

Abstract
Cerebral ischemic damage and infarction are well documented in stroke, which is presenting a foremost health concern globally with very high mortality and morbidity rates. Mechanisms that are associated with excitotoxicity, inflammation and oxidative stress are found to be critically involved in ischemic damage. Adverse effects of current therapies are imposing the need in development of neuroprotective agents that are very effective. To explore this we experimentally induced ischemic brain injury and investigated the effects of plumbagin. Induction of cerebral infarction and ischemia-reperfusion (I/R) was done by middle cerebral artery occlusion (MCAO) in Sprague-Dawley rats. Plumbagin (50, 100 or 200 mg/kg b.wt) was intragastrically administered for 9 days before ischemia induction and an hour prior on the day of ischemic insult. Plumbagin treatment attenuated pulmonary edema, neuronal apoptosis and reduced cerebral infarct volume. Cleaved caspase-3 and apoptotic cascade protein expressions were regulated. Overproduction of pro-inflammatory cytokines (TNF-α, IL-1β and IL-6) and nitric oxide (NO) following I/R were reduced. Prior plumbagin administration had down-regulated NF-κB signalling and MMP-2 and MMP-9 expression. Overall, the results reveal the potent neuroprotective efficacy of plumbagin against I/R-induced brain injury via effectively modulating apoptotic pathways, MMPs and neuro-inflammatory cascades.
Keywords: Apoptosis, Cerebral infarction, Inflammation, Matrix metalloproteinases, Nuclear factor-kappa B, Plumbagin
1. Introduction
The World Health Organization has reported that around 15 million people suffer from stroke (Hoffmann et al., 2012), which is the leading public health concern for both developing and developed countries. Stroke presents itself with very high mortality and morbidity rates and also is documented with high relapse rates (Doyle et al., 2008, Hong and Saver, 2009). Tissue-plasminogen activator (t-PA), administered within 3 h of onset of stroke, is the currently employed FDA-approved therapy. However, t-PA therapy reportedly increases the risk of haemorrhage and cerebral injury via inflammatory cascades (Guan et al., 2013). A better understanding of the underlying mechanisms would aid in the identification and development of more effective drugs. Several mechanisms have been demonstrated to be involved in cerebral ischemic injury including neuronal apoptosis, inflammatory responses, excitotoxicity, disruption of the blood brain barrier, oxidative stress and mitochondrial dysfunction (Pandya et al., 2011).
Nuclear factor kappa beta (NF-κB), important transcription factor is crucially involved in inflammatory responses (Gao et al., 2009). The NF-κB pathway is found to play pivotal roles in a various clinical conditions of CNS injury including, traumatic brain injury, transient focal and global ischemia (Williams et al., 2006). NF-κB signalling controls and regulates the expression of several genes involved in inflammatory responses as the pro-inflammatory cytokines, inducible enzymes, immune receptors and cell-adhesion molecules (Karin et al., 2002). NF-κB -induced expression of pro-inflammatory cytokines as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) are considered as the major inflammatory response mechanisms that contribute to ischemic brain injury (Barakat et al., 2014). Previous reports have illustrated the elevated levels of cytokines following sustained activation of NF-κB following 24 h of reperfusion following ischemia (Jin et al., 2015). Studies have also illustrated that matrix metalloproteinases (MMPs) exert major roles in ischemia/reperfusion induced brain injury (Rosell and Lo, 2008). MMPs as MMP-2, MMP-3 and MMP-9 were observed to be up-regulated following cerebral ischemia in animal experimental models (Chang et al., 2003). Further, TNF-α and IL-1β have been reported to induce the production of MMPs (Gottschall and Yu, 1995, Kauppinen and Swanson, 2005, Crocker et al., 2006). Thus targeting inhibition of the NF-κB and MMPs could be beneficial in ischemia/reperfusion-induced brain injury.
Recent researches have been focused on the neuroprotective effects of plant-derived compounds in stroke and ischemic injury (Tu et al., 2014, Lv et al., 2015, Antonisamy et al., 2015, Balamurugan, 2015, Rathi et al., 2015). Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone), a naphthoquinone derivative identified from the roots of Plumbago zeylanica (Sakamoto et al., 2009) has been demonstrated to possess extensive bioactive properties such as antioxidant (Tilak et al., 2004), anti-microbial (De Paiva et al., 2003); anti-cancer (Kawamura et al., 2014), anti-inflammatory (Checker et al., 2010) and neuroprotective (Luo et al., 2010) effects. Here, neuroprotective efficacy of plumbagin following ischemia/reperfusion-induced brain injury was investigated.
2. Materials and methods
2.1. Animals
Sprague-Dawley male rats that weighed 280–300 g were got from laboratory animal house of the University and were retained in sterile acrylic cages and monitored carefully under standard laboratory conditions (12 h: 12 h day/night cycle, 21–22 °C, humidity 55–60%). For 4–5 days prior experiment schedule, the animals were acclimatized to the new in-house conditions. The animals were fed on regular pellet diet and water at ad libidum. Experimental protocols were approved by the Animal Studies Committee of the University. Animal handling in this study were conducted in compliance to the guidelines of the National Animal Welfare Law of China and international guidelines for the care of laboratory animals (Garber, 2011).
2.2. Chemicals and antibodies
Plumbagin was purchased from Sigma–Aldrich (St. Louis, MO, USA). Primary antibodies against MMP-2, MMP-9, cleaved caspase-3, Bcl-2, Bax, β-actin, Bad, Bcl-xL were procured from Abcam, USA. TNF-α, NF-κBp65, IκBα, p-IκBα, p-IKKα, IKKα p-IKKβ, IKKβ, and horseradish peroxidase-labeled IgG secondary antibodies were procured from Cell Signaling Technology Inc. (Danvers, MA, USA). ELISA kits for determination of cytokines -TNF-α, IL-1β and IL-6 were obtained from Biolegend (San Diego, CA, USA). Cell lysis buffer for Western blotting analysis was purchased from Beyoime Institute of Biotechnology (Beijing, China). All the other chemicals used for analysis were from Sigma–Aldrich, if otherwise are specified.
2.3. Study design
The rats were allocated as 5 separate groups randomly (n = 18/group). Plumbagin was dissolved in saline and was administered intragastrically to rats at dose of 50, 100 or 200 mg/kg b.wt for 9 days prior ischemia/reperfusion induction. On the day of ischemic insult, a single dose of plumbagin was given 1 h prior induction of ischemia.
The rats were fasted overnight prior ischemic insult, however, had free access to water. The following day, animals were anesthetized by administering zoletil (Virbac Laboratories, Carros, France) at 25 mg/kg b.wt; i.p. The induction of cerebral infarction and ischemia/reperfusion (I/R) was performed by middle cerebral artery occlusion (MCAO) method as previously described by Leonardo et al. (2010). In brief, the left common carotid arteries (CCA) were open through a ventral midline neck incision. The internal carotid artery (ICA) was isolated and its extra cranial branch was ligated adjacent to its origin. Ischemia was induced with a 3–0 nylon monofilament suture interleaved via external carotid artery (ECA) into ICA in order to block MCA. The filament was maintained in place for 120 min. The suture was then withdrawn to reinstate the ICA-MCA blood flow. Reperfusion was allowed for about 24 h. Body temperature of the animals was kept constant at 37 °C using a thermostatically controlled heating pad until the animals recovered from surgery.
2.4. Animal grouping
Group 1 - Control (normal saline, (2 mL) intra-grastically); Group 2 - MCAO model; Group 3 - Plumbagin (50 mg/kg) + MCAO; Group 4 - Plumbagin (100 mg/kg) + MCAO; Group 5 – Plumbagin (200 mg/kg) + MCAO.
Rats that were administered equal volume of normal saline intragastrically instead of plumbagin and not subjected to MCAO, served as control. Rats that received saline and subjected to MCAO served as MCAO model. The rats from each experimental group were sacrificed following 24 h following reperfusion by transcardial perfusion of ice-cold saline followed by paraformaldehyde (4%) in 0.1 M phosphate buffer. Blood samples were collected and the brain tissues were removed immediate for analysis.
2.5. Neurobehavioral deficit evaluation
After reperfusion for 24 h the rats were evaluated for any neurobehavorial deficits. The assessment was based on - spontaneous activity, balance, symmetry of movements, symmetry of forelimbs, climbing ability, muscular co-ordination, reaction to touch, and response to vibrissae touch. The animals were scored on a five point scale as follows: 0 (no deficit) -normal; 1- mild deficit; 2- moderate deficit; 3-severe deficit; and 4- serious deficit (Longa et al., 1989).
2.6. Determination of levels of cytokines and NO
Inflammatory cytokines - TNF-α, IL-1β and IL-6 in the plasma were determined using ELISA kits (Biolegend). Levels of NO were assessed using NO assay kit (ab65328, Abcam), where utilizing nitrate reductase nitrate are converted to nitrites that gets converted to a deep purple azo compound with Griess Reagent. The intensity of the chromophore accurately reflects nitric oxide levels were measured at 540 nm using a 96-well microplate reader (Spectra MAX 340PC, Molecular Devices). The absorbance values were further analyzed using the software (Softmax Pro). The amount of NO was calculated using sodium nitrite (0–150 µM) as standard.
2.7. TTC staining
The brains were excised immediately after sacrifice and the wet weight was measured. 2,3,5-triphenyltetrazoliumchloride (TTC) staining was performed to assess brain viability and to determine the infarct size. The brain tissues were sectioned (2 mm sections) and were incubated with TTC for 30 min (at 37 °C). The tissue sections were then immersed in 4% para formaldehyde in dark (overnight). The infarct area was determined using NIH Image J software (Version1.42; National Institutes of Health, Bethesda, MD). Normal areas of the brain stain with TTC, while the infarct areas remain unstained. Infarct volume was calculated as follows - adding the infarct area of every section x thickness of the sections. The results are presented as percentage infarction/ ipsilateral hemisphere.
The brain samples (n = 6) were dried in an oven for 24 h (100 °C) to obtain the dry weight and the water content was derived (wet weight - dry weight)/wet weight × 100%.
2.8. TUNEL analysis
Neuronal apoptosis following I/R injury was assessed using Terminal transferase-mediated dUTP nickend-labeling (TUNEL) staining using DeadEnd TM fluorometric TUNEL system kit (Promega, Madision, WI, USA) as per manufacturer’s protocol. The paraffin-embedded brain tissue sections (5-μm) were subjected to analysis and the TUNEL positive cells were visualised and analyzed using NIS-Elements BR imaging processing and analysis software (Nikon Corporation, Japan).
2.9. Immunoblotting
The brain hemispheres (n = 6) following I/R were homogenized on ice using cell lysis buffer and were centrifuged at 4 °C for 15,000 rpm for 10 min. The total protein concentration of the supernatant was determined using Bicinchoninicacid (BCA) protein assay kit from Bio-Rad (Hercules, CA, USA). Further, to assess NF-κB (p65) expression both in the cytosolic and nuclear fractions, an aliquot of the homogenate was separated into fractions using an NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (Pierce Biotechnology, Rockford, IL, USA) according to the instructions specified by the manufacturer. Protein samples (50 µg) (for NF-κB (p65) from both the fractions/group) from different experimental groups were electrophoreitcally separated on SDS–PAGE (2%) and electro transferred on to polyvinylidene difluoride (PVDF) membranes (Invitrogen). The membrane was blocked (2 h at room temperature) with 5% BSA (Fetal Bovine Serum Albumin) in Tris-buffered saline containing Tween-20, 0.1% (TBST). The membranes were then incubated with respective primary antibodies, overnight at 4 °C. Following which, the blots were washed with TBST and further incubated with HRP-conjugated secondary antibodies (2 h; room temperature). The immunoreactive bands were visualized and scanned using Image Master II scanner (GE Healthcare, Milwaukee, WI, USA). The band densities were further analyzed by ImageQuant TL software (GE Healthcare, Milwaukee, WI, USA). The test protein expression levels were normalized with that of β-actin.
2.10. Statistical analysis
The data observed are expressed as mean ± SD derived from 6 individual experiments. Using SPSS software (version 21.0, SPSS Inc., Chicago, IL) all statistical analyses were performed. Multiple group comparisons were performed using ANOVA (one-way analysis of variance) followed Duncan’s Multiple Range Test (DMRT). Values at p < 0.05 were considered statistically significant.
3. Results
3.1. Plumbagin attenuated neurobehavioral deficits following MCAO
The effects of plumbagin on response and behavior of rats were examined. Prior 1 h of induction of cerebral ischemia, the rats scored normal for behavioral responses and neuromuscular coordination. Following MCAO significant (p < 0.05) neurological deficits were noticed (Fig. 1). The animals hesitated to walk and the left limbs were observed to exhibit considerably less flexibility and movement. The rats also exhibited difficulty to respond to stimuli. The neurological deficit was found to be lower substantially in rats that were treated with plumbagin as compared to MCAO controls. Plumbagin treatment markedly improved behavioral responses and motor coordination. Further 200 mg plumbagin treatment was more effective than 50 and 100 mg doses.
Fig. 1.
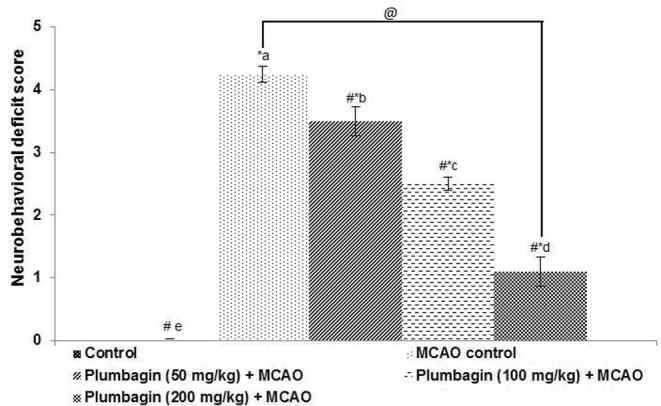
Plumbagin reduced neurobehavioral deficits following ischemia-reperfusion injury. Values are represented as mean ± SD, n = 6. p < 0.05 as determined by one-way ANOVA followed by DMRT analysis. * represents p < 0.05 vs control; # represents p < 0.05 vs MCAO control; @ represents MCAO control vs plumbagin (200 mg) + MCAO at p < 0.05. (a)–(e) represents mean values from different experimental groups that differ from each other at p < 0.05.
3.2. Plumbagin significantly reduced infarct area
TTC staining was performed to determine the extent of infarction following MCAO. The observations revealed the ischemic sections as unstained regions majorly in the fronto-parietal cortex area and in the striatum (Fig. 2). Rats that were prior treated with plumbagin for 10 days exhibited considerably (p < 0.05) lesser infarct volume as compared to MCAO controls. Also plumbagin decreased brain water content as well in a dose-dependent manner. The infarct volume decreased from 29.88 to 10.2 on treatment with 100 mg and 200 mg plumbagin, illustrating its protective effects.
Fig. 2.

Plumbagin reduced cerebral infarct area (A) brain edema following I/R injury (B). Values are represented as mean ± SD, n = 6. p < 0.05 as determined by one-way ANOVA followed by DMRT analysis. * represents p < 0.05 vs control; # represents p < 0.05 vs MCAO control; @ represents MCAO control vs plumbagin (200 mg) + MCAO at p < 0.05. (a)–(e) represents mean values from different experimental groups that differ from each other at p < 0.05.
3.3. Plumbagin inhibit neuron apoptosis following Ischemia/reperfusion injury
To further explore the neuroprotective effects of plumbagin TUNEL assay was performed. The results indicated that pre-treatment with plumbagin (50, 100 or 200 mg) significantly (p < 0.05) inhibited ischemia-induced neuronal cell death (Fig. 3a). Further, western blot analysis revealed enhanced cleaved caspase-3 expression along with elevated (p < 0.05) levels of Bax and Bad 24 h following I/R (Fig. 3b and c). Bcl-2 and Bcl-xL expressions were observed to be substantially (p < 0.05) inhibited indicating activation of apoptotic cascade. Interestingly, pre-treatment with plumbagin down-regulated cleaved caspase-3, Bax and Bad expression, while it elevated levels of Bcl-xL and Bcl-2. In line with TUNEL positive counts, the results of protein expression also illustrated significant neuroprotective effects of plumbagin against ischemia injury.
Fig. 3.

Plumbagin decreases neuronal apoptosis and regulates apoptotic pathway proteins (A and C). Expression of pro-apoptotic proteins (B and C). Values are represented as mean ± SD, n = 6. p < 0.05 as determined by one-way ANOVA followed by DMRT analysis. * represents p < 0.05 vs control; # represents p < 0.05 vs MCAO control; @ represents MCAO control vs plumbagin (200 mg) + MCAO at p < 0.05. (a)–(e) represents mean values from different experimental groups that differ from each other at p < 0.05. (L1-Control; L2-MCAO control; L3-Plumbagin (50 mg/kg) + MCAO; L4-Plumbagin (100 mg/kg) + MCAO; L5-Plumbagin (200 mg/kg) + MCAO).
3.4. Effects of plumbagin NF-κB signalling
Inflammatory responses have been well documented in I/R injury. NF-κB signalling is a key pathway in inflammatory responses (Fan et al., 2001, Tu et al., 2014). Following MACO, we noticed significantly (p < 0.05) increased NF-κB (p65) nuclear expression levels with substantial decrease in the NF-κB (p65) cytosolic fractions, indicating activation of the NF-κB pathway. Also, significantly (p < 0.05) up-regulated expression of TNF-α, IKKα, p-IKKα, IKKβ, p-IKKβ, IκBα and p-IκBα were noticed (Fig. 4) following 24 h of reperfusion. Prior administration of plumbagin caused marked (p < 0.05) inhibition of p- IKKα, p- IKKβ and p-IκBα expression vs MCAO alone group. The expression of total IKKα, IKKβ and IκBα though reduced were not significantly down-regulated. Further suppression of TNF-α and NF-κB p65 (nuclear fraction) expression were noticed on plumbagin treatment in comparison with MCAO control. These observations indicate down-regulation of the NF-κB signalling on plumbagin treatment.
Fig. 4.

Plumbagin down-regulated NF-κB activation following ischemia/reperfusion injury and regulates the inhibitor kinases of NF-κB signalling (A–C). Values are represented as mean ± SD, n = 6. p < 0.05 as determined by one-way ANOVA followed by DMRT analysis. Relative expressions of proteins from different treatment groups with control expression set at 100%. * represents p < 0.05 vs control; # represents p < 0.05 vs MCAO control; @ represents MCAO control vs plumbagin (200 mg) + MCAO at p < 0.05. (a)–(e) represents mean values from different experimental groups that differ from each other at p < 0.05. (L1-Control; L2-MCAO control; L3-Plumbagin (50 mg/kg) + MCAO; L4-Plumbagin (100 mg/kg) + MCAO; L5-Plumbagin (200 mg/kg) + MCAO).
3.5. Plumbagin reduced levels of inflammatory cytokines
NF-κB signalling pathway crucially regulates the expressions pro-inflammatory cytokines as IL-1β, IL-6 and TNF-α. These cytokines activate inflammatory response cascades and have been demonstrated to be involved in ischemia/ reperfusion - induced brain injury (Lv et al., 2015). NO is also a major inflammatory mediator. Multi-fold (p < 0.05) rise in the levels of plasma NO, IL-1β, IL-6 and TNF-α were observed in ischemia/reperfusion-induced rats as compared against normal controls (Fig. 5). Plumbagin at all the tested doses significantly (p < 0.05) reduced NO and cytokines in a dose-dependent manner.
Fig. 5.

Plumbagin - reduced the levels of inflammatory cytokines (A), reduced the levels of TNF-α (B) and also reduced the levels of NO (C). Values are represented as mean ± SD, n = 6. p < 0.05 as determined by one-way ANOVA followed by DMRT analysis. * represents p < 0.05 vs control; # represents p < 0.05 vs MCAO control; @ represents MCAO control vs plumbagin (200 mg) + MCAO at p < 0.05. (a)–(e) represents mean values for the same cytokine from different experimental groups that differ from each other at p < 0.05.
3.6. Plumbagin potentially reduced expressions of matrix metalloproteinases -2 and 9
The MMPs have been implicated in neuro inflammatory responses and play a vital role in I/R -induced brain injury. Here we determined the expression of MMP-2 and -9 after MCAO. I/R injury induced a multi-fold (p < 0.05) up-regulated expression of MMPs -2 and -9 (Fig. 4). Plumbagin (50, 100 or 200 mg) administration prior ischemic insult caused significant (p < 0.05) downregulation of MMP expression (Fig. 6), with 100 or 200 mg dose exhibiting more marked suppression than 50 mg.
Fig. 6.

Plumbagin reduced MMP-2 and -9 expression values are represented as mean ± SD, n = 6. p < 0.05 as determined by one-way ANOVA followed by DMRT analysis. Relative expression of proteins from different treatment groups with control expressions set at 100%. * represents p < 0.05 vs control; # represents p < 0.05 vs MCAO control; @ represents MCAO control vs plumbagin (200 mg) + MCAO at p < 0.05. (a)–(e) represents mean values for the same cytokine from different experimental groups that differ from each other at p < 0.05. (L1-Control; L2-MCAO control; L3-Plumbagin (50 mg/kg) + MCAO; L4-Plumbagin (100 mg/kg) + MCAO; L5-Plumbagin (200 mg/kg) + MCAO).
4. Discussion
Cerebral ischemia-induced brain injury has complex underlying pathophysiological mechanisms such as oxidative stress, inflammatory reaction, and neuronal apoptosis (Moskowitz et al., 2010). tPA therapy in stroke has limitations owing to its neurotoxic effects (Guan et al., 2013). Thus the need for development of clinically effective neuroprotective agents is high. In the present study the effects of plumbagin on experimentally-induced rodent model of cerebral infarction was explored.
Prior treatment with plumbagin (50, 100 or 200 mg) exerted substantial protective effects against I/R injury, as seen by a marked decrease in infarct area and apoptotic cell counts. Brain edema was considerably reduced as well. Plumbagin administration further improved neuromuscular coordination. Apoptosis after I/R is regarded as one of the key mechanisms that lead to neuronal cell death (Yao et al., 2012). Western blott analysis revealed significantly enhanced cleaved caspase-3 expression along with Bad and Bax proteins, suggesting activation of apoptotic pathways. The balance between pro-apoptotic proteins (Bad and Bax) and Bcl-2 and Bcl-xL, the anti-apoptotic proteins, crucially regulates the mitochondrial membrane integrity and the release of cytochrome C and apoptogenic factors that influence cell survival and cell death (Zhao et al., 2003). Interestingly, plumbagin treatment markedly improved the expression of Bcl-2 and Bcl-xL while down-regulating Bad and Bax and cleaved caspase-3. Thus observations suggest the effectiveness of plumbagin in inhibition of apoptosis.
Further, several studies have demonstrated that post-ischemic inflammation responses contribute substantially to neuronal damage and cerebral infarction (Cuartero et al., 2013). To elucidate if plumbagin had anti-inflammatory effects, the influence of plumbagin on NF-κB activation and signalling was assessed by western blotting. Also, TNF-α, IL-1β and IL-6 were determined following 24 h after reperfusion.
It is well documented that NF-κB, key transcription factor, regulates the expression of several proteins involved in immune responses (Karin et al., 2002). NF-κB (heterodimer with subunits p50 and p65) normally is bound to inhibitory proteins - inhibitors of NF-κB (IκBs) in the cytoplasm and remains inactive. In response to stimulus, IκB the inhibitor subunit gets phosphorylated and activated by IκB kinase (IKK) complex, and is rapidly degraded (Hansberger et al., 2007). The IKK complex consists of serine - threonine kinases- IKKα and β (Hayden and Ghosh, 2008). NF-κBp65 subunit then dissociates from its inhibitory protein IκBα and translocates to the nucleus and subsequently triggers the transcription of downstream target genes including- TNF-α, IL-1β and IL-6 (Hayden and Ghosh, 2008). In the present study up-regulated NF-κB activation as presented by increased expression of NF-κBp65 in the nuclear fraction was noticed following ischemia/reperfusion. Previous studies have demonstrated activation of NF-κB for following ischemic brain injury (Nurmi et al., 2004).
Enhanced expression of TNF-α and the levels of cytokines IL-1β and IL-6 in the plasma observed in the study reflect the activation of NF-kB signalling cascade following I/R. Plumbagin exposure resulted in the down-regulated phosphorylation of regulatory proteins of the pathway - IκBα, IKKα and IKK-β that lead to suppression of NF-κB activation. Reduced NF-κBp65 in the nuclear fraction reveals down-regulation of the pathway by plumbagin. Inhibition of NF-κB signalling is found to be neuroprotective as it leads to the inhibition of expression of inflammatory response genes (Jiang et al., 2011). NO, is known to be play a vital role in immune and inflammatory responses (Lv et al., 2015). Plumbagin-mediated reduced NO levels also illustrate the anti-inflammatory efficacy.
Accumulating evidences reports altered MMP activity to exert a vital role in the pathophysiology of cerebral ischemia. MMP-2 and MMP-9 are found to be activated following focal brain ischemia and are involved in the disruption of the BBB subsequently leading to brain injury in animal models (Rosell and Lo, 2008). Further, TNF-α has been found to be a potent inducer of MMP expression and also is associated with BBB disruption (Crocker et al., 2006). In the study, we observed markedly enhanced expression of MMPs- 2 and -9 following cerebral ischemia/reperfusion injury. Pre-treatment with plumbagin (50, 100 or 200 mg/kg) had effectively down-regulated the expression in a dose-dependent manner. The effective suppression of MMP-2 and -9 on plumbagin treatment thus prevents matrix degradation and BBB disruption. Plumbagin-mediated inhibition of TNF-α could have also contributed to the decrease expressions of MMPs.
Previous studies with MMP inhibitors have reported that pre and post-ischemic treatments with the inhibitors significantly reduce the infarct size and also the expressions of inflammatory mediators as TNF-α (Park et al., 2011). Thus the suppression of MMPs by plumbagin may have also possibly contributed to the decrease in cerebral infarct size observed.
5. Conclusion
The observed results indicate that plumbagin exerted neuroprotective effects against cerebral infarction-reperfusion induced brain injury via inhibition of apoptosis and effectively regulating NF-κB/TNF-α signalling and MMP -2 and -9.
Conflict of interest
The authors declare that they have no conflicts of interest.
Footnotes
Peer review under responsibility of King Saud University.
References
- Antonisamy P., Duraipandiyan V., Ignacimuthu S., Kim J.-H. Anti-diarrhoeal activity of friedelin isolated from Azima tetracantha lam. in wistar rats. South Ind. J. Biol. Sci. 2015;1:34–37. [Google Scholar]
- Balamurugan R. Smilax chinensis Linn. (Liliaceae) root attenuates insulin resistance and ameliorate obesity in high diet induced obese rat. South Ind. J. Biol. Sci. 2015;1:47–51. [Google Scholar]
- Barakat W., Safwet N., El-Maraghy N.N., Zakaria M.N. Candesartan and glycyrrhizin ameliorate ischemic brain damage through downregulation of the TLR signaling cascade. Eur. J. Pharmacol. 2014;724:43–50. doi: 10.1016/j.ejphar.2013.12.032. [DOI] [PubMed] [Google Scholar]
- Chang D.I., Hosomi N., Lucero J., Heo J.H., Abumiya T., Mazar A.P., del Zoppo G.J. Activation systems for latent matrix metalloproteinase-2 are upregulated immediately after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2003;23:1408–1419. doi: 10.1097/01.WCB.0000091765.61714.30. [DOI] [PubMed] [Google Scholar]
- Checker R., Sharma D., Sandur S.K., Subrahmanyam G., Krishnan S., Poduval T.B., Sainis K.B. Plumbagin inhibits proliferative and inflammatory responses of T cells independent of ROS generation but by modulating intracellular thiols. J. Cell. Biochem. 2010;110:1082–1093. doi: 10.1002/jcb.22620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuartero M.I., Ballesteros I., Moraga A., Nombela F., Vivancos J., Hamilton J.A., Corbí A.L., Lizasoain I., Moro M.A. N2 neutrophils, novel players in brain inflammation after stroke: modulation by the PPARγ agonist rosiglitazone. Stroke. 2013;44:3498–3508. doi: 10.1161/STROKEAHA.113.002470. [DOI] [PubMed] [Google Scholar]
- Crocker S.J., Milner R., Pham-Mitchell N., Campbell I.L. Cell and agonist-specific regulation of genes for matrix metalloproteinases and their tissue inhibitors by primary glial cells. J. Neurochem. 2006;98:812–823. doi: 10.1111/j.1471-4159.2006.03927.x. [DOI] [PubMed] [Google Scholar]
- De Paiva S.R., Figueiredo M.R., Aragão T.V., Kaplan M.A. Antimicrobial activity in vitro of plumbagin isolated from Plumbago species. Mem. Inst. Oswaldo Cruz. 2003;98:959–961. doi: 10.1590/s0074-02762003000700017. [DOI] [PubMed] [Google Scholar]
- Doyle K.P., Simon R.P., Stenzel-Poore M.P. Mechanisms of ischemic brain damage. Neuropharmacology. 2008;55:310–318. doi: 10.1016/j.neuropharm.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J., Ye R.D., Malik A.B. Transcriptional mechanisms of acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2001;281:L1037–L1050. doi: 10.1152/ajplung.2001.281.5.L1037. [DOI] [PubMed] [Google Scholar]
- Gao Y., Fang X., Tong Y., Liu Y., Zhang B. TLR4-mediated MyD88-dependent signaling pathway is activated by cerebral ischemia-reperfusion in cortex in mice. Biomed. Pharmacother. 2009;63:442–450. doi: 10.1016/j.biopha.2008.06.028. [DOI] [PubMed] [Google Scholar]
- Garber, J.C. (Chair), 2011. Committee for the update of the guide for the care and use of laboratory animals. Guide for the Care and Use of Laboratory Animals, eighth ed. National Academy of Sciences.
- Gottschall P.E., Yu X. Cytokines regulate gelatinase A and B (matrix metalloproteinase 2 and 9) activity in cultured rat astrocytes. J. Neurochem. 1995;64:1513–1520. doi: 10.1046/j.1471-4159.1995.64041513.x. [DOI] [PubMed] [Google Scholar]
- Guan T., Liu Q., Qian Y., Yang H., Kong J., Kou J., Yu B. Ruscogenin reduces cerebral ischemic injury via NF-κB-mediated inflammatory pathway in the mouse model of experimental stroke. Eur. J. Pharmacol. 2013;714:303–311. doi: 10.1016/j.ejphar.2013.07.036. [DOI] [PubMed] [Google Scholar]
- Hansberger M.W., Campbell J.A., Danthi P., Arrate P., Pennington K.N., Marcu K.B., Ballard D.W., Dermody T.S. I kappa B kinase subunits alpha and gamma are required for activation of NF-kappaB and induction of apoptosis by mammalian reovirus. J. Virol. 2007;81:1360–1371. doi: 10.1128/JVI.01860-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden M.S., Ghosh S. Shared principles in NF-kappa B signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Hoffmann A., Zhu G., Wintermark M. Advanced neuroimaging in stroke patients: prediction of tissue fate and hemorrhagic transformation. Expert Rev. Cardiovasc. Ther. 2012;10:515–524. doi: 10.1586/erc.12.30. [DOI] [PubMed] [Google Scholar]
- Hong K.S., Saver J.L. Quantifying the value of stroke disability out comes: WHO global burden of disease project disability weights for each level of the modified Rankin Scale. Stroke. 2009;40:3828–3833. doi: 10.1161/STROKEAHA.109.561365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W., Fu F., Tian J., Zhu H., Hou J. Curculigoside A attenuates experimental cerebral ischemia injury in vitro and vivo. Neuroscience. 2011;192:572–579. doi: 10.1016/j.neuroscience.2011.06.079. [DOI] [PubMed] [Google Scholar]
- Jin Z., Liang J., Wang J., Kolattukudy P.E. MCP-induced protein1 mediates the minocycline-induced neuroprotection against cerebral ischemia/reperfusion injury in-vitro and in-vivo. J. Neuroinflamm. 2015;12:39. doi: 10.1186/s12974-015-0264-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M., Cao Y., Greten F.R., Li Z.W. NF-kappa B in cancer: from innocent by stander to major culprit. Nat. Rev. Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- Kauppinen T.M., Swanson R.A. Poly (ADP-ribose) polymerase-1 promotes microglial activation, proliferation, and matrix metalloproteinase-9-mediated neuron death. J. Immunol. 2005;174:2288–2296. doi: 10.4049/jimmunol.174.4.2288. [DOI] [PubMed] [Google Scholar]
- Kawamura M., Kuriyama I., Maruo S., Kuramochi K., Tsubaki K., Yoshida H., Mizushina Y. Anti-tumor effects of novel 5-O-acyl plumbagins based on the inhibition of mammalian DNA replicative polymerase activity. PLoS One. 2014;9:e88736. doi: 10.1371/journal.pone.0088736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardo C.C., Hall A.A., Collier L.A., Green S.M., Willing A.E., Pennypacker K.R. Administration of a sigma receptor agonist delays MCAO-induced neurodegeneration and white matter injury. Translation Stroke Res. 2010;1:135–145. doi: 10.1007/s12975-009-0005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longa E.Z., Weinstein P.R., Carlson S., Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- Luo Y., Mughal M.R., Ouyang T.G., Jiang H., Luo W., Yu Q.S., Greig N.H., Mattson M.P. Plumbagin promotes the generation of astrocytes from rat spinal cord neural progenitors via activation of the transcription factor Stat3. J. Neurochem. 2010;115:1337–1349. doi: 10.1111/j.1471-4159.2010.06780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv Y., Qian Y., Fu L., Chen X., Zhong H., Wei X. Hydroxysafflor yellow A exerts neuroprotective effects in cerebral ischemia reperfusion-injured mice by suppressing the innate immune TLR4-inducing pathway. Eur. J. Pharmacol. 2015;769:324–332. doi: 10.1016/j.ejphar.2015.11.036. [DOI] [PubMed] [Google Scholar]
- Moskowitz M.A., Lo E.H., Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurmi A., Lindsberg P.J., Koistinaho M., Zhang W., Juettler E., Karjalainen-Lindsberg M.L., Weih F., Frank N., Schwaninger M., Koistinaho J. Nuclear factor-kappaB contributes to infarction after permanent focal ischemia. Stroke. 2004;35:987–991. doi: 10.1161/01.STR.0000120732.45951.26. [DOI] [PubMed] [Google Scholar]
- Pandya J.D., Sullivan P.G., Pettigrew L.C. Focal cerebral ischemia and mitochondrial dysfunction in the TNF alpha- transgenic rat. Brain Res. 2011;1384:151–160. doi: 10.1016/j.brainres.2011.01.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C.H., Shin T.K., Lee H.Y., Kim S.J., Lee W.S. Matrix metalloproteinase inhibitors attenuate neuroinflammation following focal cerebral ischemia in mice. Korean J. Physiol. Pharmacol. 2011;15:115–122. doi: 10.4196/kjpp.2011.15.2.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathi M.A., Meenakshi P., Gopalakrishnan V.K. Hepatoprotective activity of ethanolic extract of alysicarpus vaginalis against nitrobenzene-induced hepatic damage in rats. South Ind. J. Biol. Sci. 2015;1:60–65. [Google Scholar]
- Rosell A., Lo E.H. Multiphasic roles for matrix metalloproteinases after stroke. Curr. Opin. Pharmacol. 2008;8:82–89. doi: 10.1016/j.coph.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Sakamoto S., Taura F., Putalun W., Pongkitwitoon B., Tsuchihashi R., Morimoto S., Kinjo J., Shoyama Y., Tanaka H. Construction and expression of specificity-improved single-chain variable fragments against the bioactive naphthoquinone, plumbagin. Biol. Pharm. Bull. 2009;32:434–439. doi: 10.1248/bpb.32.434. [DOI] [PubMed] [Google Scholar]
- Tilak J.C., Adhikari S., Devasagayam T.P. Antioxidant properties of Plumbago zeylanica, an Indian medicinal plant and its active ingredient, plumbagin. Redox Rep. 2004;9:219–222. doi: 10.1179/135100004225005976. [DOI] [PubMed] [Google Scholar]
- Tu X.K., Yang W.Z., Chen J.P., Chen Y., Ouyang L.Q., Xu Y.C., Shi S.S. Curcumin inhibitsTLR2/4-NF-κB signaling pathway and attenuates brain damage in permanent focal cerebral ischemia in rats. Inflammation. 2014;37:1544–1551. doi: 10.1007/s10753-014-9881-6. [DOI] [PubMed] [Google Scholar]
- Williams A.J., Dave J.R., Tortella F.C. Neuroprotection with the proteasome inhibitor MLN519 in focal ischemic brain injury: relation to nuclear factor kappa B (NF-kappa B), inflammatory gene expression, and leukocyte infiltration. Neurochem. Int. 2006;49:106–112. doi: 10.1016/j.neuint.2006.03.018. [DOI] [PubMed] [Google Scholar]
- Yao R.Q., Qi D.S., Yu H.L., Liu J., Yang L.H., Wu X.X. Quercetin attenuates cell apoptosis in focal cerebral ischemia rat brain via activation of BDNF–TrkB–PI3K/Akt signaling pathway. Neurochem. Res. 2012;37:2777–2786. doi: 10.1007/s11064-012-0871-5. [DOI] [PubMed] [Google Scholar]
- Zhao H., Yenari M.A., Cheng D., Sapolsky R.M., Steinberg G.K. Bcl-2 overexpression protects against neuron loss within the ischemic margin following experimental stroke and inhibits cytochrome c translocation and caspase-3 activity. J. Neurochem. 2003;85:1026–1036. doi: 10.1046/j.1471-4159.2003.01756.x. [DOI] [PubMed] [Google Scholar]