

 Doxorubicin-induced testicular toxicity involves perturbation of microRNAs within all three of the main testicular cell types, particularly those involved in germ–Sertoli and Sertoli–Sertoli cell junctions.
Doxorubicin-induced testicular toxicity involves perturbation of microRNAs within all three of the main testicular cell types, particularly those involved in germ–Sertoli and Sertoli–Sertoli cell junctions.
Abstract
We have previously shown that in addition to its widely recognised cardiotoxicity, the chemotherapeutic doxorubicin (DOX) is able to induce transcriptional, microRNA (miRNA) and DNA methylation changes in the mouse testis. These changes perturb pathways involved in stress/cell death and survival and testicular function and lead to germ cell loss and reproductive organ damage. Here, we further investigated the differential miRNA expression induced by DOX in mouse spermatogonial (GC1), Leydig (TM3) and Sertoli (TM4) cell lines in vitro. We began by performing cell cycle analysis of the three mouse testicular cell lines to evaluate their sensitivity to DOX and thus select suitable doses for miRNA profiling. In keeping with our in vivo data, the spermatogonial cell line was the most sensitive, and the Sertoli cell line the most resistant to DOX-induced cell cycle arrest. We then further demonstrated that each cell line has a distinct miRNA profile, which is perturbed upon treatment with DOX. Pathway analysis identified changes in the miRNA-mediated regulation of specialised signalling at germ–Sertoli and Sertoli–Sertoli cell junctions following treatment with DOX. Amongst the most significant disease categories associated with DOX-induced miRNA expression were organismal injury and abnormalities, and reproductive system disease. This suggests that miRNAs play significant roles in both normal testicular function and DOX-induced testicular toxicity. Comparison of our in vitro and in vivo data highlights that in vitro cell models can provide valuable mechanistic information, which may also help facilitate the development of biomarkers of testicular toxicity and high-throughput in vitro screening methods to identify potential testicular toxicants.
Introduction
There are now millions of cancer survivors worldwide. Current statistics estimate that 2.5 million people in the UK are living with, or have had treatment for, cancer.1 This figure is estimated to increase by over three per cent each year, so that by 2030, there could be 4 million cancer survivors in the UK.1 Around 1 in 4 people in the UK suffer from poor health or disability after cancer treatment.2 The continued medical support of such survivors, particularly survivors of childhood cancers, is therefore an important Public Health issue. Childhood cancer survivors have greater life-years at risk of developing secondary cancers and chronic conditions in later life as a result of their previous treatments, and are also more likely to want to go on to have their own children. Leukaemias and lymphomas are the most common cancers diagnosed before the age of 25 in the UK.3 Treatment of these cancers normally involves DOX or another anthracycline.4 Thus, of the 35 000 survivors of childhood cancers in the UK,3 around half will have been treated with an anthracycline.4
Anthracyclines are associated with lifelong cardiotoxicity,5 and the extensive array of mechanistic studies (both from ourselves6,7 and others8–10 are helping to develop protective intervention strategies).5,9,10 In contrast, much less is known about the long term effects of DOX, or other anthracyclines, on reproduction and development. It is not only the adverse outcome(s) on the treated cancer patient themselves that is of concern, but also the potential detrimental effect(s) on their future progeny. Over the last decade, many hundreds of studies have linked a wide range of environmental factors with adverse health outcomes in later life that can be transmitted across multiple generations, giving rise to so-called multigenerational toxicity.11 Such effects are likely to involve epigenetic processes, including histone modification, DNA methylation and/or non-coding RNAs (ncRNAs) such as miRNAs. Indeed, a number of recent independent studies have implicated spermatozoal miRNAs in the transmission of stress-induced behavioural and metabolic abnormalities through subsequent generations.12–15 There is also evidence to suggest that unique epigenetic information can be transmitted in the male germline via histone modifications and DNA methylation,16–18 both of which help to activate/regulate early embryo development and can be perturbed by environmental factors.11 Furthermore, increased multigenerational genomic instability following paternal anti-cancer drug exposure has been reported in mice.19 Thus, there is concern that chemotherapeutics may induce epigenetically-mediated multigenerational reproductive and developmental toxicity via the male germline.20
The production of mature male germ cells (spermatogenesis) takes place within the seminiferous tubules of the testis and involves proliferation and differentiation of diploid spermatogonia into haploid spermatozoa.21–24 Spermatogenesis is a complex developmental process supported by the secretion of hormones and other cellular signals from Leydig and Sertoli cells.21–24 Compound-induced damage to any of these three main testicular cell types (germ, Leydig or Sertoli cells) could reduce the production of healthy spermatozoa, impair fertility and/or adversely affect the resulting embryo. While studies in rodents have associated DOX exposure with reduced testicular blood flow and volume, testicular atrophy, increased cellular apoptosis, impaired spermatogenesis, increased sperm abnormalities, sperm counts and fertility, and adverse developmental phenotypes in subsequent zygotes,25–35 the molecular mechanisms underlying these DOX-induced effects are not well understood. Therefore, we recently investigated the molecular transcriptomic and epigenetic changes in the mouse testis following DOX treatment in vivo, linking specific miRNA, DNA methylation and mRNA changes to actual phenotypic endpoints.36 Here we have further investigated the role of miRNAs within the three main testicular cell types in DOX-induced testicular toxicity by profiling miRNA expression in mouse spermatogonial, Leydig and Sertoli cell lines following treatment with DOX in vitro. We decided to focus on miRNAs since; (1) differential miRNA expression was better at separating the in vivo DOX treated samples compared to altered transcript expression,36 and (2) spermatozoal miRNAs have been implicated in the transmission of phenotypes across generations.12–15 The resulting in vitro data highlighted the roles of specific miRNAs, pathways and cell types during DOX-induced testicular toxicity.
Materials & methods
Materials
All materials, including DOX (as doxorubicin hydrochloride) were purchased from Sigma Aldrich, UK unless otherwise stated.
Cell lines
Three mouse testicular cell lines were used. The TM3 (Leydig) and TM4 (Sertoli) cell lines, originally derived from BALB/c mice testis, were gifts from the European Collection of Cell Cultures (ECACC) operated by Public Health England (PHE) (Porton Down, UK), and the GC1 (spermatogonia) cell line, also derived from BALB/c mouse testis, was obtained from the American Type Culture Collection (ATCC) via LGC Standards (Teddington, UK).
Cell culture
The TM3 and TM4 cell lines were cultured in a 1 : 1 mixture of Dulbecco's Modified Eagle Medium (DMEM) and Ham's F12 containing 17.5 mM glucose and 0.5 mM sodium pyruvate, supplemented with 5 mM glutamax, 5% horse serum and 2.5% foetal bovine serum (Life Technologies, Paisley, UK). The GC-1 cell line was cultured in DMEM containing 25 mM glucose and 1 mM sodium pyruvate, supplemented with 10% foetal bovine serum (Life Technologies, Paisley, UK). Cells were incubated in 5% CO2 at 37 °C. Media was replaced every 2 days and cells were passaged when they reach a confluency of 80%.
Treatment with DOX
Approximately 1 × 104 cells were seeded in a 6-well plate and allowed to attach overnight. The media was then replaced with fresh media containing 1 nM–5 μM DOX or vehicle-only (PBS). After a further 24 or 48 hours of incubation, cells were collected for fluorescence-activated cell sorting (FACS) analysis or RNA extraction.
FACS analysis
Following treatment, cells were washed twice with PBS, trypsined, collected in media and centrifuged at 200g for 4 minutes at RT. Supernantant was discarded and the cell pellet was resuspended in 10 ml of ice cold fixing solution (1 ml PBS and 9 ml 70% ice-cold ethanol) for 24 hours at 4 °C. Fixed cells were centrifuged at 200g for 4 minutes at RT, the supernatant was discarded and the cell pellet was resuspended in staining solution (40 μg ml–1 propidium iodide (PI) and 1 mg ml–1 RNase A in PBS) at 37 °C for 30 minutes. Cell samples were then filtered through 70 μm mesh sieves to ensure a single cell suspension and analysed using a FACSCalibur Flow Cytometer (BD Biosciences, US). Forward scatter and side scatter detectors were gated to exclude as much debris as possible and histograms of fluorescence intensity (PI) versus number of cells were analysed using ModFit LT software (Verity Software House, USA). These histograms provide a representation of DNA content and thus the percentage of cells within the G1, S and G2 phases of the cell cycle. Representative examples of cell cycle profiles for each cell line following all treatments are shown in ESI Fig. 1–6.† The proportion of cells in G2 arrest was calculated as the percentage of cells in the G2/M phase (Dip G2, ESI Fig. 1–6†). Final percentages of cells in G2 arrest were normalised by subtracting the percentages of vehicle-only treated cells in G2 arrest, and plotted (mean ± SEM, n ≥ 3) against Dox concentration (nM) to generate EC50 values and select low and high doses for RNA extraction and miRNA profiling studies.
RNA extraction
Following treatment, cells were washed twice with PBS, trypsined, collected in media and centrifuged at 200g for 4 minutes at RT. Supernantant was discarded and RNA was extracted from the cell pellet using the miRNeasy Mini kit (Qiagen, Germany) according to the manufacturer's instructions. RNA was stored at –80 °C until microarray analysis. RNA integrity was determined using a 2100 Bioanalyser and RNA 6000 LabChip® kit (Agilent Technologies, USA) according to the manufacturer's instructions. Only RNA samples with an RNA integrity number (RIN) ≥ 8.0 were used for downstream analysis.
miRNA profiling
Global changes in miRNA expression were profiled as described in our previous in vivo study36 using miRNA microarrays obtained from the Medical Research Council Toxicology Unit (Leicester, UK) with the Label IT® miRNA labelling kit (Mirus, USA), except that all samples were run against a reference sample (instead of randomly pairing control versus treated samples and incorporating a dye-swap). The reference sample was made by mixing an equal amount of RNA extracted from all treated and untreated samples. The reference sample was labelled with Cy3 and hybridised against control or treated samples labelled with Cy5. Thus, expression values represent log2[cell line control/reference], log2[DOX/control] or fold change[DOX/control].
Bioinformatic analysis
Raw data was extracted, normalised and statistically significant differentially expressed miRNAs were identified as described in our previous in vivo study36 using GenePix pro 6.0 (Molecular Devices, CA), the Bioconductor limma library package of R version 2.15.2 (; http://www.r-project.org/) and Qlucore Omics Explorer (QOE) version 3.3 (Qlucore, Sweden), respectively. Heatmaps were subjected to hierarchical clustering using the algorithms within QOE.37 Lists containing the top 100 most significantly differentially expressed miRNAs within/between the three testicular cell lines in the presence/absence of DOX were imported into Ingenuity® Pathway Analysis (IPA®) Software (Ingenuity® Systems ; http://www.ingenuity.com/products/ipa) to explore the more general functional roles of these miRNAs. In addition, lists containing the top 100 most significantly expressed miRNAs in the three testicular cell lines treated with DOX were merged and uploaded into miRwalk (; http://www.umm.uni-heidelberg.de/apps/zmf/mirwalk/) to identify experimentally validated mRNA targets. The resulting list of validated mRNA targets was then downloaded from miRwalk and imported into IPA® software to explore more specific networks and pathways involved in DOX-induced testicular toxicity.
Raw data files for all three cell lines have been submitted to GEO: GC1 (GEO***), TM3 (GEO***) and TM4 (GEO***).
Results
DOX induced cell cycle arrest
Cell cycle analysis of the three mouse testicular cell lines, GC1 (spermatogonia), TM3 (Leydig cells) and TM4 (Sertoli cells), in the presence or absence of 1 nM–5 μM DOX for 24 or 48 hours was performed to evaluate their sensitivity to DOX and thus select suitable doses for miRNA profiling. Representative examples of cell cycle profiles for each cell line following all treatments are shown in ESI Fig. 1–6.† All three cell lines demonstrated increased G2/M phase arrest following treatment with increasing doses of DOX for 24 (Fig. 1A) or 48 hours (ESI Fig. 7†). GC1 spermatogonia were the most sensitive to DOX-induced G2/M arrest, followed by TM3 Leydig cells and finally TM4 Sertoli cells (Table 1). No discernible differences were observed in the cell cycle profiles between DOX treatment for 24 and 48 hours, except for a higher proportion of post-G2/M cells from 1–5 μM at 48 hours (indicative of cell aggregation) (ESI Fig. 1–6†), and an increased response at 48 compared to 24 hours (Table 1). Thus, 24 hours exposures were selected for downstream analyses.
Fig. 1. Dose–response of (A) G2/M arrest or (B) actively dividing (S phase) cell loss following treatment with DOX for 24 hours. Cellular DNA content was analysed by FACS, and normalised by subtracting control values (percentage of cells in G2/M phase in the absense of DOX) from treated values (A) or calculated as a percentage of the total number of cells (B). Values represent mean ± SD (n = 3). *p < 0.05, **p < 0.01 and ***p < 0.001 (T-test compared to vehicle only).

Table 1. EC50 values for DOX-induced G2 arrest in the three testicular cell lines.
| Exposure time (hours) | GC1 Spermatogonia (μM) | TM3 Leydig (μM) | TM4 Sertoli (μM) |
| 24 | 1.5 | 3.5 | 6.0 |
| 48 | 1.2 | 3.2 | 5.0 |
The rate of DNA replication within each cell line, (as a measure of the percentage of S phase cells), was also compared (Fig. 1B). In the absence of DOX, TM4 Sertoli cells had the highest percentage of cells in S phase (50.5 ± 1.1%), followed by GC1 spermatogonia (44.0 ± 0.6%) and finally TM3 Leydig cells (38.0 ± 1.9%). All three cell lines demonstrated a reduction in the percentage of S phase cells following treatment with increasing doses of DOX for 24 hours. GC1 spermatogonia were the most sensitive to DOX-induced loss of S phase cells, followed by TM3 Leydig cells and finally TM4 Sertoli cells. The dose of DOX required to induce a statistically significant (p < 0.05) decrease in the percentage of S phase was 100 nM in GC1 spermatogonia, 1 μM in TM3 Leydig cells and 5 μM in TM4 Sertoli cells.
In summary, treatment with DOX induced significant G2/M arrest and loss of actively dividing (S phase) cells, with GC1 spermatogonia showing greatest sensitivity and TM4 Sertoli cells greatest resistance to DOX.
All three testicular cell lines demonstrated cell-specific miRNA profiles
We first compared the miRNA profiles of the three testicular cell lines in the absence of DOX. There was statistically significant (p < 0.01) differential expression of over 160 miRNAs, with the TM3 Leydig cells demonstrating the lowest level of change. The top 100 most statistically significant changes between the three cell lines, corresponding to 96 miRNAs, are shown in Fig. 2. Functional analysis of these 96 miRNAs using IPA® software revealed roles in various molecular and cellular functions, particularly cellular development, cell cycle, cell-to-cell signalling and interaction, and cellular assembly and organisation (covering approximately 55%, 20%, 15% and 5% of the total identified functional categories, respectively).
Fig. 2. Heatmap of the top 100 most significantly altered miRNAs (p ≤ 0.01) between the three testicular cell lines GC1 spermatogonia, and TM3 Leydig and TM4 Sertoli cells. Expression levels represent log2[cell line/reference] (Log2 R).

In summary, the three testicular cell lines demonstrated cell-specific miRNA profiles, with the most differentially expressed miRNAs predominantly functioning in cellular development and interaction.
DOX induced dose- and cell-dependent miRNA expression changes in all three testicular cell lines
Following cell cycle analysis of DOX-induced G2/M arrest, two doses (a low and high) were selected for each testicular cell line. The low dose (100 nM for GC1 spermatogonia, and 500 nM for TM3 Leydig and TM4 Sertoli cells) marked the onset of G2/M arrest, while the high dose (1 μM for GC1 spermatogonia, and 5 μM for TM3 Leydig and TM4 Sertoli cells) induced significant G2/M arrest with minimal cell death.
There was statistically significant (p < 0.01) differential expression of between 60–120 miRNAs in the three cell lines following treatment with a low or high dose of DOX for 24 hours. TM4 Sertoli cells demonstrated the greatest number (117) of changes, closely followed by GC1 spermatogonia (101), and finally TM3 Leydig cells (64). The top 100 most statistically significant changes with increasing dose, corresponding to 96 (GC1, p < 0.01), 95 (TM3, p < 0.05) or 93 (TM4, p < 0.01) miRNAs, are shown in Fig. 3. Approximately one third of these DOX-induced miRNAs were present in two or more cell lines, with only two (miR-767 and miR-3072) present in all three cell lines (ESI Fig. 8†). miR-767 was downregulated with increasing dose in all three cell lines, while miR-3072 was upregulated in GC1 spermatogonia but downregulated in TM3 Leydig and TM4 Sertoli cells with increasing dose. Indeed, only 12 of the DOX-induced miRNAs present in two or more cell lines were altered with increasing dose in the same direction (ESI Table 1†).
Fig. 3. Heatmap of the top 100 most significantly altered miRNAs (p ≤ 0.05) within the three testicular cell lines (A) GC1 spermatogonia, and (B) TM3 Leydig and (C) TM4 Sertoli cells following treatment with DOX. Expression levels represent log2[DOX/control] (Log2 R) or fold change[DOX/control] (FC), with downregulations shown in green and upregulations in red.

The top 96, 95 and 93 most statistically significant (p < 0.05) DOX-induced miRNAs in each cell line were then independently analysed using IPA® software. This revealed roles in various molecular and cellular functions, including cellular development, cell cycle, cell-to-cell signalling and cell proliferation/death, with subtle differences in DOX-induced miRNA-associated functional categories between the different cell lines (ESI Fig. 9†). Although the cell cycle accounted for between 10–20% of the total functional categories in GC1 spermatogonia and TM3 Sertoli cells, it was not identified as a functional category in TM4 Sertoli cells. In contrast, the cell movement and morphology categories together represented approximately 30% of the total functional categories in TM3 Leydig cells, but were absent in GC1 spermatogonia or TM4 Sertoli cells. There was also a decrease in the cell communication functional category in GC1 spermatogonia and TM4 Sertoli cells compared to TM3 Leydig cells. Similarly, while cell proliferation and cell death/survival accounted for approximately 50% of the total functional categories in GC1 spermatogonia and TM4 Sertoli cells, they were absent in TM3 Leydig cells. In all three cell lines, organismal injury and abnormalities and reproductive system disease were among the top three/four disease categories associated with DOX-induced miRNA changes.
Finally, the validated mRNA targets of the top 96, 95 and 93 most statistically significant (p < 0.05) DOX-induced miRNAs in each cell line were merged and analysed using IPA® software. Two signalling pathways were identified with specific functions in cell–cell interaction/communication during testicular development: germ cell–Sertoli cell junction signalling (Fig. 4A) and Sertoli cell–Sertoli cell junction signalling (Fig. 4B).
Fig. 4. The two signalling pathways identified with specific functions in cell–cell interaction/communication during testicular development (A) germ cell–Sertoli cell junction signalling and (B) Sertoli cell–Sertoli cell junction signalling. Validated mRNA targets of the most statistically significant (p < 0.05) DOX-induced miRNAs are highlighted in pink.

In summary, treatment with DOX induced dose- and cell-dependent miRNA changes in vitro that were associated with developmental disorders and reproductive system disease, and focussed around perturbation of germ cell–Sertoli cell and Sertoli cell–Sertoli cell communication.
DOX-induced miRNA changes in vitro can be correlated with those in vivo
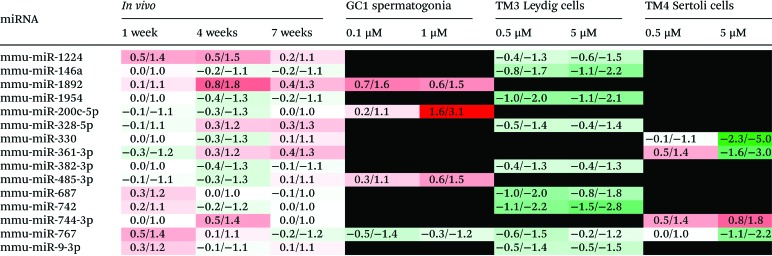
Comparison of the DOX-induced miRNA expression profiles from the three in vitro testicular cell lines with that from the in vivo testis reported in our previous study36 identified 15 miRNAs present in both the in vivo and in vitro systems (Table 2). miR-767 was present in the in vivo testis and all three in vitro testicular cell lines, being downregulated following treatment with DOX. Of the remaining 14 DOX-induced miRNAs, 3 were common to the in vivo testis and either the in vitro GC1 spermatogonia (miR-1892, 200c-5p and 486-3p) or the TM4 Sertoli cells (miR-330, 361-3p and 744-3p), while 8 were common to the in vivo testis and the in vitro TM3 Leydig cells (miR-1224, 146a, 1954, 323-5p, 382-3p, 687, 742, and 9-3p). The majority of these DOX-induced miRNAs (12/15) were altered in a direction that mechanistically correlated.
Table 2. DOX-induced miRNAs present in both the in vivo testis and one or more of the in vitro testicular cell lines.
|
In summary, DOX induced a number of miRNA changes in vitro that could be mechanistically correlated with DOX-induced miRNA changes in vivo.
Discussion
In addition to its widely recognised cardiotoxicity, DOX can cause male reproductive toxicity.25–35 We have previously shown that DOX is able to induce transcriptional, miRNA and DNA methylation changes in the mouse testis, which perturb pathways involved in stress/cell death and survival and testicular function and lead to germ cell loss and reproductive organ damage.36 This is of concern because (1) DOX is a widely used chemotherapeutic often used in children and young people who are more likely to want to go on to have their own children,3,4 (2) epigenetic changes (such as altered DNA methylation and miRNA expression) have been linked to adverse health outcomes resulting from environmental factors,11 and (3) spermatozoal miRNAs have been implicated in the transmission of phenotypes across generations.12–15 Thus, having identified DOX-induced miRNA perturbation in the mouse testis in vivo,36 we wanted to further explore the differential miRNA expression induced by DOX in mouse spermatogonia, Leydig and Sertoli cell lines in vitro to assess the role of miRNAs within the different cell types in DOX-induced testicular toxicity.
Numerous in vitro studies have demonstrated that DOX induces cell cycle arrest at the G2/M checkpoint in a wide range of cell types.38–42 Thus, we performed our own dose–response analysis of DOX-induced cell cycle arrest in three testicular cell lines representative of the cell types we investigated in vivo: germ (GC1 spermatogonia), Leydig (TM3) and Sertoli (TM4) cells. We chose these three cell lines as they were all isolated from the same mouse strain (Balb/c), and spermatogonia as the representative germ cell as these were more sensitive to DOX treatment than the more mature germ cells in vivo.36
DOX induced increasing dose-dependent G2/M arrest in all three testicular cell lines (Fig. 1A and Table 1). In keeping with our in vivo results, GC1 spermatogonia were the most sensitive to DOX-induced G2/M arrest, followed by the TM3 Leydig and finally the TM4 Sertoli cells. We have discussed potential reasons for the differing sensitivities of testicular cells in our previous publication, including the protective effects of physiological barriers, the expression of different cell membrane transporters and the varied replication rates of different testicular cell types.36 The blood-testes or blood-epididymis barriers, both of which offer protection to the more mature germ cells (spermatocytes/spermatids) in vivo, are not present within our single cell type in vitro cultures. Moreover, in vivo spermatogonia (the most DOX sensitive, but least mature germ cells), Leydig and Sertoli cells are all located outside these physiological barriers. Thus barrier protection does not contribute to the differing sensitivities of these three specific cell types to DOX, in vivo or in vitro. In contrast, differences in the types and/or levels of membrane efflux pumps expressed by different testicular cell types are likely to contribute to the final intracellular DOX levels and thus the differing sensitivities of spermatogonia, Leydig and Sertoli cells both in vivo and in vitro. Determining the exact transporter profile of these three cell types and their intracellular DOX levels would provide further insight. Likewise, differences in cell replication rates are also likely to contribute to the differing sensitivities of spermatogonia, Leydig or Sertoli cells to DOX both in vivo and in vitro. Since DOX intercalates DNA and inhibits DNA synthesis, it would be expected to have a greater effect on more proliferative cells. Prior to any in vitro treatment, TM4 Sertoli cells had the greatest proportion of cells in S (replicating) phase, followed by GC1 spermatogonia and TM3 Leydig cells (Fig. 1B). Yet, GC1 spermatogonia were the most sensitive to DOX-induced loss of S phase cells, followed by TM3 Leydig cells and finally TM4 Sertoli cells (Fig. 1B). This suggests that although there was a greater proportion of TM4 Sertoli cells in replication phase at any one time in vitro, their rate of DNA synthesis remained slower than the GC1 spermatogonia cells. Sertoli (and Leydig) cells are terminally differentiated and do not normally undergo mitotic division in the sexually mature testis. Thus, even when forced into continuous proliferation as single cell monolayers, the spermatogonia, Leydig and Sertoli cells maintained their characteristic sensitivity profiles, highlighting that simple in vitro systems can model specific in vivo endpoints.
In vitro data can also offer mechanistic insights. As already described, we were particularly interested in the differential miRNA expression within the three testicular cell lines in the absence and presence of DOX. In the absence of DOX, all three testicular cell lines demonstrated cell-specific miRNA profiles (Fig. 2), with the most differentially expressed miRNAs predominantly functioning in cellular development and interaction. This likely mimics the specific and differing roles of spermatogonia, Leydig and Sertoli cells in vivo. During spermatogenesis, Leydig and Sertoli cells support the proliferation and differentiation of spermatogonia into spermatozoa.43,44 Leydig cells provide endocrine secretions (androgens, including testosterone),43 while Sertoli cells provide endocrine, nutrient and direct physical support.44 That the lowest level of change was observed in the TM3 Leydig cells suggests miRNA-mediated regulation plays a greater role in germ and Sertoli cells, particularly with respect to cellular interaction.
Cell-specific miRNA changes were also observed in the presence of DOX, some of which showed some dose-dependence (Fig. 3). Although organismal injury and abnormalities and reproductive system disease were identified as the major disease categories associated with DOX-induced toxicity in all three cell lines, there were subtle differences in the DOX-induced miRNA-associated functional categories between the different cell lines (ESI Fig. 9†). The absence of the cell cycle as a miRNA-associated functional category within TM4 Sertoli cells treated with DOX may help to explain the increased resistance of TM4 Sertoli cells to DOX-induced G2/M arrest compared to GC1 spermatogonia and TM3 Leydig cells. The presence of cell movement and cell morphology as DOX-induced miRNA-associated functional categories within TM3 Leydig cells may be linked to the apparent change in location, packing and number of the Leydig cells observed in vivo as the seminiferous tubules collapse following treatment with DOX.36 Similarly, the presence of cell proliferation and cell death/survival as DOX-induced miRNA-associated functional categories within GC1 spermatogonia and TM4 Sertoli cells may be linked to the germ cell loss observed in vivo following treatment with DOX.36 The DOX-induced reduction of the cell communication miRNA-associated functional category in GC1 spermatonia and TM4 Sertoli cells compared to TM3 Leydig cells, together with the increased number of miRNAs common to GC1 spermatogonia and TM4 Sertoli cells (ESI Fig. 8†), highlights that communication within and between germ cells and Sertoli cells may be a key miRNA-mediated process disrupted by DOX. That almost double the number of DOX-induced miRNA changes were observed in the GC1 spermatogonia and TM4 Sertoli cells compared to the TM3 Leydig cells further supports miRNA-mediated communication between germ and Sertoli cells as a key target of DOX-induced testicular toxicity.
miRNAs are known to target multiple mRNAs and therefore form large interactive networks that regulate complex pathways of gene expression. To perform a more comprehensive analysis of the types of networks and pathways perturbed by DOX in vitro the validated mRNA targets of the top 100 most significant DOX-induced miRNAs in each cell line were merged and subjected to pathway analysis. This identified two pathways important in cellular communication between Sertoli cells and Sertoli and germ cells (Fig. 4), providing further evidence that DOX perturbs Sertoli and germ cell communications.
Comparison of the in vitro and in vivo36 miRNA profiles also provides some mechanistic insights, particularly the contribution of different cell types to DOX-induced miRNA changes and the potential roles of these miRNA changes within different cell types during DOX-induced testicular toxicity (Table 2). Of the 15 DOX-induced miRNAs present within the in vivo model and at least one of the in vitro testicular cell lines, 1 (miR-767) was common to, and downregulated across, all systems. This suggests all three cell types contribute to the loss of miR-767 within the testis following treatment with DOX in vivo. Interestingly, human miR-767 has been shown to target and repress expression of ten-eleven translocation (Tet) mRNAs in vitro.45 The TET family of enzymes are involved in the active demethylation of DNA. They catalyse the oxidation of 5-methylcytosine to 5-hydroxymethylcytosine (and then 5-formylcytosine and finally 5-carboxylcytosine), all of which can either be passively depleted through DNA replication or actively reverted to cytosine through thymine DNA glycosylase-mediated base excision repair.46 Reduction of miR-767 could therefore lead to increased TET levels and thus DNA demethylation. This may contribute to the DNA demethylation observed in the testis following DOX treatment in vivo.36 The remaining 14 miRNAs were present within the in vivo and just one of the in vitro cell line systems. While 4 miRNAs were common to the in vivo testis and either GC1 spermatogonia or TM4 Sertoli cells, 9 were common to the in vivo testis and TM3 Leydig cells. The majority (12) of these changes can be mechanistically correlated, indicating the cell type from which the DOX-induced miRNA change(s) may originate in vivo. For example, while miR-200c-5p and 485-3p were upregulated with increasing dose in GC1 spermatogonia in vitro, they were initially downregulated over 1 and 4 weeks post-treatment before returning to normal or even increasing at 7 weeks post-treatment in vivo. DOX-induced spermatogonial cell loss in the testis followed the same pattern; spermatogonia (and spermatocytes and round spermatids), absent at both 1 and 4 weeks post-treatment, began to reappear at 7 weeks post treatment. This suggests that GC1 spermatogonia are the main source of these DOX-induced miRNA changes in vivo. Overexpression of miR-485-3p has been shown to increase DNA Topoisomerase (TopII) expression in human laeukaemia cells in vitro.47 Since DOX is a TopII inhibitor, it is plausible that miR-485-3p is upregulated in an attempt to produce more TOPII in these mitotically active cells. Similarly, miR-330 and miR-744-3p were downregulated and upregulated, respectively, with increasing dose in TM4 Sertoli cells in vitro and at 4 weeks-post treatment in vivo. By 7 weeks post-treatment, both of these miRNAs returned to more normal levels, possibly signifying recovery of the Sertoli cells and/or rebuilding of Sertoli and germ cell communications as the germ cells begin to repopulate the testis. miR-744-3p has been shown to inhibit components of the DNA damage and repair response, leading to delayed DNA repair in human prostate cancer cells in vitro.48 Since Sertoli cells do not appear to undergo cell death in the presence of DOX either in vitro or in vivo, it is possible that DOX-induces the upregulation of miR-744-3p, which inhibits the DNA damage and repair response and signals a more senescent-like phenotype. Finally, Leydig cells appear to be the main source of DOX-induced changes in the expression of miR-9-3p, 146a, 382-3p, 687, 742, 767, 1224 and 1954 in vivo. All of these miRNAs were downregulated both in vivo and in vitro. DOX-induced downregulation of miR-146a has also been observed in cardiomyocytes derived from human pluripotent stem cells.49 Furthermore, the reduced expression of miR-9-3p, 687 or 1224 has been reported to protect against reactive oxygen species (ROS)-induced apoptosis/cytotoxicity in human and mouse cells in vitro,50–52 and may offer similar protection to Leydig cells following treatment with DOX (also known to generate ROS). The remaining (uncorrelated) DOX-induced in vivo miRNA changes could originate from other cell types not investigated here, including other germ cells such as spermatocytes, round spermatids and elongated spermatids, or other testicular cells including the peritubular myoid and blood-, lymphatic- and membrane-related cells.
The in vitro approach employed in this study has enabled further exploration of the potential roles for miRNAs within different cell types during DOX-induced testicular toxicity, successfully highlighting key miRNAs, pathways and cell types of interest. These putative mechanisms require further investigation in vitro, beginning with experimental validation of specific miRNA and mRNA targets within each cell line and ultimately the development of a co-culture system to examine germ cell and Sertoli cell communications. Thus, while single monolayers are very different from the in vivo testis, they do provide cost-effective and time-efficient opportunities to investigate potential mechanisms, examine specific end points of toxicity and identify potential biomarkers of toxicity.
Conclusions
miRNAs play an important role in both normal testicular function and DOX-induced testicular toxicity. Here we have built upon our previous in vivo study to show that DOX-induced testicular toxicity involves miRNA changes in all of the three main types of testicular cell (germ, Sertoli and Leydig cells), particularly miRNAs involved in specialised signalling at germ–Sertoli and Sertoli–Sertoli cell junctions. Therefore, it is not only direct DOX-induced toxicity that leads to germ cell loss in vivo. miRNA changes in the support (Leydig and Sertoli) cells, and possibly even more distant tissues such as the epididymis, are also important. An improved understanding of mechanisms underlying such testicular toxicity may help to develop biomarkers of testicular toxicity and batteries of simple, end-point specific, cost effective and high-throughput in vitro assays for screening the potential toxicity of drugs/chemicals during the compound discovery process.
Conflicts of interest
The authors have no conflicts of interest to declare.
Supplementary Material
Acknowledgments
This works was supported by Public Health England (PHE), the Medical Research Council (MRC) Toxicology Unit and the National Institute for Health Research Health Protection Research Unit (NIHR HPRU) in Health Impact of Environmental Hazards at King's College London in partnership with PHE. The authors would also like to thank Kate Dudek for printing the microarrays. The views expressed are those of the authors and do not necessarily reflect those of PHE, the MRC, the NIHR, the NHS or the Department of Health.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tx00314e
References
- Support M. C., Cancer Statistics, http://www.macmillan.org.uk/about-us/what-we-do/evidence/cancer-statistics.html#260441.
- Support M. C., Throwing light on the consequences of cancer and its treatment, 2013. [Google Scholar]
- NCIN, National Registry of Childhood Tumours Progress Report, http://www.ncin.org.uk/publications/.
- Bryant J., Picot J., Baxter L., Levitt G., Sullivan I., Clegg A. Br. J. Cancer. 2007;96:226–230. doi: 10.1038/sj.bjc.6603562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadurska E. Pediatric cardiol. 2015;36:1112–1119. doi: 10.1007/s00246-015-1176-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry J. D., Pointon A. V., Lutz U., Teichert F., Charlwood J. K., Chan P. H., Athersuch T. J., Taylor E. L., Singh R., Luo J., Phillips K. M., Vetillard A., Lyon J. J., Keun H. C., Lutz W. K., Gant T. W. Chem. Res. Toxicol. 2009;22:717–725. doi: 10.1021/tx800472z. [DOI] [PubMed] [Google Scholar]
- Pointon A. V., Walker T. M., Phillips K. M., Luo J., Riley J., Zhang S. D., Parry J. D., Lyon J. J., Marczylo E. L., Gant T. W. PLoS One. 2010;5:e12733. doi: 10.1371/journal.pone.0012733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho F. S., Burgeiro A., Garcia R., Moreno A. J., Carvalho R. A., Oliveira P. J. Med. Res. Rev. 2014;34:106–135. doi: 10.1002/med.21280. [DOI] [PubMed] [Google Scholar]
- Rochette L., Guenancia C., Gudjoncik A., Hachet O., Zeller M., Cottin Y., Vergely C. Trends Pharmacol. Sci. 2015;36:326–348. doi: 10.1016/j.tips.2015.03.005. [DOI] [PubMed] [Google Scholar]
- Mitry M. A., Edwards J. G. Int. J. Cardiol. Heart Vasc. 2016;10:17–24. doi: 10.1016/j.ijcha.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marczylo E. L., Jacobs M. N., Gant T. W. Crit. Rev. Toxicol. 2016:1–25. doi: 10.1080/10408444.2016.1175417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gapp K., Jawaid A., Sarkies P., Bohacek J., Pelczar P., Prados J., Farinelli L., Miska E., Mansuy I. M. Nat. Neurosci. 2014;17:667–669. doi: 10.1038/nn.3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers A. B., Morgan C. P., Leu N. A., Bale T. L. Proc. Natl. Acad. Sci. U. S. A. 2015;112:13699–13704. doi: 10.1073/pnas.1508347112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson N. O., Owens J. A., Fullston T., Lane M. Am. J. Physiol.: Endocrinol. Metab. 2015;308:E805–E821. doi: 10.1152/ajpendo.00013.2015. [DOI] [PubMed] [Google Scholar]
- Grandjean V., Fourré S., De Abreu D. A. F., Derieppe M.-A., Remy J.-J., Rassoulzadegan M. Sci. Rep. 2015;5:18193. doi: 10.1038/srep18193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surani M. A. Stem Cell Rep. 2015;4:955–960. doi: 10.1016/j.stemcr.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D. Cold Spring Harbor Perspect. Med. 2015;5:a023408. doi: 10.1101/cshperspect.a023408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly L., Chan D., Trasler J. M. Semin. Cell Dev. Biol. 2015;43:96–105. doi: 10.1016/j.semcdb.2015.07.006. [DOI] [PubMed] [Google Scholar]
- Glen C. D., Dubrova Y. E. Proc. Natl. Acad. Sci. U. S. A. 2012;109:2984–2988. doi: 10.1073/pnas.1119396109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csoka A. B., Szyf M. Med. Hypotheses. 2009;73:770–780. doi: 10.1016/j.mehy.2008.10.039. [DOI] [PubMed] [Google Scholar]
- Hess R. A., Renato de Franca L. Adv. Exp. Med. Biol. 2008;636:1–15. doi: 10.1007/978-0-387-09597-4_1. [DOI] [PubMed] [Google Scholar]
- O'Shaughnessy P. J. Semin. Cell Dev. Biol. 2014;29:55–65. doi: 10.1016/j.semcdb.2014.02.010. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S., Weinbauer G. F. Spermatogenesis. 2014;4:e996025. doi: 10.1080/21565562.2014.996025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griswold M. D. Physiol. Rev. 2016;96:1–17. doi: 10.1152/physrev.00013.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward J. A., Bardin C. W., Knight M., Robinson J., Gunsalus G., Morris I. D. Reprod. Toxicol. 1988;2:117–126. doi: 10.1016/0890-6238(88)90007-x. [DOI] [PubMed] [Google Scholar]
- Imahie H., Adachi T., Nakagawa Y., Nagasaki T., Yamamura T., Hori M. J. Toxicol. Sci. 1995;20:183–193. doi: 10.2131/jts.20.183. [DOI] [PubMed] [Google Scholar]
- Shinoda K., Mitsumori K., Yasuhara K., Uneyama C., Onodera H., Hirose M., Uehara M. Arch. Toxicol. 1999;73:274–281. doi: 10.1007/s002040050617. [DOI] [PubMed] [Google Scholar]
- Kato M., Makino S., Kimura H., Ota T., Furuhashi T., Nagamura Y. J. Toxicol. Sci. 2001;26:51–59. doi: 10.2131/jts.26.51. [DOI] [PubMed] [Google Scholar]
- Adachi T., Nishimura T., Imahie H., Yamamura T. J. Toxicol. Sci. 2000;25 Spec No:95–101. doi: 10.2131/jts.25.specialissue_95. [DOI] [PubMed] [Google Scholar]
- Tsunenari I., Kawachi M., Matsumaru T., Katsuki S. J. Toxicol. Sci. 2000;25 Spec No:103–115. doi: 10.2131/jts.25.specialissue_103. [DOI] [PubMed] [Google Scholar]
- Lui R. C., Laregina M. C., Herbold D. R., Johnson F. E. J. Urol. 1986;136:940–943. doi: 10.1016/s0022-5347(17)45136-6. [DOI] [PubMed] [Google Scholar]
- Bar-Joseph H., Ben-Aharon I., Tzabari M., Tsarfaty G., Stemmer S. M., Shalgi R. PLoS One. 2011;6:e23492. doi: 10.1371/journal.pone.0023492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meistrich M. L., Goldstein L. S., Wyrobek A. J. Mutat. Res. 1985;152:53–65. doi: 10.1016/0027-5107(85)90046-6. [DOI] [PubMed] [Google Scholar]
- Gol'dberg E. D., Borovskaya T. G., Poluektova M. E. Bull. Exp. Biol. Med. 2000;129:367–369. doi: 10.1007/BF02439273. [DOI] [PubMed] [Google Scholar]
- Vendramini V., Robaire B., Miraglia S. M. Hum. Reprod. 2012;27:2457–2466. doi: 10.1093/humrep/des159. [DOI] [PubMed] [Google Scholar]
- Akinjo O. O., Gant T. W., Marczylo E. L. Toxicol. Res. 2016;5:1229–1243. doi: 10.1039/c6tx00078a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen M. B., Spellman P. T., Brown P. O., Botstein D. Proc.Proc. Natl. Acad. Sci. U. S. A.Natl. Acad. Sci. U. S. A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling Y. H., el-Naggar A. K., Priebe W., Perez-Soler R. Mol. Pharmacol. 1996;49:832–841. [PubMed] [Google Scholar]
- O'Loughlin C., Heenan M., Coyle S., Clynes M. Eur. J. Cancer. 2000;36:1149–1160. doi: 10.1016/s0959-8049(00)00071-x. [DOI] [PubMed] [Google Scholar]
- Tyagi A. K., Singh R. P., Agarwal C., Chan D. C., Agarwal R. Clin. Cancer Res. 2002;8:3512–3519. [PubMed] [Google Scholar]
- He H., Wang C., Dai Q., Li F., Bergholz J., Li Z., Li Q., Xiao Z. X. Stem Cell Rep. 2016;7:1087–1098. doi: 10.1016/j.stemcr.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srdic-Rajic T., Santibañez J. F., Kanjer K., Tisma-Miletic N., Cavic M., Galun D., Jevric M., Kardum N., Konic-Ristic A., Zoranovic T. Sci. Rep. 2017;7:3763. doi: 10.1038/s41598-017-03898-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge R., Chen G. and Hardy M. P., in Molecular Mechanisms in Spermatogenesis, ed. C. Y. Cheng, Springer New York, New York, NY, 2008, pp. 255–269, 10.1007/978-0-387-09597-4_14. [DOI] [Google Scholar]
- Griswold M. D. Semin. Cell Dev. Biol. 1998;9:411–416. doi: 10.1006/scdb.1998.0203. [DOI] [PubMed] [Google Scholar]
- Loriot A., Van Tongelen A., Blanco J., Klaessens S., Cannuyer J., van Baren N., Decottignies A., De Smet C. Epigenetics. 2014;9:1163–1171. doi: 10.4161/epi.29628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli R. M., Zhang Y. Nature. 2013;502:472–479. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. F., He X., Arslan A. D., Mo Y. Y., Reinhold W. C., Pommier Y., Beck W. T. Mol. Pharmacol. 2011;79:735–741. doi: 10.1124/mol.110.069633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano K., Kumar B., Zhang Y., Coulter J. B., Hedayati M., Mears B., Ni X., Kudrolli T. A., Chowdhury W. H., Rodriguez R., DeWeese T. L., Lupold S. E. Nucleic Acids Res. 2015;43:4075–4086. doi: 10.1093/nar/gkv273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren G., Synnergren J., Andersson C. X., Lindahl A., Sartipy P. Toxicol. in Vitro. 2016;34:26–34. doi: 10.1016/j.tiv.2016.03.009. [DOI] [PubMed] [Google Scholar]
- Yang L., Mu Y., Cui H., Liang Y., Su X. PLoS One. 2017;12:e0174839. doi: 10.1371/journal.pone.0174839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt K., Wei Q., Pabla N., Dong G., Mi Q. S., Liang M., Mei C., Dong Z. J. Am. Soc. Nephrol. 2015;26:1588–1596. doi: 10.1681/ASN.2014050463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S., Bantel H., Wandrer F., Theres Schneider A., Gautheron J., Vucur M., Tacke F., Trautwein C., Luedde T., Roderburg C. J. Hepatol. 2017;67(5):966–978. doi: 10.1016/j.jhep.2017.06.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.