

Abstract
Influenza virus enters host cells by fusion of viral and endosomal membranes mediated by the influenza hemagglutinin (HA). The pathway of HA-catalyzed fusion has been widely investigated in influenza virus membrane fusion with liposomes. In this chapter we describe methodology for studying the virus-liposome fusion system using a combination of fluorescence dequenching assays and cryo-electron tomography (cryo-ET). In particular, the fluorescence dequenching is used to monitor the efficiency of membrane fusion between whole influenza viruses labeled with a lipophilic dye (DiD) in the membrane and liposomes labeled with a water-soluble dye (sulforhodamine B). By simultaneously monitoring the two fluorescent signals, we can determine the relative time scales of liposomal content leakage or transfer vs. lipid merging. In addition, cryoET offers a means of imaging 3-dimensional snapshots of different stages of virus-liposome fusion such as prefusion, fusion intermediates and postfusion.
Keywords: Influenza virus, Membrane fusion, Liposomes, Fluorescence spectrometry, Cryo-electron tomography
1 Introduction
Influenza viruses are members of the Orthomyxoviridae family of enveloped, negative-stranded, segmented RNA viruses. Similar to other enveloped viruses, influenza delivers the viral genome into host cells by carrying out protein-mediated fusion of viral envelope and host cell membranes. The influenza glycoprotein hemagglutinin (HA) is the viral fusion machinery that facilitates the entry of influenza viruses into host cells. Cell entry is initiated when HA binds to sialic acid receptors on the surface of the host cell. Once bound, influenza virus is taken into the host cell by endocytosis. During maturation of the endosome, the endosomal lumen acidifies triggering HA to undergo irreversible structural rearrangements that drive membrane fusion [1]. In some strains of influenza, these structural changes are initiated by exposure of fusion peptides that insert into host endosomal membrane [2], followed by refolding of the fusion subunit that draws the two membranes together and induces them to merge. Much of these intermediate processes and the mechanistic understanding of how HA drives membrane remodeling and fusion have been inferred from fluorescence approaches (both spectroscopy and microscopy approaches), antibody probes and biochemical assays (e.g. limited proteolysis) of epitope exposure during HA reorganization [3, 4], but until recently we have lacked the means to directly visualize protein-mediated membrane fusion with resolution of the fusion machinery and membrane structures that are populated during fusion.
Cryo-electron tomography offers a powerful approach for imaging membrane fusion and resolving the interplay of protein and membrane intermediates during this intricate process. Direct visualization of membrane fusion inside intact eukaryotic cells, which takes place deep in the cell close to the nucleus [5], is not feasible for most current-generation transmission electron microscope configurations. Instead, synthetic liposomes with diameters in the 100–200 nm range provide target membranes for influenza to fuse with that are compatible with the thin layer of vitreous ice that is required for electron beam penetration and imaging. These liposomes enable in vitro fusion reactions to be studied with exquisite control as one can generate liposomes with specified lipid compositions. A downside to using synthetic liposomes is that their lipid composition is far less complex and lacking in leaflet-asymmetry compared to the biological membranes with which the virus would fuse during endosomal entry. In addition, the curvature of the liposomal target membrane, at least initially is convex as opposed to the membrane in an endosome, which would be concave with the virus inside the endosomal lumen. Based upon what we have observed, in general the target membrane ends up conforming to the topography of the virus surface, and hence initial target membrane curvatures are believed to be secondary, relative for example to lipid composition, in terms of their influence on how the fusion reaction will progress. For in vitro fusion assays, timing and solution conditions for triggering the fusion reaction can also be carefully controlled and varied in order to parse the intermediate states traversed during fusion. As the fusion reactions are very sensitive to the experimental conditions, special attention must be paid to factors such as temperature, pH, liposome and virus concentration.
Here we provide a protocol for measuring stages of fusion kinetics and visualizing the fusion complex between influenza viruses and liposomes by fluorescence dequenching assays and cryo-electron tomography (cryo-ET). Fluorescence dequenching assays can track the overall progress of a membrane fusion reaction by monitoring liposomal content leakage and lipid mixing (Figure 1). The liposomes contain the hydrophilic fluorophore sulforhodamine-B (SRB) at self-quenching concentrations, while the membrane of influenza virus is labeled with a lipophilic fluorophore, DiD, also to a self-quenching concentration (see Note 1). When the SRB-labeled liposomes are mixed with DiD-labeled influenza virus and fusion is triggered by lowering pH, the increase in the fluorescence intensity of SRB dye resulting from dequenching reports on liposomal content leakage and transfer; distinguishing between these two scenarios is not straight-forward however, as both would give rise to some degree of dequenching. We also note that others have reportedly labeled influenza virions with SRB by incubating the virus in SRB-containing buffers for extended durations [6, 7]. In this case, it is assumed the dye crosses the viral envelope and becomes encapsulated inside of the particle. Then during the fusion reaction the SRB signal would report on viral content transfer or leakage. When the lipid mixing commences, DiD disperses over a larger membrane area and the fluorescence signal of DiD also increases.
Figure 1.

(A) Schematic diagram shows fluorescence dequenching assays. When the SRB-labeled liposomes are mixed with DiD-labeled influenza virus and fusion is triggered by low pH, the liposomal content leakage and transfer release the water-soluble SRB dyes and the weakening quenching effect results in the increase in the fluorescence intensity of SRB dye. When the lipid mixing commences, the lipophilic DiD dyes disperse over a larger membrane area and the fluorescence signal of DiD also increases. (B) Fluorescence intensity of SRB (yellow, solid curve), reporting liposomal content leakage and transfer, and DiD (blue, dashed curve), reporting membrane merging, is monitored over time. Completely dequenching is achieved in the presence of 1 % (v/v) TritonX-100 detergent.
Cryo-ET can provide unique structural insights into the influenza virus fusion system [8, 9]. This cryo-EM-based method generates three-dimensional images of samples that can be trapped and analyzed in the process of membrane fusion. The fusion reaction can be initiated by acidifying a solution containing virus and target membranes in the form of liposomes, followed by rapid plunge-freezing in liquid ethane. In the microscope, the sample is tilted about a single axis and imaged over a range of tilt angles to obtain three-dimensional information. Then the sample volume is computationally reconstructed. The major caveats of using cryo-ET to characterize specimens is that due to physical limitations of the electron microscope and sample holders, it is not possible to tilt a specimen beyond ± 60–70°; the information captured is thus incomplete, which gives rise to the so-called missing wedge artifact and relatively poor characterization of features on the top and bottom of particles as well as anisotropic resolution and a degree of distortion of three-dimensional features[10]. Despite these limitations, cryo-ET provides nanometer resolution that is sufficient to resolve individual surface protein spikes as well as membrane leaflets, disruption, and deformations during influenza virus membrane fusion. By examining fusion reactions over a time course of fusion and under different pH conditions, it becomes possible to track how the populations of various fusion intermediates wax and wane over the course of the reaction. From these population kinetics, one may infer the approximate sequence of HA-driven membrane reorganization that leads to productive membrane fusion [11].
Taken together, real time monitoring of fusion reactions in bulk by fluorescence spectroscopy and nanometer-scale imaging of fusion complexes by cryo-electron microscopy provide new insights into the physical process of protein-mediated membrane fusion. The approach is fairly generalizable to other fusion systems, though influenza virus, with its straightforward triggering by exposure to acidic pH is considerably less complex than systems where the fusion protein is triggered by receptor-mediated interactions.
2 Materials
2.1 DOPC and DOPC/cholesterol liposome preparation
1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC): 25 mg/mL dissolved in chloroform (Avanti Polar Lipids).
Cholesterol: 25 mg/mL dissolved in chloroform (Avanti Polar Lipids) (see Note 2).
Chloroform (EMD Chemicals).
Positive-displacement pipets (MICROMAN, Gilson) (see Note 3).
13 × 100 mm borosilicate glass tube.
Vortex mixer.
Compressed gas.
Vacuum desiccator (Nalgene): for drying lipids in vacuum.
HEPES buffered saline (HBS), pH 7.5: 150 mM NaCl, 10 mM HEPES, 50 mM sodium citrate (pH 7.5), 0.02 % NaN3, pH 7.5. The buffer solution is filtered through a 0.2 μm filter.
HBS-SRB: dilute sulforhodamine B sodium salt (SRB) (Invitrogen) into HBS, pH 7.5, to a final concentration of 25 mM. Adjust the pH to 7.5 after SRB is fully dissolved.
37 °C water bath.
Liquid nitrogen.
Mini-extruder set (Avanti Polar Lipids).
Flat-tip tweezers: for handling filter membranes.
Polycarbonate Membranes 0.1 μm (Avanti Polar Lipids).
PD-10 desalting column (GE Healthcare).
1.5-mL snap-cap tubes (Eppendorf).
2.2 Lipophilic dye DiD labeling of influenza viruses
X31 (H3N2) influenza A virus (2 mg/mL): grown in embryonated chicken eggs, purchased from Charles River Laboratories. Virus aliquots are stored at −80 °C.
HEPES buffered saline (HBS), pH 7.5.
1,1′-dioctadecyl-3,3,3′3′-tetramethylindodicarbocyanine (DiD), 1mM dissolved in ethanol, purchased from Life Technologies.
1.5-mL snap-cap tubes (Eppendorf).
37°C heat incubator (within shaking nutator).
Refrigerated microcentrifuge at 4 °C.
Class II biological safety cabinet and protective equipment for working with infectious influenza virus.
10 % chlorine bleach to treat the biohazardous waste.
2.3 Quantification of dye-labeled liposomes and viruses
2.3.1 Determination of liposome concentration by phosphorus assay
Distilled water (ddH2O).
Ammonium molybdate.
Sulfuric acid.
L-ascorbic acid.
Phosphate reagent: 5.04 g of ammonium molybdate in 84 mL of ddH2O; then add 33 mL of 98 % sulfuric acid to water slowly and keep gently agitating to dissipate the heat from the dilution of sulfuric acid. Store at room temperature.
Analytical solution: 0.33 g of L-ascorbic acid in 21 mL of ddH2O; then add 2 mL of phosphate reagent (step 5) into the solution. Store at room temperature.
Phosphate standard solution: 1 mM KH2PO4, pH 5.8–6.2.
70 % percholoric acid.
16 × 100 mm borosilicate glass tube.
A lab heat block.
Varian Cary 50 UV/Vis spectrophotometer (Agilent Technologies).
2.3.2 Determination of liposome size by dynamic light scattering
DynaPro NanoStar analyzer (Wyatt Tech).
MicroCuvette, 1 μL (Wyatt Tech).
2.3.3 Determination of HA concentration by Western blotting
Bromelain.
Bromelain digestion buffer: 150 mM NaCl, 10 mM HEPES (pH 7.8), 1 mM EDTA, 25 mM b-mercaptoethanol.
Nutator.
37 °C incubator.
Tabletop centrifuge with swinging bucket rotors..
Beckman ultracentrifuge with 50.2 Ti rotor.
FPLC equipped with GE-Superdex 200 analytical gel filtration column.
15 ml volume Amicon 30 kDa molecular weight cut off spin concentrator (Millipore).
0.5 ml volume Amicon 10 kDa MWCO spin concentrator (Millipore).
HEPES buffered saline (HBS), pH 7.5.
TBST buffer: 150 mM NaCl, 25 mM Tris, 0.1% (v/v) Tween 20.
Blocking buffer: 5 g of powdered milk into 100 mL of TBST buffer. Store at 4°C.
Primary antibody: anti-HA Tag Antibody (EMD Millipore) diluted 1:2,000 in the blocking buffer.
Secondary antibody: recombinant Protein G-Horseradish Peroxidase (Thermo Fisher) diluted 1:5,000 in the blocking buffer.
NuPAGE 4–12 % Bis-Tris Gel (Invitrogen).
Nitrocellulose membrane for western blotting (Thermo Fisher).
XCELL Blot Module (Invitrogen).
ECL Prime Western Blotting Detection Reagent (GE Healthcare).
2.4 Fluorescence mixing assays
SRB-labeled liposomes (from section 2.1).
DiD-labeled influenza virus particles (from section 2.2).
HEPES buffered saline (HBS), pH 7.5.
HEPES buffered saline (HBS), pH 3.0: 150 mM NaCl, 10 mM HEPES, 50 mM sodium citrate (pH 7.5), 0.02% NaN3, pH 3.0. Filtered through a 0.2 μm filter.
pH meter.
Fluorescence Micro Cell, 40 μL (Agilent Technologies).
Fluorescence Spectrophotometer with temperature controller (Varian Cary Eclipse, Agilent Technologies).
10 % (v/v) Triton-HBS: Triton X-100 diluted in HBS, pH 7.5.
Protective equipment: gloves, glasses.
10 % (v/v) chlorine bleach to treat biohazardous waste.
2.5 Grid preparation for cryo-ET
SRB-labeled liposomes (from section 2.1).
DiD-labeled influenza virus particles (from section 2.2).
HBS, pH 7.5.
HBS, pH 3.0.
C-flat holey carbon grids (e.g. Electron Microscopy Sciences): CF-2/2-2C-T (hole size: 2.0 μm, hole spacing: 2.0 μm, TEM mesh: 200, thick carbon) is recommended.
6 nm BSA gold tracer (e.g. Electron Microscopy Sciences), for use as fiducial markers in tomography.
Glow discharge system for TEM grids.
Plunge-freezing device with humidity and temperature control, for rapid and reproducible freezing of samples (Vitrobot Mark IV, FEI Company).
Vitrobot filter paper.
Liquid nitrogen.
Compressed ethane (research purity), for vitrification.
Cryo grid storage boxes.
Timer.
Water bath.
Screw driver, for opening and closing grid storage boxes.
Tweezers: (a) Fine tip tweezers (Electron Microscopy Sciences, Dumont Style L4), for handling EM grids; (b) Flat tip tweezers, for handling cryo grid storage boxes during transfer.
Protective equipment: gloves, glasses.
10% (v/v) chlorine bleach to treat the biohazardous waste.
2.6 Quantification of influenza virus-liposome contacts
Software for 3-dimensional reconstruction (IMOD).
Software for visualization of reconstructed tomograms (ImageJ with Grouped_ZProjector plugins, the Grouped_ZProjector plugins can be downloaded at https://imagej.nih.gov/ij/plugins/group.html).
3 Methods
3.1 DOPC and DOPC/cholesterol liposome preparation (flowchart shown in Fig. 2A.)
Figure 2.

Flowchart of the major steps in the preparation of (A) SRB-labeled liposomes and (B) DiD-labeled influenza viruses.
Add 200 μL of chloroform, 20 μL of DOPC and the volume of cholesterol stock solution to achieve the desired molar fraction of cholesterol into borosilicate glass tubes (see Note 2).
Mix the DOPC and cholesterol on top of a vortex mixer. The lipid film is formed by slow evaporation using a gentle stream of nitrogen.
Continuously rotate the vortex mixer to produce as thin and uniform a lipid film layer as possible (see Note 4).
Transfer the glass tubes into the vacuum desiccator. Pump the vacuum and keep the lipids under the vacuum for at least 3 h (overnight is recommended) to eliminate any residual chloroform.
The phospholipid film is hydrated with 200 μL of SRB-HBS into each glass tube.
Vortex the lipid mixture for 30 s.
Freeze the lipid mixture in liquid nitrogen.
Thaw the lipid mixture in a 37 °C water bath.
Repeat steps 4–6 (i.e. freeze/thaw/vortex cycles) 5–10 times.
Assemble the mini extruder as specified by the manufacturer (Avanti Polar Lipids). Place a single 100 nm polycarbonate membrane and filter supports in the extruder, handling them only with flat-tip tweezers to avoid tearing or puncturing the membrane.
Load the extruder with 250 μL of HBS buffer, pH 7.5 using a gas tight syringe. Buffer should be extruded back and forth several times to remove any remaining gas inside the mini extruder.
Using a gas tight syringe, load 200 μL of lipid suspension and extrude 21 times passing the filter membranes. Collect the final extruded solution from the syringe opposite to the one used to initially inject the lipid suspension.
Equilibrate the PD-10 column with 2 column volumes of HBS buffer, pH 7.5. Once the buffer has passed through and minimal to no visible amounts are left in the top reservoir of the PD-10 column, cap the bottom of the PD-10 column.
Apply the extruded liposome solution evenly and spread across the top of PD-10 column and remove the bottom cap to allow flow to resume.
Elute the PD-10 column with HBS buffer, pH 7.5 (ca. 3–4 mL).
Collect the first pink peak, which contains the SRB-labeled DOPC or DOPC/Cholesterol liposomes, with 1.5-mL snap-cap tubes (Fig. 3). The liposomes can be stored at 4 °C. Use within 1 to 5 days.
Figure 3.

The SRB-encapsulating liposomes eluted as a single band from the PD-10 gel filtration column. The black box of step 2 highlighted the first pink band, which contained the SRB-labeled liposomes.
3.2 Lipophilic dye DiD labeling of influenza viruses (flowchart shown in Fig. 2B)
Remove X31 influenza viruses (2 mg/mL) from −80 °C storage and thaw at room temperature in a class II biological safety cabinet.
Centrifuge virus stocks first at 2,320 × g for 5 min at 4 °C to remove precipitates formed by egg proteins.
Add 500 μL of approximately 2 mg/mL influenza virus supernatant into 5 μL of DiD Vybrant solution and mix thoroughly by pipetting up and down gently.
Incubate the dye-labeled influenza virus particles for 2 h at 37 °C with gentle rocking on the nutator.
Recover the influenza virus by 21,000 × g ultracentrifugation for 30 min at 4 °C. Resuspended the pellet in HBS, pH 7.5 to a final concentration of 8–10 mg/mL X31 virus solution. The DiD-labeled influenza virus can be stored away from light at 4 °C and can be used for 1–5 days.
3.3 Quantification of dye-labeled liposomes and viruses (see Note 5)
3.3.1 Determination of liposome concentration by phosphorus assay
The phosphorus assay is modified from a previously described protocol [12].
Prepare the standard solutions for a calibration curve. For instance, mix 1 mM KH2PO4 solution with ddH2O to get a 0, 0.1, 0.2, 0.5, 1 mM PO43− concentration.
Load 32 μL of standard solutions and liposomes into the 16 mm glass tubes.
Add 50 μL of 70 % percholoric acid to each tube. Vortex the tubes.
Transfer the tubes to 180 °C heat block for 1 h in the chemical hood.
After 1 h of heating, allow the tubes to cool to room temperature. Then add 0.9 mL of analytical solution to each tube. Vortex shortly.
Incubate the tubes on an 80 °C heat block for 15 min in the chemical hood.
Cool the tubes and transfer the solution to the fluorescence cuvette. Measure the absorbance at 820 nm.
Generate the standard curve using the standard solutions and determine the concentration of phosphorus in the samples. The ideal concentration of DOPC for the following fluorescence assays and cryo-ET range between 0.2 and 0.5 mM (see Note 6).
3.3.2 Determination of liposome size by dynamic light scattering
Load 3 μL of liposome samples into the microcuvette.
Transfer the microcuvette into the DynaPro NanoStar analyzer and record the dynamic light scattering readings from the equipment. The radius of the DOPC and DOPC/Cholesterol liposomes through 100 nm polycarbonate membranes usually range from 55 to 65 nm with low polydispersity (< 10 %).
3.3.3 Determination of HA concentration by Western blotting
Bromelain-cleaved HA (BHA) is used as the standard for determining HA concentration and is purified as previously described in [2].
Dissolve 30 mg of bromelain in 30 mL digestion buffer and mix with 5 mL, 6 mg/mL X31 virus sample.
Digest for 3 h at 37 °C with gentle rocking on nutator.
Pellet virus particles by 21,000 × g ultracentrifugation for 30 min at 4 °C.
Repeat additional digestions as steps 1–3 two additional times.
Pool the supernatant containing BHA and concentrate using spin filtration in tabletop swinging bucket rotor with 15ml concentrator.
Purify BHA from supernatant via size-exclusion chromatography in HBA, pH 7.5.
Concentrate BHA-containing fractions using 0.5 ml 10 kDa MWCO spin concentrator to desired concentration and volume.
Measure the concentration of BHA by UV absorbance at 280 nm.
Load 1, 2, 3 and 5 μL of 10 mg/mL DiD-labeled influenza viruses as well 1, 2, 4, 8 μg BHA into the wells of a NuPAGE gel and perform sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
Transfer the protein from the gel to a membrane using the XCELL Blot Module following the manufacturer’s instructions.
Block the membrane with blocking buffer for 1 h at room temperature or overnight at 4 °C using gentle rocking.
Incubate the membrane with primary antibody dilution for 1 h at room temperature.
Wash the membrane in TBST 3 times, 10 min each, at room temperature with gentle rocking.
Incubate the membrane with secondary antibody dilution for 1 h at room temperature.
Wash the membrane in TBST 3 times, 10 min each at room temperature with gentle rocking.
Expose blot to a film using ECL prime western blotting detection reagent in the darkroom.
The amount of HA in DiD-labeled influenza virus can be accessed through digital imaging using a related software tool (ImageJ). As shown Fig. 4, the concentration of HA in our DiD-labeled influenza virus was estimated from the standard curve at 2.1 mg/mL, taking a portion of 21 % of the overall protein concentration at 10 mg/mL (see Note 7).
Figure 4.
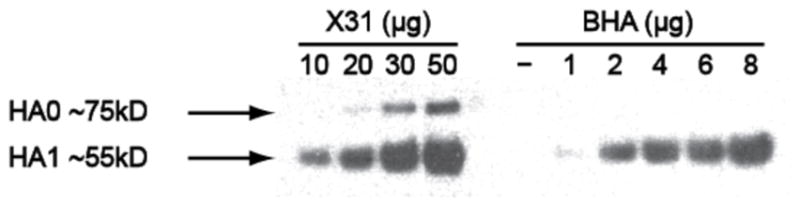
Western blot probed with antibodies against influenza HA. The amount of HA in the viral sample was estimated by comparing with HA standards. We also noted that a small fraction of uncleaved HA0 was present in the influenza virus specimen.
3.4 Fluorescence dequenching assays (see Note 8)
Set the water bath of the fluorescence spectrophotometer at 37 °C and fluorescence settings as described in Table 1.
Adjust the volumes of SRB-labeled liposomes, DiD-labeled influenza viruses and HBS, pH 7.5 and to a final volume of 70 μL (see Note 9).
Transfer the cuvette into the fluorescence spectrophotometer and start recording the baseline for 10 min. The reading at neutral pH is set to the initial level of fluorescence [I(0)].
Pause the fluorescence readings. Then add the corresponding volume of HBS, pH 3.0 into the cuvette to reach the requested pH. For example, we add 30 μL HBS, pH 3.0 to achieve a final pH of 5.25. Quickly mix by pipetting and continue recording the fluorescence readings [I(t)].
After 1 h of measurement, add 11 μL of 10 % Triton-HBS and mix by pipetting to achieve complete dequenching of the fluorescent dyes. Record the reading as the maximum intensity of fluorescence [I(max)].
- The extent of liposomal content leakage or transfer (indicated by SRB fluorescence signal) and membrane merging (indicated by DiD fluorescence signal), as the ratios of dequenching, are given by:
Table 1.
Fluorescence spectrophotometer settings for the dequenching assays
| Dye | λEx (Excitation) | Slit Widths (nm) | λEm (Emission) | Slit Widths (nm) |
|---|---|---|---|---|
| SRB | 556 | 2.5 | 578 | 2.5 |
| DiD | 644 | 2.5 | 655 | 2.5 |
3.5 Virus-liposome specimen vitrification for cryo-ET
Specimen preparation: adjust the volumes of SRB-labeled liposomes, DiD-labeled influenza viruses and HBS, pH 7.5 at the same ratio as fluorescence studies to a final volume of 15–30 μL. Add 1:10 (v/v) ratio of 6 nm BSA gold tracers. Mix well and place the tube on ice.
Glow discharge grids: place CF-2/2-2C grids on a clean glass slide with carbon side facing up, then glow discharge for 20 s with a current of 25 mA (see Note 10).
For the sample-freezing step, we use the FEI Vitrobot Mark IV. Set up the following parameters of the Vitrobot: temperature 25 °C; humidity 100 %; blot time 7–8 s; wait time 0 s; drain time 0 s; blot force 0; and blot total 1. These parameters may need to be optimized for the specific instrument being used for grid preparation (see Note 11).
Mount the filter papers on the blotting pads of the Vitrobot and leave the filter papers in the 100% humidity for at least 15 min (see Note 12).
Fill up liquid ethane and liquid nitrogen in the coolant container. When ethane reaches the proper temperature (−173 to −178 °C), a thin layer of solid ethane floating on the surface of the liquid ethane will appear.
Use the Vitrobot tweezers to carefully pick up the grids at the very edge and mount them on the Vitrobot.
Add the corresponding volume of HBS, pH 3.0 to acidify the virus-liposome sample and start the timer. Keep the tube in the water bath at 37 °C to allow the fusion reaction to progress for the desired duration.
Apply 3 μL of the virus-liposome sample on the carbon side of the grid, blot the excess solution and plunge freeze the sample into liquid ethane. Record the readings on the timer that specify the acidification time for that particular specimen (see Note 13).
Carefully remove the tweezers from the mount and quickly transfer the grids into the cryo grid storage boxes. Close the grid storage boxes with a screwdriver. The storage boxes then can be either transferred to a cryo station for tomography data collection or stored in liquid nitrogen for a long period of time without noticeable ice contamination.
3.6 Quantification of influenza virus-liposome contacts
Image the vitrified grids and collect data [11]. In brief, mount the vitrified specimen grid on a Gatan 626 single-tilt cryo transfer holder and image at 200 kV in an FEI Tecnai F20 transmission electron microscope. Capture images using a Gatan K2 Summit direct detector in counting mode at a calibrated magnification of 11,500 × (yielding a scaling of 3.2 Å per pixel). Acquire images at 2 to 4 μm underfocus, and tilt specimen in 2 ° steps from 58 ° to −58 ° using the Leginon software package [13].
Reconstruct tomograms using the back-projection method in the IMOD package [14]. Select 6 nm BSA gold beads as fiducial markers.
Load the volume of reconstructed tomogram in ImageJ by MRC reader.
De-noise the tomogram by using a real-space Gaussian filter in ImageJ, width 1.5 σ.
Perform a z-projection averaging on a stack of images via Grouped_ZProjector plugins to further reduce background noise (see Note 14).
Interactions between influenza viruses and liposomes can be classified based on important features such as deformations of target membrane, HA, width of contacts, internal features listed in Table 2.
Calculate the abundance of each type of fusion interaction as a percentage of the total number of virus-liposome contacts.
Table 2.
Quantification of influenza virus-liposome contacts, modified from [11].
| HA Bridging | Pinching | Extended Interface | Hemifusion | Postfusion | |
|---|---|---|---|---|---|
| Target Membrane | Not significantly deformed | Pinched and drawn to the virus envelope | Running parallel with the virus envelope. Proximal leaflets may be indistinguishable | Two proximal leaflets of lipid bilayers have joined and a diaphragm is composed of only the two remaining leaflets | Already merged with the virus envelope |
| HA | HAs bridge virus and liposome membrane | HAs form a fringe around the dimple | No density of HA can be observed at the contact sites | No density of HA can be observed at the contact sites | HA can adopt a variety of different conformations on the fused surface |
| Width of Contacts | Several to 100 nm | < 10 nm | 10 – 100 nm | ~100 nm | The fused complexes may grow up to 500 nm |
| Internal Features | Virus and liposome have not fused | Virus and liposome have not fused | Virus and liposome have not fused | No matrix layer is present and dense clumps formed by M1 and ribonucleoprotein are observed | Internal vesicles originate from multi-lamellar liposomes and dense clumps are formed by M1 and ribonucleoprotein |
| Example | Figure 5A | Figure 5B | Figure 5C | Figure 5D | Figure 5E |
Figure 5.

Four different categories of influenza virus-liposome interactions. (A) HA bridging: HA spikes bridge the virus and target membrane surfaces. Blue arrowhead highlights the relatively intact target membrane at the interface. (B) Pinching: the target membrane is pinched and drawn to the virus surface by HA spikes, as highlighted by yellow arrowhead. The localized dimples are usually < 10 nm wide. (C) Extended interface: the target membrane is tightly docked with the viral envelope and runs parallel with the viral envelope from 10 to 100 nm (highlighted as green arrowhead). No density that could be attributable to glycoprotein is observed in the extended contact zones. (D) Hemifusion: the two proximal leaflets of lipid bilayers have joined and hemifusion diaphragm is composed of only the two remaining leaflets. No matrix (M1) layer was present at the interface. (E) Postfusion: postfusion complexes are featured by additional encapsulated smaller vesicles and dense clumps presumably composed of M1 protein with ribonucleoproteins encapsulated inside the outer fused membrane. Scale bars; 50 nm. L: liposome, V: influenza virus particle, PF: postfusion complex.
Footnotes
Different dyes can be used for different labeling systems. Here we chose SRB and DiD because: (a) this pair of dyes exhibit minimal spectral overlap, and low efficiencies of fluorescence resonance energy transfer (FRET); (b) both dyes exhibit pH-independent fluorescence over the range of pH (5.0–7.5) examined in these studies.
A variety of lipid compositions can be used. Here we provide the specific example of a cholesterol concentration series in DOPC-based liposomes as studied in [11]. In subsequent steps, it is important to work at temperatures above the highest lipid phase transition temperature in order to maintain uniform lipid compositions across the liposomes in the preparation. The phase transition temperature (Tm) of DOPC was reported to be −18.3 °C and previous studies observed that DOPC and DOPC-cholesterol mixture were in the fluid (Lα) phase throughout the temperature range in our studies [15].
We recommend using Gilson MICROMAN positive-displacement pipets instead of regular air-displacement pipets for accurate organic solution transfers.
After the drying step, the lipid film should form a homogenous layer of lipid with no visible clumps. Preparation of the lipid mixture and the evaporation steps should be done in the chemical fume hood.
The quantification step is strongly recommended because it provides critical information such as liposome and HA concentration, which can affect efficiency and rates of fusion. Significant batch-to-batch variation can be observed for commercially produced influenza virus stocks, thus it is necessary to determine the HA content for each batch that is used, and it is ideal to carry out an entire series of experiments using material from the same batch and preparation.
The final lipid concentration of liposomes was much lower than expected. Usually the concentration of DOPC measured from the phosphorus assay range from 0.3 to 0.4 mM, while the concentration of DOPC calculated from starting materials is 0.6 to 0.8 mM. We propose several reasons for this discrepancy: (a) liposomes are lost during the extrusion step because a portion of them trapped on the 100 nm polycarbonate membranes; (b) not all of the liposomes are recovered from the PD-10 column.
The 2 mg of total protein per mL of X31 influenza viruses purchased from Charles River Laboratories was measured by the manufacturer using a Bio-Rad colorimetric protein assay and did not present the actual amount of influenza HAs in the virus specimens. Quantitative western blotting using a standard curve of bromelain-released HA was performed in order to estimate the amount of HAs and the purity of HA in the virus specimens, which varies from batch to batch.
Prior to the fluorescence dequenching assays, pH titration curves should be obtained by plotting the pH that is obtained by adding known quantities of HBS pH 3.0 to HBS pH 7.5. The curve is useful to calculate the required volume of HSB, pH 3.0 for the acidification of virus-liposome mixture. This mixture of buffers, as opposed to adding concentrated acid solution for example, is selected to minimize osmotic changes that might disrupt liposomes or virus particles.
Based on our previous experiences, conditions with HA concentrations in the range of 0.5 to 1.0 μM and lipid concentrations in the range of 0.1 to 0.5 mM produce reasonable signal intensities and measurable fusion kinetics.
This procedure increases the hydrophilicity of the carbon substrate.
A significant advantage of using an automated cryo-plunger is the availability of an environmentally controlled chamber for grid blotting that allows us to set the humidity and temperature.
This additional incubation of filter papers at 100% humidity helps to reduce the dryness of the blotting papers and makes the experiments easier to reproduce.
Our previous studies have demonstrated that different stages of fusion intermediates could be observed according to the acidification time. For instance, at pH 5.25, early fusion intermediates such as locally pinching contacts were highly populated at 3 min following acidification. However, after 30 minutes of acidification, postfusion complexes were the dominant population among the total virus-liposome interacting categories [11].
We typically average our stacks by performing a z-projection of 10 images. Given the pixel size is 0.32 nm, the thickness of final projection is 3.2 nm.
References
- 1.White JM, Delos SE, Brecher M, Schornberg K. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol. 2008;43:189–219. doi: 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garcia NK, Guttman M, Ebner JL, Lee KK. Dynamic changes during acid-induced activation of influenza hemagglutinin. Structure. 2015;23:665–676. doi: 10.1016/j.str.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 4.Harrison SC. Viral membrane fusion. Virology. 2015;479–480:498–507. doi: 10.1016/j.virol.2015.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lakadamyali M, Rust MJ. Visualizing infection of individual influenza viruses. Proc Natl Acad Sci U S A. 2003;100(16):9280–5. doi: 10.1073/pnas.0832269100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Floyd DL, Ragains JR, Skehel JJ. Single-particle kinetics of influenza virus membrane fusion. Proc Natl Acad Sci U S A. 2008;105(40):15382–7. doi: 10.1073/pnas.0807771105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ivanovic T, Choi JL, Whelan SP, et al. Influenza-virus membrane fusion by cooperative fold-back of stochastically induced hemagglutinin intermediates. eLife. 2013;2:e00333. doi: 10.7554/eLife.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Subramaniam S, Bartesaghi A, Liu J, et al. Electron tomography of viruses. Curr Opin Struct Biol. 2007;17:596–602. doi: 10.1016/j.sbi.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee KK, Gui L. Dissecting virus infectious cycles by cryo-electron microscopy. PLoS Pathog. 2016;12:e1005625–8. doi: 10.1371/journal.ppat.1005625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baumeister W, Grimm R, Walz J. Electron tomography of molecules and cells. Trends Cell Biol. 1999;9:81–85. doi: 10.1016/s0962-8924(98)01423-8. [DOI] [PubMed] [Google Scholar]
- 11.Gui L, Ebner JL, Mileant A, et al. Visualization and sequencing of membrane remodeling leading to influenza virus fusion. J Virol. 2016;90(15):6948–62. doi: 10.1128/JVI.00240-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen PS, Toribara TY, Warner H. Microdetermination of Phosphorus. Anal Chem. 1956;28:1756–1758. [Google Scholar]
- 13.Suloway C, Shi J, Cheng A, et al. Fully automated, sequential tilt-series acquisition with Leginon. J Struct Biol. 2009;167(1):11–18. doi: 10.1016/j.jsb.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 15.Karmakar S, Sarangi BR, Raghunathan VA. Phase behavior of lipid–cholesterol membranes. Solid State Commun. 2006;139:630–634. [Google Scholar]