

Abstract
Vertebrate cone photoreceptors are known to show lower light sensitivity and briefer photoresponses than rod photoreceptors. To understand the molecular mechanisms characterizing cone photoresponses, we compared some of the reactions in the phototransduction cascade between rods and cones. For this purpose, rods and cones were obtained in quantities large enough to do biochemical studies. The cells were purified from the retina of carp (Cyprinus carpio) with a stepwise Percoll gradient. The purified rod fraction contained almost no other kinds of cells besides rods, and the purified cone fraction contained a mixture of red-, green-, and blue-sensitive cones in the ratio 3:≈1:≈1. We prepared membrane preparations from the rod and the cone fraction, and in these membranes, we measured activation efficiencies of the reactions in the phototransduction cascade. The results showed that the signal amplification is lower in the cone membranes, which accounts for the lower light sensitivity in cones. Furthermore, we measured the time courses of visual pigment phosphorylation. The result showed that the phosphorylation is much faster in the cone membranes, which also explains the lower light sensitivity and, in addition, the briefer photoresponse in cones.
In the vertebrate retina, there are two types of photoreceptors, rods and cones. Rods mediate twilight vision and cones daylight vision, so that the photoresponse characteristics differ in rods and cones (for reviews, see refs. 1–3). The light sensitivity of a cone is 25–100 times lower than that of a rod, and the response is much briefer in cones than in rods (4–6). In the present study, we attempted to elucidate the molecular mechanisms characterizing cone photoresponses.
The phototransduction mechanism in rods is well documented (7–10) and is now regarded as a model system of G-protein-coupled receptor signaling. It has been known that there are rod and cone versions of phototransduction enzymes (for example, visual pigment, transducin, cGMP phosphodiesterase (PDE) and cGMP-gated channel). From this result, the phototransduction cascades in rods and cones are thought to be basically similar (for reviews, see refs. 1–3). It is therefore possible that the differences between rod and cone photoresponse characteristics are because of differences in the phototransduction reactions in rods and cones. Actually, some of the cone components were purified and studied. For example, cone visual pigment was purified and its quantum efficiency was shown to be similar to that of the rod pigment rhodopsin (11). In other studies, with the use of a purified cone protein, its interaction with a rod protein was measured (12–16). To know the mechanism characterizing the cone photoresponses, however, it is essential to measure the interaction between cone proteins and compare the result with the corresponding reactions in rods. With the use of cone-dominant retina, PDE activities and guanylate cyclase activities were actually measured (17, 18), but in these studies, the focus of the experiment was different from ours.
In the present study, we tried to measure the efficiencies of the phototransduction reactions in cones and compare the results with those in rods obtained from the same animal species. We therefore first purified rods and cones simultaneously from the retina of carp (Cyprinus carpio) and measured some of the phototransduction reactions in both types of the cells. The result showed that the signal amplification is lower in cones. Furthermore, we measured the phosphorylation of visual pigment, one of the shut-off mechanisms of the phototransduction cascade. The result showed that it is much faster in cones.
Materials and Methods
Isolation of Rod and Cone Photoreceptor Cells and Preparation of Rod and Cone Membranes.
Carp (Cyprinus carpio), 25–30 cm in length, were dark-adapted in a light-tight tank for >3 h before use, and the retina was dissected after pith. The photoreceptors were brushed off in a Ringer's solution (119.9 mM NaCl/2.6 mM KCl/0.5 mM CaCl2/0.5 mM MgCl2/0.5 mM MgSO4/1 mM NaHCO3/16 mM glucose/0.5 mM NaH2PO4/4 mM Hepes, pH 7.5), and the resultant suspension of the photoreceptors was filtered through a nylon mesh to eliminate large fragments of retinal tissue. The pass-through containing isolated photoreceptors was layered on the top of a stepwise Percoll gradient (see Fig. 1) and centrifuged for 20 min at 10,000 × g. Cells at the interfaces were collected and mixed with the same volume of the Ringer's solution to reduce the density of Percoll. After the cells were sedimented by centrifugation first at 600 × g for 12 s and then at 3,000 × g for 4 s, they were then disrupted by freeze-thaw. The resultant membranes were washed twice with and resuspended in a potassium-gluconate buffer (115 mM K-gluconate/2.5 mM KCl/2 mM MgCl2/0.2 mM EGTA/0.1 mM CaCl2/1 mM DTT/10 mM Hepes, pH 7.5) (K-gluc buffer) (19). To quantify the protein content, we measured the amount of visual pigments in the membranes (see below). The membranes thus obtained were kept at −80°C until they were used. Typically, 20–30 carp were used for a single measurement in the present study. All manipulations were carried out in complete darkness with the aid of an infrared image converter (NVR 2015; NEC) under illumination of >800-nm light. With the use of anti-human Gt1α (Santa Cruz Biotechnology), we confirmed that >90% of rod transducin α-subunit remained in the rod membranes. In addition, with the use of anti-carp GRK1 (rhodopsin kinase) and anti-carp GRK7 (cone visual pigment kinase) antibodies, we confirmed that >95% of these kinases were present in the membranes used.
Figure 1.

Purification of rod and cone photoreceptor cells. (A) Cells brushed off the retina. (B) Rod fraction. (C) Cone fraction. The scale bar indicates 20 μm. (D) Absorption spectra of the visual pigments in the carp. Only rhodopsin was found in the rod fraction (dotted curve), and red, green, and blue pigments (red, green, and blue curves) were found in the cone fraction. Spectral intensities of green and blue pigments are expressed as the values relative to that of the red pigment in a typical cone fraction. The absorption maxima of rhodopsin and red, green, and blue pigments were 522, 618, 535, and 460 nm, respectively, in agreement with a previous report (27). (E and F) Families of photoresponses at different flash intensities of a rod and a red-sensitive cone, respectively. The arrowhead indicates the timing of the delivery of a light flash. (G) Flash intensity–response relations of rods (black, two cells), green-sensitive cones (green, six cells), and red-sensitive cones (red, four cells). The sensitivity of cones was relatively constant, whereas that of rods varied more than 10 times, depending on the preparation. In the figure, the relation obtained in the most sensitive rods is shown.
The photoresponses were measured with suction electrodes (20), with the use of mechanically dissociated photoreceptors, which are different from the cells purified by Percoll gradient (see below). Because it takes a few hours to obtain the purified cells, we did not use these cells for the recording of photoresponses.
Spectroscopic Measurement.
To quantify the amount of visual pigments in the rod and the cone membranes, small portions of the membranes were solubilized in an extraction buffer [0.75% (wt/vol) 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate/1 mg/ml phosphatidylcholine/50 mM Hepes/140 mM NaCl/2 mM MgCl2/20% (wt/vol) glycerol/1 mM DTT, pH 7.5). The spectrum of each type of the visual pigment was determined by the partial bleach method with the use of cut-off filters passing >660-, >600-, or >520-nm light (21).
Light Source.
The flash light source used was a Sunpak auto 25SR equipped with a cut-off filter passing >410-nm light. The flash half-duration was 0.9 ms, and the light intensity was attenuated by neutral density filters. In the biochemical studies, we calibrated the flash intensity by measuring the amount of visual pigment bleached. In the measurement of electrical photoresponses, the intensity of a light flash at 450 nm was about half of that at 600 nm because we used a light guide. We therefore estimated the relative effectiveness of the light for each type of the visual pigment by calculating the overlapping area between the light energy distribution and the absorption spectrum of each pigment.
Transducin Activation Assay.
Radionucleotide-filter binding assay was carried out as described (22) with some modifications. Rod or cone membranes (15 μl) were mixed with 10 μl of the K-gluc buffer containing [35S]GTPγS, GDP, EGTA, and, when necessary, ATP (final concentrations: 3 μM rhodopsin or 0.3 μM cone pigment/5 μM [35S]GTPγS/5 μM GDP/0.8 mM EGTA/0.1 mM ATP). After preincubation for 30 s under this condition, the sample was irradiated with a light flash. The reaction was carried out at 20°C throughout and terminated by the addition of 200 μl of the ice-chilled K-gluc buffer containing 20 μM cold GTPγS and 10 μM GDP. The sample was then filtered immediately through a nitrocellulose membrane and washed with the K-gluc buffer containing 25 mM MgCl2. The amount of [35S]GTPγS bound to the nitrocellulose membrane was quantified with an image analyzer (BAS 2000; Fuji).
Phosphorylation Assay.
A phosphorylation assay was performed as described (23) with some modifications. Rod or cone membranes (15 μl) were mixed with 10 μl of the K-gluc buffer containing [γ-32P]ATP, GTP, and EGTA (final concentrations: 0.3 μM visual pigment/0.1 mM [γ-32P]ATP/0.5 mM GTP/0.8 mM EGTA). After preincubation for 30 s, the sample was irradiated with a light flash bleaching 1.9% rhodopsin or 4.0% cone visual pigments. The reaction was terminated by adding 150 μl of 10% (wt/vol) trichloroacetic acid. After centrifugation (14,000 × g for 10 min), the precipitate was washed with the K-gluc buffer and subjected to SDS/PAGE. The amount of 32P incorporated into the visual pigment band was quantified by an image analyzer (BAS 2000; Fuji). All manipulations were carried out at room temperature.
PDE Assay.
PDE activity was measured with the pH assay method (19). Rod or cone membranes were suspended in 100 μl of the K-gluc buffer containing (as final concentrations) 0.5–2 μM visual pigment, 0.5 mM GTP, 5 mM cGMP, and 0.8 mM EGTA. The membranes were first irradiated with a test flash in the presence or absence of 0.1 mM ATP. After a measurement of the light-induced PDE activation and the following inactivation, a bright steady light from a 100-W tungsten-halogen lamp was used to measure the maximum PDE activity. The range of the pH drop during a measurement was less than 0.1 pH unit. All manipulations were carried out at room temperature.
Results and Discussion
Purification of Rod and Cone Cells.
Rods and cones were brushed off the retina (Fig. 1A). The ratio of the number of the cells was ≈50 (rod):1 (cone) at this stage (Table 1). The cells were purified with the use of a stepwise Percoll gradient. Rods were obtained at the 45/60% (wt/vol) interface (rod fraction, Fig. 1B), and cones were obtained at the 75/90% (wt/vol) interface (cone fraction, Fig. 1C). Contamination of the other type of photoreceptor was negligible in both the rod and the cone fractions (Table 1). The cone fraction, however, contained erythrocytes, so that, with the use of the rod membranes, we always did control experiments and confirmed that erythrocytes do not affect the reactions.
Table 1.
Purification of rods and cones from carp
| Initial isolation | |
|---|---|
| No. of rod and cone cells | rod:cone = ∼50:1 |
| After purification | |
| Rod fraction | |
| Yield | ∼6,000,000 rods/retina |
| Contamination* | Cone: < 1% |
| Pigment | Rhodopsin |
| Cone fraction | |
| Yield | ∼250,000 cones/retina |
| Contamination* | Rod: < 1% |
| Erythrocyte: 10–30% | |
| Pigment | Red:green:blue = 3:∼1:∼1 |
Expressed based on the number of the cells.
With the partial bleach method, we detected only a single visual pigment, rhodopsin, in the rod fraction (Fig. 1D, dotted line). The cone fraction contained both single and double cones (see Fig. 1C) with three types of visual pigments (Fig. 1D). The ratio of the three pigments, and therefore possibly that of other phototransduction proteins, was 3 (red):≈1 (green):≈1 (blue). Based on the number of purified rods and cones and on the assumption that the ɛmax of visual pigments is 40,000 O.D./M-cm, we estimated that, on average, a rod contained 1.6 × 108 rhodopsin molecules and a cone contained 7.8 × 107 cone visual pigment molecules. Based on the average cell dimensions measured, the calculated pigment concentrations were 2.7 mM in rods and 2.2 mM in cones.
With mechanically dissociated cells but not the purified cells (see Materials and Methods), we measured the electrical photoresponses of rods (Fig. 1E) and red- (Fig. 1F) and green-sensitive cones (not shown). The results showed that the flash response was much briefer in cones than in rods: the time to peak was shorter and the recovery time course was faster in cones. Among rods and cones, the photoresponse was briefest in red-sensitive cones, in agreement with previous studies (5, 6). The flash intensity–response relation showed that the light sensitivity was 102 to 103 times lower in cones than in rods (Fig. 1G). The light sensitivity of a green-sensitive cone was ≈10 times lower than that of a red-sensitive cone, as has been reported (5, 6). In the purified cone fraction, blue cone pigment was present (Fig. 1D). However, we could not record the photoresponse of blue-sensitive cones. It might be the case that the yield of blue cones was low with the mechanical dissociation method that was used for electrophysiological measurement.
Transducin Activation.
Fig. 2A shows the transducin activation measured by the binding of GTPγS as a function of flash intensity in the rod (circles, 40-s incubation) and the cone membranes (triangles, 20-s incubation) with (filled symbols) or without (open symbols) ATP. The result showed that the flash intensity giving half-maximum activation was ≈102 times higher in the cone membranes. ATP reduced the light sensitivity of transducin activation most probably because of facilitated inactivation of light-activated visual pigment (R*) by phosphorylation (see below). The maximum transducin activation level in the cone membranes was ≈1/3 of that in the rod membranes (dashed lines), which indicated that the transducin/visual pigment ratio was not so different in the rod and the cone membranes used. In addition, because the molar ratio of transducin to rhodopsin is roughly 1:10 (2), the maximum GTPγS binding data showed that most transducin molecules were present in the membranes used. A similar conclusion was obtained in our Western blot analysis (see Materials and Methods).
Figure 2.

Transducin activation in the rod and the cone membranes. (A) Transducin activation as a function of flash intensity, expressed as the number of GTPγS incorporated per visual pigment present. The GTPγS binding reaction in the rod membranes (○, ●, n = 3) was terminated 40 s after a light flash, and that in the cone membranes (▵, ▴, n = 3–4) was terminated at 20 s. The dashed lines show the maximum GTPγS binding in the rod (0.25 ± 0.03 GTPγS bound per pigment present) and the cone (0.071 ± 0.002 GTPγS bound per pigment present) membranes. The maximum activity in the rod membranes was determined 300 s after a light flash bleaching 0.19% of rhodopsin, and that in the cone membranes was determined 80 s after a flash bleaching 95% visual pigment. For unknown reasons, GTPγS binding in the rod membranes decreased as the flash intensity increased above 0.2% bleach. (B) Time courses of GTPγS binding in the rod (○, ●, n = 3) and the cone (▵, ▴, n = 3–6) membranes with (●, ▴) and without (○, ▵) ATP. GTPγS binding is expressed as the amount of GTPγS incorporated per visual pigment bleached. The light flash used for the rod membranes bleached 0.019% of rhodopsin and that for the cone membranes bleached 0.41% of cone visual pigments. The time course of GTPγS binding in the cone membranes is shown at an expanded scale (Inset). With curve fitting, we determined the turnover number of the transducin activation and the decay time constant of R* (curves 1–4; see text). To determine the light-induced activation, background activity was always subtracted in each measurement. The dotted line shows the fitting of an extreme case when the initial rate of GTPγS binding is the same as that in the rod membranes.
To estimate the rate of transducin activation, we measured the time course of the GTPγS binding at the light intensity giving half-maximum activation (Fig. 2B). The measurements were made at first in the absence of ATP (open symbols). The result was analyzed by assuming that R* activates transducin with a turnover number of kcat and decays exponentially with a time constant of τ. Then the total amount of activated transducin at time t, T*(t), is expressed in the equation
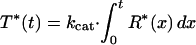 |
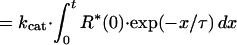 |
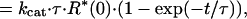 |
where R*(x) is the amount of R* at time x.
By fitting the experimental points with this equation, we estimated the values of kcat and τ in the rod and the cone membranes (curves 1 and 3). The best fitted kcat values were 53.6 T*/R*-s (transducin activated (T*) per R* per second) in the rod membranes and 1.9 T*/R*-s in the cone membranes. Thus, the ratio of the transducin activation rate is calculated to be ≈30 (rod):1 (cone). It has been known that the quantum efficiency of visual pigment activation is similar in rod and cone visual pigments (11). Therefore, the responsible step for the lower transducin activation efficiency in the cone membranes should not be at the bleaching step but at the transducin activation step.
The best fitted decay constant (τ) was 15.9 s in the rod membranes and 2.9 s in the cone membranes. The result showed that the lifetime of R* is much shorter in cone visual pigments, in agreement with a previous study (24). This shorter lifetime of R* would be one of the mechanisms that account for the reduced light sensitivity of transducin activation in the cone membranes (Fig. 2A).
The lifetime of R* is expected to be shortened by visual pigment phosphorylation. In fact, R* decayed more rapidly in the presence of ATP: with the addition of ATP, the τ values decreased from 15.9 s to 7 s in the rod membranes (curve 2) and, interestingly, from 2.9 s to 1.0 s in the cone membranes (curve 4). In the fitting of curve 2, the best fit kcat value was 57.3 T*/R*-s, which was very similar to the value obtained in the absence of ATP (curve 1). The result suggested that ATP does not affect the initial phase of transducin activation but affects the later stage. In the fitting of curve 4, we had to assume that the kcat value is the same as that in the absence of ATP, because the GTPγS binding was completed within our limit of time resolution of the reaction (filled triangles).
In Fig. 2B, the GTPγS binding time course and its steady level were affected by ATP addition (compare open and closed symbols). It is evident from this result that the phosphorylation on R* affects the time course at the later stage and thus the total amount of transducin activation.
Phosphorylation of Visual Pigment.
In Fig. 2B, the decay of R* was much faster in the cone membranes in the presence of ATP, which suggested that R* phosphorylation is very fast in the cone membranes. In fact, Fig. 3 shows that bleached rhodopsin (filled circles) was phosphorylated with an apparent time constant of ≈20 s (fitted at 0–80 s, with 1.9% bleaching flash), but bleached cone visual pigments (filled triangles) were phosphorylated fully in less than 1 s (4% bleaching). Because the phosphorylation reached the maximum level at our earliest time point (1 s), we could not determine the time constant precisely. The result, therefore, showed that the phosphorylation reaction on R* is >20 times faster in the cone membranes. The maximum phosphorylation was ≈3 per R* in the rod membranes and ≈2 in the cone membranes. Rhodopsin was phosphorylated by two phosphate groups at ≈80 s after the flash, and cone pigments were phosphorylated by two phosphate groups within 1 s. The result in Fig. 3 unequivocally showed that the phosphorylation on R* is much faster in the cone membranes.
Figure 3.

Time courses of visual pigment phosphorylation in the rod and the cone membranes. After SDS/PAGE, 32P incorporated into the visual pigment band was quantified in the rod (●, n = 3) and the cone (▴, n = 3) membranes. To determine the light-induced phosphorylation, background activity was always subtracted in each measurement.
PDE Activation.
Fig. 4 shows PDE activation measured with the pH assay method in the rod (Fig. 4A) and the cone (Fig. 4B) membranes with (bold traces) and without (thin traces) ATP. The first derivatives of the pH traces (PDE activities) were determined and are shown in Fig. 4 C and D (C from A, and D from B). Fig. 4E summarizes the relation between a peak PDE activity shown in Fig. 4 C and D as a function of flash intensity. As seen in Fig. 4E, the maximum PDE activity elicited by a saturating light in the cone membranes was similar (≈80%) to that in the rod membranes. However, the light intensity required for half-maximum activation was more than 102 times higher in the cone membranes. The half-maximum activation in the rod membranes was observed at ≈0.01% bleach, consistent with the previous study (25).
Figure 4.

PDE activation in the rod and the cone membranes. PDE activity was measured with the pH assay method. (A and B) The pH drop was monitored in either the rod (A) or the cone (B) membranes with (bold traces) or without (thin traces) ATP. An arrow indicates the timing of the test flash and an arrowhead the onset of a bright steady illumination. To measure the light-induced changes in PDE activity, dark PDE activity was always subtracted. (C and D) First derivatives determined from the data shown in A and B, respectively. (E) Peak PDE activities in the rod (○, ●, n = 3) and the cone (▵, ▴, n = 3) membranes as a function of test flash intensity. In each measurement, maximum PDE activity was measured, and the PDE peak activity as elicited by a test flash was expressed as a percentage of the maximum activity. In the rod membranes, the maximum PDE activity was 22.3 ± 3.1 cGMP hydrolyzed per pigment present per second (n = 3), and in the cone membranes it was 17.8 ± 1.5 cGMP hydrolyzed per pigment present per second (n = 3). To compare the rod and cone PDE activation directly, each PDE peak activity in the cone membranes is expressed so that the maximum cone PDE activity is 80%. (F) Half-life of activated PDE as a function of test flash intensity (n = 2–3). The PDE half-life was determined as the time required for the recovery of the PDE activity to 50% of the peak activity. Symbols are as used in E.
As can be seen in Fig. 4E, the effect of ATP on the PDE peak activity was not so large in either the rod or the cone membranes. However, the ATP affected the time course of PDE inactivation significantly in the cone membranes. The half-life of the activated cone PDE was approximately halved in the presence of ATP (Fig. 4F). At the half-saturating flash intensity, the half-life of PDE in the rod membranes was ≈15 s, and that in the cone membranes ≈4 s in the presence of ATP.
The efficiencies of PDE activation in the presence of ATP were calculated at a light intensity bleaching of 0.0078% of rhodopsin and 1.5% of cone visual pigments. Previous reports showed that the molar ratio of rhodopsin to PDE is ≈150:1 (2). Because 34% of PDE was activated by bleaching of 0.0078% of rhodopsin (Fig. 4E), the number of PDE molecules activated per R* (PDE*/R*) was calculated to be 29 PDE*/R*. Assuming the molar ratio of cone PDE to cone visual pigment is ≈150:0.8 (see below), we obtained the value of 0.11 PDE*/R* in the cone membranes. The resultant ratio was ≈260 (rod):1 (cone). Because the transducin activation ratio was ≈30 (rod):1 (cone) (Fig. 2), the above calculation strongly suggested that the efficiency of PDE activation by an activated transducin molecule is ≈10 times less effective in the cone membranes.
The specific enzyme activity of cone PDE has been reported to be similar to that of rod PDE (12). Because the maximum PDE activity in the cone membranes was ≈80% of that in the rod membranes (Fig. 4E), the amount of cone PDE molecules was ≈80% of that of rod PDE.
In a recent study, steady thermal activation of visual pigment in L (red-sensitive) cones has been suggested to occur (26). In our measurement of PDE activation, however, the PDE dark activity measured in the cone membranes (2.5 ± 2.2% of the maximum PDE activity, n = 7) was low and very similar to that in the rod membranes (1.1 ± 1.0%, n = 4). Although the steady activation of visual pigment might take place during our measurements, its contribution to our study seemed to be negligible.
Molecular Bases of the Difference in the Photoresponse Characteristics Between Rods and Cones.
The present study showed that, in the cone membranes (i) the transducin activation is ≈30 times less effective (Fig. 2) and (ii) the phosphorylation of visual pigment is >20 times faster (Fig. 3). In addition, in the cone membranes, (iii) PDE activation by transducin is ≈10 times less effective (Fig. 4) and (iv) PDE inactivation is several times faster (Fig. 4). These results are summarized in Table 2. The above results reasonably and qualitatively account well for the low light sensitivity, short time to peak, and fast recovery time course of the cone photoresponses. The lower light sensitivity could be because of the lower efficiency of transducin activation by visual pigment plus the lower efficiency of PDE activation by transducin, and the shorter time to peak could be because of the faster decay of R*. Although other mechanisms are doubtlessly present, the faster inactivation of PDE can be one of the mechanisms that contribute to the faster recovery time course in the cone photoresponse.
Table 2.
Transduction efficiencies in rods and cones†
| Rods | Cones | Reference | |
|---|---|---|---|
| Activation | |||
| Photoresponse (light sensitivity) | 102–3 | 1 | Fig. 1 |
| Pigment activation (per light) | 1 | 1 | Ref. 11 |
| Transducin activation (per R*-sec) | <30 | 1 | Fig. 2 |
| PDE activation | |||
| (per R*) | ∼260 | 1 | Fig. 4 |
| (per T*) | >10 | 1 | Fig. 4 |
| Termination and recovery | |||
| R* phosphorylation (rate constant) | 1 | ∼20 | Fig. 3 |
| PDE half-life (time) | 2∼3 | 1 | Fig. 4 |
Shown as relative values, not in actual units.
There has been a suggestion that the transduction gain is not so different between rods and cones (2). As an extreme case, therefore, we postulated that the transducin activation rate is the same between rods and cones. The dotted line in the inset in Fig. 2B shows the fitting of the data with the initial rate set at 54 T*/R*-s, the same rate in the rod membranes, and τ = 0.08 s. The result shows that the fitting is possible, which suggests that our estimation of the transducin activation in the cone membranes may be underestimated. Obviously, GTPγS binding data at faster time points are needed to obtain more quantitative results.
In previous studies by others, rod enzyme activities on cone proteins were measured. Starace and Knox (16) measured activation of rod transducin by cone visual pigment in a reconstituted system. The result showed that cone visual pigment activates rod transducin 3–5 times less effectively than rhodopsin does. In our study, we obtained a qualitatively similar result (Table 2). However, the rates they obtained were 1.44–2.1 T*/R*-min (rhodopsin) and 0.46 T*/R*-min (cone pigment) and are 200-2000 times lower than our estimate (≈54T*/R*-s in rods and ≈2T*/R*-s in cones; Fig. 2). Fukada et al. (13) measured phosphorylation by rod GRK1 (rhodopsin kinase) on bleached rhodopsin and a cone pigment. In contrast to our study, their measurement showed that the time course of phosphorylation on R* of cone pigment was slow and similar to that of rhodopsin. It is suggested that protein interaction between a rod and a cone protein is different from that between a cone and a cone protein.
In the present work, we did not examine the effects of Ca2+ or soluble proteins in photoreceptors; nor did we examine the difference in the cGMP synthesis. It would be essential to study these issues to account for the cone photoresponse in a quantitative way. In addition, we need to know whether the result obtained in this study biochemically can be applied to intact cells. Obviously, further examination of this matter is required.
Abbreviations
- PDE
phosphodiesterase
- K-gluc buffer
potassium-gluconate buffer
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Yau K-W. Invest Ophthalmol Visual Sci. 1994;35:9–32. [PubMed] [Google Scholar]
- 2.Pugh E N, Jr, Lamb T D. Biochim Biophys Acta. 1993;1141:111–149. doi: 10.1016/0005-2728(93)90038-h. [DOI] [PubMed] [Google Scholar]
- 3.Pugh E N Jr, Lamb T D, editors. Handbook of Biological Physics. Rotterdam: Elsevier Science; 2000. pp. 183–255. [Google Scholar]
- 4.Nakatani K, Yau K-W. J Physiol (London) 1989;409:525–548. doi: 10.1113/jphysiol.1989.sp017511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perry R J, McNaughton P A. J Physiol (London) 1991;433:561–587. doi: 10.1113/jphysiol.1991.sp018444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller J L, Korenbrot J I. Visual Neurosci. 1993;10:653–667. doi: 10.1017/s0952523800005356. [DOI] [PubMed] [Google Scholar]
- 7.Kawamura S. In: Neurobiology and Clinical Aspects of the Outer Retina. Djamgoz M B A, Archer S N, Vallerga S, editors. London: Chapman & Hall; 1995. pp. 105–131. [Google Scholar]
- 8.Baylor D A. Proc Natl Acad Sci USA. 1996;93:560–565. doi: 10.1073/pnas.93.2.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leskov I B, Klenchin V A, Handy J W, Whitlock G G, Govardovskii V I, Bownds M D, Lamb T D, Pugh E N, Jr, Arshavsky V Y. Neuron. 2000;27:525–537. doi: 10.1016/s0896-6273(00)00063-5. [DOI] [PubMed] [Google Scholar]
- 10.Fain G, Matthews H R, Cornwall M C, Koutalos Y. Physiol Rev. 2001;81:117–151. doi: 10.1152/physrev.2001.81.1.117. [DOI] [PubMed] [Google Scholar]
- 11.Okano T, Fukada Y, Shichida Y, Yoshizawa T. Photochem Photobiol. 1992;56:995–1001. doi: 10.1111/j.1751-1097.1992.tb09722.x. [DOI] [PubMed] [Google Scholar]
- 12.Gillespie P G, Beavo J A. J Biol Chem. 1988;263:8133–8141. [PubMed] [Google Scholar]
- 13.Fukada Y, Kokame K, Okano T, Shichida Y, Yoshizawa T, McDowell J H, Hargrave P A, Palczewski K. Biochemistry. 1990;29:10102–10106. doi: 10.1021/bi00495a013. [DOI] [PubMed] [Google Scholar]
- 14.Hamilton S E, Prusti R K, Bentley J K, Beavo J A, Hurley J B. FEBS Lett. 1993;318:157–161. doi: 10.1016/0014-5793(93)80012-j. [DOI] [PubMed] [Google Scholar]
- 15.Kawamura S, Kuwata O, Yamada M, Matsuda S, Hisatomi O, Tokunaga F. J Biol Chem. 1996;271:21359–21364. doi: 10.1074/jbc.271.35.21359. [DOI] [PubMed] [Google Scholar]
- 16.Starace D M, Knox B E. J Biol Chem. 1997;272:1095–1100. doi: 10.1074/jbc.272.2.1095. [DOI] [PubMed] [Google Scholar]
- 17.Booth D P, Hurwitz R L, Lolley R N. J Neurochem. 1991;56:1949–1956. doi: 10.1111/j.1471-4159.1991.tb03452.x. [DOI] [PubMed] [Google Scholar]
- 18.Denton T L, Yamashita C Y, Farber D B. Exp Eye Res. 1992;54:229–237. doi: 10.1016/s0014-4835(05)80212-x. [DOI] [PubMed] [Google Scholar]
- 19.Kawamura S. Nature (London) 1993;362:855–857. doi: 10.1038/362855a0. [DOI] [PubMed] [Google Scholar]
- 20.Baylor D A, Lamb T D, Yau K-W. J Physiol (London) 1979;288:589–611. [PMC free article] [PubMed] [Google Scholar]
- 21.Okano T, Fukada Y, Artamonov I D, Yoshizawa T. Biochemistry. 1989;28:8848–8856. doi: 10.1021/bi00448a025. [DOI] [PubMed] [Google Scholar]
- 22.Terakita A, Yamashita T, Tachibanaki S, Shichida Y. FEBS Lett. 1998;439:110–114. doi: 10.1016/s0014-5793(98)01340-4. [DOI] [PubMed] [Google Scholar]
- 23.Tachibanaki S, Nanda K, Sasaki K, Ozaki K, Kawamura S. J Biol Chem. 2000;275:3313–3319. doi: 10.1074/jbc.275.5.3313. [DOI] [PubMed] [Google Scholar]
- 24.Shichida Y, Imai H. Cell Mol Life Sci. 1998;54:1299–1315. doi: 10.1007/s000180050256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kawamura S, Murakami M. J Gen Physiol. 1986;87:737–759. doi: 10.1085/jgp.87.5.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rieke F, Baylor D A. Neuron. 2000;26:181–186. doi: 10.1016/s0896-6273(00)81148-4. [DOI] [PubMed] [Google Scholar]
- 27.Tomita T, Kaneko A, Murakami M, Pautler E L. Vision Res. 1967;7:519–531. doi: 10.1016/0042-6989(67)90061-2. [DOI] [PubMed] [Google Scholar]