

Abstract
Background:
Development of inhibitors to transfused factor VIII in patients with hemophilia A continues to be a challenge for professionals involved in hemophilia care. The majority of patients in India receive ‘on-demand’ rather than prophylactic therapy. The present study was done to assess the prevalence of factor VIII inhibitors in patients with hemophilia A (PWHA) receiving ‘on-demand’ therapy in a North Indian population and to study the clinicopathological parameters influencing the development of inhibitors.
Methods:
The study group comprised of 300 PWHA. Detailed clinical parameters, treatment history, bleeding profile including family history were recorded. Diagnosis of hemophilia A was confirmed by relevant coagulation tests. Inhibitors were screened using mixing based studies followed by quantification by Bethesda assay and Nijmegen modified Bethesda assay. Samples were collected from five cities in North India where a free supply of factor VIII was available and effectively used in three of these cities.
Results:
In the 300 PWHA, disease phenotype was severe in 219 (73%), moderate in 62 (20.67%) and mild in 19 (6.34%), based on the factor VIII bioassay. Inhibitor prevalence was 9.6% (n = 29) and seen only in the severe phenotype. Inhibitor titers ranged from 0.8 to 108.8 BU/ml. A total of 12 PWHA had low and 17 had high titers. Correlation of various clinicopathological parameters in inhibitor-positive versus negative PWHA showed significant correlation with age at onset of disease, severity of disease, age at first exposure to treatment, annual factor intake (IU/kg/year), intense treatment episodes and bleeding manifestations like central nervous system bleed and hematuria. The total study sample had blood group B in 33.34% PWHA, followed by O (27.34%), A (24.34%) and AB (15%), however, in inhibitor-positive samples, significant inhibitor formation was associated with the ABO subtype A (19/29, 65.51%).
Conclusions:
Factor VIII inhibitor prevalence in PWHA receiving ‘on-demand’ therapy was 9.6%. Clinicopathological correlates of inhibitor development in such PWHA have been analyzed in this novel study.
Keywords: hemophilia A, factor VIII, inhibitors, Bethesda assay, plasma derived factor VIII
Introduction
Hemophilia A is a recessively inherited X-linked bleeding disorder which results from a deficiency of blood coagulation factor VIII. It affects one in 5000 male births worldwide.1 The mainstay of treatment of hemophilia A is usually protein replacement using plasma-derived or recombinant factor VIII concentrates to achieve a prophylactic status and to prevent bleeding. A recent United Kingdom guideline recommended prophylactic administration of factor VIII or factor IX concentrate 2–3 times a week as the standard of care for persons with severe hemophilia A or B to prevent hemarthrosis.2 However, prophylactic replacement therapy is expensive and not affordable in developing countries where an on-demand factor replacement is provided to achieve hemostasis during bleeding episodes or prior to surgical procedures. The exogenous factor VIII may cause an alloantibody or inhibitor formation.3 These factor VIII inhibitors develop in approximately 25–30% of patients with hemophilia A (PWHA) with a severe phenotype. Presence of inhibitors renders replacement therapies ineffective, limits patient access to a safe and effective standard of care and predisposes them to an increased risk of morbidity and mortality.4 Factors that may increase the risk of inhibitor development comprise genetic factors including the type of mutation in the F8 gene, HLA type, ethnicity, family history and immune regulatory genes as well as nongenetic factors including early age at start of treatment, intensity of treatment and type of replacement therapy.5–9 The risk of inhibitor development appears highest during the first 50 days of exposure to factor concentrate in PWHA on prophylaxis.10 On the contrary, a study conducted by Eckhardt and colleagues suggests that in nonsevere PWHA, the risk of inhibitor development may continue beyond 100 exposure days.11 The worldwide prevalence of inhibitors in PWHA is 25–30% and in India it is estimated to be 8.2–13%.12,13 Due to the prohibitive cost of the factor and limited affordability of the population in India, PWHA usually have an on-demand schedule of factor VIII replacement. Recently, the availability of plasma-derived factor VIII (Hemofil M, Baxalta US Inc, USA) has increased with support from certain state governments in India. Though still not used on a prophylactic basis, the overall use by patients has increased for therapy on-demand. It was therefore hypothesized, that the prevalence of factor VIII inhibitors would also consequently increase. The present study was conducted to assess the prevalence of factor VIII inhibitors in various cities of North India, where factor VIII has been made available and to observe their occurrence and analyze factors associated with the development of factor VIII inhibitors in PWHA using it on-demand.
Materials and methods
Study group: The study group comprised of a series of PWHA from the population of North India. The blood samples from PWHA were collected through direct reference to our laboratory as well as in outreach camps held in various cities of North India in association with the local chapters of the Hemophilia Federation of India. The study was conducted over a period of 12 months and a total of 300 diagnosed PWHA were included in the study after prior consent as per protocol. All the PWHA were treated with plasma-derived factor VIII (Hemofil M, Baxalta US Inc, USA) which is not enriched with von Willebrand factor. Ethical clearance was obtained from the institutional ethical committee.
Inclusion/exclusion criteria: PWHA giving consent to enter the study were included. Patients of other bleeding disorders not related to factor VIII were excluded. PWHA with active infection were also excluded from the study.14
Clinical assessment: A detailed history was recorded including age at onset, age at diagnosis, disability profile, number of bleeding episodes, factor VIII requirement per annum in terms of IU/kg/year was calculated for each patient, and frequency of whole blood and cryoprecipitate transfusions was recorded. Pedigree was charted on the basis of family history to enumerate PWHA and suspected or confirmed carriers within the family. The treatment record in most PWHA revealed that factor VIII was given during bleeding episodes as well as during surgical procedures in some cases.
The dose of factor VIII was calculated as: Factor VIII (IU) required = body weight (in kg) × desired factor VIII increase (% normal) × 0.5. The therapy protocol is in Table 1.15
Table 1.
On-demand therapy protocol for PWHA.
| Type of bleed | Desired increase | Duration of therapy |
|---|---|---|
| Minor hemorrhage (superficial, early hemorrhages, hemorrhages into joints) | Therapeutically necessary plasma level of FVIII activity is 20–40% of normal | Repeated every 12–24 h as necessary until resolved |
| Moderate (bleeding into muscles, mild head trauma, bleeding into the oral cavity) | Therapeutically necessary plasma level of FVIII activity is 30–60% of normal | Repeated every 12–24 h for 3–4 days or until adequate local hemostasis is achieved |
| Major (gastrointestinal bleeding, intracranial, intraabdominal or intrathoracic bleeding, fractures) | Therapeutically necessary plasma level of FVIII activity is 60–100% of normal | Repeated every 8–24 h until bleeding is resolved, or in the case of surgery, until adequate local hemostasis and wound healing are achieved |
PWHA, patients with hemophilia A.
Collection of samples: Peripheral blood was collected from PWHA in 3.2% sodium citrate in blood collection tubes (Becton & Dickinson, NJ, USA) in a ratio of 9:1. Samples were processed within 1 h of collection for plasma separation. Plasma was separated from the cellular fraction by centrifugation at 4000 RPM for 10 min at 4°C and stored at −80°C until further testing.
Pooled normal plasma (PNP) was prepared by an identical protocol following collection of blood from 20 normal (10 male/10 female) volunteers. Each individual platelet-poor plasma sample was screened [activated partial thromboplastin time (APTT)/prothrombin time (PTs)] prior to pooling to provide a given lot of PNP.
Phenotype analysis: Phenotype of the PWHA was assessed by their bleeding profiles and factor VIII activity. The PWHA were categorized into severe, moderate and mild on the basis of factor VIII levels of <1%, 1–5%, >5–30% respectively.
Lab investigations: Investigations performed included complete blood count, platelet count (Sysmex XN-1000, Japan), PT, international normalized ratio (INR), APTT, mixing studies, factor VIII bioassay, inhibitor screening and vWF antigen assay. The vWF antigen assay was performed by enzyme-linked immunosorbent assay usinga vWF assay kit (Diagnostic Stago, France). PT and APTT were performed as per the manufacturer’s protocols on Diagnostic StagoSTart 4 Coagulation Analyzer using Diagnostic Stago reagents (Diagnostic Stago, S.A.S, France).
Correction test: A mixing study was performed in samples showing a prolonged APTT to determine the cause. This mixing test was performed by mixing 1:1 volume of patient’s plasma with the PNP. If the APTT corrected by more than 50% of the difference between the clotting times of the PNP and test plasma, a factor deficiency was indicated.16
Factor VIII assay: Standard one stage assay for factor VIII based on ability of dilutions of patient’s plasma and standard plasma to correct the APTT of known factor VIII-deficient plasma was done. Dilutions of 1/10, 1/20, 1/40, 1/80 of standard plasma were prepared in imidazole buffer. Calibration curves were drawn using values obtained from normal reference plasma (STA-Unicalibrator, Diagnostic Stago). The factor VIII level for the test plasma was derived from the calibration curve.
Inhibitor assay: An inhibitor assay was performed in patients with prolonged APTT when a mixing study after 2 h incubation at 37°C showed poor correction.16 Inhibitor titers in PWHA were quantified by classical Bethesda assay (CBA) and Nijmegen modified Bethesda assay (NBA) in an attempt to quantify low titer inhibitors.17,18 In the Bethesda assay, a unit is defined as the amount of inhibitor that will neutralize 50% of 1 unit of factor VIII in normal plasma after 2 h of incubation at 37°C. Dilutions of test plasma were incubated with an equal volume of the PNP at 37°C and residual factor VIII was assayed. The normal plasma pool was taken to represent 1 unit of factor VIII. An equal volume of normal plasma mixed with buffer was taken to represent the 100% value and was incubated for 2 h and the residual factor VIII was assayed. Inhibitor strength was calculated from a standard table of residual factor VIII activity versus inhibitor units.
Factor VIII activity (control)
Statistical analysis: The results have been presented as frequency, percentage and mean ± standard deviation. The Chi-square test was used to compare categorical variables. A p-value <0.05 was considered significant. All the analyses were carried out on SPSS version 16.0 (IBM Chicago Inc, USA).
Results
Patient demographics: A total of 300 PWHA were included in the study. The mean age was 18.54 ± 10.39 years (range: 2–60, median: 16 years) and mean age at onset of bleeding was 3.23 ± 1.28 years (range: 0–10, median: 4 years). Analysis of pedigree profile was done in all PWHA and 195 out of 300 PWHA (65%) had a family history of bleeding. Severe hemophilia A was present in 219 (73%) PWHA while 62 (20.67%) had moderate and 19 (6.34%) had mild hemophilia A. The demographic and clinicopathological parameters of the study group are detailed in Table 2.
Table 2.
Clinicopathological characteristics of study population.
| Clinicopathological parameters | Severity of disease |
||
|---|---|---|---|
| Severe (factor VIII <1%) (n = 219) |
Moderate (factor VIII 1–5%) (n = 62) |
Mild (factor VIII >5%) (n = 19) |
|
| Mean age of PWHA | 18.79 ± 12.92 Range: 2–58 years |
18.93 ± 10.28 Range: 3–35 years |
17.83 ± 9.79 Range: 6–45 years |
| Mean age at onset | 1.98 ± 1.5 Range: 6 days–7 years |
5.9 ± 3.23 Range: 1–14 years |
9.2 ± 5.23 Range: 1–17 years |
| Family history of bleeding present | 173 | 14 | 8 |
| Mean factor VIII levels (%) | 0.64 ± 0.19 Range: 0.1–0.9 |
2.36 ± 0.94 Range: 1.1–4.9 |
8.99 ± 5.29 Range: 8.55–24.0 |
| Mean annual factor intake (IU/kg/year) | 2329.74 ± 1826.93 | 1598.66 ± 1028.62 | 1097.64 ± 527.99 |
| Mean age at first transfusion | 4.92 ± 3.87 Range: 16 days–17 years |
7.23 ± 5.44 Range: 1–18 years |
8.13 ± 5.42 Range: 3–22 years |
PWHA, patients with hemophilia A.
Inhibitors in PWHA: A total of 29 of the 300 PWHA showed inhibitors and the overall prevalence of factor VIII inhibitors was 9.67%. Inhibitors were seen to develop only in the severe phenotype of PWHA [Figure 1(a)]. The mean level of inhibitors was 22.3 BU/ml (Range 0.8–108.8 BU/ml). A total of 17 of the 29 inhibitor-positive PWHA had high titers (>5 BU/ml) which were detected by CBA. Overall, 12 PWHA had a low titer of inhibitors (⩽5 BU/ml), of which 6 were detectable only by NBA while 6 were detected by CBA (Figure 2).
Figure 1.

Comparison of inhibitor-positive versus inhibitor-negative patients with reference to: (a) phenotype of patients with hemophilia A (b) blood products and factor administered, and (c) city-level distribution in relation to the annual factor intake.
Figure 2.

Figure showing distribution of levels of inhibitors in PWHA; six patients show black bars, where the inhibitor was detected by Nijmigen modified bethesda asssay; others were detected by classical Bethesda assay.
Clinicopathological parameters and development of inhibitors: Table 3 compares the clinicopathological parameters in inhibitor-positive versus negative PWHA in the full study group (n = 300) as well as in the severe subgroup (n = 219). The following parameters were analyzed:
Table 3.
Comparison of clinicopathological parameters in inhibitor-positive versus negative PWHA.
| S. no. | Clinicopathological parameters | Total PWHA (n =
300) |
Severe PWHA (n =
219) |
||||
|---|---|---|---|---|---|---|---|
| Inhibitor positive (n = 29) | Inhibitor negative (n = 271) |
p-value | Inhibitor positive (n = 29) |
Inhibitor negative (n = 190) |
p-value | ||
| 1. | Mean age of patients at the time of study (years) | 18.72 ± 10.17 | 17.85 ± 10.37 | 0.66 | 18.72 ± 10.17 | 17.38 ± 10.62 | 0.506 |
| 2. | Mean factor VIII level (%) | 0.64 ± 0.19 | 1.35 ± 2.18 | 0.03* | 0.64 ± 0.19 | 0.68 ± 0.17 | 0.277 |
| 3. | Mean age at bleeding onset (years) | 1.00 ± 0.82 | 3.46 ± 3.05 | 0.203 | 1.00 ± 0.82 | 2.71 ± 3.78 | 0.328 |
| 4. | Presence of family history | 17 (55.6%) | 178 (70.3%) | 0.453 | 17 (55.6%) | 124 (65.3%) | 0.48 |
| 5. | Type of treatment | ||||||
| Factor VIII only | 13 (44.8%) | 108 (39.9 %) | 0.109 | 13 (44.8%) | 77 (40.5%) | 0.186 | |
| Both factor VIII and blood/blood products | 16 (55.2%) | 136 (50.2%) | 16 (55.2%) | 93 (48.9%) | |||
| Blood/blood products only | 0 (0%) | 27 (10%) | 0 (0%) | 20 (10.5%) | |||
| 6. | Average factor VIII treatment courses/year | ||||||
| 1–5/year | 0 (0%) | 146 (55.0%) | 0.002* | 0 (0%) | 94 (49.5%) | <0.001* | |
| 6–10/year | 17 (55.6%) | 79 (27.5%) | 17 (55.6%) | 54 (28.4%) | |||
| >10/year | 12 (44.4%) | 46 (17.5 %) | 12 (44.4%) | 42 (22.1%) | |||
| 7. | Factor VIII replacement therapy | ||||||
| (a) | Mean age at 1st replacement therapy (years) | 2.08 ± 0.83 | 6.17 ± 3.45 | 0.0055* | 2.08 ± 0.83 | 4.24 ± 5.27 | 0.017* |
| (b) | Age at 1st replacement therapy | ||||||
| ⩽5 years | 26 (89.66%) | 178 (65.68%) | 0.001* | 26 (89.66%) | 107 (56.3%) | 0.019* | |
| >5 years | 3 (10.34%) | 93(34.0%) | 3 (10.34%) | 83 (43.7%) | |||
| 8. | Annual factor intake (IU/kg/year) | 4353.45 ± 3713.82 |
1636.35 ± 1352.29 |
0.0001* | 4353.45± 3713.82 |
2179.6 ± 1835.5 | 0.025* |
Applied unpaired Student’s t test for significance of scale/continuous data and Chi-square test for categorical data. *Significant (p-value <0.05).
PWHA, patients with hemophilia A.
Mean factor VIII level (%)
The mean factor VIII level (%) of the full study group was 3.99 ± 2.59 while in the severe subgroup it was 0.64 ± 0.19. Inhibitor-positive PWHA had a mean factor VIII level of 0.64 ± 0.19 which when compared with the full study group was significantly lower (p = 0.03) however with reference to the severe subgroup no significant difference in mean factor VIII levels was observed.
Mean age of onset of bleeding
Mean age of onset of bleeding for the full study group was 5.69 ± 3.73 years while in the severe subgroup it was 1.98 ± 1.5 years. Inhibitor-positive PWHA had a mean age of onset of bleeding of 1.00 ± 0.82 years which when compared with the full study group as well as the severe subgroup did not show any significant difference.
Family history and pedigree analysis
Investigation of the family history and pedigree analysis in the full study group revealed a positive family history in 195/300 patients (65%) while in the severe subgroup it was 141/219 (64.3%). Inhibitor-positive PWHA had a family history in 17/29 (58.6%) patients which when compared with the full study group as well as the severe subgroup did not show any significant difference.
Type of treatment
The patients were treated with plasma-derived factor VIII when available and also with blood products including cryoprecipitate or fresh frozen plasma. A total of 121 patients were treated with factor VIII only, 152 with both blood products and factor VIII, and 27 received blood products only. Table 3 compares the type of treatment given to the full study group and to the severe PWHA in inhibitor-positive versus negative patients. With relationship to type of treatment, inhibitors were seen to develop in PWHA taking plasma-derived factor VIII replacement therapy, whereas PWHA taking only blood products did not develop inhibitors [Figure 1(b), Table 3].
Average number of treatments administered
PWHA were grouped on the basis of mean number of treatment courses given annually and were categorized into PWHA receiving 1–5, 6–10 and >10 courses per year. The average number of treatment courses, in other words the frequency of treatment course given to the PWHA significantly correlated with the development of inhibitors both in the full study (p = 0.02) group and in the severe subgroup (p = 0.001; Figure 3, Table 3).
Figure 3.
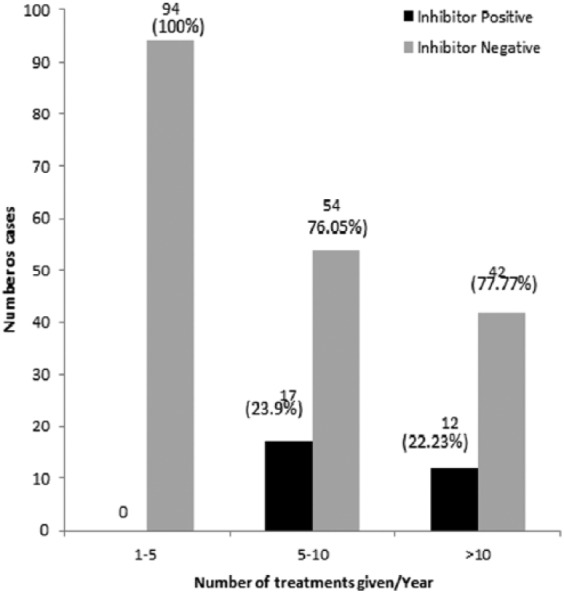
Comparison of frequency of average number of bleeds per year in inhibitor-positive and inhibitor-negative severe patients.
Mean age at first exposure to factor VIII
The mean age at exposure to factor VIII for the full study group was 6.17 ± 3.45 years while in the severe subgroup it was 4.24 ± 5.27 years. Inhibitor-positive PWHA had a mean age of 2.08 ± 0.83 years at the time of exposure to factor VIII which when compared with the full study group, as well as the severe subgroup, was significantly lower (p = 0.0055 and p = 0.017).
Age at first factor VIII replacement therapy
PWHA with and without inhibitors were further categorized into age groups above and below 5 years. Development of inhibitors correlated significantly with exposure at <5 years of age in both the full group (p = 0.001) and the severe subtype (p = 0.019; Table 3).
City-level distribution of factor VIII intake and development of inhibitors
Samples were collected from PWHA from five cities in which three cities (A, B, D) had a well-used free government supply of factor VIII for on-demand therapy to resident PWHA, while city C and E had poor availability of factor VIII. It was interesting to note on analysis that the prevalence of inhibitors was higher in city A, B and D at 10.2%, 12% and 11.55% respectively and lower at 4.87% and 5.88% in city C and E [Figure 2(c)]. The mean factor consumption in city A, B, C, D and E was 2818.27, 2925, 1129.2, 1921 and1518 (IU/kg/year) respectively. The annual mean factor intake in inhibitor-positive PWHA was found to be significantly higher at 4353.45 ± 3713.82 IU/kg/year, as compared with a lower annual factor intake in the full study group at 1636.35 ± 1352.29 IU/kg/year (p = 0.0001) and 2179.6 + 1835.5 IU/kg/year in the severe subgroup (p = 0.025).
Bleeding manifestations
The frequency of bleeding manifestations is shown in Table 4. Bleeding in joints and ecchymoses was the most common manifestation found in the study population whereas when inhibitor-positive and negative PWHA were compared, central nervous system (CNS) bleeds and hematuria were found to be significantly higher in inhibitor-positive PWHA (p = 0.0001 and p = 0.001 respectively). Subanalysis of bleeding manifestations in severe PWHA also showed significant bleeding in inhibitor-positive PWHA in the CNS as well as the occurrence of hematuria (p = 0.003 and p = 0.001 respectively).
Table 4.
Bleeding manifestations in inhibitor-positive and negative patients.
| Clinical symptoms | Total PWHA
(n=300) |
Severe PWHA
(n=219) |
||||
|---|---|---|---|---|---|---|
| Inhibitor positive (n = 29) |
Inhibitor negative (n = 271) |
p-value | Inhibitor positive (n = 29) |
Inhibitor negative (n = 190) |
p-value | |
| Hemarthrosis | ||||||
| Present | 27 (93.1%) | 242 (89.3%) | 0.52b | 27 (93.1%) | 172 (89.47%) | 0.918b |
| Absent | 2 (6.9%) | 29 (10.7%) | 2 (6.9%) | 18(10.53%) | ||
| Gum bleed | ||||||
| Present | 17 (58.6%) | 113 (41.7%) | 0.08b | 17 (58.6%) | 69 (36.3%) | 0.369b |
| Absent | 12 (41.4%) | 158 (58.3%) | 12 (41.4%) | 121 (63.7%) | ||
| CNS bleed | ||||||
| Present | 6 (20.7%) | 8 (3.0%) | 0.0001*b | 6 (20.7%) | 8 (4.2%) | 0.003*b |
| Absent | 23 (79.3%) | 263 (97.0%) | 23 (79.3%) | 182 (95.8%) | ||
| Epistaxis | ||||||
| Present | 10 (34.5%) | 64 (23.6%) | 0.19b | 10 (34.5%) | 43 (22.6%) | 0.248b |
| Absent | 19 (65.5%) | 207 (76.4%) | 19 (65.5%) | 147 (77.4%) | ||
| GI bleed | ||||||
| Present | 6 (20.7%) | 44 (16.2%) | 0.54b | 6 (20.7%) | 38 (20%) | 0.931b |
| Absent | 23 (79.3%) | 227 (83.8%) | 23 (79.3%) | 152 (80%) | ||
| Hematuria | ||||||
| Present | 14 (48.3%) | 55 (20.3%) | 0.001*b | 14 (48.3%) | 38 (20%) | 0.001*b |
| Absent | 15 (51.7%) | 216 (79.7%) | 15 (51.7%) | 152 (80%) | ||
| Melena | ||||||
| Present | 9 (31.0%) | 56 (20.7%) | 0.20b | 9 (31.0%) | 39 (20.5%) | 0.301b |
| Absent | 20 (69.0%) | 214 (79.3%) | 20 (69.0%) | 151 (79.5%) | ||
| Ecchymoses | ||||||
| Present | 15 (51.7%) | 156 (57.6%) | 0.54b | 15 (51.7%) | 107 (56.3%) | 0.792b |
| Absent | 14 (48.3%) | 115 (42.4%) | 14 (48.3%) | 83 (43.7%) | ||
Chi-square//Fisher’s exact test; *significant (p-value <0.05).
CNS, central nervous system; GI, gastrointestinal; PWHA, patients with hemophilia A.
Risk factors associated with inhibitor development: The various nongenetic risk factors associated with inhibitor development were analyzed. Intense treatment episodes were defined as administration of plasma-derived factor VIII at required doses for 3 consecutive days following trauma or spontaneous bleed. PWHA undergoing intense treatment episodes as well as those undergoing surgery were found to have significantly higher risk of developing inhibitors (p = 0.05 and p = 0.02 respectively). These risks were also evident in the analysis of the severe subgroup (p = 0.04 and p = 0.02; Table 5).
Table 5.
Analysis of risk factors associated with inhibitor development.
| Risk factors | Total PWHA (n =
300) |
Severe PWHA (n =
219) |
||||
|---|---|---|---|---|---|---|
| Inhibitor positive | Inhibitor negative | p-value | Inhibitor positive | Inhibitor negative | p-value | |
| Intense treatment (n = 19) | 5/29 (17.2%) | 14/271 (5.1%) | 0.05*b | 5/29 (17.2%) | 14/219 (7.36%) | 0.04* |
| Vaccination in PWHA (⩽5 years age near factor VIII replacement therapy) (n = 19) | 1/9 (11.12%) | 2/10 (20%) | 1.000b | 1/9 (11.12%) | 2/10 (20%) | 1.000b |
| Surgery (n = 10) | 3/29 (10.34%) | 7/271 (2.5%) | 0.02*b | 3/29 (10.34%) | 7/219 (3.19%) | 0.02*b |
| Immune disorders (n = 0) | 0/29 (0%) | 0/29 (0%) | – | 0/29 (0%) | 0/219 (0%) | – |
| Malignancy (n = 1) | 1/29 (3.45%) | 0/271 (0%) | – | 1/29 (3.45%) | 0/219 (0%) | – |
Chi-square/Fisher’s exact test; *significant (p-value <0.05).
PWHA, patients with hemophilia A.
Association of blood groups with inhibitor development: Blood group B was present in 100/300 (33.34%) patients in the full study group of which 98 were inhibitor-negative and 2 were inhibitor-positive. In the severe subgroup, blood group B was present in 69/219 (31.5%) PWHA. In the inhibitor-positive PWHA, blood group A was present in 19/29 (65.5%). The association of inhibitor development with blood group A was statistically significant (p = 0.005; Table 6).
Table 6.
(a): ABO blood group and inhibitor development. (b) Statistical significance between ABO blood groups.
(a).
| Parameters | ABO blood group |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Inhibitor positive
(n = 29) |
Inhibitor negative
(n = 271) |
||||||||
| O (n = 6) |
A (n = 19) |
B (n = 2) |
AB (n = 2) |
O (n = 76) |
A (n = 54) |
B (n = 98) |
AB (n = 43) |
||
| Inhibitor titer | Low (mean titer) |
3 (2.05) |
9 (2.9) |
0 | 0 | – | – | – | – |
| High (mean titer) |
3 (28.8) |
10 (30.16) |
2 (6.4) |
2 (35.2) |
– | – | – | – | |
| Phenotype | Severe (219) | 6 | 19 | 2 | 2 | 57 | 34 | 69 | 30 |
| Moderate +Mild (81) | – | – | – | – | 19 | 20 | 29 | 13 | |
| Mean dose (IU/kg/year) | 4083.3 | 3652.6 | 5250 | 4972.2 | 1768.7 | 1569.4 | 1919.4 | 1207.1 | |
(b).
| ABO blood groups | p-value1 |
|---|---|
| O versus A | 0.02* |
| O versus B | 0.12 |
| O versus AB | 0.52 |
| A versus B | 0.005* |
| A versus AB | 0.49 |
| B versus AB | 0.05 |
Chi-square test, *Significant.
Discussion
In the current case series of 300 PWHA, we have analyzed clinicopathological parameters associated with inhibitor development. The study is novel in the estimation of inhibitor occurrence in a study population which has received plasma-derived factor VIII on demand. Since the factor VIII supplied was through a government supply system, all PWHA received the same plasma-derived factor VIII and hence were a homogenous group with respect to the type of factor used. Most studies in literature describe the occurrence of inhibitors in PWHA on prophylactic therapy. Our study aimed at estimating the prevalence of inhibitors and evaluating various risk factors associated with the development of inhibitors in PWHA taking on-demand therapy in the North Indian population. It may be interesting to note here that the North Indian population is genetically closer to Eurasians and Central Asians than populations of other geographical regions of India.19,20 In terms of the prevalence of hemophilia A, no significant difference exists between the various states in India.21 In contrast with other studies on inhibitor development which have been performed in developed countries where the recombinant factor VIII was also available to the patients, our patients received only plasma-derived factor VIII. This is an important difference as Peyvandi and Mannucci and colleagues in the SIPPET (survey of inhibitors in plasma-product exposed toddlers) study22,23 concluded that patients treated with plasma-derived factor VIII which contains vWF had a lower incidence of inhibitors than those treated with recombinant factor VIII. Peyvandi and colleagues22 and Mannucci and colleagues23 speculated that epitope masking by vWF causes the protection of the factor VIII molecule from endocytosis by antigen-presenting cells, thereby preventing immunologic response against it in a fraction of patients. The plasma-derived factor used in our study was not enriched with von Willebrand factor.
Etiology of development of inhibitors is multifactorial and includes environmental and genetic factors. In the present research, we have studied in addition to conventional clinicopathological parameters, the effect of age at start of therapy, type of replacement therapy and the annual factor consumption. A total of 300 PWHA from the North Indian population were included in the study and an inhibitor prevalence of 9.67% was observed. A detailed analysis of correlates of inhibitor development in PWHA receiving on-demand therapy schedules may contribute to generating novel information. We have adhered to the guidelines for reporting inhibitors in manuscripts recommended by Iorio and colleagues.24 All PWHA in our study who developed inhibitors had a severe deficiency of factor VIII. In a systematic review Wight and colleagues have reported an overall inhibitor prevalence of 5–7% which when limited to patients with severe disease was much higher at 12–13%.10 We compared the age at first exposure to factor concentrates and observed that inhibitor development occurred in PWHA where the age at first exposure was <5 years (p = 0.0055). This parameter was also found to be significant when the analysis was done in the severe subgroup alone. Earlier studies have suggested that a higher risk of inhibitor development is associated with younger age of commencement of replacement therapy as compared with exposure at later age;6–9,25–30 however, this finding has not been consistent in all studies. Our study confirms a significant association of early age at exposure to plasma-derived factor VIII in PWHA receiving on-demand therapy. This association may be explained on the basis of the fact that immune responsiveness of children at a younger age is substantial and the development of the immune system continues through the first 5 years of life. It was also observed in our study that when the age at first exposure was <5 years a lower inhibitor titer is present (14.1 BU/ml) as compared with a higher titer when the age at exposure was >5 years (42.6 BU/ml). The increasing titers with age could also be related to multiple exposures to plasma-derived factor VIII.
The intensity and duration of factor VIII treatment covers a wide spectrum of clinical practices, ranging from single prophylactic infusions of factor VIII concentrate 2–3 times a week to frequent administration of high doses of factor VIII for several consecutive days in the case of major bleeding or surgical procedures. A number of studies have investigated the possible link between intensity (high dosage or prolonged) of treatment and increased inhibitor risk.25 It has been established in some studies that factor intake and intensity of treatment is an important risk factor for inhibitor development. We have also observed a significant increase in inhibitors in PWHA who received intense therapy. Some studies have demonstrated an increased rate of inhibitor formation after intensive treatment (i.e. surgical procedure or high-frequency treatment);20,28–30 however, a case-control study by Santagostino and colleagues did not confirm these findings.9
The mean annual factor intake in inhibitor-positive PWHA in our study was higher at 4353.45 ± 3713.82 IU/kg/year as compared with a lower annual factor intake of 1636.35 ± 1352.29 IU/kg/year in inhibitor-negative PWHA. This difference was statistically significant (p < 0.001), indicating that inhibitor development is linked to a higher factor intake in the population as a whole. This finding was further corroborated in our study by an interesting comparative study of the percentage of inhibitor-positive PWHA in different cities from where the samples were collected. Our study included PWHA from same ethnic region of North India (city A, B, C, D and E). Since 2009 the state government has started financing factors, making them freely available at some centers. After initial distribution and information difficulties, some regions successfully established a good availability of on-demand therapy for PWHA while others were not able to efficiently ensure a supply chain for factor VIII for PWHA. In our study the higher prevalence of development of factor VIII inhibitors in city A, B and D was directly proportional to the availability of plasma-derived factor VIII.
In our study, inhibitors to factor VIII developed in 29 PWHA of which all patients were exposed to factor VIII concentrates and 16 had also taken blood products. A small group of PWHA who had taken only blood products did not develop inhibitors. Further we also observed that inhibitor formation is associated with high intensity exposure to factor VIII protein. The CANAL cohort study addresses treatment-related risk factors of inhibitor development in previously untreated patients with hemophilia A. The authors conclude that treatment increases the risk of inhibitor formation and this may be explained by the intensity of treatment. Peak treatment moments may trigger inhibitor formation and have been considered the most significant determinant of inhibitor development.29 In a study, Kurnik and colleagues observed that standard prophylaxis started at or after the first joint or other severe bleed, led to the formation of inhibitors in 47% of patients, compared with only 3.8% in patients given a low-dose prophylactic regimen started at manifested bleeding tendency, with no long or intensive treatment.31 The presence of severe bleeds, trauma, surgery are associated with the use of high-dose factor VIII or prolonged treatment which leads to up-regulation of the cellular T and B-cell lymphocyte response and an increased risk of inhibitor development. In contrast, a lower dose of factor VIII protein with regular prophylaxis carries a decreased risk of inhibitor development.29
During data analysis we observed that blood group A was most common in inhibitor-positive PWHA whereas in the study group as a whole the ABO subtype B was most frequent. This association of blood group A with inhibitor development needs validation in larger studies from different geographic regions. In a recent multivariate logistic regression analysis by Franchini and colleagues, blood group O was shown to have a lower (approximately twofold) inhibitor risk, but the sample size was too small for conclusive results and further the patients were confined to the Italian population.32 In our study, the proportion of inhibitor-positive PWHA was higher in blood group A (n = 19/29); however, the distribution of titers did not vary across blood groups. The proportion of PWHA with high and low titers is shown in relation to blood group type in Table 6(a). Further, within the blood groups, variation in dose of factor VIII replacement was not statistically significant, hence dose effect was not a confounder. Development of inhibitors in our study group was also found to correlate with the availability of factor VIII concentrates in the studied region. In a study conducted by Pinto and colleagues in Indian PWHA between 2011 and 2013, the overall prevalence reported in India was 6.07% with the highest prevalence of 13.04% in South India. In North India, the percentage of inhibitor-positive PWHA was relatively low.33 During the past few decades the supply of factor VIII has been erratic and had a prohibitive cost, hence many PWHA did not have access to factor concentrates. With the detected increasing incidence of inhibitors in our study we feel that there is an urgent need for development of laboratories which are better equipped to detect and diagnose development of factor VIII inhibitors so that these PWHA can be managed well.
Evaluation of test methods for inhibitor development is difficult, because there is no gold standard against which to compare them. In practice, both laboratory and clinical evidence is used to determine whether a patient has an inhibitor. The inhibitor-negative population is defined largely by the screening test, although non-neutralizing antibodies that are not detected in clot-based assays may be suspected in some patients.34 Quantification of inhibitor titers is important for deciding the therapy for PWHA. The Bethesda assay, which is the most frequently used method, lacks sensitivity especially in the lower titer range resulting in unreliable data. In our study those samples which were found to be inhibitor-positive after the screening assay were confirmed for presence of inhibitors and their titers quantified by the Bethesda assay. In cases with suspected low titers the Nijmegen modification was used. This modification involves replacement of the imidazole buffer in the control mixture by immunodepleted factor VIII-deficient plasma and allows better discrimination between positive and negative samples with improvement in reliability. The quantification of inhibitor titers by CBA and NBA went through rigorous standardization processes and results were validated by external quality assurance. Association with factor VIII replacement therapy near the time of vaccination, presence of comorbid conditions like immune disorders and malignancy was attempted. However, the number of PWHA with these associations was low or nil and conclusions could not be derived.
The current study has comprehensively evaluated clinicopathological parameters and risk factors involved in the development of inhibitors in PWHA on-demand therapy in a large cohort with interesting results. It is paradoxical that while the availability of factor VIII has helped PWHA overcome bleeding complications, it has introduced this new complication in therapy with development of inhibitors in one tenth of patients, a number which is fast growing.
Supplemental Material
Supplemental material, Supplementary_Material for Clinicopathological parameters influencing inhibitor development in patients with hemophilia A receiving on-demand therapy by Sanya Arshad, Anshima Singh, Namrata Punit Awasthi, Swati Kumari and Nuzhat Husain in Therapeutic Advances in Hematology
Footnotes
Funding: Authors wish to acknowledge Department of Science and Technology (DST), New Delhi, India for providing SERB grant support (Ref. no SB/SO/HS/025/2014).
Conflict of interest statement: The authors declare that there is no conflict of interest.
Supplementary material: Supplementary material for this article is available online.
Contributor Information
Sanya Arshad, Dr. Ram Manohar Lohia Institute of Medical Sciences, Lucknow, Uttar Pradesh, India.
Anshima Singh, Dr. Ram Manohar Lohia Institute of Medical Sciences, Lucknow, Uttar Pradesh, India.
Namrata Punit Awasthi, Department of Pathology, Dr. Ram Manohar Lohia Institute of Medical Sciences, Vibhuti Khand, Gomti Nagar, Lucknow 226010, India.
Swati Kumari, Dr. Ram Manohar Lohia Institute of Medical Sciences, Lucknow, Uttar Pradesh, India.
Nuzhat Husain, Dr. Ram Manohar Lohia Institute of Medical Sciences, Lucknow, Uttar Pradesh, India.
References
- 1. Soucie J, Evatt B, Jackson D. Occurrence of hemophilia in the United States. The Hemophilia Surveillance System Project Investigators. Am J Hematol 1998; 59: 288–294. [DOI] [PubMed] [Google Scholar]
- 2. Richards M, Williams M, Chalmers E, et al. ; Paediatric Working Party of the United Kingdom Haemophilia Doctors’ Organisation. A United Kingdom Hemophilia Centre Doctors’ Organization guideline approved by the British Committee for Standards in Haematology: guideline on the use of prophylactic factor VIII concentrate in children and adults with severe hemophilia A. Br J Haematol 2010; 149: 498–507. [DOI] [PubMed] [Google Scholar]
- 3. Di Minno M, Di Minno G, Di Capua M, et al. Cost of care of hemophilia with inhibitors. Hemophilia 2010; 16: e190–e201. [DOI] [PubMed] [Google Scholar]
- 4. Astermark J, Santagostino E, Keith Hoots W. Clinical issues in inhibitors. Haemophilia 2010; 16(Suppl. 5): 54–60. [DOI] [PubMed] [Google Scholar]
- 5. Schwaab R, Brackmann HH, Meyer C, et al. Hemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost 1995; 74: 1402–1406. [PubMed] [Google Scholar]
- 6. Goudemand J, Rothschild C, Demiguel V, et al. Influence of the type of factor VIII concentrate on the incidence of factor VIII inhibitors in previously untreated patients with severe hemophilia A. Blood 2006; 107: 46–51. [DOI] [PubMed] [Google Scholar]
- 7. Lorenzo JI, Lopez A, Altisent C, Aznar JA. Incidence of factor VIII inhibitors in severe hemophilia: the importance of patient age. Br J Haematol 2001; 113: 600–603. [DOI] [PubMed] [Google Scholar]
- 8. van der Bom JG, Mauser-Bunschoten EP, Fischer K, et al. Age at first treatment and immune tolerance to factor VIII in severe hemophilia. Thromb Haemost 2003; 89: 475–479. [PubMed] [Google Scholar]
- 9. Santagostino E, Mancuso ME, Rocino A, et al. Environmental risk factors for inhibitor development in children with hemophilia A: a case-control study. Br J Haematol 2005; 130: 422–427. [DOI] [PubMed] [Google Scholar]
- 10. Wight J, Paisley S. The epidemiology of inhibitors in hemophilia A: a systematic review. Hemophilia 2003; 9: 418–435. [DOI] [PubMed] [Google Scholar]
- 11. Eckhardt CL, van Velzen AS, Peters M, et al. Factor VIII gene (F8) mutation and risk of inhibitor development in nonsevere hemophilia A. Blood 2013; 122: 1954–1962. [DOI] [PubMed] [Google Scholar]
- 12. Ghosh K, Shetty S, Kulkarni B, et al. Development of inhibitors in patients with hemophilia from India. Hemophilia 2001; 7: 273–278. [DOI] [PubMed] [Google Scholar]
- 13. Mathews V, Nair SC, David S, et al. Management of hemophilia in patients with inhibitors: the perspective from developing countries. Semin Thromb Hemost 2009; 35: 820–826. [DOI] [PubMed] [Google Scholar]
- 14. Ghosh K, Shetty S. Immune response to FVIII in hemophilia A: an overview of risk factors. Clin Rev Allergy Immunol 2009; 37: 58–66. [DOI] [PubMed] [Google Scholar]
- 15. Treatment Guidelines Working Group. Guidelines for the management of hemophilia. 2nd ed. Montreal, Quebec: World Federation of Hemophilia, 2012. [Google Scholar]
- 16. Kitchen S, McCraw A, Echenagucia M. Diagnosis of hemophilia and other bleeding disorders. 2nd ed. Quebec: World Federation of Hemophilia, 2010, p. 41. [Google Scholar]
- 17. Kasper CK, Aledort LM, Counts RB, et al. A more uniform measurment of factor VIII inhibitors. Thromb Diath Haemorrh 1975; 34: 869–872. [PubMed] [Google Scholar]
- 18. Verbruggen B, Novakova I, Wessels H, et al. The Nijmegen modification of the Bethesda assay for factor VIII:C inhibitors: improved specificity and reliability. Thromb Haemost 1995; 73: 247–251. [PubMed] [Google Scholar]
- 19. Bamshad M, Kivisild T, Watkins WS, et al. Genetic evidence on the origins of Indian caste populations. Genome Res 2001; 11: 994–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Basu A, Mukherjee N, Roy S, et al. Ethnic India: a genomic view, with special reference to peopling and structure. Genome Res 2003; 13: 2277–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maclean P, Richards M, Williams M, et al. Treatment related factors and inhibitor development in children with severe hemophilia A. Hemophilia 2011; 17: 282–287. [DOI] [PubMed] [Google Scholar]
- 22. Peyvandi F, Mannucci PM, Garagiola I, et al. , A randomized trial of factor VIII and neutralizing antibodies in hemophilia A. N Engl J Med 2016; 374: 2054–2064. [DOI] [PubMed] [Google Scholar]
- 23. Mannucci PM, Shi Q, Bonanad S, et al. Novel investigations on the protective role of the FVIII/VWF complex in inhibitor development. Hemophilia 2014; 20 (Suppl. 6): 2–16. [DOI] [PubMed] [Google Scholar]
- 24. Iorio A, Barbara M, Bernardi, et al. Recommendations for authors of manuscripts reporting Inhibitor cases developed in previously treated patients with hemophilia: communication from the SSC of the ISTH. J Thromb Haemost 2006; 14: 1668–1672. [DOI] [PubMed] [Google Scholar]
- 25. Chalmers EA, Williams MD, Richards M, et al. Early FVIII exposure and subsequent inhibitor development in children with severe hemophilia A. J Thromb Haemost 2005; 3: abstract OR092. [DOI] [PubMed] [Google Scholar]
- 26. Fontes E, Carvalho S, Amorim L. Age at the beginning of factor VIII replacement is not a risk factor for inhibitor development in hemophilia A. Hemophilia 2004; 10(Suppl. 3): abstract 12 OC 14. [Google Scholar]
- 27. Coppola A, Santoro C, Tagliaferri A, et al. Understanding inhibitor development in hemophilia A: towards clinical prediction and prevention strategies. Hemophilia 2010; 16(Suppl. 1): 13–19. [DOI] [PubMed] [Google Scholar]
- 28. Gouw S, van den Berg H, le Cessie S, et al. Treatment characteristics and the risk of inhibitor development: a multicenter cohort study among previously untreated patients with severe hemophilia A. J Thromb Haemost 2007; 5: 1383–1390. [DOI] [PubMed] [Google Scholar]
- 29. Gouw S, van der Bom J, Marijke van den Berg H. Treatment-related risk factors of inhibitor development in previously untreated patients with hemophilia A: the CANAL cohort study. Blood 2007; 109: 4648–4654. [DOI] [PubMed] [Google Scholar]
- 30. Ragni M, Ojeifo O, Feng J, et al. Risk factors for inhibitor formation in hemophilia: a prevalent case-control study. Hemophilia 2009; 15: 1074–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kurnik K, Bidlingmaier C, Engl W, et al. New early prophylaxis regimen that avoids immunological danger signals can reduce FVIII inhibitor development. Haemophilia 2010; 16: 256–262. [DOI] [PubMed] [Google Scholar]
- 32. Franchini M, Coppola A, Mengoli C, et al. Blood group O protects against inhibitor development in severe hemophilia A patients. Semin Thromb Hemost 2017; 43; 69–74. [DOI] [PubMed] [Google Scholar]
- 33. Pinto P, Shelar T, Nawadkar V, et al. The epidemiology of FVIII inhibitors in Indian haemophilia A patients. Indian J Hematol Blood Transfus 2014; 30: 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gilles JG, Amout J, Vermylen J, et al. Anti-factor VIII antibodies of hemophiliac patients are frequently directed towards nonfunctional determinants and do not exhibit isotypic restriction. Blood 1993; 82: 2452–2461. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Supplementary_Material for Clinicopathological parameters influencing inhibitor development in patients with hemophilia A receiving on-demand therapy by Sanya Arshad, Anshima Singh, Namrata Punit Awasthi, Swati Kumari and Nuzhat Husain in Therapeutic Advances in Hematology