

Abstract
Homozygous loss-of-function mutations in optineurin (OPTN) are a rare cause of amyotrophic lateral sclerosis (ALS), whereas heterozygous loss-of-function mutations have been suggested to increase ALS disease risk. We report a patient with ALS and frontotemporal dementia (FTD) from the Clinical Research in ALS and Related Disorders for Therapeutic Development (CReATe) Consortium carrying compound heterozygous loss-of-function variants in OPTN. Quantitative real-time mRNA expression analyses revealed a 75–80% reduction in OPTN expression in blood in the OPTN carrier as compared to controls, suggesting at least partial nonsense-mediated decay of the mutant transcripts. This case report illustrates the diverse inheritance patterns and variable clinical presentations associated with OPTN mutations, and underscores the importance of complete OPTN gene screening in patients with ALS and related disorders, especially in the context of clinical genetic testing.
Keywords: Amyotrophic lateral sclerosis, frontotemporal dementia, optineurin, compound heterozygous, mutation
INTRODUCTION
Mutations in the optineurin gene (OPTN) were first reported as a rare cause of autosomal recessive amyotrophic lateral sclerosis (ALS) (1). In this study, we report compound heterozygous loss-of-function mutations in OPTN in a patient with familial ALS and frontotemporal dementia (FTD) enrolled in the CReATe Consortium.
CASE REPORT
Our patient is a male who first presented with right hand weakness at the age of 58, followed within two months by cognitive (language) symptoms. Left arm weakness developed two months later and he was diagnosed five months after symptom onset with ALS-FTD. His ALS Functional Rating Scale-Revised (ALSFRS-R) at initial assessment was 36 with an estimated progression rate since symptom onset of 1.09 points/month (average to fast). Longitudinal monitoring using the Edinburgh Cognitive and Behavioral ALS Screen (ECAS) (2) identified progressive cognitive decline, which initially manifested in an isolated domain (verbal fluency), but after 12 months involved multiple areas (language, and executive functioning, and memory). Full neuropsychological testing confirmed this impression, with severe impairment (<1st percentile) in language-mediated tasks, (confrontational naming, verbal learning, and sematic/phonemic fluency), and moderate impairments (<5th percentile) in executive function (mental set-shifting, social cognition). Over the ensuing 16 months, ALSFRS-R declined at a rate of 1.67 points/month (fast). He died 21 months after symptoms onset.
The patient’s father died without neurological disease at 65 years, while his mother is unaffected at 83. He has one sister who died of ALS at the age of 45, one sibling with multiple sclerosis, and two living siblings without neurological diseases. He has three living and healthy children.
GENETIC AND EXPRESSION ANALYSES
Illumina whole-genome sequencing in our patient identified in OPTN one nonsense variant p.Ser262* (NM_001008211.1:c.785C>A) and one frameshift deletion p.Leu430Argfs*16 (NM_001008211.1:c.1289_1290del); both were validated by Sanger sequencing. Mutations in other known familial and recessive ALS genes were excluded(3). PCR amplification using a c.785C>A wild-type or mutant specific primer revealed that these mutations are inherited in trans (Figure 1A). Quantitative real-time PCR using RNA isolated from PAXgene Blood RNA kits obtained from the patient and two unrelated controls, showed ~75–80% reduction in OPTN expression in our patient compared to controls suggesting that both mutant transcripts undergo (at least partial) nonsense-mediated decay (Figure 1B). cDNA amplification of OPTN in a multiplex PCR with an unrelated control gene fragment (CSF1R) confirmed the quasi absence of OPTN (Figure 1C).
Figure 1: Reduction of OPTN mRNA expression in blood.
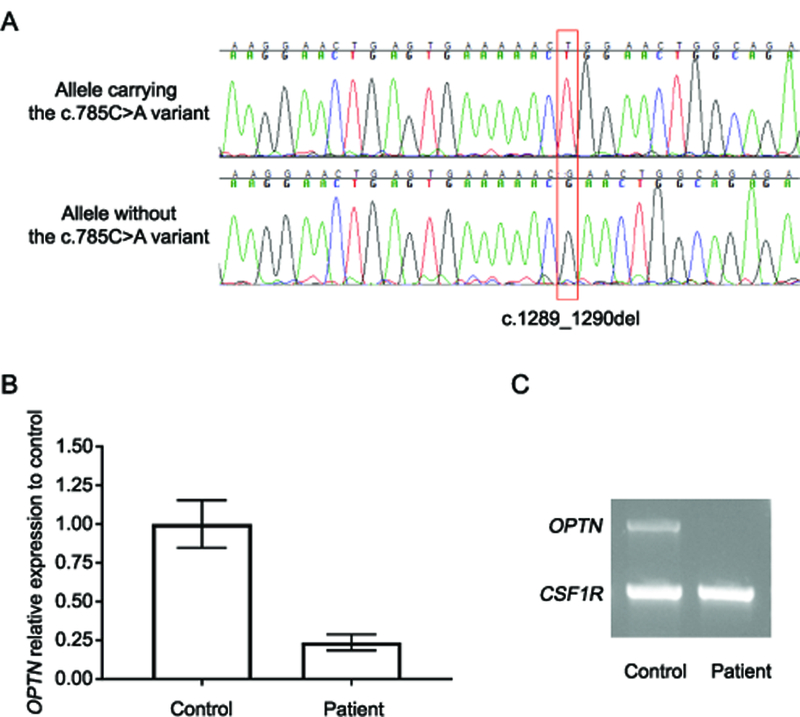
A) Sanger sequencing was performed using cDNA generated from the OPTN patient and sequence traces around the c.1289_1290del mutation (red rectangle) are shown. In the top panel, a primer specific to the mutant allele of the second mutation (c.785C>A) was used, whereas in the bottom panel, a PCR primer only able to amplify the wild-type sequence at c.785C>A was used. Together these traces show that the two mutations are inherited in trans. B) Relative expression of OPTN mRNA as measured by TaqMan gene expression assay (probe Hs00184221_m1) in the OPTN patient and two controls after normalization to the reference gene RPLPO (probe Hs00420895_gh) and further normalization to the average expression in controls (100%). Data from three independent experiments (+/− standard deviation) are shown. C) Migration profile of the multiplex PCR product from OPTN (exons 6–16) and CSF1R cDNA is presented for one healthy control and the patient with OPTN mutations.
DISCUSSION
We report a patient with novel compound heterozygous loss-of-function mutations in OPTN as the probable cause of ALS-FTD. Lack of DNA samples from family members prohibited segregation analysis; however, blood expression studies showed strong reduction in OPTN mRNA to ~20% of normal levels. The OPTN protein, encoded by OPTN, is a multifunctional ubiquitin-binding adaptor protein known to regulate numerous cellular processes including autophagy, inflammation and necroptosis(4). Homozygous loss-of-function mutations in OPTN were first reported in Japanese families with autosomal recessive ALS(1). Heterozygous OPTN mutations were subsequently reported as either causative for ALS or as ALS risk factors; however, the exact contribution of these mutations to disease pathogenesis remains uncertain(4–6). The strongest support for an increased ALS risk in individuals heterozygous for loss-of-function variants in OPTN is provided by a founder mutation in Moroccan and Ashkenazi Jews (7) and by a recent exome-based genome-wide association study(8). Whether these patients carry undetected second mutations in OPTN or mutations in another gene affecting the same functional pathway is not known. Interestingly, we recently reported a patient with FTD and TDP-43 pathology (FTLD-TDP) with heterozygous loss-of-function mutations in OPTN and in the gene encoding the TANK Binding Kinase 1 (TBK1), an important binding partner of OPTN, supporting a role for OPTN in causing disease through an oligogenic mechanism(9).
Compound heterozygous mutations in OPTN have previously been reported in a Dutch family with three patients with progressive muscular atrophy without cognitive impairment (10) and in a single case of FTLD-TDP(9). Our findings expand the phenotypic spectrum of compound heterozygous OPTN loss-of-function mutations to include ALS-FTD with potentially unknown genetic and environmental factors influencing the ultimate phenotypic presentation in each patient.
Together with the published reports, this new case illustrates that the contribution of OPTN mutations to ALS and related disorders is complex. This poses a major challenge for genetic counseling in OPTN families, where single heterozygous rare loss-of-function mutations could be considered genetic risk-factors; however, if an additional rare coding variant is located either within OPTN or in another gene from the same pathway such as TBK1, these mutations could be causative. To identify all possible contributing genetic factors, including partial and full gene deletions, we recommend ALS genetic testing at the exome or genome level, combined with quantitative mRNA analyses, where possible.
ACKNOWLEDGEMENTS
The authors would like to thank the patient and his family. The CReATe consortium (U54NS092091) is part of Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), NCATS. This consortium is funded through collaboration between NCATS, and the NINDS. CReATe is also supported by the ALS Association (grants 17-LGCA-331 and 16-TACL-242).This work was further supported by NIH/NINDS grant R35NS097261.
Footnotes
DECLARATION OF INTEREST
The authors report no conflicts of interest.
REFERENCES
- 1.Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010;465(7295):223–6. [DOI] [PubMed] [Google Scholar]
- 2.Abrahams S, Newton J, Niven E, Foley J, Bak TH. Screening for cognition and behaviour changes in ALS. Amyotroph Lateral Scler Frontotemporal Degener 2014;15(1–2):9–14. [DOI] [PubMed] [Google Scholar]
- 3.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011;72(2):245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Markovinovic A, Cimbro R, Ljutic T, Kriz J, Rogelj B, Munitic I. Optineurin in amyotrophic lateral sclerosis: Multifunctional adaptor protein at the crossroads of different neuroprotective mechanisms. Prog Neurobiol 2017;154:1–20. [DOI] [PubMed] [Google Scholar]
- 5.Tumer Z, Bertelsen B, Gredal O, Magyari M, Nielsen KC, Lucamp, et al. Novel heterozygous nonsense mutation of the OPTN gene segregating in a Danish family with ALS. Neurobiol Aging 2012;33(1):208 e1–5. [DOI] [PubMed] [Google Scholar]
- 6.Weishaupt JH, Waibel S, Birve A, Volk AE, Mayer B, Meyer T, et al. A novel optineurin truncating mutation and three glaucoma-associated missense variants in patients with familial amyotrophic lateral sclerosis in Germany. Neurobiol Aging 2013;34(5):1516 e9–15. [DOI] [PubMed] [Google Scholar]
- 7.Goldstein O, Nayshool O, Nefussy B, Traynor BJ, Renton AE, Gana-Weisz M, et al. OPTN 691_692insAG is a founder mutation causing recessive ALS and increased risk in heterozygotes. Neurology 2016;86(5):446–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015;347(6229):1436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pottier C, Bieniek KF, Finch N, van de Vorst M, Baker M, Perkersen R, et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol 2015;130(1):77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beeldman E, van der Kooi AJ, de Visser M, van Maarle MC, van Ruissen F, Baas F. A Dutch family with autosomal recessively inherited lower motor neuron predominant motor neuron disease due to optineurin mutations. Amyotroph Lateral Scler Frontotemporal Degener 2015;16(5–6):410–1. [DOI] [PubMed] [Google Scholar]