

Abstract
Mitochondria are the metabolic powerhouses of cells. In addition to generating adenosine triphosphate (ATP), they play important roles in cell survival pathways such as apoptosis and necrosis. Mitochondrial size and shape are dynamically regulated by a process known as mitochondrial dynamics. The significance of this process in metabolically active cells such as skeletal and cardiac muscle are only now beginning to be elucidated. In cardiac muscle, mitochondrial dynamics plays an important role in mitochondrial quality control and defects in regulatory pathways that govern these processes and leads to heart failure. In response to nutrient excess such as lipid overload, as occurs in diabetes, mitochondrial shape and morphology are altered by effects of nutrient stress on mitochondrial dynamics signaling pathways, which have important implications for understanding mitochondrial dysfunction in diabetic cardiomyopathy. Moreover, crosstalk between mitochondria and other organelles such as the endoplasmic reticulum can regulate generation of hormones such as fibroblast growth factor 21, with potent anti-diabetic and anti-obesity effects.
INTRODUCTION
Mitochondria are complex organelles that possess exquisite metabolic machinery that mediates the conversion of multiple metabolic substrates to adenosine triphosphate (ATP), which provides energy for most cellular functions. They are double-membrane organelles, with an outer membrane through which metabolites traverse either out of the mitochondrion or into the mitochondrial matrix, which is surrounded by the inner mitochondrial membrane (Figure 1). The inner mitochondrial membrane is folded into lamellar or tubular structures known as mitochondrial cristae. The mitochondrial inner and outer membranes harbor multiple protein complexes which serve as channels for metabolic intermediates, reactive oxygen species, and ions. The inner membrane also contains the electron transport chain which transfers electrons derived from metabolic intermediates, while pumping protons into the intermembrane space to generate the mitochondrial membrane potential which drives ATP synthesis. In addition to its central role in metabolism, mitochondria also regulate cell death pathways such as apoptosis and necrosis and in contractile cells such as muscle; they buffer intracellular calcium. Thus, mitochondria represent a hub, not only for cellular energy production, but also for cell survival and signal transduction.
Fig. 1.
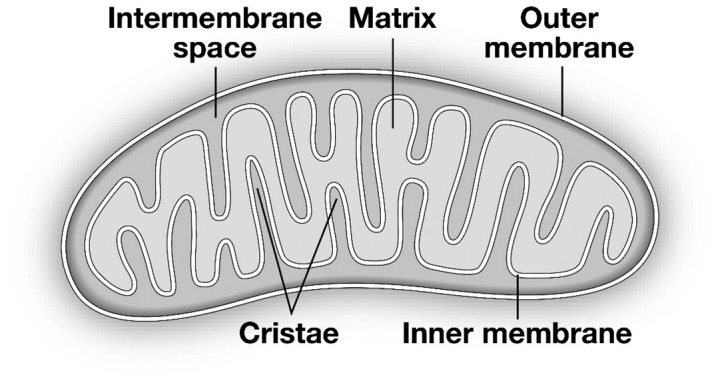
Schematic representation of mitochondrial structure.
For most of us, the images of mitochondria that stick in our minds are beautiful electron micrographs of intracellular organelles that appear frozen in place. Studies performed initially in live yeast and single cells revealed that mitochondria do not sit still, but are dynamically regulated, undergoing repeated rounds of fusion and fission and even moving around the cell (1). This phenomenon of repeated cycles of mitochondrial fusion and fission is known as mitochondrial dynamics. This is a highly regulated process that is mediated by specific cellular proteins (Figure 2). Mitochondrial membrane fusion is mediated by a family of dynamin-related proteins (DRP) with guanosine triphosphatase activity. Outer mitochondrial membrane fusion is mediated by members of the mitofusin family (mitofusin 1 and mitofusin 2, also known as MFN1 and MFN2). Inner membrane mitochondrial fusion is mediated by the protein optic atrophy 1 (OPA1). OPA1 also regulates crista morphology by forming oligomers at the base of mitochondrial cristae (2). Naturally occurring mutations in MFN2 lead to Charcot Marie Tooth syndrome type 2A and mutations in OPA1 are associated with autosomal dominant optic atrophy. Mitochondrial fission is mediated by the interactions between another family of dynamin related proteins DRP1/DLP1, which are usually resident in the cytosol, but in response to various stimuli migrate to the mitochondrial outer membrane where they interact with an outer mitochondrial membrane protein Mitochondrial fission 1 (Fis-1), to form oligomers, which constrict the mitochondria leading to a fission event that generates two (or more) daughter mitochondria (Figure 2). The proteins that regulate mitochondrial dynamics are subject to post-translational modifications that modify their function. For example, mitofusins are subject to ubiquitination that regulate their interactions with molecules involved in autophagy (3,4). Proteolytic processing of OPA1 by the proteases OMA1 and YMEL1 (both are ATP-dependent metalloproteases) generates inactive fragments that reduce its ability to promote inner mitochondrial membrane fusion (5). DRP1 is subject to phosphorylation on distinct sites (6,7). Phosphorylation on serine 637 (S637) by protein kinase A inhibits DRP1 by promoting a cytosolic localization, whereas the phosphatase calcineurin dephosphorylates S637 to promote translocation of DRP1 to the mitochondria to promote fission. Phosphorylation of DRP1 on a separate site serine 616, by calcium/calmodulin-dependent protein kinase II (CaMKII), Cyclin-dependent kinase 1 (Cdk1), and mitogen activated protein kinases, promotes mitochondrial localization and increases mitochondrial fission.
Fig. 2.

(A) Schematic representation of mitochondrial fusion and molecular mediators of mitochondrial outer membrane fusion (mitofusin 1/2 [MFN1/2]) and optic atrophy 1 (OPA1). (B) Schematic representation of mitochondrial fission and its molecular mediators Mitochondrial fission 1 (Fis-1) and dynamin-related protein 1 (DRP1). The schematic shows DRP1 phosphorylation on serine 637 (p-DRP1) which promotes cytosolic localization, whereas dephosphorylation increases association with fis1 to promote mitochondrial fission.
Mitochondrial dynamics is believed to play an important role in mitochondrial biogenesis and provides a mechanism by which mitochondrial DNA can be exchanged to maintain the mitochondrial genomes (8). Conversely, mitochondrial fission represents an important mechanism for removal of damaged mitochondria, as shown by studies that indicate that following mitochondrial damage, impaired mitochondrial elements might be physically removed and targeted for degradation by autophagy, a process that is termed mitophagy (8). Mitofusins also mediate important interorganellar communication between the outer mitochondrial membrane and the endoplasmic reticulum (ER), which promotes interorganellar signaling and exchange of ions and metabolites (9), whereas OPA1 also regulates crista morphology and efficiency of the mitochondrial electron transport chain (2).
A question that has intrigued us and others working in the field is: what is the role of mitochondrial dynamics in differentiated skeletal and cardiac muscle cells, which are packed with sarcomeres and, at face value, could have a limited repertoire of surrounding neighbors, with which a given mitochondria could fuse? Limited studies in human heart failure samples have provided some evidence of increased mitochondrial fragmentation that correlates with reduced expression of OPA1 (10). Recent studies have also suggested that perturbations in mitochondrial dynamics in skeletal muscle (and liver) may contribute to the pathophysiology of insulin resistance (11,12). The general concept is that caloric excess promotes mitochondrial fission via incompletely understood mechanisms. If prolonged, this exacerbates reactive oxygen species (ROS) overproduction and impairs insulin signaling. Moreover, this pathophysiology might be exacerbated by mutations in or altered expression of mitochondrial dynamics proteins that impairs fusion or promotes mitochondrial fission. Insulin resistance is associated with mitochondrial dysfunction, characterized by decreased mitochondrial biogenesis and reduced bioenergetic responses to insulin stimulation. Mechanisms include transcriptional repression of mitochondrial genes, lipotoxicity, and direct effects of insulin resistance (13). It is debated whether skeletal muscle mitochondrial dysfunction is a cause or a consequence of insulin resistance. What is less controversial is that once mitochondrial dysfunction occurs, this can lead to additional defects such as overproduction of ROS, incomplete oxidation of fatty acids, and reduced energy expenditure, which exacerbates insulin resistance (13). However, in skeletal muscle, mitochondrial dysfunction can occur without insulin resistance and conversely, insulin resistance has been shown in the absence of mitochondrial dysfunction (14). A small number of studies have investigated expression levels of mitochondrial dynamics proteins in animal models and humans with obesity and insulin resistance. Most studies focused on MFN2 and revealed reduced levels of MFN2 mRNA and protein in skeletal muscles of Zucker fatty rats and in humans with type 2 diabetes. Transcriptional repression by circulating cytokines or decreased transcriptional regulation by peroxisome proliferator gamma coactivator 1 alpha (PGC-1α) has been implicated in the repression of MFN2. Decreased MFN2 levels correlate with skeletal muscle insulin resistance and increase following bariatric surgery–induced weight loss (15).
More insights have been gained from various mouse models with targeted mutations of all of these regulatory proteins either in cardiac or skeletal muscle. Germline and conditional/tissue-specific mutations have been generated for all of these regulators of mitochondrial dynamics (16). In general, disruption of either mitochondrial fusion or mitochondrial fission are deleterious for mitochondrial homeostasis indicating that a healthy balance between fusion and fission of mitochondria is essential for maintaining cellular and mitochondrial homeostasis (16–18). In the heart, disruption of mitochondrial fusion by targeting MFN1 and MFN2 leads to eccentric cardiac remodeling and early mortality, whereas loss of DRP1 precipitates dilated heart failure (17). Intriguingly, when both mitochondrial fusion and fission were conditionally inhibited in the heart, although these animals survived longer than animals with disruption of fission or fusion pathways, respectively, they manifested a phenotype of accelerated myocardial senescence on the basis of impaired mitochondrial quality control. Knockout of MFN2 in liver (19) or in muscle/heart/brain significantly perturbs metabolic homeostasis, characterized by insulin resistance that may result from increased ER or oxidative stress (19) with negligible effects on body weight. Although various defects in oxidative phosphorylation (OXPHOS) subunit expression and respiration rates were noted, ATP synthesis was normal despite increased ROS. Thus, loss of MFN2 in skeletal muscle drives phenotypes that may be due to oxidative stress versus impaired mitochondrial bioenergetics. In vitro studies also suggest that lipid overload will increase DRP1-mediated mitochondrial fission partially on the basis of oxidative stress, which further exacerbates insulin resistance (11).
Expression levels of the rhomboid protease Presenilin Associated Rhomboid Like (PARL) are reduced in obese animals and in humans with type 2 diabetes. Although initially discovered as an OPA1protease, PARL regulates mitochondria structure and function in complex ways, which include transcriptional effects on OPA1 and MFN2 gene expression by the PARL C-terminal product of β-cleavage (PACT). Thus, siRNA-mediated silencing of PARL in mouse skeletal muscle or in cultured human myotubes repressed OPA1 and MFN2 expression, reduced crista integrity, and induced oxidative stress, insulin resistance, and lipid accumulation (20). Reduced levels of PARL, OPA1, and MFN2 have been described in humans with insulin resistance (elderly subjects, patients with type 2 diabetes) and obese primates (1). Germline deficiency of PARL leads to premature mortality from cachexia (21). Given its multiple targets, the ability of the PARL-deficient models to evaluate specific contributions of OPA1 to insulin regulation of mitochondrial bioenergetics is limited. Germ line deletion of OMA, the OPA1 protease, reveals a complex metabolic phenotype characterized by obesity, hyperleptinemia, and liver steatosis following high-fat feeding, but unexpectedly without insulin resistance (12). These studies underscore the complex interactions between mitochondrial dynamics, metabolic homeostasis, and insulin resistance, but leave unresolved a precise understanding of the tissue-specific role of OPA1 in muscle to regulate insulin sensitivity in vivo.
Insulin increases mitochondrial metabolism in cardiac and skeletal muscle. Our laboratory reported that this increase is associated with increased mitochondrial fusion and increased expression of OPA1 in vivo and in vitro (22). Importantly, when OPA1 expression was genetically reduced in cultured muscle cells, the ability of insulin to increase mitochondrial metabolism was prevented (22). Thus, mitochondrial fusion represents a previously unrecognized action of insulin that is essential for its short-term metabolic effects. It is not currently known if the OPA1-mediated increase in mitochondrial metabolism is a direct consequence of increased mitochondrial fusion, increased mitochondrial cristae density, or both. Therefore, we hypothesized that if OPA1 expression was impaired in skeletal muscle in vivo, impaired insulin action or insulin resistance would develop. As expected, examination of mitochondria with reduced OPA1 expression in skeletal muscle revealed impaired mitochondrial cristae morphology and reduced mitochondrial bioenergetics. However, in contrast to our hypothesis, mice with inducible skeletal muscle reduction in OPA1 (smOPA1-/-) expression did not manifest insulin resistance (23).
smOPA1-/- mice did not develop age-related obesity or insulin resistance. Feeding mice, a high-fat diet is commonly used to model obesity, insulin resistance, and type 2 diabetes. Interestingly, when smOPA1-/- mice were placed on a high-fat diet they did not develop obesity or diabetes. Body composition analysis revealed a striking reduction in fat mass that accounted for the reduction in adiposity. We also induced skeletal muscle OPA1 deficiency in mice following high-fat feeding, which induced obesity and type 2 diabetes. Intriguingly, reduction in OPA1 in skeletal muscle of obese and diabetic mice led to a dramatic reversal in obesity and diabetes (23). These observations led to the hypothesis that OPA1-deficient skeletal muscle was secreting a factor that influenced systemic metabolic homeostasis. We screened for potential secreted factors that could regulate metabolic homeostasis and observed a large induction in the mRNA and protein levels of a recently described hormone fibroblast growth factor 21 (FGF21). We also confirmed that circulating concentrations of FGF21 were increased (23).
Under physiological conditions, FGF21 is predominantly a liver-derived hormone that may also be released from adipose tissue. In mammals, it has pleiotropic effects that include increasing energy expenditure, improving glucose homeostasis in some species, reducing lipids, and reducing the taste preference for sugar (24). Thus, FGF21 was a plausible candidate to explain the metabolic effects that we observed. However, given that skeletal muscle was not believed to be an important source of circulating FGF21, we had to prove that muscle-derived FGF21 was indeed responsible for the phenotypes observed. To address this, we generated mice in which the FGF21 and the OPA1 genes were simultaneously and inducibly reduced in skeletal muscle (23). These animals were no longer protected from age-dependent or diet-induced obesity, insulin resistance, and type 2 diabetes. Having proven that skeletal muscle was the source of OPA1, we then sought to understand the mechanism linking OPA1 deficiency in skeletal muscle with induction of FGF21 gene expression. Reduced expression of OPA1 in skeletal muscle led to activation of the unfolded protein response, which is also known as the ER stress pathway. Activation of this pathway, which is conserved in all eukaryotic cells, primarily serves to maximize the ability of the endoplasmic reticulum to maintain protein folding under conditions of increased protein synthesis. The activation of this pathway leads to the generation of transcriptional regulators such as spliced XBP1 and ATF4, which can bind the promoter of the FGF21 gene. To prove that this mechanism was in play in our model, smOPA1-/- mice were treated with an inhibitor of the ER stress pathway, which prevented the increase in FGF21 from skeletal muscle. Thus, mitochondrial stress in skeletal muscle, which could be associated with impaired skeletal muscle and subsequently whole-body metabolism, induces a hormetic response that maintains insulin sensitivity in the entire animal (Figure 3). We are now pursuing experiments to understand at a mechanistic level how a defect in OPA1 function actually leads to activation of the ER stress pathway. Given the relative mass of skeletal muscle, we believe that an understanding of how this pathway is regulated could enable us to harness skeletal muscle to produce FGF21 as a novel strategy to counteract the metabolic consequences of obesity and insulin resistance. Recent studies in humans with mitochondrial myopathy have revealed increased circulating levels of FGF21. Thus, the notion that skeletal muscle could be harnessed as a potential endogenous source of FGF21 is plausible if a mechanistic target that regulates FGF21 in skeletal muscle is identified that can be safely manipulated by pharmacological means (25).
Fig. 3.
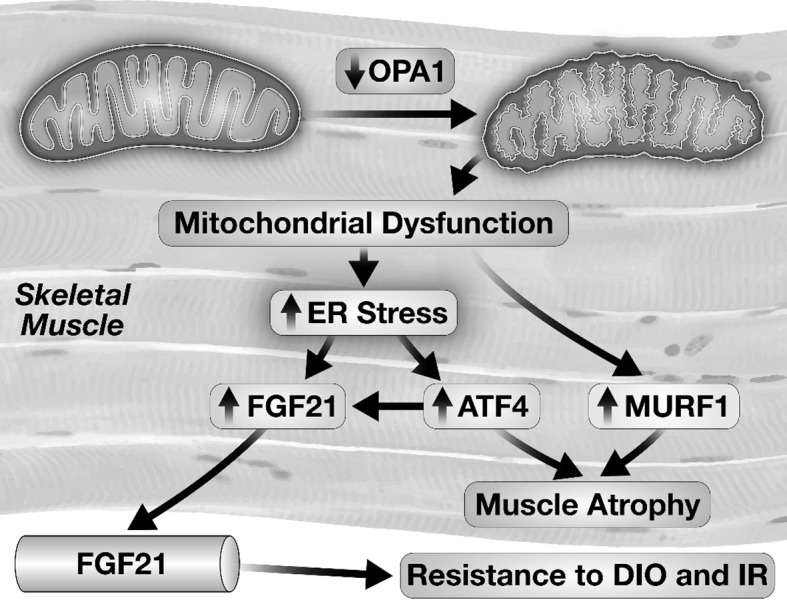
Schematic representation of mechanisms by which a reduction of optic atrophy 1 (OPA1) levels in skeletal muscle leads to the activation of the endoplasmic reticulum (ER) stress pathway and induction of fibroblast growth factor 21 (FGF21) gene expression and FGF21 release. Abbreviations: ATF, Activating Transcription Factor 4; MURF, muscle ring finger protein 1; DIO, diet-induced obesity; IR, insulin resistance.
Studies in mutant mice have suggested that mitochondrial dynamics play an important role in mitochondrial quality control and mitochondrial energy metabolism. Less is known about the impact of environmental or hemodynamic stress on the regulation of mitochondrial dynamics in the heart. Diabetes mellitus is associated with many changes in myocardial metabolism and mitochondrial function, including an increase in fatty acid uptake and use, increased mitochondrial ROS generation, and impaired mitochondrial bioenergetics (26). However, the impact of diabetes on mitochondrial dynamics in the heart was incompletely understood.
To evaluate the consequence of increased myocardial fatty acid use on mitochondrial dynamics, we studied a mouse model with low-level overexpression of the enzyme acyl CoA synthetase (ACStg) that increased myocardial fatty acid use by 2-fold (27). This increase in myocardial lipid uptake induced developmental remodeling of the mitochondrial network. Immediately following birth mitochondrial diameter is reduced, and over the course of maturation there is a progressive increase in mitochondrial width that is associated with inhibition of DRP1, as evidenced by increased phosphorylation of DRP1 on Ser 637. In contrast, in ACStg mice the developmental increase in mitochondrial size failed to occur and correlated with decreased phosphorylation of DRP1 on Ser 637. This phosphorylation pattern of DRP1 is consistent with increased DRP1 activity that would increase mitochondrial fission and reduce mitochondrial fusion. Indeed, when we genetically reduced the levels or activity of DRP1 in the context of lipid overload, the degree of mitochondrial network fragmentation was attenuated. The mitochondrial protein A-kinase anchor protein 121 (AKAP121) is a scaffold protein that facilitates the interaction of protein kinase A (PKA) with substrates such as DRP1. Our studies revealed lower levels of AKAP121, which were reduced on the basis of increased proteasomal degradation. In addition to post-translational modifications of DRP1, lipid overload also impaired the proteolytic cleavage of OPA1, leading to increased abundance of relatively inactive isoform (27). Taken together, these studies reveal that lipid excess modifies signaling pathways that regulate mitochondrial dynamics in the heart (Figure 4).
Fig. 4.

(A) Two-dimensional electron micrographs of cardiac tissue from healthy hearts and from the hearts of mice with lipid overload that develops as a result of overexpression of Acyl-CoA synthetase. (B) Schematic representation of the mechanisms by which increased mitochondrial lipid overload induced by overexpression of Acyl-CoA synthetase generates reactive oxygen species (ROS) that leads to decreased phosphorylation of DRP1 and inactivation of OPA1 that ultimately results in increased mitochondrial fission. Abbreviations: OPA1, optic atrophy 1; DRP1, dynamin-related protein; ATP, adenosine triphosphate; PKA, protein kinase A; Acyl-CoA, acyl Coenzyme A synthetase; ACSL1, Acyl-CoA synthetase isoform 1; AKAP 121, A Kinase anchoring protein 121; NADH, reduced nicotinamide adenine dinucleotide.
Mechanistically, the modifications in signaling proteins that regulate mitochondrial dynamics were induced by oxidative stress and ROS overproduction. Indeed, when we crossed ACStg mice with those harboring a transgene that overexpressed the endogenous ROS scavenger, mitochondrial superoxide dismutase (Sod2tg), mitochondrial network fragmentation was reduced. Strongly scavenging ROS under ambient conditions, as is the case in Sod2tg mice, resulted in enlarged mitochondria, potentially on the basis of increased mitochondrial fusion (27). Thus, mitochondrial size in cardiac muscle is dynamically regulated by the REDOX state underscoring the notion that mitochondria are dynamic organelles who adjust their size and shape, even in differentiated, but metabolically active tissues such as cardiac muscle. Typical electron micrographs generate a 2D view of tissue ultrastructure and analysis of 2D micrographs in ACStg mouse hearts generated an appearance of multiple fragmented mitochondria (Figure 4). However, 3D tomography of these hearts launched us into a virtual reality experience, whereby the pattern that we observed reflects numerous narrow and tortuous mitochondria that entered the plane of the myocardial section multiple times (27). Thus mitochondrial “fission” in the heart is characterized by thinning, prolongation and increased tortuosity of mitochondria as opposed to fragmentation per se.
In summary, mitochondria are dynamic organelles that change their shape and size in cells in response to environmental stimuli. Moreover, there is a close relationship between physiological changes in mitochondrial size and critical metabolic functions of mitochondria such as redox regulation and ATP generation. Finally, alterations in mitochondrial structure and function modulates interorganellar communication leading to novel endocrine responses in muscle cells with important consequences for systemic metabolic homeostasis. Thus, mitochondria in metabolically active tissues such as muscle should no longer be viewed as static organelles that quietly generate ATP, but rather dynamic organelles whose form and shape determines their function.
ACKNOWLEDGMENTS
Studies in the Abel laboratory are supported by grants from the National Institutes of Health and the American Heart Association.
Footnotes
Potential Conflicts of Interest: None disclosed.
DISCUSSION
Schreiner, Los Altos: A very nice demonstration of the role of altered bioenergetics in heart failure. Could you explain the relationship between the three-dimensional structure in either fission or fusion affected mitochondria and oxygen efficiency utilization.
Abel, Iowa City: Sure. So basically what we believe happens is that the redox state of mitochondria directly regulates the fusion and fission process and initially when you increase reactive oxygen species (ROS), the increase in mitochondrial fission is actually somewhat protective so by making the mitochondria smaller and thinner, you might in fact become relatively more energetically efficient; however, if the insult persists over time, then what we see is a gradual accumulation of ROS-mediated mitochondrial injury and a decline in mitochondrial respiratory function.
Ziegelstein, Baltimore: That was a terrific talk. Very quick question: Jeremy Walston and Peter Abadir at John’s Hopkins have been looking at the mitochondrial angiotensin 2 receptor in aging and frailty and, given some of the similarities between diabetes and aging, I’m wondering if anyone has looked at this in diabetes?
Abel, Iowa City: People have looked at the interaction between diabetes and aging. There is a nice study out of Germany looking at age-related mitochondrial energetics in young individuals and then in older individuals and, in fact, what they saw was that the interaction of aging and diabetes does increase the evidence of mitochondrial oxidative stress and evidence of impaired mitochondrial oxidative capacity.
Ziegelstein, Baltimore: But has anyone looked at angiotensin 2?
Abel, Iowa City: No, not in the context of diabetes as I am aware.
Ziegelstein, Baltimore: Okay, thank you.
Epstein, Philadelphia: Do the mice still have the dramatic increase in fission in mitochondria — do they get heart failure? And do you try putting them on a high-fat diet?
Abel, Iowa City: The animals I showed you with acyl CoA synthase overexpression, they do develop cardiac hypertrophy, but with preserved ventricular function at a young age. They develop LV contractile dysfunction around about 15 or so weeks of age.
Epstein, Philadelphia: Did you put them on a high-fat diet?
Abel, Iowa City: Yes, we did, and it turns out funny enough that there’s this whole thing about high-fat diet in mice. It turns out that in mice, actually, their hearts are fairly capable of tolerating high-fat feeding for extended periods of time. So, for short-term high-fat feeding, we didn’t see any additional exacerbation but then for these studies — you have to decide what kind of fat, duration of the diet, and so forth. So, I would say it’s not completely clear at this point.
Wolf, Boston: Diabetes causes animals to lose magnesium through their kidneys, and I’m wondering if you give your diabetic animals supplemental magnesium since magnesium deficiency has major adverse effects on mitochondria metabolism.
Abel, Iowa City: Yes, that’s a very good question. In terms of the animal model I described earlier, which is a cardiomyocyte-specific transgenic, the defect in terms of increased fat utilization is restricted to cardiomyocytes. So, we never had reason to believe their systemic metabolism is abnormal and that they would in fact be losing magnesium. In our studies in diabetic animals, we have not supplemented magnesium, so I guess that could be a potential confounder.
Wolf, Boston: Well, you sort of wonder whether that could be one of the reasons why their electron transport is deficient.
Abel, Iowa City: Potentially.
REFERENCES
- 1.Liesa M, Palacin M. Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev. 2009;89:799–845. doi: 10.1152/physrev.00030.2008. [DOI] [PubMed] [Google Scholar]
- 2.Pernas L. Scorrano L. Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu Rev Physiol. 2016;78:505–31. doi: 10.1146/annurev-physiol-021115-105011. [DOI] [PubMed] [Google Scholar]
- 3.Anton F, Dittmar G, Langer T. Escobar-Henriques M. Two deubiquitylases act on mitofusin and regulate mitochondrial fusion along independent pathways. Mol Cell. 2013;49:487–98. doi: 10.1016/j.molcel.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Gegg ME, Cooper JM, Chau KY, Rojo M, et al. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19:4861–70. doi: 10.1093/hmg/ddq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wai T. Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. 2016;27:105–17. doi: 10.1016/j.tem.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Chang CR. Blackstone C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein DRP1. Ann N Y Acad Sci. 2010;1201:34–9. doi: 10.1111/j.1749-6632.2010.05629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu S, Wang P, Zhang H, et al. CaMKII induces permeability transition through DRP1 phosphorylation during chronic beta-AR stimulation. Nat Commun. 2016;7:13189. doi: 10.1038/ncomms13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ni HM, Williams JA, Ding WX. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015;4:6–13. doi: 10.1016/j.redox.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zorzano A, Liesa M, Sebastian D, Segales J. Palacin M. Mitochondrial fusion proteins: dual regulators of morphology and metabolism. Semin Cell Dev Biol. 2010;21:566–74. doi: 10.1016/j.semcdb.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 10.Chen L, Gong Q, Stice JP, Knowlton AA. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res. 2009;84:91–9. doi: 10.1093/cvr/cvp181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jheng HF, Tsai PJ, Guo SM, et al. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol. 2012;32:309–19. doi: 10.1128/MCB.05603-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quiros PM, Ramsay AJ, Sala D, et al. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J. 2012;31:2117–33. doi: 10.1038/emboj.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pagel-Langenickel I, Bao J, Pang L, Sack MN. The role of mitochondria in the pathophysiology of skeletal muscle insulin resistance. Endocr Rev. 2010;31:25–51. doi: 10.1210/er.2009-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pulliam DA, Bhattacharya A, Van Remmen H. Mitochondrial dysfunction in aging and longevity: a causal or protective role? Antioxid Redox Signal. 2013;19:1373–87. doi: 10.1089/ars.2012.4950. [DOI] [PubMed] [Google Scholar]
- 15.Zorzano A, Hernandez-Alvarez MI, Palacin M, Mingrone G. Alterations in the mitochondrial regulatory pathways constituted by the nuclear co-factors PGC-1alpha or PGC-1beta and mitofusin 2 in skeletal muscle in type 2 diabetes. Biochim Biophys Acta. 2010;1797:1028–33. doi: 10.1016/j.bbabio.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 16.Dorn GW. 2nd. Mitochondrial dynamics in heart disease. Biochim Biophys Acta. 2013;1833:233–41. doi: 10.1016/j.bbamcr.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song M, Franco A, Fleischer JA, Zhang L, Dorn GW. 2nd. Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence. Cell Metab. 2017;26:872–83. doi: 10.1016/j.cmet.2017.09.023. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wai T, Garcia-Prieto J, Baker MJ, et al. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science. 2015;350:aad0116. doi: 10.1126/science.aad0116. [DOI] [PubMed] [Google Scholar]
- 19.Sebastian D, Hernandez-Alvarez MI, Segales J, et al. Mitofusin 2 (MFN2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci U S A. 2012;109:5523–8. doi: 10.1073/pnas.1108220109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Civitarese AE, MacLean PS, Carling S, et al. Regulation of skeletal muscle oxidative capacity and insulin signaling by the mitochondrial rhomboid protease PARL. Cell Metab. 2010;11:412–26. doi: 10.1016/j.cmet.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cipolat S, Rudka T, Hartmann D, et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell. 2006;126:163–75. doi: 10.1016/j.cell.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 22.Parra V, Verdejo HE, Iglewski M, et al. Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt-mTOR-NFkappaB-Opa-1 signaling pathway. Diabetes. 2014;63:75–88. doi: 10.2337/db13-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pereira RO, Tadinada SM, Zasadny FM, et al. OPA1 deficiency promotes secretion of FGF21 from muscle that prevents obesity and insulin resistance. EMBO J. 2017;36:2126–45. doi: 10.15252/embj.201696179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markan KR, Potthoff MJ. Metabolic fibroblast growth factors (FGFs): mediators of energy homeostasis. Semin Cell Dev Biol. 2016;53:85–93. doi: 10.1016/j.semcdb.2015.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lehtonen JM, Forsstrom S, Bottani E, et al. FGF21 is a biomarker for mitochondrial translation and mtDNA maintenance disorders. Neurology. 2016;87:2290–9. doi: 10.1212/WNL.0000000000003374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–23. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 27.Tsushima K, Bugger H, Wende AR, et al. Mitochondrial reactive oxygen species in lipotoxic hearts induces post-translational modifications of AKAP121, DRP1 and OPA1 that promote mitochondrial fission. Circ Res. 2018;122:58–73. doi: 10.1161/CIRCRESAHA.117.311307. [DOI] [PMC free article] [PubMed] [Google Scholar]