

 Di-(2-ethylhexyl) phthalate (DEHP) is widely used as a plasticizer and people are exposed to various amounts on a daily basis.
Di-(2-ethylhexyl) phthalate (DEHP) is widely used as a plasticizer and people are exposed to various amounts on a daily basis.
Abstract
Di-(2-ethylhexyl) phthalate (DEHP) is widely used as a plasticizer and people are exposed to various amounts on a daily basis. This study was designed to evaluate the genotoxic, histologic, immunohistochemical, morphometric and hormonal effects of DEHP (100, 200 and 400 mg kg–1 per day DEHP) administered daily to rats by oral gavage for 28 days. The rats were divided into five groups including oil control, positive control (MMS) and treatment groups (100, 200 and 400 mg kg–1 per day DEHP). They were euthanized at the end of the experiment, organ and body weights were recorded and serum was collected for biochemical and hormone analysis. The genotoxic effect was measured in blood and sperm using the Comet assay. The testes, epididymis, prostate gland and seminal vesicle were collected and stained with hematoxylin and eosin for histopathologic analysis. Epithelial height, luminal and tubular diameters (μM) in seminiferous tubules were also measured. Moreover, the study revealed an increase in the DNA damage level in both blood lymphocytes and sperm. At the end of the experiment, the tail intensity showed a significant increase in the 100 mg kg–1 per day (p = 0.032), 200 mg kg–1 per day (p = 0.019) and 400 mg kg–1 per day (p = 0.012) dose groups compared to the control group in blood. Furthermore, testosterone was decreased in all treatment groups compared to the control group. Besides, DEHP caused a significant decrease in the leukocyte levels (p = 0.017) and hemoglobin content, as well as an increased mean cell volume (MCV) count (p = 0.029) in the 400 mg kg–1 per day group when compared to the control values. It is important to indicate that there were apoptotic cells seen in the lumen of testes in the 200 and 400 mg kg–1 per day dose groups using the Tunel method. Therefore, with this study, it has been illustrated that DEHP caused DNA damage in blood and sperm and concrete negative effects on the reproductive system in rats from the pre-pubertal period to the pubertal period. This is a unique study since there has not been any other study that presents the indicated level of DNA damage while considering the genotoxic, histologic, immunohistochemical, morphometric and hormonal effects of DEHP.
1. Introduction
Phthalate esters (PAEs) are a class of synthetic chemicals that are used for numerous industrial applications (e.g. polyvinyl chloride plasticizers for food contact or medical devices, personal care products, residential construction and automotive industries). Di-(2-ethylhexyl)-phthalate (DEHP) is the most widely used congener. Humans are exposed to these chemicals through transcutaneous absorption, inhalation, medical transfusions and ingestion. Despite their rather rapid turnover, phthalates and their metabolites are consistently detected in human body fluids such as plasma, urine, amniotic fluid and breast milk, thus reflecting substantial and constant exposure.1–5
The potential public health risks associated with phthalate exposure not only include carcinogenesis6 but also metabolic and endocrine disruption. Adverse effects of PAEs on humans depend upon their potential toxicity as well as the levels and exposure period. DEHP is the most commonly used phthalate plasticizer in the production of polyvinyl chloride (PVC). It is suspected to be an endocrine disrupting chemical that exhibits carcinogenic action.7,8 Animal studies provide consistent evidence that certain phthalates target the developing male reproductive system. These effects in animal studies have been termed Phthalate Syndrome (PS), and the effects mirror a set of reproductive symptoms seen in human males, termed Testicular Dysgenesis Syndrome (TDS). TDS and PS include symptoms that arise from insufficient testosterone production during the in utero development, including undescended testes, malformations of the penis, reduced anogenital distance (AGD), decreased sperm motility and mobility, infertility, and testicular cancer.9 Phthalates are thought to cause their toxic effects on male reproductive development by interrupting testosterone and insl3 production in the testes during the sensitive in utero masculinization programing window. The Carcinogen Assessment Group classified DEHP as a probable human carcinogen (group B2).10 The daily exposure level of DEHP in women was estimated to be 41.7 μg kg–1 body toxicity. The value obtained in the present study for DEHP in women was higher than obtained in other studies. Based on these data, hazard indices (HIs) were calculated to be 1.12 (41.7/37 TDI) for women and 0.33 (12.4/37 TDI) for children. DEHP and MEHP levels were higher in women than in children. A higher DEHP exposure level for women may be due to higher food consumption, more air inhaled, or the use of phthalate-containing consumer products. The data suggest that women are exposed to significant levels of DEHP, and indicate that these should be reduced to as low levels as are technologically feasible.11–14
Phthalate levels are generally higher in fatty foods, including dairy products, meats, and vegetable oils. Recent testing of 72 foods purchased from a U.S. supermarket demonstrated that various phthalates were detectable in all classes of food with DEHP being the highest of the phthalates tested in most food categories, particularly in pork, dairy products, vegetable oils and grains, although the sample sizes were limited.15 This is largely consistent with concentrations found in European foods, reviewed in a scenario-based model that found that diet had a major influence on DEHP exposure.16 The sporadic contamination of food was seen in the three U.S. cooking oil samples, which had concentrations of benzyl butyl phthalate (BBzP) of 459, 2.20 and 0.35 nanograms per gram (ng g–1), with the highest value detected in virgin olive oil from a glass container, although the source of phthalates found in that sample was unknown.15 Exposure to phthalates other than DEHP and its substitutes are primarily from non-dietary sources, possibly from consumer goods in the indoor environment and personal care products. A study that paired indoor and outdoor air measurements found that phthalates were the most abundant chemicals in indoor air and house dust, with maximum levels of dibutyl phthalate (DBP) in air of 1.1 microgram per cubic meter (1 μg m–3) and levels in house dust approaching 1 mg g–1.17,18 A Danish study of children aged 3–6 estimated that the dermal absorption of diethyl phthalate (DEP), di-n-butyl phthalate (DnBP) and diisobutyl phthalate (DiBP) was the dominant exposure pathway of these phthalates in the indoor environment. More research on the dermal exposure pathway from ambient indoor air is forthcoming.19 Although there are now some restrictions on phthalates in children's toys in the EU and U.S., high levels have been reported in PVC-based toys, some containing up to 40% DINP or DEHP.20,21 The use of phthalate-containing medical devices can also affect potential exposure. For instance, infants in a neonatal intensive care unit showed elevated urinary levels of DEHP, DBP, and BBzP metabolites, with the highest levels observed in conjunction with the most intensive use of medical devices that contained or came in contact with PVC.22
The typical human exposure to DEHP ranges from 3 to 30 μg kg–1 per day but it can be exceeded in specific medical conditions, reaching 1.5 mg kg–1 per day exposure in hemodialysis patients, or as high as 10–20 mg kg–1 per day during neonatal transfusion or parenteral nutrition.23–25 The mechanisms by which phthalates and specifically DEHP exert their toxic effects in the reproductive system are not yet fully elucidated. Some of the effects of phthalates are related to their anti-androgenic potential.26,27 Sertoli cells and Leydig cells were thought to be the primary targets of phthalate exposure in testes.26,28 Also, DEHP and its metabolites were reported to produce oxidative DNA damage, thereby inducing apoptosis in testicular cells.29,30
Increased germ cell apoptosis is associated with abnormal spermatogenesis in men.31 In rodents, heat and irradiation as well as xenohormones and testis toxicants like DEHP, 25-hexanedione, nitrobenzene, deltamethrin, and hydroxyurea are known inducers of germ cell apoptosis.32–35
DEHP is a well-known peroxisome proliferator, and is regarded as a non-classic type endocrine disruptor, i.e., in contrast to the classical endocrine disrupters that interfere with the endocrine process at the receptor level, altering the reproductive function by affecting hormone synthesis.36,37 Recent data have also shown that phthalates were able to produce free radicals by several pathways in germ cells including the activation of peroxisome proliferator-activated receptor alpha (PPARα), suggesting the possibility that oxidative stress and mitochondrial dysfunction in germ cells may contribute to the phthalate-induced disruption of spermatogenesis.
Genotoxicity includes molecular changes in deoxyribonucleic acid (DNA), such as adduct formation or oxidative damage and biological responses that can be attributed to molecular changes in DNA. Reactive oxygen species (ROS) production and related DNA damage can result in apoptosis.38 The reduction of oxygen by one electron at a time produces relatively stable intermediates. The superoxide anion (O2–), the product of a one-electron reduction of oxygen, is the precursor of most ROS and a mediator in oxidative chain reactions. Dismutation of O2– (either spontaneously or through a reaction catalysed by superoxide dismutases) produces hydrogen peroxide (H2O2), which in turn may be fully reduced to water or partially reduced to the hydroxyl radical (˙OH), one of the strongest oxidants in nature. The formation of ˙OH is catalysed by reduced transition metals, which in turn may be re-reduced by O2–, propagating this process.39 The mitochondrial electron transport chain contains several redox centres that may leak electrons to oxygen, constituting the primary source of O2– in most tissues. In addition, O2– may react with other radicals including nitric oxide (NO) in a reaction controlled by the rate of diffusion of both radicals. The balance of internal signals to protect against apoptosis and those inducing apoptosis are altered with internal damage to the cell. Several forms of sperm DNA damage are caused by ROS (chromatin cross-linking, chromosome deletion, DNA strand breaks and base oxidation) with seminal oxidative stress, sperm DNA damage, and apoptosis interlinked.40 One role of ROS in DEHP treatment was reported to provoke oxidative stress, as measured by increases in ROS in subsequently isolated rat spermatocytes.41 ROS are shown to contribute to cellular damage, apoptosis and cell death, but are also involved in the regulation of gene expression by controlling signal transduction through direct participation in cell signaling, and/or the modulation of cell redox states.41 ROS have also been suspected of being involved in the formation of testicular atrophy in phthalate-exposed rats.42In vivo exposure to a single gavage dose of DEHP (2000 mg kg–1 per day) increased apoptosis in Sprague–Dawley rat testis as indicated by the TUNEL assay, but not by histopathological alteration.43 According to Garaj-Vrhovac and Zeljezic,44 DNA damage, revealed by the Comet assay, could originate from DNA single-strand breaks, the repair of DNA double-strand breaks, DNA adducts, DNA–DNA and DNA–protein cross-links. This assay, also called the single-cell gel electrophoresis assay, is a rapid and sensitive method for the detection of DNA damage in individual cells, induced by a variety of genotoxic agents. A wide range of methods can be used to assess if low-level environmental exposure to (potential) human carcinogens is able to induce the loss of DNA integrity or DNA damage. These include well-established biomarkers such as the alkaline Comet assay on peripheral blood cells or urinary concentrations of 8-hydroxydeoxyguanosine (8-OHdG), both reflecting short-term (days) DNA damage, and the micronucleus test in peripheral blood lymphocytes as a measure of long-term damage.45,46 DEHP-induced developmental toxicity, endocrine disruption, and testicular targets have led to several studies that have implications for cancer. The epigenome is particularly susceptible to deregulation during gestation, neonatal development, puberty, and old age. It is most vulnerable to environmental factors during embryogenesis because the DNA synthetic rate is high, and the elaborate DNA methylation pattern and chromatin structure required for normal tissue development is established during early development.47 Similarly, there is mounting evidence that “mutagenic” as well as “non-mutagenic” carcinogens have greater potency from early life exposures compared to exposure in the mature organism,48 and there is an increasing appreciation that carcinogens may act by multiple mechanisms, including non-mutagenic ones.49,50
The main purposes of the present study are to evaluate the DEHP influence on the following: weight, relative and absolute organ weight, and the food and water intake of rats; its influence on blood parameters; determine the morphometric parameters in testes and its effects on the genotoxic potential in blood and sperm (with the Comet assay) from the pre-pubertal period to the pubertal period in male rats. Also, this study aims to evaluate apoptosis with DEHP treatments at different concentrations using the terminal deoxynucleotidyl transferase (Tdt) deoxyuridine triphosphate nick end-labeling (TUNEL) in testes tissues of male rats.
There have been many studies on DEHP but there are not enough comprehensive, short-term in vivo animal experiments on this phthalate. Children are more sensitive to the toxicants compared to adults and among the published papers about DEHP, there are no studies in the literature related to the impact of DEHP from the prepubertal to the pubertal stage. This study was designed for the pre-pubertal period to the pubertal period and the results reflect the adverse effects in this period.
2. Materials and methods
2.1. Chemicals
Di-(2-ethylhexyl) phthalate (DEHP), CAS No. 117-81-7 EC No 204-211-0, purity 99.7%, was purchased from Sigma-Aldrich. MMS and NaCl were purchased from Sigma-Aldrich. DMSO (CAS 67-68-5), NaOH (CAS 1310-73-2), Tris (CAS 77-86-1), EDTA (CAS 6381-92-6), Triton X-100 (CAS 9002-93-1), low melting agarose (CAS 9012-36-6), normal melting agarose (CAS 9012-36-6), EtBr (CAS 1239-45-8) were obtained from Applichem. PBS (CAS L1825) and Biocoll (CAS L6115) were obtained from Biochrom AG. The ApopTag PlusPeroxidase In Situ Apoptosis Detection Kit was purchased from Chemicon International, Inc., Temecula, California, USA. The testosterone hormone kit was purchased from Cayman Chemical (Cayman Chemical, Ann Arbor, MI, USA). The water used to prepare aqueous buffers was deionized and purified using a Milli-Q water purification system (Millipore, Molsheim, France). All other chemicals were of analytical grade and obtained from Sigma-Aldrich, USA.
2.2. Animals and housing
Thirty healthy pre-pubertal male Wistar albino rats (Rattus norvecigus), [6 weeks of age and average body weight of 200–220 g] were used in this study. The rats were obtained from the Experimental Animal Center, University of Hacettepe, Turkey. All experimental procedures and animal use were approved by the Approval of Ethics Committee of Hacettepe University (2013/55-03). They were maintained in a 12 h dark/light cycle in a controlled atmosphere of 22 ± 2 °C and 50–70% humidity. Rats were housed in polycarbonate cages and fed with the standard rat diet and tap water ad libitum. The animals were allowed to acclimate for 7 days before treatment.
2.3. Doses and administration of chemicals
The rats were randomly divided into 5 groups, each consisting of 6 male rats. These groups were oil control, MMS (ethyl methanesulphonate) control, 100, 200 and 400 mg kg–1 per day DEHP treatment groups. The age of the animals and the duration of exposure were used according to the recommendations of the U.S. EPA Endocrine Disrupter Screening and Testing Advisory Committee. The low-dose level was chosen according to the no observed adverse effect level and the high-dose level was also chosen according to the lowest-observed-adverse-effect level for androgen inhibition in pubertal rats and would induce adverse effects on rats without causing systemic toxicity.51,52 The MMS group was a positive control for genotoxicity and it was administered intraperitoneally (60 mg kg–1 per day) to the rats and after 24 hours of administration, the rats in this group were sacrificed and the Comet assay was performed on their lymphocyte and sperm. After acclimatization for 1 week, the rats were orally exposed to DEHP or to the vehicle control at 8:30 AM each day for 28 consecutive days. DEHP was dissolved in corn oil and was introduced into the back of the mouth using a gavage needle. In the oil control group, corn oil (1 mL) was administered to the rats. The dosing solution was prepared by mixing the compound with corn oil to the desired concentration of 100, 200 and 400 mg kg–1 per day of DEHP.
2.4. Food and water intake, body and organ weights
Every day, before the treatment, the food and water consumption of the rats were measured and recorded. Also, their body weights were recorded daily and the dose administered each day was adjusted for body weight. At the end of the experiment, the final body weights and weight gain (%) for all animals were calculated and recorded. The comparison of the organ weights of treated animals with untreated animals is often complicated by differences in body weights between groups; therefore, another parameter that is commonly used for the analysis of organ weight is the ratio of the organ weight to body weight. The testes, epididymis, seminal vesicle and prostate gland were dissected and weighed in order to calculate the organ/body weight ratio for each animal. For each treatment group, the ratio of organ weight (Y) to body weight (X) is μ:Y/X = μ
Also, the absolute and relative weights of the right and left testes, right and left epididymis, prostate gland and seminal vesicle were measured and recorded for all animals. The relative organ weight of each animal was then calculated as follows: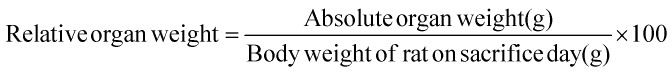
2.5. Comet assay
At the end of every week, blood was taken from the tails of the rats and the Comet assay was performed with blood lymphocytes. This study took 28 days and the measurements were calculated at the end of days 7, 14, 21 and 28.
The alkaline version of the Comet assay was performed.53,54 Lymphocytes were isolated using Biocoll separating solution. Isolated lymphocytes (10 μL) were immediately added to 120 μL of 0.5% low-melting-point agarose at 37 °C and layered on pre-coated slides with 1.5% normal-melting-point agarose in duplicate. The slides were immersed in 1% lysing solution for 1 h. After electrophoresis, the slides were fixed in absolute alcohol and stored until the moment of analysis. The slides were stained with 100 μL of ethidium bromide and analyzed using a fluorescence microscope connected to a camera with an image-analysis system (Comet Assay II, Perceptive Instruments, Suffolk, Haverhill, UK). A total number of 100 cells (Comets) per slide were analyzed.55 Three parameters were taken into account in order to estimate DNA damage: tail moment (DNA product contained by the tail), the intensity of the Comet tail (% of migrated DNA) and tail length (μm).
The sperm cells were taken from seminal vesicles and washed in saline, then were centrifuged and after discarding 900 μL of the supernatant, the sperm cells were suspended in 2% low melting point agarose, spread on slides and allowed to gel and topped up with a layer of 1.5% low melting point agarose. The slides were immersed in cold lysis solution consisting of 2.5 M NaCl, 100 mM EDTA, 10 mM Trizma base, 10% DMSO and 1% Triton X-100. Lysis of sperm was performed in two steps. In the first step, 10 mM dithiothreitol (DTT), in a lysing solution consisting of 2.5 M NaCl 100 mM EDTA, 10 mM Trizma base, 0.225 M NaOH and 1% Triton® X-100 was used and in the second step, instead of 10 mM DTT, 0.125 mg mL–1 proteinase K was used with the same lysing solution. The total incubation period was 2 h. After lysis, the unwinding of DNA was allowed in the electrophoresis buffer and 1 mM Na2 EDTA (pH 13.5) for a period of 30 min and electrophoresis was performed at a constant voltage of 25 V at 4 °C. Sperm DNA unwinding was carried out for 20 min. The slides were then neutralized with three washes of cold Tris buffer (pH 7.5) and stained with 20 g mL–1 ethidium bromide. After this treatment, the slides were analyzed with the same system and the same parameters as used for blood above. The results for sperm were measured and recorded.
2.6. Histopathological analysis
After weighing all the tissue samples, they were fixed for 8 h with Bouin solutions. Samples were then dehydrated using increasing concentrations of ethanol, cleared in xylene and embedded in paraffin and serially sectioned at 5 μm. The tissues were stained with Harris hematoxylin and eosin (H&E) for microscopic observation. Pathologic abnormalities and potential treatment-related effects and abnormalities/lesions were noted. All slides were examined using an Olympus BX51 light microscope. The photographs were captured using Bs200prop software and all histopathologic changes were recorded for each animal.
2.7. Measurement of seminiferous tubules
Seminiferous round tubules were evaluated for tubule diameter, lumen diameter, and height of tubule epithelium with an Olympus BX51 light microscope equipped with a Pixera Pro 150ES camera and Bab Bs200prop software. For seminiferous tubule measurement, tubules at stages VII and VIII were selected. The values obtained for the 10 tubules for each animal were averaged and used for calculating means and performing statistics.
2.8. Blood sampling and serum hormone measurement
At the end of the study, blood samples were collected from the animals by cardiac puncture for serum preparation for each group and after the completion of the treatments, on the same day and at the same time, from 9 to 10 a.m. The serum was separated after centrifugation at 3000g for 30 minutes, then the serum was pipetted into silicon microcentrifuge tubes and stored at –20 °C until hormone analysis. Testosterone (the sensitivity of the assay was 20 pg mL–1) was measured by using commercially available ELISA kits for rats according to the manufacturer's instructions. All samples were measured in duplicate in the same assay, the intra- and inter-assay coefficients of variation were less than 9.1%.
2.9. TUNEL method
Preparations on APS-coated glass slides were dehydrated in xylene and 100%, 95%, 80%, 70% ethanol and rinsed with phosphate buffer (PBS). Then, they were incubated with 20 mg mL–1 proteinase K solution in 10 mM Tris/HCl, pH 7.4–8 (Roche) in a humidified chamber for 10 min at 37 °C and rinsed in PBS, incubated with a blocking solution (3% H2O2 solution in methanol). The sections were rinsed twice with PBS, the areas around the selected sections were dried and loaded with TdT-terminal deoxynucleotidyl transferase, 50 mL per section. Slides were rinsed twice in PBS and dried. Then, 50 mL of horseradish peroxidase was added per sample and the sections were incubated in a humidified chamber for 30 min at 37 °C. Samples were rinsed in PBS three times and 50 mL of diaminobenzidine (0.05% DAB) per section was added. Preparations were incubated for 10 min at room temperature, rinsed with PBS, dehydrated with alcohol and xylene and closed with DePeX. The number of tubules, in which >10 degenerated cells were observed per the number of tubules visible in the cross-section, was determined. The apoptotic cells of testicular sections were counted by randomly selecting five areas for each slide.
2.10. Haematologic analysis
For the haematological examination, blood samples were collected and transferred to EDTA tubes. Haematological parameters, namely leukocytes (mm3), lymphocytes (%), monocytes (%), neutrophil granulocytes (N-granulocyte;%), erythrocytes (mm3), mean cell volume (MCV), hematocrit (%), mean cell hemoglobin (MCH), MCH concentration (MCHC) were measured with an auto cell counter for veterinary purposes, specific for rat (MS9-5 of Melet Schloesing Lab, France). For analysis, each group had six samples of collected blood.
2.11. Statistical analysis
All data were presented as the mean ± S.D. initial and final body weights, absolute and relative organ weights at necropsy, and serum hormones were analyzed by Analysis of Variance (ANOVA). All statistical analyses were performed using the SPSS for Windows 11.5 package program. Multiple comparisons were performed by the Tukey test. For histologic analysis, the animals were sacrificed and the samples were obtained. Also, Fisher's exact test was used to compare the frequencies of histopathological lesions in tissues. P < 0.05 was considered evidence of significance.56,57
3. Results
3.1. Food and water intake, body and organ weight results
The food and water consumption of rats in the control and treatment groups are given in Table 1. There were no significant effects of DEHP on the food and water intake at the end of the experiment. The body and absolute and relative organ weights of male rats in the control and treatment groups are given in Table 2. There was no significant difference between the control and treatment groups with respect to the percentage change in body weight from the baseline. Absolute right testis weight was decreased in the 200 and 400 mg kg–1 per day DEHP treatment group, as compared to the control and 100 mg kg–1 per day DEHP group. Otherwise, the relative weights of the right testes were increased in the 200 and 400 mg kg–1 per day DEHP treatment group compared to the control. Also, absolute left testis weight was decreased in all treatment groups, which was statistically significant compared to the control group. However, the relative weights of the left testes in male rats given 100 and 400 mg kg–1 per day of DEHP showed significant (P < 0.05) increase compared to that in the control group. For the absolute and relative weights of the left and right epididymis, there were no significant differences among the groups. The absolute and relative prostate gland weights decreased significantly in the 400 mg kg–1 per day of DEHP dose group when compared with the control group. Otherwise, in all DEHP given dose groups, the absolute seminal vesicle weight was decreased compared to the control, but in the relative weight of the seminal vesicle, a statistically significant decrease was observed only in the 400 mg kg–1 per day of DEHP treatment group.
Table 1. Food and water consumption of rats in the control and treatment groups.
| Oil control | DEHP |
|||
| 100 mg kg–1 per day | 200 mg kg–1 per day | 400 mg kg–1 per day | ||
| Food (g) | 11.70 ± 3.60 | 12.43 ± 4.37 | 14.06 ± 6.51 | 15.05 ± 5.72 |
| Water (mL) | 11.82 ± 2.76 | 12.75 ± 2.95 | 12.12 ± 2.39 | 13.34 ± 3.71 |
Table 2. Body weight, absolute and relative organ weights of rats in the control and treatment groups.
| Oil control | DEHP |
|||
| 100 mg kg–1 per day | 200 mg kg–1 per day | 400 mg kg–1 per day | ||
| Initial body weight (g) | 220.8 ± 1.0 | 222.3 ± 0.9 | 217.8 ± 0.1 | 218.9 ± 0.1 |
| Final body weight (g) | 310.9 ± 1.2 | 333.2 ± 0.7 | 340.5 ± 0.4 | 348.7 ± 1.1 |
| Weight gain (%) | 50.02 ± 0.2 | 51.09 ± 0.8 | 55.01 ± 0.3 | 54.06 ± 0.4 |
| Right testes | ||||
| Absolute (g) | 1.784 ± 0.1 | 1.780 ± 0.9 | 1.662 ± 0.2a,b | 1.540 ± 0.3a,b |
| Relative (mg g–1) | 4.761 ± 0.2 | 4.882 ± 0.7 | 5.031 ± 0.4a | 5.040 ± 0.2a |
| Left testes | ||||
| Absolute (g) | 1.932 ± 0.7 | 1.520 ± 0.1a | 1.401 ± 0.1a | 1.532 ± 0.4a. |
| Relative (mg g–1) | 5.222 ± 0.1 | 5.990 ± 0.2a | 4.101 ± 0.1a,b | 5.780 ± 0.2a,b |
| Left epididiymis | ||||
| Absolute (g) | 0.546 ± 0.03 | 0.544 ± 0.03 | 0.602 ± 0.04 | 0.578 ± 0.06 |
| Relative (mg g–1) | 1.651 ± 0.01 | 1.622 ± 0.02 | 1.643 ± 0.02 | 1.623 ± 0.03 |
| Right epididiymis | ||||
| Absolute (g) | 0.571 ± 0.01 | 0.582 ± 0.02 | 0.580 ± 0.02 | 0.576 ± 0.04 |
| Relative (mg g–1) | 1.671 ± 0.07 | 1.698 ± 0.03 | 1.572 ± 0.03 | 1.613 ± 0.02 |
| Prostate gland | ||||
| Absolute (g) | 0.667 ± 0.02 | 0.660 ± 0.09 | 0.578 ± 0.01 | 0.522 ± 0.04a,b |
| Relative mg (g–1) | 1.691 ± 0.04 | 1.640 ± 0.02 | 1.706 ± 0.01 | 1.569 ± 0.02a,c |
| Seminal vesicle | ||||
| Absolute (g) | 0.702 ± 0.01 | 0.650 ± 0.01d | 0.618 ± 0.05a | 0.549 ± 0.04a,b |
| Relative mg (g–1) | 1.704 ± 0.04 | 1.752 ± 0.01 | 1.677 ± 0.03 | 1.578 ± 0.03a,b |
3.2. Comet analysis results
Comet parameters in groups before the application are presented in Table 3. There was a significant difference between the positive control (MMS) and other groups. Weekly measurements of the tail lengths of the control and treatment groups are presented in Fig. 1. At the end of the 7th, 14th and 21st days, tail length was increased in a dose-dependent manner in the 100, 200 and 400 mg kg–1 per day DEHP dose groups, respectively, and it was statistically significant compared with the oil control group. At the end of the experiment (28 days), the tail length showed a statistically significant increase in the 100, 200 and 400 mg kg–1 per day DEHP groups, compared with the oil control.
Table 3. Comet parameters for the control and treatment groups of the male rats before the application.
| Groups | Dose | Tail length (μm) | Intensity of tail (%) | Tail moment (μm) |
| Oil control | 1 mL | 38.02 ± 7.17b | 0.68 ± 6.32b | 0.10 ± 1.61b |
| Positive control (MMS) | 60 mg kg–1 | 72.23 ± 9.87a,c,d,e | 9.07 ± 1.22a,c,d,e | 1.49 ± 1.02a,c,d,e |
| DEHP | 100 mg kg–1 per day | 38.55 ± 5.6b | 0.71 ± 0.7b | 0.12 ± 0.9b |
| DEHP | 200 mg kg–1 per day | 38.87 ± 5.63b | 0.75 ± 0.4b | 0.11 ± 0.1b |
| DEHP | 400 mg kg–1 per day | 38.13 ± 0.1b | 0.77 ± 5.5b | 0.13 ± 2.63b |
Fig. 1. Weekly measurements of the tail lengths of the control and treatment groups of the male rats. Data are mean ± S.E. *P < 0.05 is significantly different from the control group.

Weekly measurements of the intensity of the Comet tails of the control and treatment groups of the male rats are presented in Fig. 2. The intensity of the Comet tail was increased in all DEHP dosed groups statistically according to the oil control. However, at the end of the 21st day, this increase was much higher than the oil control and other dose groups. Weekly measurements of the moments of the Comet tails of the control and treatment groups in blood samples of the male rats are given in Fig. 3. The tail moment was statistically increased in all DEHP measurements except for the 7th and 14th days of the experiment in the 100 mg kg–1 per day DEHP groups, compared to the oil control. The 200 and 400 mg kg–1 per day of DEHP dose groups showed an increase in the tail moment for the weekly measurements, compared with other groups.
Fig. 2. Weekly measurements of the intensity of the Comet tails of the control and treatment groups of the male rats. Data are mean ± S.E. *P < 0.05 is significantly different from the control group.

Fig. 3. Weekly measurements of the moment of the Comet tails of the control and treatment groups of the male rats. Data are mean ± S.E. *P < 0.05 is significantly different from the control group.

Comet parameters of sperm of the control and treatment groups of the male rats at the end of the experiment are given in Fig. 4. For sperm samples, the tail length was increased in the 100, 200 and 400 mg kg–1 per day of DEHP dose groups compared with the oil control. Also, the intensity of the tail and tail moment values were increased statistically in the 200 and 400 mg kg–1 per day of DEHP, compared to the 100 mg kg–1 per day of the DEHP and oil control.
Fig. 4. Comet parameters for the sperm of the control and treatment groups of the male rats at the end of the experiment (n = 6). Data are mean ± S.E. *P < 0.05 is significantly different from the control group.

3.3. Histopathologic analysis
The incidence of exposure-related histopathologic lesions of male rats in the control and treatment groups is given in Table 4. Combined and atrophic tubules were increased in the 400 mg kg–1 per day DEHP dose group in the testes, which was statistically significant compared to other groups. The pycnotic nucleus was shown in the 200 and 400 mg kg–1 per day DEHP dose groups and the congestion and degeneration of the tubules were increased in all DEHP dose groups compared to the control group in the testes. With the vacuolization of Sertoli cells, the cells in the lumen, tubules without sperm were increased in the 200 and 400 mg kg–1 per day DEHP groups, and were statistically significant compared to the 100 mg kg–1 per day DEHP and control groups in the testes (Fig. 5). In the epididymis, the tight lumens of the tubules and the sperm in the tubule increased, and these were statistically significant in all treatment groups. Atrophic tubules and spermatogenic cells are shown in groups but were not statistically significant (Fig. 6). In the prostate, atrophic tubules and prostatic intraepithelial neoplasia shown in the 200 and 400 mg kg–1 per day of DEHP group were increased compared to other groups (Fig. 7). In the seminal vesicle, the decrement of secretion was increased in all treatment groups but cells in the lumen were not statistically significant in the 400 mg kg–1 per day DEHP group (Fig. 8).
Table 4. Incidence of exposure-related histopathologic lesions in male rats in the control and treatment groups.
| Tissue and lesion | Control | DEHP |
||
| 100 mg kg–1 per day | 200 mg kg–1 per day | 400 mg kg–1 per day | ||
| Testes | ||||
| Combined tubules | 0/6 | 1/6 | 1/6 | 5/6a |
| Atrophic tubules | 0/6 | 2/6 | 3/6 | 5/6a |
| Pycnotic nucleus | 0/6 | 1/6 | 5/6a | 6/6a |
| Congestion | 0/6 | 6/6a | 6/6a | 6/6a |
| Vacuolization of Sertoli cell | 0/6 | 3/6 | 6/6a | 6/6a |
| Cells in the lumen | 0/6 | 2/6 | 5/6a | 6/6a |
| Tubules without sperm | 0/6 | 3/6 | 6/6a | 5/6a |
| Degeneration of tubules | 0/6 | 6/6a | 6/6a | 6/6a |
| Epididymis | ||||
| Atrophic tubules | 0/6 | 2/6 | 1/6 | 2/6 |
| Spermatogenic cell | 0/6 | 0/6 | 0/6 | 1/6 |
| Tight lumens of tubules | 0/6 | 6/6a | 6/6a | 5/6a |
| Tubules without sperm | 0/6 | 2/6 | 6/6a | 6/6a |
| Less sperm in the lumen | 0/6 | 6/6a | 6/6a | 6/6a |
| Prostate gland | ||||
| Mononuclear cell infiltration | 0/6 | 3/6 | 2/6 | 6/6a |
| Atrophic tubules | 0/6 | 6/6a | 6/6a | 6/6a |
| Prostatic intraepithelial neoplasia | 0/6 | 5/6a | 6/6a | 6/6a |
| Seminal vesicle | ||||
| Decrement of secretion | 0/6 | 6/6a | 6/6a | 6/6a |
| Cells in the lumen | 0/6 | 5/6a | 5/6a | 3/6 |
Fig. 5. Photomicrographs showing testis tissues of control and treatment groups. (A) is from the control group. (B) is from the 100 mg kg–1 per day DEHP treatment group and it shows congestion (black arrow) and pycnotic nucleus (asterisk). (C) is from the 200 mg kg–1 per day DEHP treatment group and it shows congestion (black arrow) and cells in the lumen (asterisk). (D) is from the 400 mg kg–1 per day DEHP and it shows pycnotic nucleus (asterisk) and congestion (black arrow) (all were stained with contrast by H&E, ×200). In TUNEL staining, (A) is from the control group. (B) is the 100 mg kg–1 per day of DEHP, (C) is the 200 mg kg–1 per day of DEHP and (D) is the 400 mg kg–1 per day of DEHP. Black arrows show apoptotic cells in testes (×400).

Fig. 6. Photomicrographs showing the epididymis tissues of the control and treatment groups. (A) is from the control group. (B) is from the 100 mg kg–1 per day DEHP treatment group and it shows tubules without sperm (black arrow). (C) is from the 200 mg kg–1 per day DEHP treatment group and it shows tight lumens of tubules (black arrow). (D) is from the 200 mg kg–1 per day DEHP treatment group and it shows atrophic tubules (black arrow) and less sperm in the lumen (asterisk). (E) is from the 400 mg kg–1 per day DEHP treatment group and it shows tight lumen of tubules (black arrow) and less sperm in the tubule (asterisk). (F) is from the 400 mg kg–1 per day DEHP treatment group and it shows atrophic tubules (black arrow) and less sperm in the tubule (asterisk) and there is a decrease in the tubule epithelial length (all were stained with contrast by H&E, ×200).

Fig. 7. Photomicrographs showing prostate tissues of the control and treatment groups. (A) is from the control group. (B) is from the 100 mg kg–1 per day DEHP treatment group and it shows atrophic tubules (black arrow) and mononuclear cell infiltration (asterisks). (C) is from the 200 mg kg–1 per day DEHP treatment group and it shows congestion (black arrow) and prostatic intraepithelial neoplasia (asterisk). (D) is from the 200 mg kg–1 per day DEHP treatment group and it shows atrophic tubules (black arrow) and a decrement of secretion (asterisk). (E) is from the 400 mg kg–1 per day DEHP treatment group and it shows congestion (black arrow) and atrophic tubules (asterisk). (F) is from the 400 mg kg–1 per day DEHP treatment group and it shows atrophic tubules (asterisks) (all were stained with contrast by H&E, ×200).

Fig. 8. Photomicrographs showing seminal vesicle tissues of the control and treatment groups. (A) is from the oil control group. (B) is from the 100 mg kg–1 per day DEHP treatment group and it shows increasing connective tissue (black arrow) and the decrement of secretion (asterisk). (C) is from the 200 mg kg–1 per day DEHP treatment group and it shows the decrement of secretion (asterisk). (D) is from the 200 mg kg–1 per day DEHP treatment group and it shows cells in the lumen (black arrow) and the decrement of secretion (asterisk). (E) is from the 400 mg kg–1 per day DEHP treatment group and it shows the decrement of secretion (black arrow) and cells in the lumen (asterisk). (F) is from the 400 mg kg–1 per day DEHP treatment group and it shows the decrement of secretion (black arrow) (all were stained with contrast by H&E, ×200).

3.4. Measurement of seminiferous tubules
Epithelial height, luminal and tubular diameters (μM) in the seminiferous tubules of male rats in the control and treatment groups are given in Fig. 9. The epithelial height was significantly increased in the 400 mg kg–1 per day DEHP treatment groups compared with the control group. The luminal diameter was increased in 100 mg kg–1, 200 mg kg–1 and 400 mg kg–1 DEHP dose groups compared to the oil control group. Also, the tubular diameter had a significant increase in the 100 mg kg–1 per day DEHP treatment group compared with all the other groups, and it was 371.7 ± 1.1 μm; however, in the 400 mg kg–1 per day DEHP group, it was slightly decreased.
Fig. 9. Epithelial height, luminal diameter and tubular diameter (μM) in the seminiferous tubule of male rats in the control and treatment groups. Data are mean ± S.E. *P < 0.05 is significantly different from the control group.

3.5. Hormone analysis
In Fig. 10, the testosterone hormone levels are shown in the control and treatment groups. The 100, 200 and 400 mg kg–1 dose groups showed an important decrease compared to the control group but in the 200 mg kg–1 per day DEHP group, the lowest level was shown compared to the others.
Fig. 10. Testosterone (pg mL–1) hormone levels of male rats in control and treatment groups. Data are mean ± S.E. *P < 0.05 is significantly different from the control group.

3.6. TUNEL method results
Histologic photomicrographs of testes tissues that were stained using the TUNEL method are shown in Fig. 5. In the oil control group (Fig. 5A), testes tissue cells showed normal morphology and complete arrangement. However, in the 100, 200 and 400 mg kg–1 per day DEHP dose groups, the apoptotic cells were observed.
3.7. Haematologic analysis
The results of the haematologic analysis parameters of the control and treatment groups of the pre-pubertal male rats are presented in Table 5. Leukocyte, haemoglobin and monocyte levels were decreased in the 400 mg kg–1 per day DEHP dose group but MCV levels were increased in the same group compared with other groups. There was no statistically significant difference between for the other haematological parameters in any of the treatment groups compared to the control.
Table 5. Effects of di-(2-ethylhexyl) phthalate on the hematological parameters of the control and treatment groups of the pubertal male rats.
| Parameters | Oil control (1 mL) | DEHP (100 mg kg–1 per day) | DEHP (200 mg kg–1 per day) | DEHP (400 mg kg–1 per day) |
| Leukocyte (mm3) | 2.35 ± 1.54 | 2.98 ± 2.34 | 3.13 ± 1.11 | 1.19 ± 0.39a,b,c,d |
| Lymphocyte (%) | 69.60 ± 16.17 | 70.83 ± 21.34 | 65.42 ± 8.39 | 78.41 ± 16.90 |
| Monocyte (%) | 11.17 ± 5.40 | 11.40 ± 9.41 | 10.30 ± 1.97 | 2.40 ± 0.10a,b,c,d |
| Neutrophil/granulocyte (%) | 19.27 ± 11.06 | 17.73 ± 14.22 | 24.28 ± 6.89 | 17.48 ± 12.79 |
| Erythrocyte (mm3) | 13.22 ± 3.53 | 11.6 ± 2.68 | 13.3 ± 1.76 | 8.26 ± 0.68 |
| MCV (fL) | 27.8 ± 5.34 | 29.42 ± 2.13 | 25.78 ± 3.89 | 53.12 ± 2.89a,b,c,d |
| Hematocrit (%) | 35.27 ± 8.18 | 33.6 ± 6.06 | 27.1 ± 6.29 | 39.27 ± 3.65 |
| MCH (pg) | 7.87 ± 4.52 | 8.93 ± 3.39 | 11.6 ± 11.24 | 14.9 ± 1.24 |
| MCHC (g dL–1) | 26.75 ± 8.78 | 29.8 ± 9.16 | 23.05 ± 6.05 | 28.07 ± 1.06 |
| Hemoglobin (g dL–1) | 8.92 ± 1.26 | 9.5 ± 1.16 | 9.02 ± 1.6 | 6.33 ± 0.93a,b,c,d |
4. Discussion
This study was conducted in order to investigate the genotoxic, hematological, histopathological, immunohistochemical, morphometric and hormonal effects of DEHP on the reproductive systems of the growing male rats exposed to this estrogenic compound. A lot of synthetic substances have been produced to facilitate our lives but their dangerous and biological effects are not fully known. It is necessary and important to determine the effects of these chemicals on environmental and human health. For years, phthalates were classified as epigenetic carcinogens, but recent evidence suggests that these chemicals also possess genotoxic properties.58,59 For this purpose, in vivo and in vitro experiments were performed and toxic and adverse effects of DEHP were determined.
In the current study, DEHP was applied to pre-pubertal male rats via oral gavage for 28 days, and the Comet assay was applied in blood lymphocytes and sperm samples to evaluate possible genotoxic effects. We noted an increase in body weight in all the treatment groups, but none of the increments reached statistical significance. However, in many studies with the application of 1000 mg kg–1 per day of DEHP to rodents for five days or more, weight loss was observed, but in these studies there was no food consumption during the experiment so it was not determined whether this loss was caused by the decrease in food intake or the increase in metabolic rate.60 Kurahashi and colleagues61 reported that there was no significant increase in the body weight of rats given 50 and 100 mg kg–1 per day of DEHP. Rusyn et al.62 applied 1000 mg kg–1 DEHP per day for 6 weeks to male rats and at the end of the study, an increase in the body weight was observed like in other studies and it was connected to the increase in cell proliferation. Our findings are in accordance with the findings of Kurahashi et al.61 and Rusyn et al.62
Testosterone plays a dominant role in the growth of these organs; several other hormones and growth factors can influence sex organ weights.63 Cardoso et al.64 reported that the adverse effects of DEHP in male rats exposed during the pre-and early postnatal periods showed that the testosterone levels were significantly decreased. In the current study, the serum testosterone concentration of adult males exposed to DEHP showed a significant decrease. The reported effects of DEHP and MEHP exposure on CYP19 enzyme activity in human tumor cell lines are relevant to cancer risk as well. DEHP exposure is associated with Leydig cell tumor induction in the rat.65 The Leydig cell's primary function is the production of testosterone, and that production is stimulated by luteinizing hormone (LH). Agents that increase LH levels or Leydig cell responsiveness to LH will also induce hyperplasia and Leydig cell tumors in the rat. Estradiol is synthesized via CYP19 from testosterone and provides negative feedback on the production of LH, as does testosterone. Hence, decreased levels of testosterone or estrogen can stimulate LH production and stimulate Leydig cell tumor induction. Testosterone, estradiol, and LH are regulated through the hypothalamic-pituitary-testis (HPT) axis in both rats and humans, and agents that induce Leydig cell tumors in rats by disruption of that axis are thought to pose a hazard to humans.66
Also, in this study, the right and left absolute testes weights were decreased, especially with medium and high treatment doses. Absolute prostate weights were decreased only for high treatment doses, but absolute seminal vesicle weights were decreased for all DEHP dosed groups compared to the control; therefore, the weights of these organs were affected by the decrease in testosterone hormone levels. Several studies have reported the adverse effects of DEHP on the development of the male reproductive tract when animals were perinatally exposed to DEHP.67,68 These effects include reduced testes size, decreased sperm production, cryptorchidism, and reduced reproductive organ weights. In one study,69 adult male Fischer rats were exposed subcutaneously for 1 or 2 months to DEHP. The study revealed statistically significant reductions in the absolute and relative weights of the testes and epididymis. Some studies used multiple dose levels, giving the opportunity to define dose–response relationships. The investigations were single-dose-level studies focusing on modes of action for developmental reproductive toxicity. In one study, Sprague–Dawley rats were orally dosed with DEHP at 0, 375, 750, or 1500 mg kg–1 per day from gestation day (GD) 3 to postnatal day (PND) 21, and endpoints related to sexual development were studied through puberty and adulthood in male and female offspring. In the two highest dose groups, developmental effects were observed and these effects persisted until adulthood.70 Oral exposure to DEHP from GD 7 to 18 resulted in increased levels of multinucleated germ cells (125 mg kg–1 per day) and interstitial hyperplasia at 250 or 500 mg kg–1 per day in rat offspring.71
For the measurements with the Comet assay in blood lymphocytes, the tail length and tail intensity increased in a dose-dependent manner for the 100, 200 and 400 mg kg–1 per day DEHP treatment groups compared to the oil control groups. In the same way, the tail moment increased in the 200 and 400 mg kg–1 per day DEHP treatment groups, compared with the oil control group. These measurements are very important for the strong effects between genotoxicity and carcinogenicity, but previous results showed inconsistencies. In the current study, for the tail length in sperm, there was an increase in the damage to DNA in all treatment doses. However, the intensities of tail and tail moment were increased for medium and high doses (200 and 400 mg kg–1 per day) of DEHP compared to the 100 mg kg–1 per day and oil control. Few published human studies have examined the effect of environmental chemicals, such as phthalates, on DNA integrity in sperm as measured by the Comet assay.72–75 These results showed that exposure to DEHP at high doses caused adverse effects on sperm and blood samples. Ahbab et al. showed DNA damage of DCHP and DHP on testicular cells in male rats.76 DEHP caused increases in Comet assay parameters (tail intensity, tail moment), cytotoxicity and oxidant/antioxidant status in LnCAP (human prostatic cell line) and MA-10 Leydig cells.77
In this study, the TUNEL assay was performed to ascertain the mode of cell death. It was based upon the principle that TdT binds to the exposed 3-OH ends of the DNA fragments generated in response to apoptotic signals and catalyzes the addition of labeled deoxynucleotides. This assay can detect early-stage apoptosis in systems where chromatin condensation has begun and DNA strand breaks are fewer, even before the nucleus undergoes major morphological changes.78 An advantage of the TUNEL staining is that it identifies cell apoptosis in situ. DEHP was also correlated with increased DNA damage in a group of men exposed to doses comparable to those reported for the U.S. general population.79 It was reported that male rats were orally administered DEHP (200 mg kg–1 per day) for 30 or 60 days and it was found that there was adversely influenced sperm morphology as well as weights and the histological structure of the testes and seminal vesicles. In testes tissues, apoptosis increased and testosterone levels were decreased.80 The current study showed the same results. Serum analyses showed a significant decrease in testosterone, indicating the presence of Leydig cell dysfunction. In the 400 mg kg–1 per day DEHP treatment group, the apoptotic cells were increased compared to the control group, showing a dose-dependent increase. Recent data have also shown that phthalates were able to produce free radicals by several pathways in germ cells including activation of PPARα, suggesting the possibility that oxidative stress and mitochondrial dysfunction in germ cells may contribute to phthalate-induced disruption of spermatogenesis.81 One of the mechanisms underlying the reproductive toxicity of DEHP might be the induction of intracellular ROS and/or alterations of intracellular enzymatic and non-enzymatic antioxidants, thereby producing oxidative stress. Phthalates have the capacity to induce change in the mitochondrial membrane potential and generate ROS, and lysosomal destabilization has been recognized as a feature of oxidative stress-induced cell damage. GSH is the most important intracellular anti-oxidative defense against oxidative stress.82 The depletion of GSH indicates that oxidative stress has occurred,83 and it is also related to the augmentation of a pro-inflammatory signal by upregulating ROS.84 DEHP also significantly decreased SOD activity and increased MDA content in rats.85 The literature reports that part of the mechanism of MEHP-induced germ cells apoptosis is mediated through the Fas-signaling system57 Data on the time duration between Fas-stimulation and caspase-3 activation are sparse, but one study reports that an agonistic Fas antibody can induce caspase-3 activation 2.5 h after exposure in vitro, indicating a short interval. Numerous studies have confirmed that DEHP induces reproductive toxicity mainly by inducing ROS and MDA production and by disrupting the activity of antioxidant enzymes (SOD, CAT).86–88
In the testes, atrophic and pycnotic cells, congestion, and the disorder of the tubule cells increased in the 400 mg kg–1 per day DEHP group compared to the oil control. The arrangements of spermatogenic cells were irregular and disordered in the treatment groups. The germinal cell debris and atrophic tubules were observed in DEHP treatment groups, which may be due to the apoptotic effects, depending on the antiandrogenic effects of DEHP. Apoptosis plays an important role in the regulation of the production of sperm. In the epididymis, there were confined lumens of tubules and less sperm in the tubules, which increased statistically in all the treatment groups. It was concluded that DEHP, at a dose of 400 mg kg–1 per day, could reduce the size and function of the entire male gametogenic and accessory reproductive organs in an estrogenic manner.
The tubular diameter and the epithelium height can indicate spermatogenesis activity in the experimental and toxicological analysis.89 In the morphometric evaluation of seminiferous tubules, epithelial heights were significantly increased in the 400 mg kg–1 per day DEHP treatment groups compared with the control and 100, 200 mg kg–1 per day group. However, luminal diameters were significantly increased in all treatment groups compared to the control. Soleimani-Mehranjani et al.90 reported that reduction in the diameter and epithelium height of seminiferous tubules may be because of a decrease in the number of type A spermatogonia, spermatocyte, spermatid and Sertoli cells. Also, this increment may result from the reduced serum testosterone levels of rats in these groups. In our previous study,91 we found that in dose groups given 125 and 250 mg kg–1 per day of bisphenol A and 125 and 250 mg kg–1 per day of octylphenol, the testosterone levels were decreased and epithelial heights and tubule diameters were increased significantly compared to the control group. DEHP is also an endocrine disrupter like OP and BPA, so the effect may be similar.
The obtained data show that treatment with DEHP caused a significant decrease in the leukocyte levels, especially monocyte% and Hb content, as well as an increased MCV count in the 400 mg kg–1 per day group, when compared to the control values. These parameters might be a result of an increase in the rate of erythrocyte destruction and inhibition of erythropoiesis, given the impairment in haematopoietic organ function. Also, lymphocytes are important cells of the immune system so exposure of the DEHP caused a negative impact on blood-forming organs. Usually, the MCV value is increased in vitamin B12 deficiency. Moreover, an increase in the MCV count may indicate activation of the immune response.
5. Conclusion
In DEHP dose groups DNA damage was observed as statistically significant when compared to the oil control and positive control. According to the doses in previous studies, we chose 3 doses, namely, 100, 200 and 400 mg kg–1 per day of DEHP. These doses were high compared to the other studies because we studied an earlier period for the rats and we wanted to see how that could have different effects. So far, there has not been any other study with male rats that was started in the prepubertal period and ended at the pubertal period. Also, this study is the only one to demonstrate direct DNA damage to sperm cells in pubertal male rats. It therefore, sheds light on this period in the lives of rodents, and these findings will reveal the possibilities for humans in cases of exposure.
In recent years, there have been many studies showing increasing interest in phthalates, but none have investigated the genotoxic, histopathologic, morphometric and haematological effects of DEHP from the pre-pubertal period into puberty. This study is therefore important for showing the DNA damage in blood and sperm caused by the often-used DEHP. These results will be used for the regulation of the usage levels in the environment and human health and it will lead to other studies on DEHP.
Conflicts of interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Acknowledgments
We would like to thank the authors for their commitment to this project. Development of this study financial supported by the Hacettepe University Scientific Research Projects Coordination Unit (Project number is 1183).
References
- Koch H. M., Drexler H., Angerer J. Int. J. Hyg. Environ. Health. 2004;207(1):15–22. doi: 10.1078/1438-4639-00270. [DOI] [PubMed] [Google Scholar]
- Gaudin R., Marsan P., Ndaw S. Int. Arch. Occup. Environ. Health. 2011;84:523–531. doi: 10.1007/s00420-010-0566-7. [DOI] [PubMed] [Google Scholar]
- Sidorkiewicz I., Zareba K., Wołczynski S., Czernieck J. Toxicol. Ind. Health. 2017;33(7):601–609. doi: 10.1177/0748233717695160. [DOI] [PubMed] [Google Scholar]
- Saleh T. A., Advanced nanomaterials for water engineering, treatment, and hydraulics, IGI Glob., 2017. [Google Scholar]
- Saleh T. A. and Gupta V. K., Nanomaterial and polymer membranes, Elsev., 2016. [Google Scholar]
- Ito Y., Nakajima T. PPAR Res. 2008:759–716. doi: 10.1155/2008/759716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calafat A. M., Mckee R. H. Environ. Health Perspect. 2006;114(11):1783–1789. doi: 10.1289/ehp.9059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice D. C., Phthalates: Maine Chemicals of High Concern A Review of the Science on Toxicity and Exposure, 2014. [Google Scholar]
- Voss C., Zerban H., Bannasch P., Berger M. R. Toxicol. 2005;206:359–371. doi: 10.1016/j.tox.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Meeker J. D. and Ferguson K. K., Dioxins and health: including other persistent organic pollutants and endocrine disruptors, in Phthalates: human exposure and related health effects, Wiley, Hoboken, 2012, ch. 13. [Google Scholar]
- Adibi J. J., Perera F. P., Jedrychowski W., Camann D. E., Barr D., Jacek R., Whyatt R. M. Environ. Health Perspect. 2003;111:1719–1722. doi: 10.1289/ehp.6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppin J. A., Brock J. W., Davis B. J., Barid D. D. Environ. Health Perspect. 2002;110:515–518. doi: 10.1289/ehp.02110515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doull Q., Qian J., Xu L. Food Chem. Toxicol. 1999;44:1355–1361. doi: 10.1016/j.fct.2006.02.014. [DOI] [PubMed] [Google Scholar]
- Poon R., Lecavalier P., Mueller R., Valli V. E., Procter B. G., Chu I. Food Chem. Toxicol. 1997;35:225–239. doi: 10.1016/s0278-6915(96)00064-6. [DOI] [PubMed] [Google Scholar]
- Schecter A., Lorber M., Guo Y. Environ. Health Perspect. 2013;121(4):473–479. doi: 10.1289/ehp.1206367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wormuth M., Scheringer M., Vollenweider M. Risk Anal. 2006;26(3):803–824. doi: 10.1111/j.1539-6924.2006.00770.x. [DOI] [PubMed] [Google Scholar]
- Rudel R. A., Camann D. E., Spengler J. D. Environ. Sci. Technol. 2003;37(20):4543–4553. doi: 10.1021/es0264596. [DOI] [PubMed] [Google Scholar]
- Rudel R. A., Dodson R. E., Perovich L. J. Environ. Sci. Technol. 2010;44(17):6583–6590. doi: 10.1021/es100159c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekö G., Weschler C. J., Langer S. PLoS One. 2013;8(4):e62442. doi: 10.1371/journal.pone.0062442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer R., Labunska I., Santillo D. Environ. Sci. Pollut. Res. Int. 2000;7(1):27–36. doi: 10.1065/espr199910.007. [DOI] [PubMed] [Google Scholar]
- Schettler T. Int. J. Androl. 2006;29(1):134–139. doi: 10.1111/j.1365-2605.2005.00567.x. [DOI] [PubMed] [Google Scholar]
- Weuve J., Sanchez B. N., Calafat A. M. Environ. Health Perspect. 2006;114(9):1424–1431. doi: 10.1289/ehp.8926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loff N., Pandolf M., Lavalle J. J. Physiol. Biochem. 2000;67:559–567. doi: 10.1007/s13105-011-0102-6. [DOI] [PubMed] [Google Scholar]
- Kavlock D., Yanagiba Y., Duan Z. Toxicol. Lett. 2005;194:16–25. doi: 10.1016/j.toxlet.2010.02.002. [DOI] [PubMed] [Google Scholar]
- Ge M. K., Simon L., Akingbemi B. T. Biol. Reprod. 2007;86:1–12. doi: 10.1095/biolreprod.111.095349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noriega K. V., Sultana T., Soder O. Mol. Cell. Endocrinol. 2009;2:193–201. doi: 10.1016/s0303-7207(01)00554-8. [DOI] [PubMed] [Google Scholar]
- Grasso C. B., Wanzhu J., Watanabe G. Endocrine. 2003;25:163–172. [Google Scholar]
- Dalgaard S., Kavitha C., Ramesh M. J. Appl. Toxicol. 2001;31:752–776. doi: 10.1002/jat.1629. [DOI] [PubMed] [Google Scholar]
- Awal K. L., Rider C. V., Wilson V. S. Environ. Res. 2004;108:168–176. doi: 10.1016/j.envres.2008.08.009. [DOI] [PubMed] [Google Scholar]
- Lin K., Asklund C., Skakkebaek N. E. Best Pract. Res., Clin. Endocrinol. Metab. 2007;20:77–90. doi: 10.1016/j.beem.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Lee B. C., Silva M. J., Caudhill S. P. Environ. Health Perspect. 1997;108:972–982. [Google Scholar]
- Shinoda H. B., Garside D. A., Liu R. Exp. Mol. Pathol. 1998;58:179–193. doi: 10.1006/exmp.1993.1016. [DOI] [PubMed] [Google Scholar]
- El-Gohary D., Srivastava S. P., Singh G. B. Hum. Toxicol. 1999;37:310–313. [PubMed] [Google Scholar]
- Shin C., Bornman M. S., Oosthuizen J. M. Andrology. 1999;31:107–113. doi: 10.1046/j.1439-0272.1999.00246.x. [DOI] [PubMed] [Google Scholar]
- Akingbemi B. T., Sottas C. M., Koulova A. I. Endocrinology. 2004;145:592–603. doi: 10.1210/en.2003-1174. [DOI] [PubMed] [Google Scholar]
- Wilson C., Zerban H., Bannasch P. Toxicol. 2004;206:359–371. doi: 10.1016/j.tox.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Liochev S. I., Fridovich I. IUBMB Life. 1999;48:157–161. doi: 10.1080/713803492. [DOI] [PubMed] [Google Scholar]
- Agarwal A., Said T. M. BJU Int. 2005;95:503–507. doi: 10.1111/j.1464-410X.2005.05328.x. [DOI] [PubMed] [Google Scholar]
- Puppel K., Kapusta A., Kuczyńska B. J. Sci. Food Agric. 2015;95(11):2179–2184. doi: 10.1002/jsfa.7015. [DOI] [PubMed] [Google Scholar]
- Kasahara K. W., Hensley J. B., Liu D. Toxicol. Sci. 2002;97:491–503. doi: 10.1093/toxsci/kfm049. [DOI] [PubMed] [Google Scholar]
- Dalton R. T., Brown T. R., Doan L. L. Endocrinology. 2009;153:4097–4110. doi: 10.1210/en.2012-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel M. Horm. Behav. 2011;55:621–632. [Google Scholar]
- Kijima S. K., Lee H. J., Yang H. Arch. Androl. 2014;50:427–441. doi: 10.1080/01485010490475093. [DOI] [PubMed] [Google Scholar]
- Garaj-Vrhovac V., Zeljezic D. Mutat Res. 2000;469:279–285. doi: 10.1016/s1383-5718(00)00092-9. [DOI] [PubMed] [Google Scholar]
- Andreazza A. C. J. Psychiatry Neurosci. 2008;33:516–524. [PMC free article] [PubMed] [Google Scholar]
- Maluf S. W., Erdtmann B. Mutat Res. 2000;471:21–27. doi: 10.1016/s1383-5718(00)00107-8. [DOI] [PubMed] [Google Scholar]
- Dolinoy D. C., Weidman J. R., Jirtle R. L. Reprod. Toxicol. 2007;23:297–307. doi: 10.1016/j.reprotox.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Barton H. A., Cogliano V. J., Flowers L., Valcovic L., Setzer R. W., Woodruff T. J. Environ. Health Perspect. 2005;113:1125–1133. doi: 10.1289/ehp.7667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cal EPA (California EPA), Technical Support Document for Cancer Potency Factors Methodologies for Derivation, Listing of Available Values, and Adjustments to Allow for Early Life Stage Exposures, California Environmental Protection Agency, Office of Environmental Health Hazard Assessment, Air Toxicology and Epidemiology Branch, 2009
- Guyton K. Z., Kyle A. D., Aubrecht J., Cogliano V. J., Eastmond D. A., Jackson M., Keshava N., Sandy M. S., Sonawane B., Zhang L., Waters M. D., Smith M. T. Mutat Res. 2009;681:230–240. doi: 10.1016/j.mrrev.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Wittassek M., Koch H. M., Angerer J., Brüning T. Mol. Nutr. Food Res. 2011;55:7–31. doi: 10.1002/mnfr.201000121. [DOI] [PubMed] [Google Scholar]
- Laura N. V., Colborn T., Tyrone B. H., Jerrold J. H., David R. J. Endocr. Rev. 2012;33:378–455. doi: 10.1210/er.2011-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N. P., Mccoy M. T., Tice R. R. Exp. Cell Res. 1988;175:184–191. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- Tice R. R., Agurell E., Anderson D. Environ. Mol. Mutagen. 2000;35:206–221. doi: 10.1002/(sici)1098-2280(2000)35:3<206::aid-em8>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Hartmann A., Agurell A., Beevers C. Mutagen. 2003;18(1):45–51. doi: 10.1093/mutage/18.1.45. [DOI] [PubMed] [Google Scholar]
- Saleh T. A., Adio S. O., Asif M., Dafalla H. J. Cleaner Prod. 2018;182:960–968. [Google Scholar]
- Saleh T. A. Environ. Sci. Pollut. Res. 2015;22(21):16721–16731. doi: 10.1007/s11356-015-4866-z. [DOI] [PubMed] [Google Scholar]
- Erkekoglu P., Rachidi W., De Rosa V. Free Radicals Biol. Med. 2010a;49:559–566. doi: 10.1016/j.freeradbiomed.2010.04.038. [DOI] [PubMed] [Google Scholar]
- Kleinsasser N. H., Wallner B. C., Kastenbauer E. R. Teratog., Carcinog., Mutagen. 2001;21:189–196. doi: 10.1002/tcm.1007. [DOI] [PubMed] [Google Scholar]
- Dostal L. A., Chapin R. E., Stefanski S. A., Harris M. W., Schwetz B. A. Toxicol. Appl. Pharmacol. 1987;95:104–121. doi: 10.1016/s0041-008x(88)80012-7. [DOI] [PubMed] [Google Scholar]
- Kurahashi N., Kondo T., Omura M. J. Occup. Health. 2005;47(5):437–444. doi: 10.1539/joh.47.437. [DOI] [PubMed] [Google Scholar]
- Rusyn I., Peters J. M., Cunningham M. L. Crit. Rev. Toxicol. 2006;36:459–479. doi: 10.1080/10408440600779065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke Y., Coffey N. Clin. Chim. Acta. 1994;361:20–29. [Google Scholar]
- Cardoso R. S., Chen G. R., Dong Q. J. Androl. 2011;28:513–520. [Google Scholar]
- Jager V. S., Lambright C., Furr J. Toxicol. Lett. 1999;146:207–215. doi: 10.1016/j.toxlet.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Cook J. C., Klinefelter G. R., Hardisty J. F., Sharpe R. M., Foster P. M. Crit. Rev. Toxicol. 1999;29:169–261. doi: 10.1080/10408449991349203. [DOI] [PubMed] [Google Scholar]
- Yoshida V., Castillo C., Ariznavarret C. Toxicol. 2001;205:131–137. [Google Scholar]
- Boockfor F. R., Blake C. A. Biol. Reprod. 1997;57:267–277. doi: 10.1095/biolreprod57.2.267. [DOI] [PubMed] [Google Scholar]
- Moore R., Rudy W., Lin T. A., Ko K., Peterson R. E. Environ. Health Perspect. 2001;109:229–237. doi: 10.1289/ehp.01109229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirota M., Saito Y., Imai K., Horiuchi S., Yoshimura S., Sato M., Nagao T., Ono H., Katoh M. J. Toxicol. Sci. 2005;30:175–194. doi: 10.2131/jts.30.175. [DOI] [PubMed] [Google Scholar]
- Duty S. M., Singh N. P., Silva M. J. Environ. Health Perspect. 2003;111:1164–1169. doi: 10.1289/ehp.5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser R., Meeker J. D., Singh N. P. Hum. Reprod. 2007;22:688–695. doi: 10.1093/humrep/del428. [DOI] [PubMed] [Google Scholar]
- Shamsi M. B., Venkatesh S., Tanwar M. Indian J. Med. Res. 2010;131:675–681. [PubMed] [Google Scholar]
- Vickers A. E. M., Lucier G. W. Carcinogenesis. 1996;17:1235–1242. doi: 10.1093/carcin/17.6.1235. [DOI] [PubMed] [Google Scholar]
- Cook J. C., Klinefelter G. R., Hardisty J. F., Sharpe R. M., Foster P. M. Crit. Rev. Toxicol. 1999;29:169–261. doi: 10.1080/10408449991349203. [DOI] [PubMed] [Google Scholar]
- Ahbab M. A., Undeger U., Barlas N., Basaran N. Hum. Exp. Toxicol. 2014;33(3):230–239. doi: 10.1177/0960327113494903. [DOI] [PubMed] [Google Scholar]
- Erkekoglu P., Rachidi W., Yuzugullu O. G. Toxicol. Appl. Pharmacol. 2010b;248:52–62. doi: 10.1016/j.taap.2010.07.016. [DOI] [PubMed] [Google Scholar]
- Migheli A., Attanasio A., Schiffer D. J. Pathol. 1995;176:27–35. doi: 10.1002/path.1711760106. [DOI] [PubMed] [Google Scholar]
- Barlow K., Foster T. Nippon Rinsho. 2003;58:2527–2532. [Google Scholar]
- Gazouli M., Yao Z. X., Boujrad N., Corton J. C., Culty M., Papadopoulos V. Endocrinol. 2002;143:2571–2583. doi: 10.1210/endo.143.7.8895. [DOI] [PubMed] [Google Scholar]
- Suna S., Yamaguchi F., Kimura S., Tokuda M., Jitsunari F. Toxicol. Lett. 2007;173:107–117. doi: 10.1016/j.toxlet.2007.06.015. [DOI] [PubMed] [Google Scholar]
- Cao J., Jiang L., Zhang X., Yao X., Geng C., Xue X., Zhong L. J. Trace Elem. Med. Biol. 2008;22:189–195. doi: 10.1016/j.jtemb.2008.03.005. [DOI] [PubMed] [Google Scholar]
- Macdonald J., Galley H. F., Webster N. R. Br. J. Anaesth. 2003;90:221–232. doi: 10.1093/bja/aeg034. [DOI] [PubMed] [Google Scholar]
- Haddad J.J., Land S.C., Tarnow-Mordi W.O., Zembala M., Kowalczyk D., Lauterbach R. J. Pharmacol. Exp. Ther. 2002;300:567–576. doi: 10.1124/jpet.300.2.567. [DOI] [PubMed] [Google Scholar]
- Zhang C., Zhang M., Sun Y., Li J., Fang M., Zhu X., Liu C. Nanfang Yike Daxue Xuebao. 2012;32:160–164. [Google Scholar]
- Erkekoglu P., Rachidi W., Yuzugullu O. G., Giray B., Favier A., Ozturk M., Hincal F. Toxicol. Appl. Pharmacol. 2010;248:52–62. doi: 10.1016/j.taap.2010.07.016. [DOI] [PubMed] [Google Scholar]
- Wang W., Craig Z. R., Basavarajappa M. S., Gupta R. K., Flaws J. A. Toxicol. Appl. Pharmacol. 2012;258:288–295. doi: 10.1016/j.taap.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambruosi B., Uranio M. F., Sardanelli A. M., Pocar P., Martino N. A., Paternoster M. S., Amati F., Dell'Aquila M. E. PLoS One. 2011;6:e27452. doi: 10.1371/journal.pone.0027452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richburg I. K., Hallmark N., McKinnell C. Endocrinology. 2009;146:613–623. doi: 10.1210/en.2004-0671. [DOI] [PubMed] [Google Scholar]
- Soleimani-Mehranjani M., Noorafshan A., Momeni H. R., Abnosi M. H., Mahmoodi M., Anvari M. Asian J. Androl. 2009;11:508–516. doi: 10.1038/aja.2009.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahbab M. A., Barlas N., Karabulut G. Toxicol. Ind. Health. 2017;33(2):133–146. doi: 10.1177/0748233715603847. [DOI] [PubMed] [Google Scholar]