve endothelial dysfunction in polycystic kidney disease. Kidney Int 87: 857, 2015 [DOI] [PubMed] [Google Scholar]
- 28.Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi BS, et al.: Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol 22: 317–326, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takakura A, Contrino L, Zhou X, Bonventre JV, Sun Y, Humphreys BD, et al.: Renal injury is a third hit promoting rapid development of adult polycystic kidney disease. Hum Mol Genet 18: 2523–2531, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mrug M, Zhou J, Woo Y, Cui X, Szalai AJ, Novak J, et al.: Overexpression of innate immune response genes in a model of recessive polycystic kidney disease. Kidney Int 73: 63–76, 2008 [DOI] [PubMed] [Google Scholar]
- 31.Bauchart-Thevret C, Cui L, Wu G, Burrin DG: Arginine-induced stimulation of protein synthesis and survival in IPEC-J2 cells is mediated by mTOR but not nitric oxide. Am J Physiol Endocrinol Metab 299: E899–E909, 2010 [DOI] [PubMed] [Google Scholar]
- 32.Changou CA, Chen YR, Xing L, Yen Y, Chuang FY, Cheng RH, et al.: Arginine starvation-associated atypical cellular death involves mitochondrial dysfunction, nuclear DNA leakage, and chromatin autophagy. Proc Natl Acad Sci USA 111: 14147–14152, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.García-Navas R, Munder M, Mollinedo F: Depletion of L-arginine induces autophagy as a cytoprotective response to endoplasmic reticulum stress in human T lymphocytes. Autophagy 8: 1557–1576, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weber C, Telerman SB, Reimer AS, Sequeira I, Liakath-Ali K, Arwert EN, et al.: Macrophage infiltration and alternative activation during wound healing promote MEK1-induced skin carcinogenesis. Cancer Res 76: 805–817, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krischel V, Bruch-Gerharz D, Suschek C, Kröncke KD, Ruzicka T, Kolb-Bachofen V: Biphasic effect of exogenous nitric oxide on proliferation and differentiation in skin derived keratinocytes but not fibroblasts. J Invest Dermatol 111: 286–291, 1998 [DOI] [PubMed] [Google Scholar]
- 36.van Rooijen N, van Kesteren-Hendrikx E: Clodronate liposomes: Perspectives in research and therapeutics. J Liposome Res 12: 81–94, 2002 [DOI] [PubMed] [Google Scholar]
- 37.van der Tuin SJL, Li Z, Berbée JFP, Verkouter I, Ringnalda LE, Neele AE, et al.: Lipopolysaccharide lowers cholesteryl ester transfer protein by activating F4/80(+)Clec4f(+)Vsig4(+)Ly6C(−) Kupffer cell subsets. J Am Heart Assoc 7, pii: e008105, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abdelkawy KS, Lack K, Elbarbry F: Pharmacokinetics and pharmacodynamics of promising arginase inhibitors. Eur J Drug Metab Pharmacokinet 42: 355–370, 2017 [DOI] [PubMed] [Google Scholar]
- 39.Zhang X, Chen Y, Hao L, Hou A, Chen X, Li Y, et al.: Macrophages induce resistance to 5-fluorouracil chemotherapy in colorectal cancer through the release of putrescine. Cancer Lett 381: 305–313, 2016 [DOI] [PubMed] [Google Scholar]
- 40.Murray-Stewart T, Wang Y, Goodwin A, Hacker A, Meeker A, Casero RA Jr: Nuclear localization of human spermine oxidase isoforms - possible implications in drug response and disease etiology. FEBS J 275: 2795–2806, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khobta A, Anderhub S, Kitsera N, Epe B: Gene silencing induced by oxidative DNA base damage: Association with local decrease of histone H4 acetylation in the promoter region. Nucleic Acids Res 38: 4285–4295, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watanabe S, Ichimura T, Fujita N, Tsuruzoe S, Ohki I, Shirakawa M, et al.: Methylated DNA-binding domain 1 and methylpurine-DNA glycosylase link transcriptional repression and DNA repair in chromatin. Proc Natl Acad Sci USA 100: 12859–12864, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fiori LM, Turecki G: Genetic and epigenetic influences on expression of spermine synthase and spermine oxidase in suicide completers. Int J Neuropsychopharmacol 13: 725–736, 2010 [DOI] [PubMed] [Google Scholar]
- 44.Kövamees O, Shemyakin A, Pernow J: Effect of arginase inhibition on ischemia-reperfusion injury in patients with coronary artery disease with and without diabetes mellitus. PLoS One 9: e103260, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shemyakin A, Kövamees O, Rafnsson A, Böhm F, Svenarud P, Settergren M, et al.: Arginase inhibition improves endothelial function in patients with coronary artery disease and type 2 diabetes mellitus. Circulation 126: 2943–2950, 2012 [DOI] [PubMed] [Google Scholar]
- 46.Krstic J, Trivanovic D, Jaukovic A, Santibanez JF, Bugarski D: Metabolic plasticity of stem cells and macrophages in cancer. Front Immunol 8: 939, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rowe I, Chiaravalli M, Mannella V, Ulisse V, Quilici G, Pema M, et al.: Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med 19: 488–493, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rowe I, Boletta A: Defective metabolism in polycystic kidney disease: Potential for therapy and open questions. Nephrol Dial Transplant 29: 1480–1486, 2014 [DOI] [PubMed] [Google Scholar]
- 49.Rodríguez-Prados JC, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P, et al.: Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J Immunol 185: 605–614, 2010 [DOI] [PubMed] [Google Scholar]
- 50.O’Neill LA, Pearce EJ: Immunometabolism governs dendritic cell and macrophage function. J Exp Med 213: 15–23, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roiniotis J, Dinh H, Masendycz P, Turner A, Elsegood CL, Scholz GM, et al.: Hypoxia prolongs monocyte/macrophage survival and enhanced glycolysis is associated with their maturation under aerobic conditions. J Immunol 182: 7974–7981, 2009 [DOI] [PubMed] [Google Scholar]
- 52.Zhu L, Zhao Q, Yang T, Ding W, Zhao Y: Cellular metabolism and macrophage functional polarization. Int Rev Immunol 34: 82–100, 2015 [DOI] [PubMed] [Google Scholar]
- 53.He C, Carter AB: The metabolic prospective and redox regulation of macrophage polarization. J Clin Cell Immunol 6, pii: 371, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Odegaard JI, Chawla A: Alternative macrophage activation and metabolism. Annu Rev Pathol 6: 275–297, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Raptis V, Loutradis C, Sarafidis PA: Renal injury progression in autosomal dominant polycystic kidney disease: A look beyond the cysts [published online ahead of print February 21, 2018]. Nephrol Dial Transplant 10.1093/ndt/gfy023 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.