

Abstract
The transcription factor GATA-3, highly expressed in many cutaneous and peripheral T-cell lymphomas, confers resistance to chemotherapy in a cell-autonomous manner. As GATA-3 is transcriptionally regulated by NF-κB, we sought to determine the extent to which proteasomal inhibition impairs NF-κB activation and GATA-3 expression and cell viability in malignant T cells. Proteasome inhibition, NF-κB activity, GATA-3 expression, and cell viability were examined in patient-derived cell lines and primary T-cell lymphoma specimens ex vivo treated with the oral proteasome inhibitor ixazomib. Significant reductions in cell viability, NF-κB activation, and GATA-3 expression were observed pre-clinically in ixazomib-treated cells. Therefore, an investigator-initiated, single-center, phase II study with this agent in patients with relapsed/refractory CTCL/PTCL was conducted. Concordant with our pre-clinical observations, a significant reduction in NF-κB activation and GATA-3 expression was observed in an exceptional responder following one month of treatment with ixazomib. While ixazomib had limited activity in this small and heterogeneous cohort of patients, inhibition of the NF-κB/GATA-3 axis in a single exceptional responder suggests that ixazomib may have utility in appropriately selected patients or in combination with other agents.
Keywords: peripheral T-cell lymphoma, cutaneous T-cell lymphoma, GATA-3, proteasome, ixazomib
Introduction
The T-cell lymphomas are a molecularly and clinically heterogeneous group of non-Hodgkin’s lymphomas (NHL) that are often resistant to conventional chemotherapeutic regimens. Mycosis fungoides (MF), the most common cutaneous T-cell lymphoma (CTCL) subtype, commonly presents with limited-stage disease and is managed with skin-directed therapies.1 Survival in these patients is measured in years to decades, with survival for those with <10% body surface area involvement comparable to that observed in age-matched subjects without CTCL.1 In contrast, patients with advanced-stage disease, or limited-stage disease unresponsive to skin-directed therapies, require systemic therapy. While responses to conventional chemotherapeutic agents may be achieved, these responses are short-lived, likely secondary to inherent resistance mechanisms that are incompletely understood.2, 3 In North America, the most common peripheral T-cell lymphoma (PTCL) remains “not otherwise specified (NOS).”4 Among PTCL, NOS patients with relapsed/refractory disease who are not eligible for hematopoietic stem-cell transplantation, novel therapies are not curative, rarely achieve a durable remission, and are effective in only a subset of patients.5, 6 Therefore, novel therapies are needed for the T-cell lymphomas, and clinical trial participation is encouraged.
The mechanisms underlying primary and acquired resistance to conventional or novel chemotherapeutic agents in CTCL and PTCL are poorly understood. Recurrent somatic alterations [e.g. PLC-γ1 mutations,7 and NF-κB truncations,8, 9], genomic losses [e.g. including p531, 9] and the expression of multidrug resistance proteins likely contribute to resistance.10 Constituents of the tumor microenvironment (TME), particularly lymphoma-associated macrophages, may also promote resistance to chemotherapy,11 likely due to the provision of cell surface ligands or cytokines that activate signaling pathways implicated in chemotherapy resistance [reviewed in 12]. A limited number of transcriptional regulators known to regulate the differentiation, proliferation, and survival of normal T cells have also been implicated in disease pathogenesis and chemotherapy resistance, chief among these is the NF-κB family of transcription factors.13–15 Constitutive NF-κB activation is frequently observed among the T-cell lymphomas,15 and its inhibition overcomes resistance to apoptosis.13, 14 Furthermore, factors with a known or suspected role in chemotherapy resistance are transcriptionally regulated by NF-κB, including the transcription factor GATA-3.16 GATA-3, while frequently described as the “master regulator” of Th2 differentiation, is widely expressed, albeit to varying degrees, in most T-cell subsets, where it not only regulates the expression of a vast number of genes in a cell-type specific manner,17 but also promotes T-cell survival.18 Notably, GATA-3 is highly expressed in both CTCL and in a recently described subset of PTCL, NOS [19, 20, reviewed in 21]. In these T-cell lymphomas, GATA-3 promotes the production of Th2-associated cytokines,19 including those that regulate the growth and survival of malignant T cells.22 More recently, GATA-3 was shown to promote resistance to chemotherapy in a cell-autonomous manner, possibly explaining the high rate (>50%) of primary refractory disease observed among GATA-3+ PTCL, NOS.16 Therefore, the development of therapeutic strategies targeting NF-κB and its target genes, including GATA-3, are warranted. It is with that goal in mind that we sought to investigate the novel proteasome inhibitor ixazomib (MLN9708).
The ubiquitin-proteasome pathway (UPP) plays a critically important role in cell homeostasis and its inhibition has emerged as a promising therapeutic approach in hematologic malignancies. Proteasomal inhibition, among its other effects, culminates in the inhibition of NF-κB activity in T-cell lymphomas.14 Limited experience with the proteasome inhibitor bortezomib in CTCL is encouraging, as an overall response rate of 67% was observed in a small phase II study.23 Furthermore, more recent evidence suggests that novel agents currently approved for the treatment of CTCL or under investigation also target the UPP.24 Therefore, we sought to investigate the safety and efficacy of ixazomib, an oral proteasome inhibitor, in relapsed/refractory cutaneous and peripheral T-cell lymphomas.
Materials and Methods
Cell culture, viability assays, and reagents
Primary T-cell lymphoma (TCL) cells were obtained from patients at the University of Michigan Comprehensive Cancer Center, Ann Arbor, MI upon approval by the Institutional Review Board of the University of Michigan, in accordance with U.S. federal regulations and the Declaration of Helsinki. Primary TCL cells were purified, as previously described 16. The TCL cell lines (T8ML-1, H9, MyLa) used in this study were mycoplasma free and independently validated, as previously described 16, 19, 25. Primary cells and cell lines were grown at 37oC in 5% CO2 in RPMI 1640 (Life Technologies) supplemented with 10% fetal bovine serum (FBS; Atlanta biologicals), 10 mM HEPES, 1 mM L-glutamine, 50 I.U./ml penicillin and 50 µg/ml streptomycin. For T8ML-1, 100 IU/ml recombinant human IL-2 was also added. To engage T-cell receptor signaling, anti-CD3/CD28 Dynabeads (Life Technologies) were cocultured with TCL cells. Ixazomib was obtained from Selleckchem. Proteasomal inhibition was examined by western blotting for total ubiquitin and β-catenin, as previously described 16. Nuclear localization of NF-kB was determined by western blot and DNA binding examined by ELISA, as previously described 16. GATA-3 expression was examined by western blot or flow cytometry, as previously described 19. Cell viability and apoptosis were examined by Annexin V/propidium iodide staining, as previously described 11
Immunohistochemistry
Formalin-fixed, paraffin embedded (FFPE) diagnostic biopsy specimens were immunohistochemically stained, as previously described 16, 19, using the DAKO Autostainer (DAKO, Carpinteria, CA) and the FLEX HRP EnVision System with diaminobenzadine (DAB) as the chromogen. Heat induced epitope retrieval was performed with FLEX TRS High pH Retrieval Buffer for 20 minutes. An appropriate positive control [H9 xenograft, 16] was stained in parallel. Serial tissue sections were incubated with anti-GATA3 antibody (Santa Cruz Biotechnology, clone HG3–35, 1:50) or anti-NF-κB (p65) antibody (Cell Signaling Technology, clone D14E12, 1:800) overnight at 4oC after peroxidase blocking. Cases were reviewed in a blinded fashion by a dermatopathologist (A.H.) or hematopathologist (N.B., N.G.B.). Slides were viewed with an Olympus BX51 microscope and pictures taken with an Olympus DP71 camera. Olympus BSW with DP Controller software was used for image acquisition and storage.
Patients
This study was an open-label, single-center phase II study of ixazomib in patients with relapsed or refractory cutaneous and peripheral T-cell lymphomas (NCT02158975). Eligible patients were age ≥18 years with histologically confirmed T-cell lymphoma, including PTCL, NOS, angioimmunoblastic T-cell lymphoma (AITL), ALK+ anaplastic large cell lymphoma (ALCL, ALK+), ALK- ALCL (ALCL, ALK-), mycosis fungoides, or Sezary syndrome. CTCL patients had stage IIb-IV disease 26. Patients had relapsed or refractory disease after ≥1 prior systemic therapy (including extracorporeal photopheresis) and were ≥90 days from autologous stem-cell transplantation. Performance status 0–2, and adequate hematologic, hepatic, and renal function, including an absolute neutrophil count ≥1,000/µL, were also required. Patients previously treated with bortezomib or with any serious medical comorbidity, including ≥grade 3 peripheral neuropathy (or grade 2 peripheral neuropathy with pain on physical examination), or active infection were excluded.
Treatment and Clinical Endpoints
Patients received ixazomib monotherapy (4 mg) orally on a weekly schedule (days 1, 8, and 15) every 28 days. Treatment was continued until disease progression, unacceptable toxicity, or patient or investigator decision. Two dose reductions for toxicity were permitted. Patients who failed to complete a cycle of treatment for any reason other than disease progression were replaced.
The primary endpoint was best objective response, encompassing complete response (CR), unconfirmed complete response (CRu), or partial reseponse (PR), by the end of cycle 6, based on the investigators’ response assessment. Patients unable to receive at least 2 cycles of ixazomib with an accompanying response assessment were considered treatment failures. Response assessments were determined using the revised response criteria in malignant lymphoma for PTCL patients 27. Radiographic response assessments (PET/CT) were performed after cycle 2, cycle 4, and cycle 6, and every 3 months thereafter until 24 months after initiation of study treatment. Secondary response endpoints included patient survival from start of treatment, both progression-free and overall, and duration of response.
Mycosis fungoides/Sezary syndrome response and progression were assessed in accordance with the response criteria recommended by the International Society of Cutaneous Lymphoma (ISCL), the United States Cutaneous Lymphoma Consortium (USCLC) and the Cutaneous Lymphoma Task Force of the European Organization for Research and Treatment of Cancer (EORTC) 28. Response in the skin, blood (for patients with blood involvement at enrollment), lymph nodes and viscera were performed after cycle 2, cycle 4, and cycle 6, and every 3 months thereafter until 24 months after initiation of study treatment. Secondary endpoints included safety, tolerability, duration of response (DoR), progression-free survival (PFS), and overall survival (OS). GATA-3 expression, determined by immunohistochemistry 19, was explored as both a predictive and pharmacodynamic biomarker.
Secondary endpoints were safety assessments, including history and physical examination, and laboratory and adverse event (AE) monitoring. Toxicity and AE were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.0), and regularly monitored by the University of Michigan Comprehensive Cancer Center Data and Safety Monitoring Board.
Trial Design and Statistical Analysis
We used a two-stage design with an interim futility analysis 29. The posited ‘null’ best objective response rate (ORR) was 30%. Given the previously reported ORR with bortezomib the trial was powered to detect an improved ORR of 60% with probability 0.90, based upon a two-sided type I error equal to 0.10 23. Subject to these statistical constraints and a pre-planned enrollment of 25 patients in total, we identified appropriate rules for the interim and final analyses. Specifically, the interim analysis would occur after an initial 11 patients were enrolled and followed for response (the first stage); the trial would continue only if at least 4/11 patients exhibit objective response. In this case, the trial would accrue an additional 14 patients (the second stage), and declare in favor of an improved ORR if at least 12/25 patients exhibit objective response.
The study protocol was approved by the institutional review board (IRB) and was conducted in accordance with Good Clinical Practice, including written informed consent and data monitoring.
Results
Ixazomib inhibits the proteasome and impairs cell viability in T-cell lymphomas
CTCL (H9 and MyLa) and PTCL, NOS (T8ML-1) cell lines were incubated with ixazomib and expression of ubiquitin and β-catenin examined in total cell lysates (Fig. 1A). Not surprisingly, brief exposure to ixazomib was associated with a robust increase in total ubiquitin and β-catenin, consistent with significant inhibition of the proteasome. Cell viability following ixazomib treatment was examined in a standard MTT assay (data not shown) and by Annexin V/propidium iodide staining (Fig. 1B, C). A significant reduction in cell viability and the induction of apoptosis in treated cells was observed. Similarly designed experiments were performed in primary CTCL (Sezary Syndrome) specimens ex vivo (Fig. 1D, E), and a time- and dose-dependent reduction in cell viability was noted.
Figure 1.
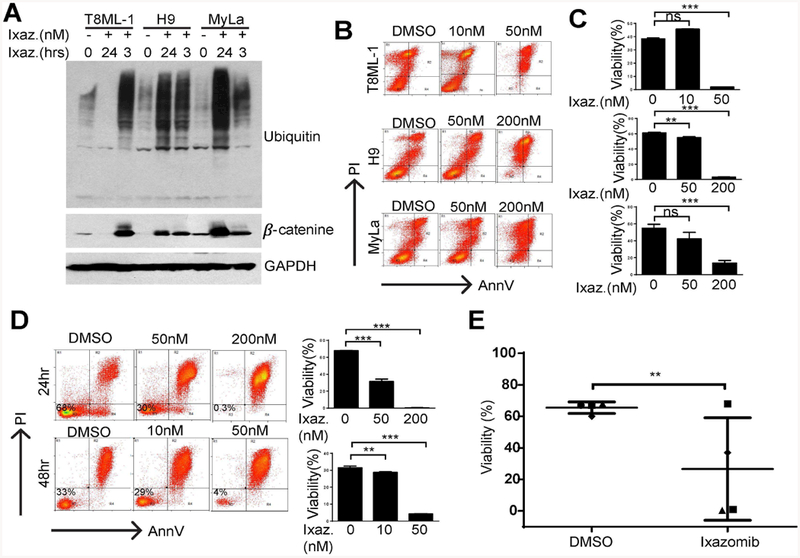
Ixazomib impairs viability in patient-derived and primary T-cell lymphoma cells. (A) A PTCL, NOS cell line (T8ML-1) and two CTCL cell lines (H9, MyLa) were cultured with ixazomib (200 nM) or vehicle control for 3 or 24 hours, as indicated. Accumulation of total ubiquitinated protein and β-catenin were determined in whole cell lysates as a measure of proteasomal inhibition. (B, C) Cell viability was determined by Annexin V/propidium iodide staining in the cell lines indicated treated with ixazomib (48 hours) at the concentrations shown. Representative data from at least 3 independently performed experiments is shown. (D) Cell viability was similarly examined ex vivo in purified malignant T cells obtained from a Sezary Syndrome patient after exposure (24 or 48 hours) to ixazomib at the concentrations shown (10–200 nM). Data obtained from technical replicates is summarized in the bar graphs shown. (E) Primary malignant T cells purified from independent patients (n=4) were cultured for 48 hours with ixazomib (200 nM) or vehicle control and cell viability similarly determined. (**p<0.01, ***p<0.001)
Nuclear translocation of NF-kB is facilitated by proteasome-dependent degradation of cytoplasmic IκB. Therefore, we examined the extent to which ixazomib impaired NF-κB nuclear translocation (Supplementary Fig. 1A) and DNA binding (Supplementary Fig. 1B) in CTCL cell lines. A significant reduction in NF-κB activation was observed. We have previously demonstrated that the T-cell transcription factor GATA-3 is expressed in CTCL and PTCL,19 including a molecularly defined subset of PTCL, NOS.19, 30 Furthermore, GATA-3 confers resistance to chemotherapy in these TCL in a cell-autonomous manner and its expression is, at least partially, NF-κB dependent.16 Therefore, we hypothesized that ixazomib-mediated NF-κB inhibition may be associated with diminished GATA-3 expression. Within 3 hours of ixazomib exposure a modest increase in GATA-3 expression was observed (Supplementary Fig. 1C), consistent with its UPP-mediated degradation [31, and data not shown]. However, within 24 hours of ixazomib treatment, a time point at which NF-kB activation is significantly impaired (Supplementary Fig. 1A, B), a significant reduction in GATA-3 expression was observed (Supplementary Fig. 1C). GATA-3 expression was examined by intracellular flow cytometry in primary CTCL (Sezary Syndrome) samples. A significant reduction in GATA-3 expression was observed, particularly among specimens that highly expressed GATA-3 (Supplementary Fig. 1D, E). Collectively, this data demonstrates that ixazomib impairs NF-κB activation and GATA-3 expression and is directly cytotoxic to malignant T cells at clinically achievable concentrations. Therefore, we launched an investigator-initiated phase II study with this agent in relapsed/refractory T-cell lymphomas.
Patient Characteristics
Between November 2014 and July 2016, 13 patients with relapsed or refractory CTCL or PTCL were enrolled. Per protocol, two patients who enrolled but did not finish at least one cycle were replaced; however, one of the replaced patients received >1 dose of therapy and was thus included for response assessment, leaving a total of 12 analyzable patients. All patients had histologically confirmed CTCL (n=5) or PTCL (n=7, Table I). A majority (10/12) of patients were Caucasian, 9/12 were men, and the median age was 70 years (range, 55–74 years). Evaluable patients received a median of 1 (range 1–3) prior systemic therapies, and 4/12 received prior radiotherapy. Additional patient characteristics are summarized in Table I.
Table I.
Patient Characteristics.
| Patient | Histology | Gender | Stage |
Age > 60 |
LDH (>ULN) |
CLIPI | IPI |
Prior Radiation |
Lines Prior Rx (#) |
|---|---|---|---|---|---|---|---|---|---|
| 001 | CTCL (SS) | M | IVA1 | Y | Y | hi | N/A | N | 12 |
| 002 | CTCL (SS) | f | IVA1 | Y | N | int | N/A | N | 1 |
| 003 | CTCL (SS) | M | IVA1 | Y | Y | hi | N/A | N | 1 |
| 004 | CTCL (SS) | M | IVA1 | Y | Y | hi | N/A | N | 1 |
| 005 | PTCL TFH phenotype | M | IVA | Y | N | N/A | 2 | N | 1 |
| 006 | PTCL, NOS | M | IIIA | N | N | N/A | 1 | Y | 1 |
| 007 | PTCL TFH phenotype | M | IVA | N | N | N/A | 1 | N | 3 |
| 008 | ALCL, ALK− | M | IVA | N | Y | N/A | 3 | Y | 2 |
| 009 | ALCL, ALK− | M | IVA | Y | Y | N/A | 4 | N | 3 |
| 010 | PTCL TFH phenotype | f | IVA | Y | Y | N/A | 4 | N | 1 |
| 011 | CTCL (SS) | M | IVA1 | Y | Y | hi | N/A | N | 2 |
| 012 | PTCL, NOS | f | IVA | Y | Y | N/A | 4 | Y | 1 |
| 013 | CTCL (MF) | M | IIb | Y | Y | int | N/A | Y | 2 |
Note: The CTCL International Prognostic Index (CLIPI) for patients with advanced-stage disease is comprised of 5 variables: male gender, age >60, B1/B2, N2/N3 and visceral metastases. Patients are risk-stratified into low (0–1 risk factors), intermediate (2 risk factors), or high-risk (≥3 risk factors) groups. The International Prognostic Index (IPI) was utilized for PTCL risk-stratification and the number of risk factors (age >60, elevated LDH, ECOG performance status >1, >1 extranodal site of disease, Ann Arbor Stage III/IV) reported.
Efficacy
The observed ORRs at the interim analysis were 0% (0/5; 95% CI 0%, 43%) and 14% (1/7; 95% CI 2.6%, 51%) for CTCL and PTCL patients, respectively (Fig. 2). One PTCL, NOS patient (patient #006) achieved a complete response. Combining histologies, the ORR was 8% (95% CI 1.5%, 35%). Stable disease was observed in 6 patients (50%). Five patients (four with CTCL and stable disease) withdrew from the study due to patient and/or investigator decision secondary to a perceived lack of benefit and/or mild toxicity. The trial was stopped for futility at the planned interim analysis. The single responding patient (patient #006) achieved a PR at the time of the first response assessment (Fig. 3A), and ultimately achieved a CR (Fig. 3A). Intranuclear NF-κB (p65) and GATA-3 were examined semi-quantitatively by immunohistochemistry in paired pre- and post-treatment biopsies. A loss of intranuclear NF-κB and partial inhibition of total GATA-3 expression was observed in this patient (Fig 3B). A similar reduction in intranuclear NF-κB or GATA-3 was not observed in the remaining patients who underwent a biopsy (n=9) while on treatment (data not shown). Six patients were on study for less than 2.5 months, and two patients had follow-up exceeding five months. Five patients were censored for PFS at their time of withdrawal; for OS, patients who were alive at the time of database lock (March 31, 2017) were administratively censored (Supplementary Fig 2). The estimated median PFS time was 3.2 months (95% CI 1.6, not reached). Five patients died between start of treatment and database lock. A median OS time could not be estimated; the lower bound of the 95% CI was 8.1 months. The estimated overall survival rate at 12 months was approximately 60%.
Figure 2.
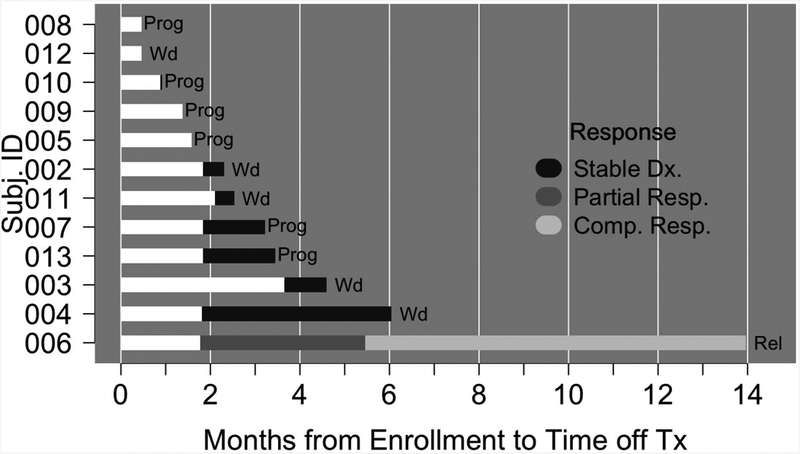
Bar diagram of the response and outcome for patients (n=12) treated with ixazomib. Prog, progression; Wd, patient withdrew from study; Rel, relapse.
Figure 3.
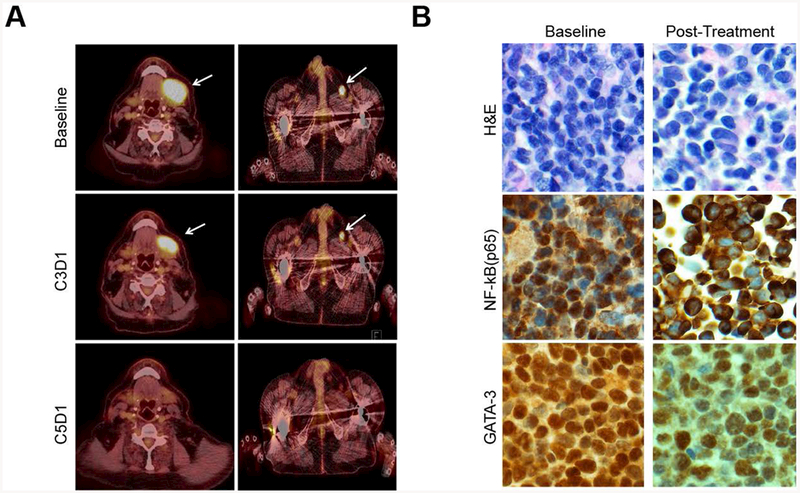
Inhibition of NF-κB and GATA-3 expression in an exceptional responder treated with ixazomib (patient 006). (A) PET/CT images are shown for patient 006 at baseline, at the time of first response (C3D1) and best response (a CR, C5D1). (B) Intranuclear NF-κB (p65) and GATA-3 expression were determined by immunohistochemistry both at baseline and following one month of ixazomib treatment (200x).
Safety
Overall, ixazomib was well tolerated, as few serious, drug-related adverse events were observed (Supplementary Table 1). No significant myelosuppression was observed, and hematologic toxicity was rare, occurring in <10% of patients. As anticipated, the most common adverse events were nausea/vomiting and peripheral neuropathy, all of which were mild (<grade 3). A single CTCL patient developed secondary impetiginization (Staphylococcus aureaus and Pseudomonas aeruginosa) that was subsequently complicated by sepsis (grade 4) and respiratory failure (grade 5) within 30 days of treatment discontinuation. Adverse events by patient are graphically summarized in Supplementary Fig. 3.
Discussion
Novel therapeutic strategies are needed for patients with relapsed or refractory T-cell lymphomas, as few complete and durable remissions are achieved with currently available agents.2–6 Histone deacetylase (HDAC) inhibitors are approved for use in both CTCL and PTCL. Among 74 patients with stage Ib-IVA MF/SS treated with vorinostat, a single CR was observed (1.4%). Patients treated with the approved HDAC inhibitors romidepsin and belinostat may anticipate CR rates of 15% and 10.8%, respectively.32, 33 Among patients achieving a CR/CRu with romidepsin (n=19), 53% were durable (≥12 months)34. Pralatrexate, a novel antifolate that is efficiently internalized by the reduced folate carrier and then retained within the cell upon being polyglutamylated, was associated with a CR rate of 11% in the PROPEL study.35 In contrast to the PROPEL study which was comprised of largely PTCL patients, a modified dosing schedule was subsequently identified, and among patients treated in the expansion cohort of that study (n=29), a single CR (3.4%) was observed.36 In the phase II setting, 69 evaluable CTCL patients (excluding those with only lymphomatoid papulosis) were treated with brentuximab vedotin, and a collective CR rate of 19% observed.37, 38 In the recently reported phase III ALCANZA trial, which randomized patients with CD30+ CTCL to either brentuximab vedotin or a physician’s choice (i.e. methotrexate or bexarotene) of therapy, 16% of patients treated with brentuximab vedotin achieved a CR. While CR rates exceeding 50% are anticipated among ALCL patients treated with brentuximab vedotin,39 among the more common PTCL subtypes response rates are more modest, with CR rates of 14% and 38% being observed among PTCL, NOS and AITL patients, respectively.40 Studies using novel combinations of these agents with either conventional anthracycline-based chemotherapy in the upfront setting or with other novel agents in the relapsed/refractory setting are ongoing, and these results are eagerly awaited.
Key regulatory proteins involved in cell growth and survival, including those conferring resistance to DNA damage or conventional chemotherapeutic agents, are proteasomal substrates and susceptible to inhibition of the proteasome. For example, NF-kB activation promotes T-cell lymphomagenesis and confers resistance to chemotherapy in malignant T cells.13, 14, 16 GATA-3 expression is not only NF-kB dependent,16, 18 but is also required for the homeostatic survival,18, 41 differentiation,42 and migration43 of conventional T-cell subsets. Consistent with its role in conventional T cells, we have previously shown that GATA-3 is an NF-κB target gene in malignant T cells and confers their resistance to chemotherapy in a cell-autonomous manner.16 Not surprisingly, GATA-3 expression has both diagnostic and therapeutic implications,19, 30 and its expression has successfully identified high-risk PTCL patients who are unlikely to achieve a remission with conventional chemotherapeutic agents.19, 44, 45 Therefore, the observation that ixazomib led to significant inhibition of both NF-κB and GATA-3 in primary TCL specimens ex vivo and following one month of treatment in the single responder treated in this study is noteworthy. In contrast to this exceptional responder, we failed to observe significant inhibition of NF-κB and GATA-3 in the remaining patients examined. While the mechanism(s) for this difference is unknown, it is tempting to speculate that IκB and/or NF-κB independent mechanisms of NF-kB and GATA-3 activation, respectively, may be involved. Consistent with our findings, Ravi et. al. observed that ixazomib induced apoptosis in multiple T-cell lymphoma cell lines and was associated with widespread changes in gene expression, including a significant reduction in c-myc expression. While c-myc expression was not examined in this study, this observation is noteworthy, as recently performed next-generation sequencing and molecular profiling studies have implicated c-myc in T-cell lymphomagenesis,9 including the GATA-3+ subset of PTCL, NOS.30 Therefore, future studies exploring c-myc as a therapeutic target in these lymphomas may be warranted.
Our experience with ixazomib adds to a preexistent, albeit limited, clinical experience with proteasome inhibition in T-cell lymphomas. Zinzani et. al. treated CTCL patients with single-agent bortezomib in a small phase II study. Among the 12 evaluable patients, 2 (17%) achieved a CR. Ixazomib is orally bioavailable and has been associated with increased proteasome inhibition and cytotoxicity when compared with bortezomib in murine NHL models.46 In a phase I study with intravenous ixazomib, 4 PTCL patients were treated, and a single partial response observed.47 In a pharmacokinetic study with oral ixazomib, a single PTCL, NOS patient was treated and achieved a PR by cycle 8, but subsequently progressed after 12 months of treatment.48
While CR rate is a more clinically meaningful endpoint, the ORR was selected as the primary endpoint in this study. As only a single response, albeit an exceptional CR, was observed, this study was closed to further accrual at the time of a pre-specified interim analysis for futility. Therefore, it is difficult to commend single-agent ixazomib in unselected CTCL/PTCL patients, thus highlighting the need for the identification of predictive biomarkers that will improve our ability to select the “right treatment” for the “right patient”. Therefore, it is notable that TNFR2 activates the non-canonical NF-κB pathway, as either chromosomal gains involving the TNFR2 locus or recurrent mutations involving a conserved threonine residue within its TRAF2 regulatory domain are observed in 18% of CTCL.8 Gains effecting the TNFR2 locus are associated with increased receptor expression, and expression of the recurrent (Thr377Ile) mutation in Jurkat cells led to increased proteolytic processing and activation of NF-κB2 p100, and a commensurate increase in NF-κB target gene expression.8 Loss-of-function studies using CRISPR-Cas9 technology were employed in cell lines, demonstrating that TNFR2 expression and NF-kB activation rendered cells more sensitive to bortezomib. Conversely, cell lines harboring a truncated NF-kB2 mutant which is not subject to IκB-dependent cytoplasmic sequestration and is observed in ≈6% of CTCL [8, reviewed in 9], were relatively resistant to bortezomib. These findings suggest that examination of appropriate biomarkers and the genetic landscape in future studies is certainly warranted, as this approach may improve our understanding of the mechanisms conferring susceptibility (or resistance) to novel therapeutic strategies, including proteasome inhibition.
Ixazomib was generally well tolerated and was not associated with significant hematologic toxicity. This is noteworthy, as proteasome inhibition is synergistic when combined with other agents, including HDAC inhibitors49. The extent to which the synergy observed with these agents and proteasome inhibitors may be attributed to the effect of proteasome inhibition on the NF-kB/GATA-3 axis, as our findings may suggest, c-myc,50 or some other proteasome-dependent mechanism, is unknown, but may warrant scrutiny in future studies.
In summary, we have demonstrated that ixazomib significantly impairs NF-κB activation and downregulates GATA-3 expression in malignant T cells. In a small and heterogeneous group of patients with relapsed/refractory PTCL, a single CR was observed. A phase I/II study combining ixazomib and romidepsin in relapsed/refractory PTCL is planned.
Supplementary Material
Acknowledgements
The authors gratefully acknowledge Ms. Tina Fields, Dr. Michael T. Goldfarb, and Dr. Ye Lu for technical assistance and the clinical trial staff who made this study possible. We also wish to thank the patients and their families who participated in this study. This work was supported in part by grants from the Leukemia & Lymphoma Society (6270–13 and 6503–16), the V Foundation for Cancer Research, Takeda Oncology, CTSA grants UL1TR000433 and UL1TR002240, and the NIH-NCI (K08CA172215, P30CA046592).
Footnotes
Conflict of interest:
R.A.W. has received research funding from Takeda Oncology. The authors have no additional conflicts of interest to declare.
References
- 1.Wilcox RA. Cutaneous T-cell lymphoma: 2011 update on diagnosis, risk-stratification, and management. Am J Hematol 2011;86:928–948. [DOI] [PubMed] [Google Scholar]
- 2.Hughes CF, Khot A, McCormack C, et al. Lack of durable disease control with chemotherapy for mycosis fungoides and Sezary syndrome: a comparative study of systemic therapy. Blood 2015;125:71–81. [DOI] [PubMed] [Google Scholar]
- 3.Hanel W, Briski R, Ross CW, et al. A retrospective comparative outcome analysis following systemic therapy in Mycosis Fungoides and Sezary Syndrome. Am J Hematol 2016. [DOI] [PubMed] [Google Scholar]
- 4.Briski R, Feldman AL, Bailey NG, et al. The role of front-line anthracycline-containing chemotherapy regimens in peripheral T-cell lymphomas. Blood cancer journal 2014;4:e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mak V, Hamm J, Chhanabhai M, et al. Survival of patients with peripheral T-cell lymphoma after first relapse or progression: spectrum of disease and rare long-term survivors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2013;31:1970–1976. [DOI] [PubMed] [Google Scholar]
- 6.Briski R, Feldman AL, Bailey NG, et al. Survival in Patients with Limited-stage Peripheral T-cell Lymphomas. Leukemia & lymphoma 2014:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vaque JP, Gomez-Lopez G, Monsalvez V, et al. PLCG1 mutations in cutaneous T-cell lymphomas. Blood 2014;123:2034–2043. [DOI] [PubMed] [Google Scholar]
- 8.Ungewickell A, Bhaduri A, Rios E, et al. Genomic analysis of mycosis fungoides and Sezary syndrome identifies recurrent alterations in TNFR2. Nature genetics 2015;47:1056–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elenitoba-Johnson KS, Wilcox R. A new molecular paradigm in mycosis fungoides and Sezary syndrome. Seminars in diagnostic pathology 2016. [DOI] [PubMed] [Google Scholar]
- 10.Jillella AP, Murren JR, Hamid KK, et al. P-glycoprotein expression and multidrug resistance in cutaneous T-cell lymphoma. Cancer investigation 2000;18:609–613. [DOI] [PubMed] [Google Scholar]
- 11.Wilcox RA, Wada DA, Ziesmer SC, et al. Monocytes promote tumor cell survival in T-cell lymphoproliferative disorders and are impaired in their ability to differentiate into mature dendritic cells. Blood 2009;114:2936–2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilcox RA. A three-signal model of T-cell lymphoma pathogenesis. Am J Hematol 2016;91:113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sors A, Jean-Louis F, Begue E, et al. Inhibition of IkappaB kinase subunit 2 in cutaneous T-cell lymphoma down-regulates nuclear factor-kappaB constitutive activation, induces cell death, and potentiates the apoptotic response to antineoplastic chemotherapeutic agents. Clin Cancer Res 2008;14:901–911. [DOI] [PubMed] [Google Scholar]
- 14.Sors A, Jean-Louis F, Pellet C, et al. Down-regulating constitutive activation of the NF-kappaB canonical pathway overcomes the resistance of cutaneous T-cell lymphoma to apoptosis. Blood 2006;107:2354–2363. [DOI] [PubMed] [Google Scholar]
- 15.Izban KF, Ergin M, Qin JZ, et al. Constitutive expression of NF-kappa B is a characteristic feature of mycosis fungoides: implications for apoptosis resistance and pathogenesis. Hum Pathol 2000;31:1482–1490. [DOI] [PubMed] [Google Scholar]
- 16.Wang T, Lu Y, Polk A, et al. T-cell receptor signaling activates an ITK/NF-kappaB/GATA-3 axis in T-cell lymphomas facilitating resistance to chemotherapy. Clin Cancer Res 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei G, Abraham BJ, Yagi R, et al. Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types. Immunity 2011;35:299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Misumi I, Gu AD, et al. GATA-3 controls the maintenance and proliferation of T cells downstream of TCR and cytokine signaling. Nat Immunol 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang T, Feldman AL, Wada DA, et al. GATA-3 expression identifies a high-risk subset of PTCL, NOS with distinct molecular and clinical features. Blood 2014;123:3007–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iqbal J, Weisenburger DD, Greiner TC, et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood 2010;115:1026–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iqbal J, Wilcox R, Naushad H, et al. Genomic signatures in T-cell lymphoma: How can these improve precision in diagnosis and inform prognosis? Blood reviews 2015. [DOI] [PubMed] [Google Scholar]
- 22.Geskin LJ, Viragova S, Stolz DB, et al. Interleukin-13 is overexpressed in cutaneous T-cell lymphoma cells and regulates their proliferation. Blood 2015;125:2798–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zinzani PL, Musuraca G, Tani M, et al. Phase II trial of proteasome inhibitor bortezomib in patients with relapsed or refractory cutaneous T-cell lymphoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2007;25:4293–4297. [DOI] [PubMed] [Google Scholar]
- 24.Fotheringham S, Epping MT, Stimson L, et al. Genome-wide loss-of-function screen reveals an important role for the proteasome in HDAC inhibitor-induced apoptosis. Cancer cell 2009;15:57–66. [DOI] [PubMed] [Google Scholar]
- 25.An J, Fujiwara H, Suemori K, et al. Activation of T-cell receptor signaling in peripheral T-cell lymphoma cells plays an important role in the development of lymphoma-associated hemophagocytosis. Int J Hematol 2011;93:176–185. [DOI] [PubMed] [Google Scholar]
- 26.Olsen E, Vonderheid E, Pimpinelli N, et al. Revisions to the staging and classification of mycosis fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood 2007;110:1713–1722. [DOI] [PubMed] [Google Scholar]
- 27.Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol 2007;25:579–586. [DOI] [PubMed] [Google Scholar]
- 28.Olsen EA, Whittaker S, Kim YH, et al. Clinical end points and response criteria in mycosis fungoides and Sezary syndrome: a consensus statement of the International Society for Cutaneous Lymphomas, the United States Cutaneous Lymphoma Consortium, and the Cutaneous Lymphoma Task Force of the European Organisation for Research and Treatment of Cancer. J Clin Oncol 2011;29:2598–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simon R Optimal two-stage designs for phase II clinical trials. Controlled clinical trials 1989;10:1–10. [DOI] [PubMed] [Google Scholar]
- 30.Iqbal J, Wright G, Wang C, et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood 2014;123:2915–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shinnakasu R, Yamashita M, Kuwahara M, et al. Gfi1-mediated stabilization of GATA3 protein is required for Th2 cell differentiation. J Biol Chem 2008;283:28216–28225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2012;30:631–636. [DOI] [PubMed] [Google Scholar]
- 33.O’Connor OA, Horwitz S, Masszi T, et al. Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015;33:2492–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coiffier B, Pro B, Prince HM, et al. Romidepsin for the treatment of relapsed/refractory peripheral T-cell lymphoma: pivotal study update demonstrates durable responses. Journal of hematology & oncology 2014;7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011;29:1182–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horwitz SM, Kim YH, Foss F, et al. Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood 2012;119:4115–4122. [DOI] [PubMed] [Google Scholar]
- 37.Kim YH, Tavallaee M, Sundram U, et al. Phase II Investigator-Initiated Study of Brentuximab Vedotin in Mycosis Fungoides and Sezary Syndrome With Variable CD30 Expression Level: A Multi-Institution Collaborative Project. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015;33:3750–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duvic M, Tetzlaff MT, Gangar P, et al. Results of a Phase II Trial of Brentuximab Vedotin for CD30+ Cutaneous T-Cell Lymphoma and Lymphomatoid Papulosis. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015;33:3759–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pro B, Advani R, Brice P, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2012;30:2190–2196. [DOI] [PubMed] [Google Scholar]
- 40.Horwitz SM, Advani RH, Bartlett NL, et al. Objective responses in relapsed T-cell lymphomas with single-agent brentuximab vedotin. Blood 2014;123:3095–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, Su MA, Wan YY. An essential role of the transcription factor GATA-3 for the function of regulatory T cells. Immunity 2011;35:337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 1997;89:587–596. [DOI] [PubMed] [Google Scholar]
- 43.Wohlfert EA, Grainger JR, Bouladoux N, et al. GATA3 controls Foxp3(+) regulatory T cell fate during inflammation in mice. The Journal of clinical investigation 2011;121:4503–4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang W, Wang Z, Luo Y, et al. GATA3 expression correlates with poor prognosis and tumor-associated macrophage infiltration in peripheral T cell lymphoma. Oncotarget 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manso R, Bellas C, Martin-Acosta P, et al. C-MYC is related to GATA3 expression and associated with poor prognosis in nodal peripheral T-cell lymphomas. Haematologica 2016;101:e336–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee EC, Fitzgerald M, Bannerman B, et al. Antitumor activity of the investigational proteasome inhibitor MLN9708 in mouse models of B-cell and plasma cell malignancies. Clin Cancer Res 2011;17:7313–7323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Assouline SE, Chang J, Cheson BD, et al. Phase 1 dose-escalation study of IV ixazomib, an investigational proteasome inhibitor, in patients with relapsed/refractory lymphoma. Blood cancer journal 2014;4:e251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gupta N, Hanley MJ, Venkatakrishnan K, et al. The Effect of a High-Fat Meal on the Pharmacokinetics of Ixazomib, an Oral Proteasome Inhibitor, in Patients With Advanced Solid Tumors or Lymphoma. Journal of clinical pharmacology 2016;56:1288–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heider U, Rademacher J, Lamottke B, et al. Synergistic interaction of the histone deacetylase inhibitor SAHA with the proteasome inhibitor bortezomib in cutaneous T cell lymphoma. Eur J Haematol 2009;82:440–449. [DOI] [PubMed] [Google Scholar]
- 50.Ravi D, Beheshti A, Abermil N, et al. Proteasomal Inhibition by Ixazomib Induces CHK1 and MYC-Dependent Cell Death in T-cell and Hodgkin Lymphoma. Cancer research 2016;76:3319–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.