

 Sulfanilamide based nonionic surfactant can self-assemble into haemocompatible and non-toxic niosomes that are promising for drug delivery.
Sulfanilamide based nonionic surfactant can self-assemble into haemocompatible and non-toxic niosomes that are promising for drug delivery.
Abstract
Biocompatible surfactants are of diverse pharmaceutical interest due to their ability to self-assemble into nano-particulate systems which can be used for single-step drug loading, based upon the hydrophobic–hydrophobic interaction between a hydrophobic drug and the lipophilic part of a surfactant molecule. However, surfactants are associated with cytotoxicity and hemolysis due to their amphiphilic interaction with cellular membranes. This study reports a novel membrane-compatible surfactant, synthesized from sulfanilamide and its self-micellization into niosomes. The surfactant was synthesized in a single step reaction via the introduction of an alkyl chain in the sulfanilamide moiety by conjugation with deconyl chloride. The synthesized surfactant (S-SDC) was characterized by 1H and 13C NMR, mass spectrometry and single crystal XRD. The S-SDC niosomes were explored for drug delivery with clarithromycin as a model drug. The biocompatibility of the surfactant was investigated through hemolysis and cytotoxicity. The surfactant presented a very low critical micellar concentration (CMC) of 0.04 mM and entrapped 65% of the drug which was released in a sustained manner, over 12 h, at acidic and physiological pH. The vesicles were spherical in shape with 234 ± 3.61 nm mean diameter and a narrow size distribution. Niosomes were hemocompatible and nontoxic to cellular membrane. The results suggested the sulfanilamide based surfactant can be applied as a novel and cell membrane compatible niosomal drug delivery vehicle.
Introduction
Surfactants are physiologically significant and versatile biological molecules. Being amphiphilic in structure, they have a lipophilic tail and hydrophilic head that enable them to interact with both non-polar and polar compounds.1 Surfactants are the major construction blocks of several chemical, physical and biological systems. They are attractive to the scientific community due to their unique solubilization and surface wetting capabilities with diverse applications such as the mining, petroleum, pharmaceutical and cosmetic industries.2–4 Nonionic surfactants are the most important type of surfactants that do not dissociate upon contact with an aqueous environment, owing to the absence of ionizable moieties. Their dispersion capability is directly linked with the length of their lipophilic tail. They are versatile solubilizing agents due to their low critical micelle concentration (CMC) and present low toxicity, cost effectiveness and biocompatibility.5,6 Nonionic surfactants with an amine group in their structure are highly soluble in water and used in hand washing products and liquid soaps with positive effects on skin.6–8 They have been reported to have antibacterial action against both Gram positive, negative bacteria and different fungal strains.9 Furthermore, in vivo plasma proteome association of non-ionic niosomes is low as compared to metal or polymeric nanomaterials which can help in achieving in vivo drug delivery targets.10–12
Recently, nonionic surfactants have been of great pharmaceutical interest due to their use in nano-vesicular drug delivery systems. Niosomes are nonionic-surfactant based vesicles that have been widely explored as an alternative to liposomes. Liposomes are formulated from expensive phospholipids and possess lower microbiological activity and chemical stability as compared to niosomes. Nonionic surfactants spontaneously self-assemble into vesicles in aqueous media and encapsulate water insoluble drugs in their lipid bilayers or water soluble drugs in the aqueous pool of the vesicles.13–15 Non-ionic lipid based niosome systems also show biocompatibility and safe delivery across the blood brain barrier.16,17 Niosomes share a number of similarities with liposomes such as preparation methods, microscopic morphologies and biophysical properties.18 The exceptional properties of niosomes are biocompatibility, ability to enhance the solubility of lipophilic drugs and permeability enhancement, supporting the potential use of nonionic surfactants as nano-carrier systems.19 Niosomes have also been reported to effectively inhibit P-glycoproteins (P-gp), thus resulting in the enhanced bioavailability of anticancer and antiviral drugs.20
Here, we report the synthesis and characterization of a novel, sulfanilamide-based nonionic surfactant (S-SDC) (Fig. 1) for encapsulation and biocompatible delivery of hydrophobic drugs. The surfactant was characterized through NMR, mass spectrometry, FTIR and XRD for its structural properties. The CMC and biocompatibility of S-SDC were explored using cell culture and blood hemolysis assays. The drug loaded niosomal vesicles of the S-SDC were characterized for shape, size, size distribution, and drug loading capacity, stability and in vitro drug releasing behavior.
Fig. 1. Physicochemical stability of the drug entrapped inside S-SDC niosomes. The niosomes retained the original functional groups of the drug after entrapment as corroborated by FTIR (A). DSC analysis revealed suppression of the crystalline peaks of the drug and S-SDC after niosomal formulation (B). TGA analysis presents the weight loss and thermal stability of the excipients (C). The ORTEP drawing of the X-ray structure of surfactant S-SDC was obtained from single crystal XRD analysis (D).

Experimental
Materials
All the solvents used were of HPLC grade. Sulfonilamide, deconyl chloride, polylysine (PLL), Dulbecco's Modified Eagle's medium (DMEM), fetal bovine serum (FBS) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich, Germany. Cholesterol and Tween 80 were purchased from BDH, UK, and Merck, Germany, respectively.
Synthesis of S-SDC
Potassium thiocyanate KSCN (194 mg, 2 mM) was dissolved in 25 mL HPLC grade acetone inside a 100 mL round bottom flask. Deconyl chloride (0.47 mL, 2 mM) was added to the reaction mixture and refluxed for 2 h at 80 °C. Then sulfanilamide (344 mg, 2 mM) was added to the reaction flask and refluxed for a further 16 h at 80 °C. The reaction was monitored through thin layer chromatography (TLC) using ethyl acetate and n-hexane (3 : 7, v/v) as the solvent system. Upon completion of the reaction, the mixture was cooled to room temperature and excess of acetone was evaporated using a rotary evaporator (BUCHI, 131 Rotavapor, Switzerland). The resulting mixture was added to water, filtered, dried and purified via column chromatography, using ethyl acetate and n-hexane (2 : 8, v/v) as the solvent system. The product was obtained as a dry solid. The synthesis scheme is presented in Scheme 1.
Scheme 1. Synthesis scheme of surfactant S-SDC.

Yield
80%, m.p. 143.8–144.5 °C.
FT-IR (KBr)
3388 cm–1 (–NH2), 3274.9 cm–1 (NH), 2922 cm–1 (CH2 asymmetric), 2854 cm–1 (CH2 symmetric), 1600 cm–1 (C C aromatic), 1324.8 cm–1 (C–N), 1156.9 cm–1 (C–S).
EI-MS
Observed M+, m/z 384.7 and calculated value for the formula C17H27N3O3S2, m/z 384.70, Fig. S1.†
1H NMR (400 MHz, DMSO, TMS)
δ: 0.845 (t, 3H, CH3, J = 6.8 Hz), 1.255 (m, 12H, CH2), 1.670 (t, 2H, CH2, J = 7.2 Hz), 4.992 (s, 2H, CH2), 7.878 (q, 4H, CH, J = 8.8 Hz), Fig. S2.†
13C NMR (300 MHz, MeOD)
δ: 3. 14.4 (CH3), 23.6 (CH2), 25.8 (β-CH2), 37.5 (α-CH2), 124.5 (o-aromatic carbon), 127.7 (m-aromatic carbon), 176.9 (carbonyl carbon), 180.0 (–NHCSNH– carbon), Fig. S3.†
Single-crystal X-ray diffraction
Single-crystal X-ray diffraction data was collected on a Bruker APEXII D8 Venture diffractometer fitted with a PHOTON 100 detector (CMOS technology) and a fine-focus sealed tube having X-ray source [Cu Kα radiation, α = 1.54178 Å]. Reflection intensities were integrated using SAINT software. Absorption 14.4 (CH3), 23.6 (CH2), 25.8 (β-CH2), 37.5 (α-CH2), 124.5 (o-aromatic carbon), 127.7 (m-aromatic carbon), 176.9 (carbonyl carbon), 180.0 (–NHCSNH– carbon). Correction was done on a Multi-scan (SADABS; Bruker, 2009). The structure was solved using SHELXTL. Empirical formula C17H27N3O3S2; formula weight 770.06; crystal system triclinic; space group P1[combining macron]; unit cell dimensions a = 4.947(2) Å, b = 11.412(4) Å, c = 18.159(5) Å, β = 92.23(3)°; volume 1016.9(6) A3; Z = 2; calculated density 1.257 mg m–3; F(000) 411; crystal size 0.33 × 0.12 × 0.0 mm; θ range for data collection 2.44 to 48.65°; total 17 873 reflections were collected, out of which 3859 were judged observed (Rint = 0.0354). Final R indices; R1 = 0.1899 for [I > 2σ(I)], wR2 = 0.4838, R indices were R1 = 0.2040, wR2 = 0.5043 for all data, goodness of fit; 5.031, largest diff. peak and hole; 5.464 and –0.736 e Å–3.
Critical micellar concentration
The CMC of S-SDC in methanol was determined by UV-visible spectrophotometry via monitoring the discontinuity in absorbance, upon micellization, against concentration. S-SDC was dissolved in methanol in a concentration range of 0.005 to 0.1 mM. All the concentrations were read spectrophotometrically (Shimadzu UV-240, Japan) and the CMC was determined by plotting the maximum absorbance versus the respective concentrations.9
Niosome preparation and characterization
Preparation of drug loaded niosomes
Drug loaded niosomes were prepared via the thin film rehydration method, using clarithromycin as a model drug. The S-SDC (15 mg) and cholesterol (5 mg) were dissolved in a solvent system of methanol and chloroform (4 : 6 v/v, 30 mL). The drug (7 mg) was dissolved in methanol (12 mL) and then added into the S-SDC/cholesterol solution. The evaporation of all the organic solvents resulted in the formation of a thin lipid film that was further dried under vacuum. The lipid film was rehydrated with PBS (7 mL, pH 7) at 45 °C for 25 min. The vesicles obtained were further sonicated (LABSONIC L, B. Braun Biotech International, USA) for 4 min at 25 °C. The niosomal suspension was stored at 4 °C for further experiments.
Shape, size and size distribution
The shape, size and size distribution of clarithromycin loaded niosomes were investigated using AFM (Agilent, 5500, USA). For imaging, a drop of the formulation was placed on a freshly cleaved mica surface, air-dried at 25 °C and visualized under a microscope. The imaging was performed in non-contact mode. The mean diameter and size distribution of the vesicles were obtained from the AFM data. Transmission electron microscopy (TEM) was performed by drying a drop of the niosomes on a copper grid and bloating with 1% ammonium molybdate. The samples were dried overnight under vacuum and imaging was performed at 15 kV with JEOL JEM 1400.
Drug entrapment efficiency
The drug entrapment efficiency of S-SDC niosomes was determined using a UV-visible spectrophotometer (Shimadzu UV-240, Japan). The vesicle suspension (1 mL containing 1 mg of the drug) was centrifuged (Universal 16, Hettich, Germany) at 5000 rpm for 10 min to remove the free drug. The free insoluble drug settled as a pellet. Next, the suspension was centrifuged at 12 000 rpm for 30 min at 4 °C to pellet down the vesicles. The clear supernatant was analyzed by DLS and no noticeable vesicles were found to be suspended. The isolated pellet of the vesicles was further washed twice with PBS. The vesicles were dissolved in methanol and diluted up to a specific volume. The sample was read spectrophotometrically for the quantification of clarithromycin. The following formula was used to determine the drug entrapment efficiency: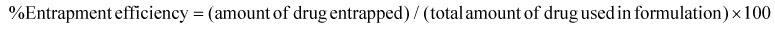
DLS and zeta potential measurement
A DTS0012 cell was used to measure the average hydrodynamic size distribution. The number of runs was 5 cycles and the time duration was 10 s. A dip cell was used for the measurement of zeta potential; number of runs: 5, measurements: 3.
FT-IR study
To investigate the possible interactions of the drug with niosomal excipients, the IR spectra of clarithromycin, cholesterol, S-SDC and drug loaded niosomes were recorded from 400 to 4000 cm–1 using an IR spectrometer (Shimadzu, Kyoto, Japan). To obtain self-supporting disks, all samples were diluted with KBr powder at 1% (w/w) and pressed.
DSC analysis
To investigate the physical state of the drug inside the niosomes, the thermal behavior of pure clarithromycin, S-SDC, cholesterol and drug loaded S-SDC niosomes was analyzed using TA instruments SDT Q600 under a nitrogen atmosphere at 50 kPa pressure with a heating rate of 10 °C min–1. Each sample (2 mg) was heated from room temperature up to 800 °C at a rate of 10 °C min–1.
TGA study
A TGA study of S-SDC, pure clarithromycin, cholesterol and drug loaded vesicles was carried out using a TA instruments SDT Q600 system. Samples (10 mg) were placed in a crucible and scanned with a thermal ramp over a temperature range of 25–800 °C at a heating rate of 10 °C min–1 in a nitrogen atmosphere at 50 kPa pressure.
In vitro release study
An in vitro drug release study of the niosomes was performed at two different pH values (pH 7.4 and 1.2) using a UV-Vis spectrophotometer. Drug loaded niosomes containing 3 mg of clarithromycin were taken in 4 mL of phosphate buffer (pH 7.4 and 1.2), packed into the dialysis membrane (12 000 kDa MWCO) and placed in a beaker containing 50 mL phosphate buffer at the respective pH. The beaker was placed on shaker at 100 rpm at 37 °C. At specific time intervals, 2 mL media was withdrawn which was subsequently replaced with fresh media. Clarithromycin released into the media was quantified at 288 nm.
Biocompatibility studies
Blood hemolysis
S-SDC niosomes were tested for hemolysis and RBC compatibility using fresh human blood. Blood was centrifuged at 700 g for 10 min to separate the red blood cells (RBCs) from the plasma. The isolated RBCs were washed three times with PBS (pH 7.4) and centrifuged again as above. The RBC pellet was suspended in PBS in a 1 : 10 ratio (RBC : PBS, w/v). To the RBC suspension (200 μL), test samples (4 mL) were added in different concentrations (62.5–1000 mg mL–1). Tween 80 was used as a positive control. After 4 h incubation at 37 °C, the non-lysed RBCs were removed from the samples through centrifugation (700g for 10 min). The supernatant was collected and analyzed spectrophotometrically at 540 nm to quantify the hemolysis. RBCs added to PBS were used as negative control. The percentage hemolysis was determined by the following equation,Hemolysis % = (Abt – Ab0)/(Ab100 – Ab0) × 100where Abt, Ab0 and Ab100 are the absorbance of the test samples, a solution of 0% hemolysis and a solution of 100% hemolysis, respectively.21
1 mL of blood was collected from the human subject. Written informed consent was obtained from the subject. The study is approved by the Institutional Review Board of Dow University of Health Sciences with the protocol number: IRB-864/DUHS/Approval/2017/51. The experiments were performed in accordance with ethical guidelines of Ethical Review Committee HEC Research Institute of Chemistry, University of Karachi.
Cell culture study
NIH/3T3 cells were cultured in DMEM containing FBS (10%) and antibiotics (streptomycin and penicillin, 50 units per mL–1 of each) in 5% CO2 humidified atmosphere at 37 °C. Cells were seeded into 96-well plates at a density of 8.0 × 103 cells per well in culture medium (200 mL). After incubation for 24 h, the medium was replaced with fresh medium (200 mL) containing various concentrations (30–90 mM) of S-SDC niosomes. For the negative control, cells were incubated with the media only. The cells were grown for a further 24 h. MTT solution (20 μL of 5 mg mL–1 in PBS) was added into each well. After 4 h incubation, the medium containing unreacted dye was removed. The obtained purple formazan crystals were dissolved in 200 μL per well DMSO and the absorbance was measured in a microplate reader (ELx808, BioTek, USA) at 570 nm. Tween 80 and poly (L) lysine (PLL) were used as negative and positive controls, respectively. The following formula was used to calculate the cells viability for the test samples after 24 h,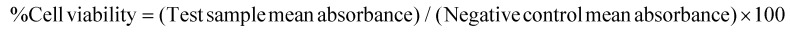
Statistical analysis
All the experiments were carried out in triplicate. The data of the study is expressed as mean ± SEM. Two-way ANOVA followed by a Bonferroni post-test was used for finding statistical significance. P < 0.05 were considered statistically significant.
Results and discussion
Synthesis and characterizations of S-SDC
An EI-MS molecular ion peak was observed at m/z 384.7 that corresponds to the molecular formula C17H27N3O3S2, Fig. S1.† The 1H NMR shows a 3 H triplet of methyl protons at 0.845 ppm with a coupling constant of 6.8 Hz, a 12H multiplet of methylene protons at 1.25 ppm, a 2 H triplet of methylene protons at 1.67 ppm with a coupling constant of 7.2 Hz, a 2 H singlet methylene proton at 4.99 ppm, a 4 H quartet of Ar–H protons at 7.87 ppm with a coupling constant of 8.8 Hz as shown in Fig. S2.† The 13C NMR shows chemical shifts at 14.4 (CH3), 23.6 (CH2), 25.8 (β-CH2), 37.5 (α-CH2), 124.5 (o-aromatic carbon), 127.7 (m-aromatic carbon), 176.9 (carbonyl carbon), 180.0 (–NHCSNH– carbon) Fig. S3.†
The FT-IR spectrum showed characteristic peaks at 3390 cm–1 for –NH2, at 3273 cm–1 for –NH, at 2922 cm–1 for CH2 asymmetric, at 2852 cm–1 for CH2 symmetric, 1697.2 cm–1 for C O, at 1598 cm–1 for C C aromatic, at 1317 cm–1 for C–N, at 1157 cm–1 for C–S which confirm the formation of the desired compound as shown in Fig. 1A.
Compound S-SDC was crystallized from ethyl acetate and n-hexane as solvents. The ORTEP view (Fig. 1D) shows the structure is composed of a planer benzene ring (C-1–C-6) with sulfonyl amide (S3/O2–O3/N3) and N-decanone thiourea (S4/O1/N1–N2/C-8–C-17) moieties attached at C3 and C6 respectively.
Physicochemical stability of the drug in S-SDC niosomes
Clarithromycin showed a characteristic peak of the C O stretching vibration from the ketone group in the lactone ring at 1692.1 cm–1, a O–C O stretching vibration in the lactone ring at 1731.2 cm–1, –O– ether function bands at 1171.5–1052.8 cm–1, alkyl-CH3 substitution bands at 2974.8–2939.5 cm–1 and OH bands at 3471.1 cm–1 as shown in Fig. 1A. S-SDC showed absorption peaks at 3388 cm–1 for –NH2, at 3274.9 cm–1 for –NH, at 2922 cm–1 for CH2 asymmetric, at 2854 cm–1 for CH2 symmetric, 1690 cm–1 for C O, at 1600 cm–1 for C C aromatic, at 1324 cm–1 for C–N, at 1157 cm–1 for C–S. The IR spectra of S-SDC based niosomes showed absorptions at 3391.8 and 3277.0 cm–1 that correspond to –NH2 and –NH stretching of the synthesized surfactant S-SDC. –CH2 asymmetric and symmetric stretching was observed at 2927.4 and 2854.3 cm–1. The O–C O stretching vibration of the lactone ring of the drug is shifted from 1731.2 to 1698.2 cm–1 and the stretching vibration of C O is shifted to 1596.8 cm–1 which could be due to the change in the environment of these groups.22 The alkyl-CH3 substitution and OH absorption bands of the drug were covered by strong absorption bands of the –CH2 asymmetric absorption at 2927.4 cm–1 and –NH2 absorption in the 3390 cm–1 region. The absorption of the ether functional group (–O–) in the drug is observed as a strong peak at 1151.2 cm–1. All the major absorption bands of the drug and carrier were present in the spectra of the drug loaded S-SDC niosomes which indicates that there is no chemical interaction and the drug is present intact in the niosomes.
In order to investigate the physical state of the drug inside the niosomes, a DSC study was performed for clarithromycin, S-SDC, cholesterol and the drug loaded S-SDC niosomes. The DSC thermogram of clarithromycin showed an endothermic peak at 225 °C as shown in Fig. 1B. The surfactant S-SDC showed an endothermic peak at 139.14 °C. Cholesterol showed two endothermic peaks at 143.17 and 297.94 °C, respectively. The drug loaded niosomal vesicles showed an endothermic peak at 137 °C.
TGA was used to assess the thermal stability of the clarithromycin loaded niosomal formulation. Surfactant S-SDC started losing weight at 28.60 °C (2.86%) and lost almost 100% of its weight at 359.78 °C (Fig. 1C). Cholesterol was shown to be thermally stable as it started losing weight at 248 °C. Clarithromycin was also stable as it started weight loss at 239.37 °C (6.55%). The drug loaded vesicles showed good stability against the applied heat and started weight loss at 175.95 °C (2.15%) and 90% of weight was lost at 790.67 °C. The DSC behavior of drug loaded S-SDS niosomes revealed the amorphous nature of surfactant and drug after niosome formation. The endothermic peak at 137 °C in drug loaded niosomes corresponds to the endothermic peak of surfactant S-SDC. It is slightly decreased and indicates a decrease in the crystallinity of the surfactant in the niosomes. The endothermic peaks of the drug did not appear in the drug loaded niosomes. The DSC results confirmed the FT-IR data showing the drug in intact form inside the niosomes. The results suggested that the synthesized surfactant and the encapsulated drug remained stable in the niosome formulation.
Shape, size and entrapment efficiency of S-SDC niosomes
AFM imaging revealed the drug loaded S-SDC niosomes to be rounded in shape as shown in Fig. 2A and B. The vesicles were found to be distributed in a size range of 178–263 nm with a mean diameter of 234 ± 3.61 nm (Fig. 2C). The size distribution of drug loaded vesicles showed the homogeneous nature of their population. TEM images showed monodispersed niosomes with thick shells (Fig. 2D).
Fig. 2. AFM images of S-SDC based drug loaded niosomal vesicles (A, B), size distribution of vesicles (C) and TEM images of S-SDC niosomes (D).

The homogeneous size of the vesicles in the nano range contributes to the increased permeability of the drug across biological membranes, avoids enzymatic inactivation, prevents rapid or abrupt drug release in the physiological environment, and reduces adverse effects.23,24 Drug entrapment efficiency of S-SDC niosomes was 65 ± 2% (Table S1†). This indicates that 0.29 mg of the drug was entrapped against each mg of the lipid phase (S-SDC and cholesterol). Increased drug entrapment efficiency is a vital parameter for drug cargo systems as it indicates the likelihood of a sustained release pattern of the drug and increased accumulation in the target sites, if functionalized for targeted delivery.19,25 High drug entrapment of the S-SDC can be linked to the lipophilic environment in the surfactant layer of the vesicles by virtue of its alkyl chain and cholesterol. Secondarily, the presence of cholesterol (25% of the total lipid phase in the case of S-SDC based niosomes in this study) also enhances the entrapment of the drug and avoids drug leaching from the vesicles.26–28 The role of cholesterol in the mixed vesicular system of niosomes, is to improve the monodispersity of the vesicles and enhance the membrane integrity by imparting stability, decreasing the fluidity and resisting the destabilization of bilayer vesicles into monolayer micelles.29
DLS and zeta potential measurement
The average zeta size distribution by intensity was 270.1 nm for S-SDC and the value of the zeta potential was noted at about –31 mV using a dip cell (Table S1†). The high negative value of the zeta potential can be attributed to the amide functionality of the sulfanilamide head group of surfactant. The mean diameter of >200 nm can be attributed to the hydrophobic nature of the niosomal surfaces, the drug and its interaction with the surfactant head groups. This, in turn, increases the mutual repulsion of the surfactant vesicles, which may provide space for more drug molecules to be entrapped.30 The difference in actual and hydrodynamic size of the niosomes, as studied by AFM and DLS respectively, is around 35 nm which can be explained as being due to hydrophilic nature of the niosomes. The additional water molecules concentrated around the nanoparticle surfaces and Brownian motion imparts an increase in their hydrodynamic size.
Critical micellar concentration (CMC)
The synthesized surfactant S-SDC was screened for the CMC. A low CMC value of around 0.04 mM was observed by analyzing both peaks from the spectrum of absorbance vs. concentration (Fig. 3A). It has been reported that a discontinuity in absorbance against concentration occurs due to micellization, thus this method provides an easy and effective way for determination of the CMC.9 The UV-visible spectrum of the surfactant in methanol, at room temperature, is given in Fig. 3B. The lower CMC value of the S-SDC can be attributed to the alkyl chain length (C10 for S-SDC). The CMCs of the non-ionic surfactants are inversely related to the hydrophobic alkyl chain while directly related to the molecular weight of the hydrophilic part of the surfactant molecule.31,32 A lower CMC is advantageous for amphiphilic molecules when they are exploited for drug delivery purposes. It reduces the surface tension, emulsification behavior and increases the drug entrapment by the surfactant. A lower value of CMC is also promising for drug delivery vehicles as it predicts an efficient in vivo performance and integrity of the drug loaded micelles or vesicles, even after dilution in the bloodstream.33
Fig. 3. Change in linearity of absorption vs. concentration of S-SDC shows a CMC value for S-SDC of 0.4 mM (A) and the corresponding UV-visible spectrum of S-SDC and its CMC determination at 307 nm and 252 nm (B). In vitro drug release study of clarithromycin loaded S-SDC based niosomal vesicles at pH 7.4 and 1.2 (C). Data is represented as mean ± SEM, n = 3.

In vitro release study
The in vitro drug release study was carried out at 7.4 and 1.2 pH respectively. The results are summarized in Fig. 3C. No abrupt release of the drug was observed from the formulation at any stage of the study. The study revealed that the S-SDC based niosomes are stable at physiological (7.4) as well as acidic pH (1.2).
However, the drug release pattern at acidic pH was not sustained and 50% drug release was achieved at the 4th h of the study. In the case of pH 7.4, the drug release was sustained as 50% drug release from the vesicles was achieved at the 8th h. The drug release continued in the same pattern up to 18 h for both pH values. Drug release patterns like this have been reported for other niosomal nanocarriers, attributing the slow release of drugs at physiological pH to the integrity of niosomal membrane.34,35 The sustained release, up to 12 h, of the drug from the S-SDC based niosomal vesicles can be related to its increased drug entrapment efficiency as the more drug is retained inside the vesicle, the release profile will be more sustained.36,37 Furthermore, 50% of the drug was released in 4 h at acidic pH, however, oral absorption of the nanoparticles can occur within 1 h and drug release will be sustained subsequently after absorption into the blood stream.34,38,39 The in vitro drug release profile of S-SDC niosomes reveal their suitability for both oral and intravenous administration.
Biocompatibility studies
Blood hemolysis
S-SDC was screened for blood hemolysis and it was found to be hemocompatible, causing negligible release of hemoglobin from RBCs even at the highest concentration of 1000 μg mL–1. It caused 5.78 ± 0.32 and 7.91 ± 0.56% hemolysis at 500 and 1000 μg mL–1 concentrations as compared to 15.34 ± 0.89 and 19.67 ± 1.29% hemolysis caused by Tween 80 at 500 and 1000 μg mL–1 concentrations, respectively (Fig. 4A). The longer alkyl chain length from C14–C18 and amphiphilic nature of free surfactant molecules also interact with RBCs membrane and contribute in hemolysis. The hemolysis assay of S-SDC niosomes revealed significantly less hemolysis as compared to the control and indicates the suitability of S-SDC niosomes for in vivo applications.40,41 The negligible blood hemolysis of our newly synthesized surfactant can be attributed to its nonionic nature.
Fig. 4. RBCs hemolysis (A) and cytotoxicity (B) of S-SDC micelles.

Cell culture study
The in vitro cytotoxicity of S-SDC was investigated in mouse embryonic fibroblast cells NIH/3T3 by MTT assay. The surfactant showed improved cell viability at the highest concentration as compared to the negative control of Tween 80. It showed 93.45 ± 3.78, 86.21 ± 2.41 and 78.84 ± 2.06% cell viability in a concentration range of 30–90 mM, respectively. While Tween 80 showed 88.71 ± 2.50, 74.66 ± 3.39 and 62.89 ± 2.65% cell viability in a concentration range of 30–90 mM, respectively as presented in Fig. 4B. The lower cytotoxicity and increased cell viability of the surfactant can be attributed to its nonionic nature and saturation in the lipophilic part of its structure, as reported in the literature.27 The results indicate that the novel nonionic surfactant is hemocompatible and presented no toxicity to the cells in MTT assay. These results support further exploration of the novel sulfanilamide based surfactants for their biomedical and drug delivery applications.
Conclusions
Surfactants are versatile amphiphilic molecules having a wide range of applications. Novel and cheap nonionic surfactants have been the subject of great scientific interest for both synthetic chemists and drug delivery scientists. Here, we synthesized a sulfanilamide based biocompatible surfactant to formulate drug loaded niosomes by a single step thin-film rehydration. The surfactant showed decent drug entrapment in the form of niosomes. The niosomes were spherical in shape with 234 nm mean diameter, with homogeneous size distribution, presented hemocompatibility and no toxicity when subjected to blood hemolysis and cytotoxicity assays. The drug was released in a sustained manner from the niosomes. i.e., 80% of the drug was released in 12 h at pH 7.4. Based on these results, the newly synthesized surfactant is safe, stable, biocompatible and shows promising potential for drug entrapment and sustained release over a 12 h period. S-SDC niosomes can be further explored for oral bioavailability and pharmacokinetics studies of hydrophobic drugs.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tx00108a
References
- Kumar R. S., Arunachalam S. Biophys. Chem. 2008;136:136–144. doi: 10.1016/j.bpc.2008.05.007. [DOI] [PubMed] [Google Scholar]
- Söderlind E., Wollbratt M., von Corswant C. Int. J. Pharm. 2003;252:61–71. doi: 10.1016/s0378-5173(02)00599-9. [DOI] [PubMed] [Google Scholar]
- Rahate A. R., Nagarkar J. M. J. Dispersion Sci. Technol. 2007;28:1077–1080. [Google Scholar]
- Neto V., Granet R., Krausz P. Tetrahedron. 2010;66:4633–4646. [Google Scholar]
- Imran M., Shah M. R., Ullah F., Ullah S., Elhissi A. M., Nawaz W., Ahmad F., Sadiq A., Ali I. Drug Delivery. 2016:1–12. doi: 10.1080/10717544.2016.1214991. [DOI] [PubMed] [Google Scholar]
- Ullah I., Shah A., Badshah A., Rana U. A., Shakir I., Khan A. M., Khan S. Z. J. Surfactants Deterg. 2014;17:1013–1019. [Google Scholar]
- Aguiar J., Carpena P., Molina-Bolıvar J., Ruiz C. C. J. Colloid Interface Sci. 2003;258:116–122. [Google Scholar]
- Flores M. V., Voutsas E. C., Spiliotis N., Eccleston G. M., Bell G., Tassios D. P., Halling P. J. J. Colloid Interface Sci. 2001;240:277–283. doi: 10.1006/jcis.2001.7627. [DOI] [PubMed] [Google Scholar]
- Ullah I., Shah A., Badshah A., Shah N. A., Tabor R. Colloids Surf., A. 2015;464:104–109. [Google Scholar]
- Wang M., Siddiqui G., Gustafsson O. J., Käkinen A., Javed I., Voelcker N. H., Creek D. J., Ke P. C., Davis T. P. Small. 2017;13:1701528. doi: 10.1002/smll.201701528. [DOI] [PubMed] [Google Scholar]
- Javed I., Sun Y., Adamcik J., Wang B., Kakinen A., Pilkington E. H., Ding F., Mezzenga R., Davis T. P., Ke P. C. Biomacromolecules. 2017;18:4316–4322. doi: 10.1021/acs.biomac.7b01359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., Gustafsson O. J., Siddiqui G., Javed I., Kelly H. G., Blin T., Yin H., Kent S. J., Creek D. J., Kempe K. Nanoscale. 2018;10:10863–10875. doi: 10.1039/c8nr00835c. [DOI] [PubMed] [Google Scholar]
- Uchegbu I. F., Vyas S. P. Int. J. Pharm. 1998;172:33–70. [Google Scholar]
- Imran M., Shah M. R., Ullah F., Ullah S., Elhissi A. M., Nawaz W., Ahmad F., Sadiq A., Ali I. Int. J. Pharm. 2016;505:122–132. doi: 10.1016/j.ijpharm.2016.03.042. [DOI] [PubMed] [Google Scholar]
- Carafa M., Santucci E., Alhaique F., Coviello T., Murtas E., Riccieri F., Lucania G., Torrisi M. R. Int. J. Pharm. 1998;160:51–59. [Google Scholar]
- Sohail M. F., Sarwar H. S., Javed I., Nadhman A., Hussain S. Z., Saeed H., Raza A., Bukhari N. I., Hussain I., Shahnaz G. Toxicol. Res. 2017;6:814–821. doi: 10.1039/c7tx00146k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman M., Madni A., Shi D., Ihsan A., Tahir N., Chang K., Javed I., Webster T. Nanoscale. 2017;9:15434–15440. doi: 10.1039/c7nr05216b. [DOI] [PubMed] [Google Scholar]
- Valdés K., Morilla M. J., Romero E., Chávez J. Colloids Surf., B. 2014;117:1–6. doi: 10.1016/j.colsurfb.2014.01.029. [DOI] [PubMed] [Google Scholar]
- Ullah S., Shah M. R., Shoaib M., Imran M., Elhissi A. M., Ahmad F., Ali I., Shah S. W. A. Drug Delivery. 2016:1–41. doi: 10.1080/10717544.2016.1196768. [DOI] [PubMed] [Google Scholar]
- Mahale N., Thakkar P., Mali R., Walunj D., Chaudhari S. Adv. Colloid Interface Sci. 2012;183:46–54. doi: 10.1016/j.cis.2012.08.002. [DOI] [PubMed] [Google Scholar]
- Cenni E., Granchi D., Avnet S., Fotia C., Salerno M., Micieli D., Sarpietro M. G., Pignatello R., Castelli F., Baldini N. Biomaterials. 2008;29:1400–1411. doi: 10.1016/j.biomaterials.2007.12.022. [DOI] [PubMed] [Google Scholar]
- Mohammadi G., Nokhodchi A., Barzegar-Jalali M., Lotfipour F., Adibkia K., Ehyaei N., Valizadeh H. Colloids Surf., B. 2011;88:39–44. doi: 10.1016/j.colsurfb.2011.05.050. [DOI] [PubMed] [Google Scholar]
- Hao Y., Zhao F., Li N., Yang Y., Li K. a. Int. J. Pharm. 2002;244:73–80. doi: 10.1016/s0378-5173(02)00301-0. [DOI] [PubMed] [Google Scholar]
- Tavano L., Gentile L., Rossi C. O., Muzzalupo R. Colloids Surf., B. 2013;110:281–288. doi: 10.1016/j.colsurfb.2013.04.017. [DOI] [PubMed] [Google Scholar]
- Muzzalupo R., Tavano L., La Mesa C. Int. J. Pharm. 2013;458:224–229. doi: 10.1016/j.ijpharm.2013.09.011. [DOI] [PubMed] [Google Scholar]
- Abdelkader H., Alani A. W., Alany R. G. Drug Delivery. 2014;21:87–100. doi: 10.3109/10717544.2013.838077. [DOI] [PubMed] [Google Scholar]
- Aramaki K., Yamada J., Tsukijima Y., Maehara T., Aburano D., Sakanishi Y., Kitao K. Langmuir. 2015;31:10664–10671. doi: 10.1021/acs.langmuir.5b02454. [DOI] [PubMed] [Google Scholar]
- Manconi M., Sinico C., Valenti D., Lai F., Fadda A. M. Int. J. Pharm. 2006;311:11–19. doi: 10.1016/j.ijpharm.2005.11.045. [DOI] [PubMed] [Google Scholar]
- Nematollahi M. H., Pardakhty A., Torkzadeh-Mahanai M., Mehrabani M., Asadikaram G. RSC Adv. 2017;7:49463–49472. [Google Scholar]
- Essa E. Asian J. Pharm. 2010;4:227. [Google Scholar]
- Zhang T., Marchant R. E. J. Colloid Interface Sci. 1996;177:419–426. [Google Scholar]
- El Achouri M., Alehyen S., Assioui A., Chami R., Bensajjay F., Pérez L., Infante M. R. J. Surfactants Deterg. 2013;16:473–485. [Google Scholar]
- de Medeiros Modolon S., Otsuka I., Fort S. b., Minatti E., Borsali R., Halila S. Biomacromolecules. 2012;13:1129–1135. doi: 10.1021/bm3000138. [DOI] [PubMed] [Google Scholar]
- Bayindir Z. S., Yuksel N. J. Pharm. Sci. 2010;99:2049–2060. doi: 10.1002/jps.21944. [DOI] [PubMed] [Google Scholar]
- Imran M., Shah M. R., Ullah F., Ullah S., Sadiq A., Ali I., Ahmed F., Nawaz W. Artif. Cells, Nanomed., Biotechnol. 2016:1–12. doi: 10.1080/21691401.2016.1246451. [DOI] [PubMed] [Google Scholar]
- El-Laithy H. M., Shoukry O., Mahran L. G. Eur. J. Pharm. Biopharm. 2011;77:43–55. doi: 10.1016/j.ejpb.2010.10.011. [DOI] [PubMed] [Google Scholar]
- Uchegbu I. F., Florence A. T. Adv. Colloid Interface Sci. 1995;58:1–55. [Google Scholar]
- Calleja P., Espuelas S., Vauthier C., Ponchel G., Irache J. M. J. Pharm. Sci. 2015;104:2877–2886. doi: 10.1002/jps.24354. [DOI] [PubMed] [Google Scholar]
- Javed I., Hussain S. Z., Ullah I., Khan I., Ateeq M., Shahnaz G., Rehman H. ur, Razi M. T., Shah M. R., Hussain I. J. Mater. Chem. B. 2015;3:8359–8365. doi: 10.1039/c5tb01258a. [DOI] [PubMed] [Google Scholar]
- Javed I., Hussain S. Z., Shahzad A., Khan J. M., Rehman M., Usman F., Razi M. T., Shah M. R., Hussain I. Colloids Surf., B. 2016;141:1–9. doi: 10.1016/j.colsurfb.2016.01.022. [DOI] [PubMed] [Google Scholar]
- Tao L., Uhrich K. E. J. Colloid Interface Sci. 2006;298:102–110. doi: 10.1016/j.jcis.2005.12.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.