

Abstract
Non-covalent interactions of organic moieties with Lewis acidic alkali cations can greatly affect structure and reactivity. Herein, we describe the effects of interactions with alkali metal cations within a series of reduced iron complexes bearing a redox-active formazanate ligand, in terms of structures, magnetism, spectroscopy, and reaction rates. In the absence of a crown ether to sequester the alkali cation, dimeric complexes are isolated wherein the formazanate has rearranged to form a five-membered metallacycle. The dissociation of these dimers is dependent on the binding mode and size of the alkali cation. In the dimers, the formazanate ligands are radical dianions, as shown by X-ray absorption spectroscopy, Mössbauer spectroscopy, and analysis of metrical parameters. These experimental measures are complemented by DFT calculations that show the spin density on the bridging ligands.
Synopsis
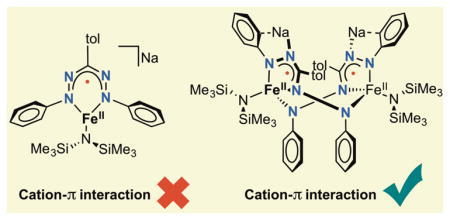
Cation-π interactions in reduced iron complexes bearing formazanate radical dianions induce a ligand rearrangement that results in the formation of dimeric complexes. The nature of the alkali cation affects both the structure and reactivity of the dimers.
Introduction
Alkali metal cations play essential roles in both biological chemistry1 and synthetic chemistry2 where they stabilize complex structures or act as cofactors in (bio)catalytic transformations. Detailed studies have shown that non-covalent interactions of organic moieties with redox-inactive metal cations can result in changes in electronic structure that affect geometry,3 redox potentials,4 N2 cleavage5, reaction rates6 and selectivity of chemical transformations.7 The nature of the interaction with s-block cations is greatly influenced by their charge (z) : size (r) ratio. Cations with a high z/r ratio have the capacity for bond polarization, inducing bond covalency.8 When descending the s-block metal groups, the radii of the metals significantly increase,9 resulting in predominantly ionic bonding and an increased tendency for ion dissociation. Noncovalent interactions in the form of cation-π contacts are weaker than covalent interactions but can play a significant role in metal cation association. The electrostatic nature of cation-π contacts causes the strength of this interaction to rise with increasing z/r ratio.8 Typically, these interactions are observed when the distance is below the sum of the van der Waals radii of the independent elements.10 In the absence of neutral co-ligands such as THF, Et2O or HMPA, aggregates can form resulting in low solubility or concentration dependent solution speciation. Multidentate ligands such as DME, TMEDA and crown ethers can sequester/stabilize the cation, thereby preventing aggregate formation, and at times drastically affect reactivity.11 The right cation and crown ether or cryptand combination can give rise to fully separated ion pairs, which is often used to facilitate crystallization. However, allowing cation-π interactions to persist could be a useful strategy to tune the electronic structure and corresponding reactivity of transition metal complexes.
In many examples where alkali metal cations are involved in homogeneous catalysis and activating strong chemical bonds, the alkali metal cations result from using the corresponding zerovalent alkali and alkaline earth metals for reductive activation of pre-catalysts.12 In addition to classical metal-centered reduction, electrons can be stored in redox-active ligands. These ligands have been shown to facilitate multi-electron processes in systems where the transition metal changes oxidation states by less than two electrons,13 and enable the formation of substrate-centered radicals.14 Given the increasing popularity of redox-active ligands15 that is demonstrated in this Forum, it is important to consider whether the reducing electron from the alkali metal goes into a metal- or ligand-centered orbital. Since redox-active ligands often contain extended π-systems and/or multiple heteroatoms that are potentially capable of Coulomb interactions with alkali cations, it is evident that cation-π interactions must be considered when evaluating redox-active ligands, especially in reductive reactions. Indeed, cation-π interactions have been observed for well recognized redox-active ligands, including catecholates,16 amidophenolates,17 o-phenylenediamides,18 salophens,19 dithiolenes20 and bis(imino)pyridines.21 Despite this link, there have been few systematic studies into the interplay of cation-π interactions and redox-active ligands. A recent example that demonstrates the effect that different alkali cations can have was reported by Mazzanti and coworkers, who showed that one-electron reduction of a (salophen)cobalt(II) complex results in either metal or ligand-centered reduction, depending on the identity of the alkali metal.22
As part of our interest in redox-active ligands and low-coordinate iron chemistry, we recently reported that Fe[N(SiMe3)2]223 gives access to low-coordinate iron formazanate complexes that undergo ligand-centered reduction at the formazanate ligand (Scheme 1).24 Formazanates have been established as good one-electron acceptors, particularly from the work of Otten and coworkers, who demonstrated ligand-centered reduction in various formazanates bound to redox-inert boron25 or zinc.26 Interestingly, a recently reported homoleptic formazanate iron(II) complex adopted a low spin iron(I) configuration upon one-electron reduction rather than generating a ligand-centered radical.27 We found that one-electron reduction of complex 1 gives a complex with a formazanate radical dianion (S = 1/2) that is strongly antiferromagnetically coupled to a high-spin iron(II) (S = 2) to give an overall S = 3/2 electronic configuration. When Na was used as the reductant in the presence of two equivalents of 12-crown-4, a mononuclear anionic iron complex 2 with a sequestered sodium cation was obtained. Herein, we report that one-electron reduction of 1 with alkali metals in the absence of a crown ether forms dimeric complexes in which alkali cation binding is connected to an isomerization of the formazanate into an unprecedented bridging mode, which changes the electron localization. Importantly, we show that the choice of alkali metal cation has a substantial effect on the structure and reactivity of these dimeric complexes.
Scheme 1.
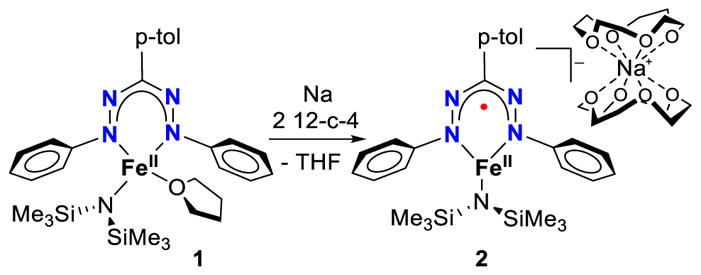
Previously reported ligand centered reduction of complex 1.
Results
The addition of Na sand to a purple THF solution of 1 at −78 °C gives a dark green solution upon warming to room temperature, similar to what is observed in the synthesis of complex 2. However, concentrating the mixture and addition of pentane, followed by cooling to −40 °C affords a differently colored complex 3-Na, which is a black crystalline solid in 71% yield (Scheme 2).
Scheme 2.
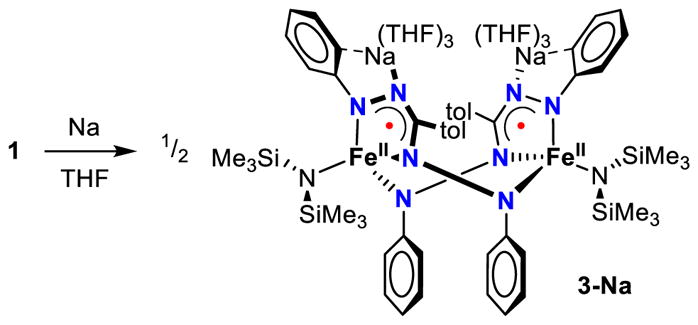
Reduction of complex 1 in the absence of 12-crown-4 to form dimer 3-Na.
Crystals suitable for single crystal X-ray diffraction were obtained by cooling a solution of 3-Na in a THF/pentane mixture to −40 °C. The solid state structure of 3-Na (Figure 1) revealed a dimeric structure wherein the formazanate ligand has rearranged to coordinate through N1 and N3 to form a five-membered chelate. The exocyclic nitrogen (N4) coordinates to the other iron center in the dimer. Ligand rearrangements to bind N1 and N3 have been observed in non-reduced formazanates bound to boron25 and zinc,26 but we are not aware of any previous examples where the exocyclic N4 atom coordinates to another metal. The two [Na(THF)3]+ ions have interactions with the internal N2 atoms of the five membered chelates and with nearby phenyl groups. The N–Na distances of 2.373(3) Å and 2.372(3) Å are comparable to those observed for non-isomerized formazanates bound to B (2.35 – 2.39 Å)28 and Zn (2.41 – 2.52 Å)29. The formazanate Fe–N bond lengths (2.006(3) – 2.067(3) Å) are all elongated compared to the solid state structure of 2 (1.935(5) – 1.958(5) Å), consistent with the higher coordination number of iron in 3-Na, which is four-coordinate in contrast to three-coordinate 2.
Figure 1.
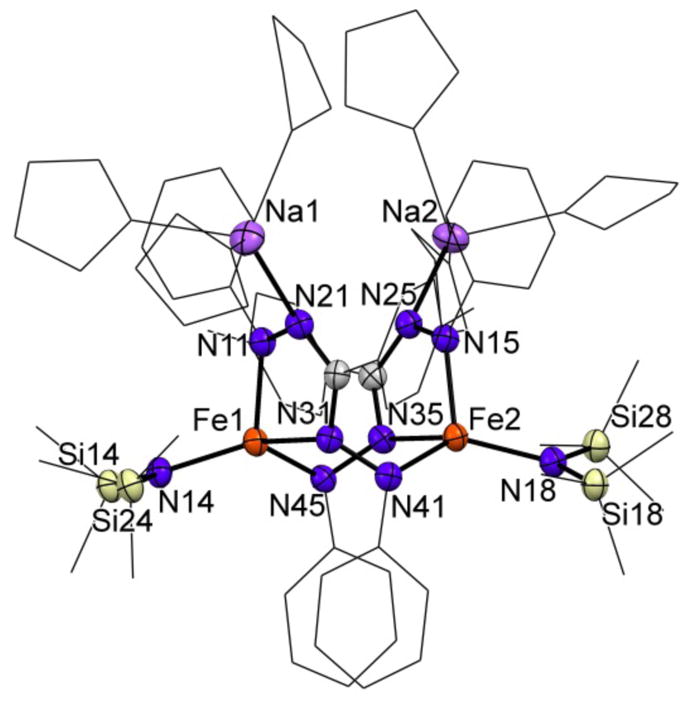
Displacement ellipsoid plot (50% probability) of complex 3-Na. Hydrogen atoms and a THF molecule have been omitted, and part of the structure is depicted as a wireframe for clarity. See Tables 1 and 2 for metrical parameters.
Metal complexes with multiple redox-active ligands can display ligand-based mixed valence, which is often reflected in distinct differences in intraligand metric parameters.29,30 Although the two formazanate ligands in 3-Na are not crystallographically related, there are no significant differences in bond lengths and angles (Table 1), indicating that both ligands are in the same oxidation state. Analysis of the intraligand bond lengths of the formazanate core shows an elongation of the N–N bonds (1.372(4) – 1.393(4) Å) compared to 1 (1.317(2) – 1.325(2) Å), similar but more pronounced to what is observed in 2 (1.377(6) and 1.345(6) Å), suggestive of a ligand-centered reduction (Chart 1). The C–N bonds within the formazanate core of complex 2 (1.343(7) and 1.339(8) Å) show no significant changes when compared to 1 (1.339(2) – 1.350(2) Å). However, in 3-Na a contraction of the N2–C1 bonds (both 1.323(5) Å) and an elongation of the C1–N3 bonds (1.370(5) and 1.363(5) Å) are observed, indicating more localized double bond character for the N2–C1 bond. A comparable observation was made for a similarly rearranged but non-reduced formazanate bound to Zn, which showed double bond localization for N2–C1 and N3–N4.26 In contrast, the N3–N4 bonds in 3 are significantly longer (1.393(4) and 1.378(4) Å), and are comparable to the N1–N2 bonds (1.372(4) and 1.375(4) Å), indicating that there is no double bond character for the N3–N4 bond as was observed for the non-reduced formazanate bound to Zn. The relative bond distances within the rearranged formazanate in 3-Na are very similar to DFT calculated values for a diamidoborane complex – formed upon intramolecular hydride reduction of the formazanate in a boron dihydride complex – with an identical binding mode as in 3-Na, yet protonated at N4.25a The possibility of ligand-centered reduction is further supported by reported examples of five-membered metallacycles consisting of isothiosemicarbazide π-radical anions bound to nickel(II)31 and iron(III)32 with the same N-N-C-N motive as in 3-Na. In these examples elongation of the N-N bonds and contraction of the C-N bonds was also observed upon reduction of a neutral ligand to the corresponding radical monoanion. The mononanionic radical form of these ligands is characterized by intraligand bond distances of 1.333(1) – 1.350(1) Å for the N–N bond. This is significantly elongated compared to N–N bond in the neutral form of the ligand (1.254(7) – 1.267(2) Å), similar to the difference between 1 and 3-Na. However, in the reported isothiosemicarbazide π-radical anions both C–N bonds are comparable (1.322(1) – 1.355(1) Å), and less localized than in 3-Na. Overall, the metrical analysis implies that the rearranged formazanate ligands in 3-Na bind iron as radical dianions.
Table 1.
Selected bond lengths (Å) for the dimers.
| 3-Na | 3-K | 3-Rb | 3-Cs | |
|---|---|---|---|---|
| Fe1–N14 | 1.993(3) | 1.987(4) | 1.9910(2) | 1.985(5) |
| Fe2–N18 | 1.981(3) | 1.991(4) | 1.984(2) | 1.983(5) |
|
| ||||
| Fe1–N11 | 2.014(3) | 2.051(3) | 2.043(2) | 2.025(5) |
| Fe2–N15 | 2.006(3) | 2.046(3) | 2.040(2) | 2.028(4) |
|
| ||||
| Fe1–N31 | 2.067(3) | 2.053(4) | 2.055(2) | 2.051(4) |
| Fe2–N35 | 2.063(3) | 2.055(4) | 2.044(2) | 2.066(4) |
|
| ||||
| Fe1–N45 | 2.032(3) | 2.055(3) | 2.053(2) | 2.030(5) |
| Fe2–N41 | 2.031(3) | 2.047(3) | 2.046(2) | 2.039(5) |
|
| ||||
| N11–N21 | 1.372(4) | 1.384(5) | 1.380(2) | 1.377(6) |
| N15–N25 | 1.375(4) | 1.387(5) | 1.380(2) | 1.367(6) |
|
| ||||
| N21–C11 | 1.323(5) | 1.329(5) | 1.317(2) | 1.324(7) |
| N25–C15 | 1.323(5) | 1.313(5) | 1.323(3) | 1.332(7) |
|
| ||||
| C11–N31 | 1.370(5) | 1.377(5) | 1.369(2) | 1.370(7) |
| C15–N35 | 1.363(5) | 1.378(5) | 1.359(2) | 1.358(7) |
|
| ||||
| N31–N41 | 1.378(4) | 1.390(5) | 1.382(2) | 1.369(6) |
| N35–N45 | 1.393(4) | 1.384(5) | 1.389(2) | 1.365(6) |
| M = Na | M = K | M = Rb | M = Cs | |
|
| ||||
| N21–M | 2.373(3) | 2.767(3) | 2.924(2) | 3.117(5) |
| N25–M | 2.372(3) | 2.766(3) | 2.889(2) | 3.092(5) |
Chart 1.
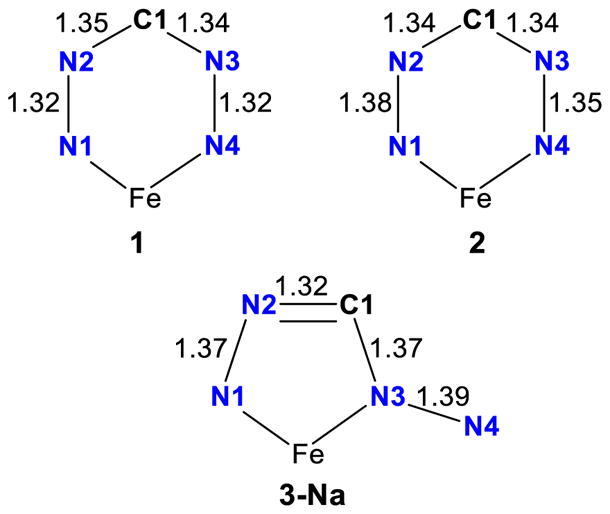
Averaged metrical details of the formazanate ligands in complexes 1, 2 and 3-Na. The average error of a given bond distance is ± 0.01 Å (3σ).
The zero-field Mössbauer spectrum of a solid sample of 3-Na at 80 K shows a slightly asymmetric doublet with isomer shift δ = 0.74 mm s−1 and quadrupole splitting |ΔEQ| = 2.30 mm s−1 (Figure 2). The observation of only one doublet indicates identical electronic structures and environments for the two iron sites in 3-Na. The observed isomer shift of 3-Na is similar to that of complex 1 (δ = 0.71 mm s−1), suggesting that the oxidation state at iron is the same in both cases. For additional insight, 3-Na was also analyzed by X-ray absorption spectroscopy at the Fe K-edge. The spectra of 1, 2 and 3-Na overlaid in Figure 3 are characterized by electric dipole forbidden but quadrupole-allowed 1s → 3d pre-edge transitions that appear at the base of the dipole-allowed 1s → np rising-edge that dominates a K-edge spectrum. Since the ligand field typically increases with increasing oxidation state for a given ligand set, the pre-edge energy often provides a useful marker for oxidation state.33 The centers of mass for pre-edge peaks in spectra obtained for 1, 2 and 3-Na are identical within experimental error at 7112.2 ± 0.1 eV. Moreover the pre- and rising-edge spectral profile for 1 and 3 are almost superimposable, clearly indicating the same effective nuclear charge and coordination geometry (tetrahedral) about the Fe ions in these complexes. Thus, both Mössbauer and XAS analysis indicate that 1, 2, and 3-Na have the same oxidation state at iron. Since 1 is clearly iron(II), this suggests an iron(II) oxidation state in 3-Na, which in turn implies that the formazanates are reduced to radical dianions - in agreement with the metrical analysis described above.
Figure 2.
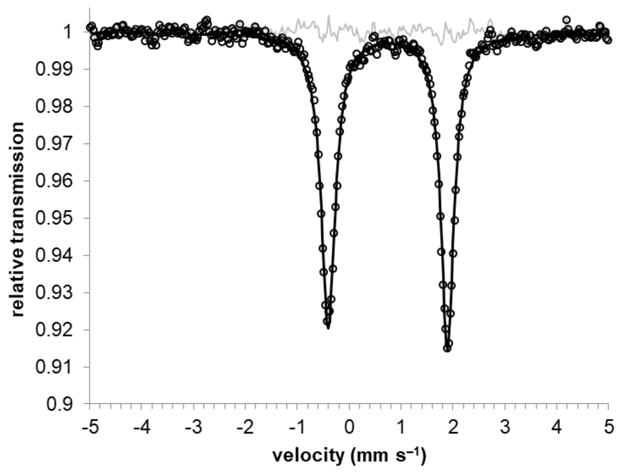
Zero-field Mössbauer spectrum of a solid sample of 3-Na at 80 K. The black circles are the data, the black line is a fit with two Lorentzians of equal intensity but slightly different line widths (γL = 0.328 and γR 0.304); the thin grey line is the residual from the fit.
Figure 3.
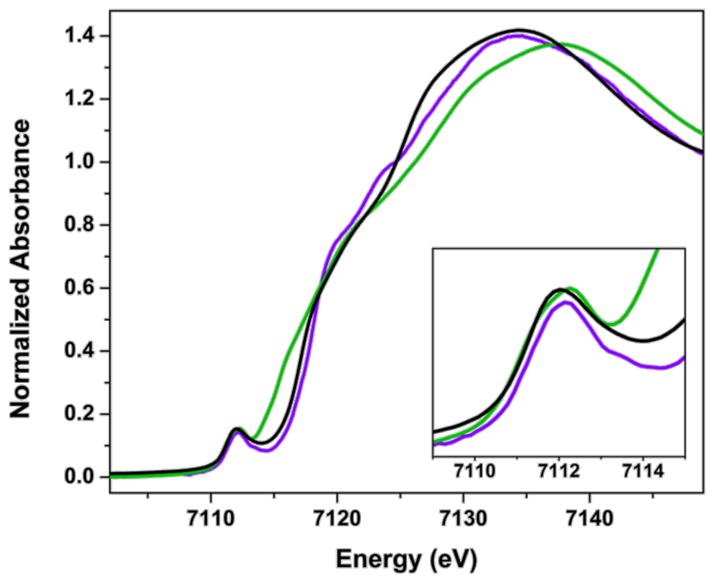
Overlay of the normalized Fe K-edge XAS spectra of 1 (violet), 2 (green), and 3-Na (black) at 10 K. Inset shows expansion of the pre-edge region.
The room temperature magnetic moment (μeff) in THF-d8 solution using the Evans method was 6.95 ± 0.3 μB, which is comparable to the value obtained from SQUID measurements of a solid sample in the temperature range 50–300 K, μeff = 7.6 μB (Figure S19). The experimental values are close to the spin-only (s.o.) value for four uncoupled spin centers, two SFe = 2 and two ligand radicals Srad = 1/2, namely μeff, s.o. = ge√[2(2 + 1) + 2(2 + 1) + 1/2(1/2 + 1) + 1/2(1/2 + 1)] = 7.3 μB. The temperature independence of μeff(T) in this temperature range indicates is consistent with this model requires that the various pair exchange coupling constants |Jij| between the four spins would need to be less than a few wavenumbers (Figure S19). Unfortunately, the low-temperature magnetic data are severely obscured by steep overshooting of μeff(T) below 50 K, presumably due to intermolecular interactions, which prevented us from further characterizing J values or zero-field splitting. Qualitative, rough fits (adopting grad = ge for the radicals) suggest weak to moderate zero-field splitting for iron, iron-radical coupling of |JFe-rad| < 5 cm−1 (Ĥex = −2J S1·S2), and gFe = 2.07.
It is not a priori clear why exchange coupling between iron(II) centers and their coordinated ligand radicals in 3-Na should be weak, whereas the monomeric ferrous compound 2 shows strong local spin coupling.24 We presume that the reason is small overlap and near-orthogonality of magnetic orbitals in the quasi-tetrahedral coordination. We also considered a model of very strong antiferromagnetic exchange coupling between each iron(II) centers and the ligand radical coordinated to it (as in 2), but weak coupling between the two sides. This model is made up of two local site spins S′= 3/2, and predicts a spin-only magnetic moment of μeff, s.o. = ge√[3/2(3/2 + 1) + 3/2(3/2 + 1)] = 5.5 μB, much lower than observed. Thus, the iron g-value would have to be gFe = 2.64 to match the experimental magnetic data. Since this value is far above the (geometrically averaged) g-value for 2 obtained from EPR simulations (gav = 2.35),24 this model of strong iron-radical coupling for 3-Na is not defensible, particularly as the corresponding large zero-field splitting of 2 (D = 16 cm−1) is not consistent with the magnetic data of 3-Na (Figure S19).
The 1H NMR spectrum of 3-Na in THF-d8 shows resonances in the range from +160 to −70 ppm, and the number and integration of the peaks indicates that the two SiMe3 groups are chemically inequivalent (Figure S1). Several protons on the formazanate are also split into pairs, suggesting that the dinuclear structure, with its lowered symmetry, is retained in solution. However, over time it fully converts to a new species (Figure S2), which has a similar 1H NMR spectrum and green color as complex 2 (Figure S3), consistent with dissociation of the dimer to a mononuclear species (4-Na, Scheme 3). The rate of the dissociation of 3-Na to 4-Na in THF-d8 was monitored between 273–313 K by 1H NMR spectroscopy (Figure 4), and the concentrations followed an exponential time dependence indicating a rate law rate = kobs[3-Na] (see SI for more details). Fitting the decay curves using Eq. 1 afforded rate constants, which were used to generate an Eyring plot (Figure S10) that gave the activation parameters ΔH‡ = 28.0 ± 1.1 kcal mol−1 and ΔS‡ = −14.8 ± 3.7 cal mol−1 K−1. The loss in entropy in the transition state indicates an ordered early transition state that could involve binding of exogenous THF molecules to the Na atoms. This is consistent with the observation that the addition of two equiv of 12-crown-4 to complex 3-Na significantly increased the rate of dimer dissociation, and gives mononuclear complex 2 as previously reported. Moreover, in the solid state structure of the K analogue of 4-Na (see below) four THF molecules are bound to K, supporting the idea that incorporation of an additional THF molecule is feasible.
Scheme 3.
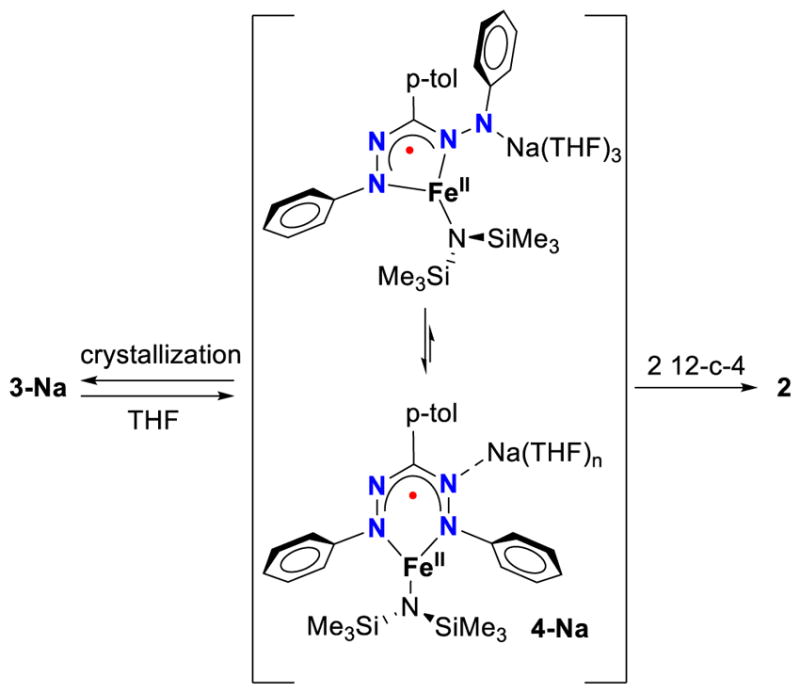
Dissociation of complex 3-Na in solution into a monomeric complex 4-Na in which the different formazanate binding modes are in equilibrium, and irreversible conversion into 2 by addition of a crown ether.
Figure 4.
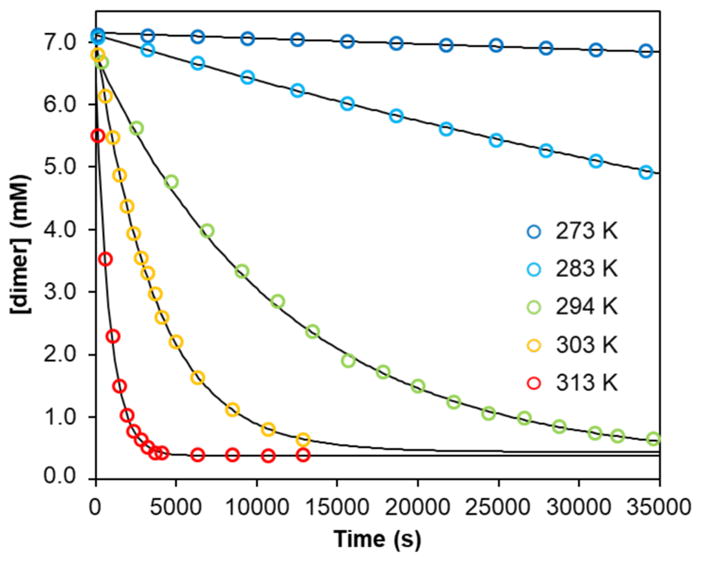
Concentration of dimer 3-Na vs time in THF solution at various temperatures. Open circles are data points and solid lines are fitted curves. Log plots of the data are in the Supporting Information (Fig. S11–S16).
| (1) |
Cooling mixtures of dissociated 4-Na gives no increase in dimer concentration as judged by 1H NMR spectroscopy, indicating that the dissociation of the dimer is irreversible in dilute solution. However, crystallization from THF/pentane mixtures where 3-Na has fully converted to 4-Na affords solely black crystals of the dimer 3-Na. Analysis of a frozen THF solution of 3-Na that has partially dissociated into 4-Na by Mössbauer spectroscopy showed two signals that have isomer shift and quadrupole splitting parameters within the error margin of those for pure samples of 2 and 3-Na (Figure S17). Although dissociation of the dimer is irreversible in THF solution, we propose that an equilibrium involving the different coordination modes of the formazanate is present that enables crystallization as a dimer (Scheme 3). The room-temperature 1H NMR spectra of 2 and 4-Na in THF-d8 both show a single species with a number of peaks indicating idealized C2v symmetry. However, the observed resonances broaden and start to deviate from Curie behavior below −50 °C (Figure S6 and S7), which could be due to intermediate exchange between formazanate binding modes on the NMR time scale. However, we were unable to resolve details because the spectra remained difficult to interpret at −90 °C, which was the lower temperature limit of the NMR spectrometer.
Intrigued by the change in formazanate binding mode induced by a coordinating Na cation, we investigated the effect of using different alkali metal reductants. The addition of MC8 (M = K, Rb or Cs) to a THF solution of 1 at −78 °C gives a dark green solutions upon warming to room temperature. Addition of pentane and cooling to −40 °C gave complexes 3-K, 3-Rb and 3-Cs as dark crystalline solids (Scheme 4). Interestingly, rapid crystallization of 3-K from a THF/pentane mixture afforded two different crystal morphologies. 1H NMR spectroscopy in THF-d8 and Mössbauer spectroscopy of the obtained solids, and single crystal X-ray diffraction revealed a mixture of dimer 3-K (see below) and monomer 4-K. Although monomer 4-K was a minor component in the mixture, its solid state structure can be analyzed (Figure 5). The FeN and intraligand bond lengths in 4-K are within the uncertainty limits of the solid state structure of complex 2. A notable difference is that the potassium cation is bound to N21, three THF molecules and has close contacts to C22 and C31. Crystallization of 5 from a THF solution with less pentane at −40 °C, thereby slowing down the crystallization process, gives a crystalline material comprising >95% 4-K based on 1H NMR and Mössbauer spectroscopies.
Scheme 4.
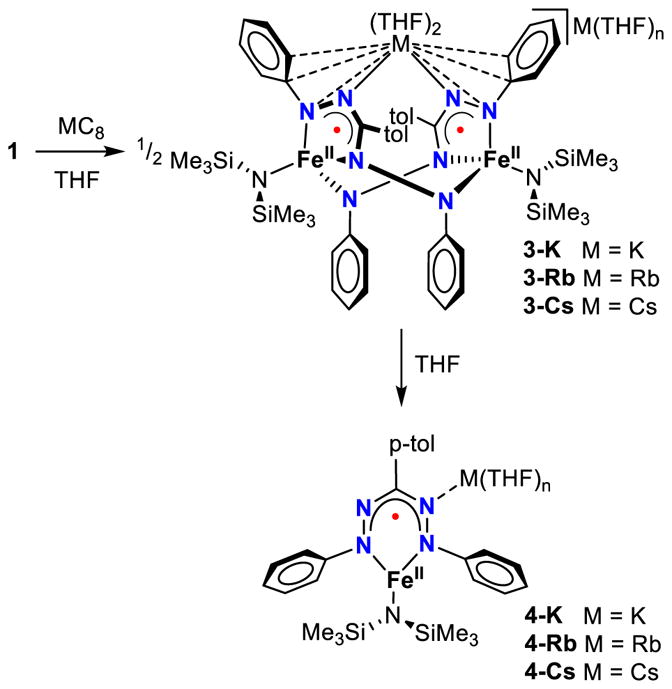
Synthesis of complexes 3-K, 3-Rb and 3-Cs and their decay to monomers in THF solution.
Figure 5.
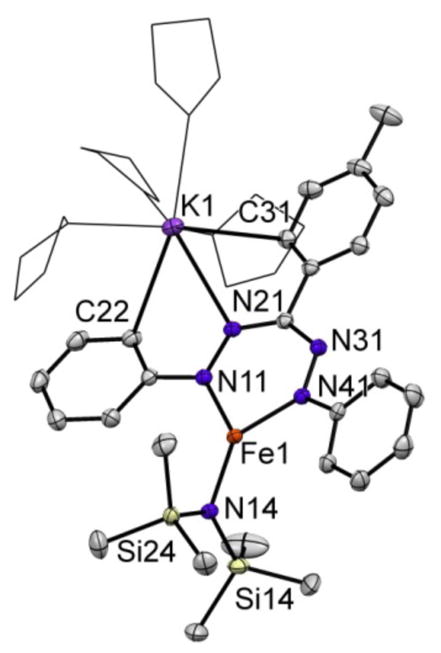
Displacement ellipsoid plot (50% probability) of 4-K. Hydrogen atoms are omitted and THF molecules are depicted in wireframe for clarity. Selected bond lengths (Å) and angles (°): Fe1–N11 1.933(1); Fe1–N41 1.937(1); Fe1–N14 1.944(1); N11–N21 1.362(2); N31–N41 1.358(2); N21–K1 2.865(1); C22–K1 3.344(1); C31–K1 3.213(1); N11–Fe1–N41 96.78(5); N11–Fe1–N14 133.75(5); N41–Fe1–N14 129.46(5).
Although we obtained crystals of 3-K, 3-Rb and 3-Cs suitable for single crystal X-ray diffraction, the rapid desolvation of the crystalline material, when transferred from the mother liquor to the immersion oil, resulted in poorly diffracting crystals. Fortunately, handling at temperatures below −40 °C in combination with cryo-mounting of the crystals (see SI for more details), led to solid state structures of all dimers (Figure 6). In agreement with the observed rapid desolvation of the crystals, several solvent molecules are in the asymmetric units. The quality of the structures of 3-K and 3-Cs was of lower quality than the other dimers (3-K diffracted weakly, and 3-Cs appears to have whole molecule disorder; see SI for details), so the structural detail is limited. Overall, it is clear that all complexes have a similar dimeric core structure as 3-Na where the formazanate has isomerized. No significant deviations in Fe-ligand bond distances are observed between the four dimers (Table 1). However, the manner in which the alkali cations are bound varies greatly. Whereas each formazanate in complex 3-Na is bound to a single Na cation, in 3-K, 3-Rb and 3-Cs one alkali cation bridges between the N21 and N25 of two separate formazanates. Close interactions with several other atoms in the N-phenyl substituent are also observed, which is in agreement with the softer nature of the larger cations favoring interaction with delocalized π systems over the harder N atom.34 The other alkali cation is separated from the dimer and has only THF solvent molecules coordinated in the solid state structures of 3-K and 3-Rb. Interestingly, in the solid state structure of complex 3-Cs the second Cs cation is also bound to the N21 and N25 to yield a repeating polymeric structure (Figure S18).
Figure 6.

Displacement ellipsoid plots (50% probability) of the complexes 3-Na, 3-K, 3-Rb and 3-Cs showing the core structures. Hydrogen atoms, select organic fragments and solvent molecules are omitted for clarity.
The zero-field Mössbauer spectra of 3-K, 3-Rb and 3-Cs show doublets with isomer shifts that are indistinguishable from that of 3-Na (Table 3, Figure S8). The observed quadrupole splittings of 3-K, 3-Rb and 3-Cs range from 2.16 – 2.18 mm s−1, which is slightly different from the quadrupole splitting of solid 3-Na. This is likely due to the different binding mode of the alkali cations, which is also reflected N–Fe–N bond angles of the dimers (Table 2).
Table 3.
Mössbauer parameters for solid samples of 3; experimental uncertainty is ±0.02 mm s−1.
| Complex | δ (mm s−1) | |ΔEQ| (mm s−1) |
|---|---|---|
| 3-Na | 0.74 | 2.30 |
| 3-K | 0.72 | 2.16 |
| 3-Rb | 0.71 | 2.17 |
| 3-Cs | 0.72 | 2.18 |
Table 2.
Selected bond angles (°) for the dimers.
| 3-Na | 3-K | 3-Rb | 3-Cs | |
|---|---|---|---|---|
| N14–Fe1–N45 | 124.8(1) | 122.6(1) | 122.8(1) | 121.2(2) |
| N18–Fe2–N41 | 122.7(1) | 123.0(1) | 123.2(1) | 120.5(2) |
|
| ||||
| N14–Fe1–N11 | 110.1(1) | 108.4(1) | 107.9 (1) | 107.2(2) |
| N18–Fe2–N15 | 109.4(1) | 108.6(1) | 107.9 (1) | 108.1(2) |
|
| ||||
| N11–Fe1–N45 | 116.1(1) | 121.4(1) | 121.1(1) | 124.4(2) |
| N15–Fe2–N41 | 117.3(1) | 120.5(1) | 121.2 (1) | 123.8(2) |
|
| ||||
| N45–Fe1–N31 | 90.9(1) | 89.7 (1) | 89.5 (1) | 88.9(2) |
| N41–Fe2–N35 | 90.3(1) | 89.5(1) | 89.5(1) | 88.7(2) |
|
| ||||
| N14–Fe1–N31 | 128.4(1) | 130.0(1) | 130.9(1) | 131.4(2) |
| N18–Fe2–N35 | 132.2(1) | 130.2(1) | 130.1(1) | 132.8(2) |
|
| ||||
| N11–Fe1–N31 | 77.2(1) | 77.1(1) | 77.2(1) | 77.5(2) |
| N15–Fe2–N35 | 77.4(1) | 77.3(1) | 77.1(1) | 77.1(2) |
The 1H NMR spectra and solution magnetic moments of 3-K, 3-Rb and 3-Cs in THF-d8 are similar to that of 3-Na in THF-d8, indicating that the dimeric structures are retained in solution. Similarly, 3-K, 3-Rb and 3-Cs also convert to the corresponding monomers as judged by 1H NMR spectroscopy. However, the rate at which the dimer breaks up in solution is highly dependent on the cation that is present (Figure 7). Complex 3-Cs is the most rapid (kobs = 5.89 ± 0.03 × 10−4 s−1) after 3-Rb (kobs = 1.66 ± 0.01 × 10−4 s−1) and 3-K (kobs = 6.37 ± 0.04 × 10−5 s−1). Though these follow a trend where the heavier alkali metal cation is fastest, the decay rate of 3-K is slower than that of 3-Na (kobs = 8.50 ± 0.08 × 10−5 s−1), breaking the periodic trend.
Figure 7.
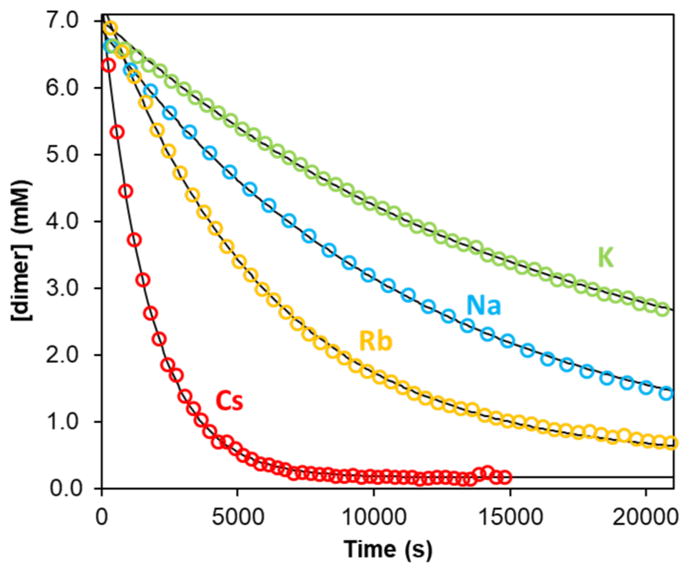
Concentration of dimers 3-Na, 3-K, 3-Rb and 3-Cs vs time in d8-THF at 21 °C. Open circles are data points and solid lines are fitted curves.
Discussion
Interaction of Alkali Metals with Reduced Formazanate-Iron Complexes: Influences on Structure and Dynamics
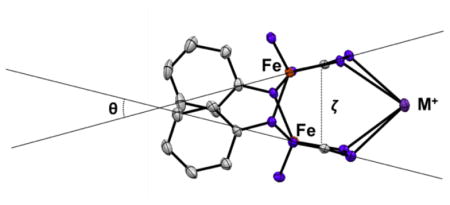
The reduction of complex 1 in THF solution by different alkali metals gives dimeric complexes (3) featuring a rearranged formazanate. The formazanate binding mode in the dimeric complexes is stabilized by cation-π interactions with alkali metal cations. Although the different cations do not significantly alter the intraligand bond lengths of the rearranged formazanate, the interplanar angle (θ) and centroid-centroid distance (ζ) between the five-membered metallacycles significantly increases with the larger cations (Table 4). This trend is consistent with increasing ionic radii of the corresponding alkali cations, and likely results in the observed differences in the N–Fe–N bond angles (Table 2).
Table 4.
Distances and angles between the 5-membered metallacycles and N4 atoms in of 3.
| |||
|---|---|---|---|
| Complex | Angle θ (°) | Distance ζ (Å) | N4-N4 (Å) |
| 3-Na | 18.3 | 3.11 | 3.046(4) |
| 3-K | 23.3 | 3.21 | 2.999(5) |
| 3-Rb | 25.9 | 3.23 | 2.998(2) |
| 3-Cs | 33.7 | 3.37 | 2.989(5) |
Although the dimeric nature is retained in solution as judged by initial 1H NMR spectra, each dimer converts completely into the corresponding monomeric species in THF solution. Interestingly, the rate at which this process occurs varies greatly between the dimers. The slow rate of dissociation of 3-K shows that the energy barrier for dimer/monomer interconversion is higher than for the other complexes. This could explain why 4-K was the only one that could be crystallized as a monomer, because the higher barrier would also slow the reverse reaction, dimerization upon crystallization.
A periodic trend for the rates of monomer formation is observed with K < Rb < Cs, which correlates with weaker cation-π interactions as the z/r ratio of the cations decreases. This may arise from a stronger cation-π interaction with smaller alkali cations, which stabilizes the isomerized binding mode of the formazanate, and increases the activation energy for isomerization and splitting of the dimer. The deviation of the periodic trend for the Na analogue could be ascribed to the different binding of the alkali cations in 3-Na, where each rearranged formazanate has an interaction with a separate alkali cation in contrast with the bridging alkali cation observed in 3-K, 3-Rb and 3-Cs. The difference in 3-Na also influences the iron center, as judged by the subtly different quadrupole splitting in the Mössbauer spectrum of 3-Na in comparison to 3-K, 3-Rb and 3-Cs. The rate of dimer cleavage is faster for 3-Na than expected from the periodic trend, suggesting that the Na cations hold the core into the dimeric structure more weakly, which is consistent with the more central position of the alkali metal cations in 3-K, 3-Rb and 3-Cs. Alternatively, the relative rates of monomer formation could be due to the need for THF to access the alkali metal cation, which is suggested by the negative activation entropy for 3-Na going to 4-Na. In this alternative explanation, the faster rates for the larger cations would result from the larger size and faster expected exchange rate for solvent.
Analysis of the chemical shifts in the 1H NMR spectra of the dimers shows another trend between the chemical shift with the size of the alkali metal for 3-K, 3-Rb and 3-Cs, but the trend does not fit well for all resonances of 3-Na (Figure S4), suggesting that the structural differences noted above are also retained in solution. After dissociation into the monomeric complexes, the chemical shifts in the 1H NMR spectra also vary greatly by alkali cation (Figure 8). Comparison of the chemical shifts with complex 2, which has a fully separated ion pair in solution, shows that the chemical shifts move towards a narrower range going from 2 to 4-Na, 4-K, 4-Rb and 4-Cs. In this case the Na monomer does fit the periodic trend. Assuming that in solution the Na cation is fully sequestered by the crown ethers for 2, this trend in chemical shifts could be interpreted by a strengthening cation-π interaction between the formazanate and alkali cation in the series 4-Na, to 4-K, to 4-Rb to 4-Cs. However, this would be opposite to the typical trend in magnitudes of cation-π interactions, which are known to become weaker as the ion becomes larger.8 Therefore, a more likely explanation is that the alkali cation and THF ligands (Figure 5) perturb the geometry of the monomeric complex in solution to accommodate the cation-π interaction. This effect would be more pronounced with increasing alkali cation size, as observed in the solid state structures of dimers 3-K, 3-Rb and 3-Cs, in agreement with the biggest change in chemical shifts between 2 and 4-Cs.
Figure 8.
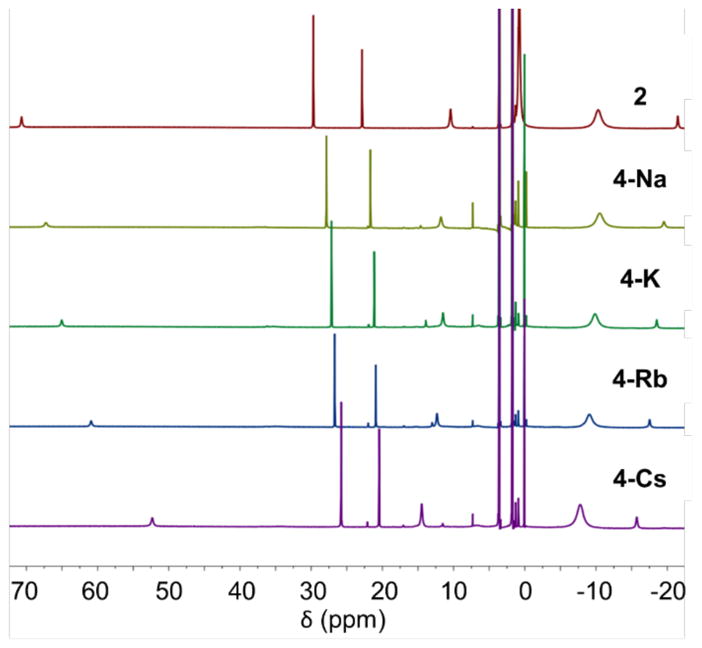
Stacked 1H NMR spectra of complex 2 (top) and dissociated complexes 4-Na, 4-K, 4-Rb and 4-Cs, showing the trends in chemical shifts upon binding of alkali cations.
Electronic structure of 3-Na
Mössbauer and X-ray absorption spectra show experimentally that two high-spin iron(II) centers are present in 3, which implies that the two formazanate ligands are not in their usual closed-shell state but rather are reduced, open-shell radicals. The intraligand distances are distorted from that in conventional iron(II)-formazanate complexes, further supporting the assignment of a reduced formazanate.
The magnetic susceptibility data for 3-Na indicate very little magnetic coupling between the radical ligands and the iron(II) centers. It is notable that there is a short distance between the two N4 atoms of each formazanate ligand in 3 (2.989(5) – 3.046(4) Å, Table 4). In addition, the two five-membered metallacyles are parallel alignment and are separated by only 3.11 – 3.37 Å in this series, indicating a π-π interaction like those that have been reported for several chelating π radical ligands.35 For example, two o-diiminosemiquinonate ligand radicals coordinated to zirconium(IV) are aligned parallel, and the short interligand separation of 3.42 Å gives rise to intramolecularly antiferromatic coupling between this two spin centers in this complex.36 Moreover, in the previous forum on redox-active ligands, Scarborough and Wieghardt described a similar intramolecular π•-π• antiferromagnetic interaction in a dimeric complex between two bipyridine radicals.37 In the system described here, details of weak coupling were obscured by low-temperature deviations in the SQUID magnetometry data, so we were unable to glean further experimental evidence.
To gain insight into the electronic structure of the dimeric complexes, the ground state electronic structure of 3-Na was calculated using DFT calculations at the B3LYP/ZORA level of theory, using the broken symmetry (BS) formalism to give localized spins in an overall singlet state (MS = 0). The electronic structure derived from a BS(5,5) calculation has five pairs of magnetic orbitals (the members of each pair are related to the other half of the dimer by symmetry), which are antiferromagnetically coupled to give the singlet ground state (Figure 9). The high-spin MS = 5 solution is 15.9 kcal mol−1 less favored than the BS solution.
Figure 9.

Qualitative MO scheme for 3-Na as derived from broken symmetry DFT calculations (B3LYP/ZORA). The Fe and formazanate (fz) content and spatial overlaps of the corresponding pairs of magnetic orbitals are given.
The DFT methodology is limited because BS calculations can only partition a molecule into two orbital fragments, one with spin-up electrons (α-spin) and the other spin-down electrons (β-spin). Of the five pairs of magnetic orbitals, three can be described as Fe-based dxz, dyz, dz2 orbitals of one Fe ion in the α-spin manifold that are antiferromagnetically coupled to the corresponding β-spin of matching symmetry on the other Fe ion. The computed overlap integrals (S) between these d orbitals in 3-Na are all very small, underscoring the essentially non-existent coupling between these well-separated spin centers. The highest doubly-occupied MOs are based on the dx2-y2 orbital for each Fe ion. The remaining two pairs of magnetic orbitals share Fe and formazanate character. The remaining d orbital (dxy) has the correct symmetry to overlap with the highest ligand-based π* orbital (two N–N π* interactions) previously assigned with a2 symmetry for the formazanate ligand.24 The bonds between the dxy and formazanate are highly polarized, giving rise to an antiferromagnetic interaction between this symmetry-matched pair. The most unusual aspect comes from the exocyclic nitrogen (N4), which has a bonding interaction with the other Fe dxy orbital that is visible in the lowest magnetic orbital with S = 0.59 (Figure 9). This four-spin ensemble is challenging to compute because of the aforementioned limitations of BS-DFT, and the result here is one where the calculation mixes two pairs of SOMOs that are very close in energy, with polarized metal-ligand bonds at one end and covalent bonds at the other. The most appropriate description is that the first pair are predominantly ligand-centered with significant Fe dxy content that produces a large overlap integral. The second pair can be considered almost exclusively the dxy orbital of each Fe ion, who have a significantly smaller overlap integral (Figure 9).
This description is consistent with the Mössbauer and Fe K-edge data that diagnose two high-spin iron(II) (S = 2) ions, which are each coordinated by a dianionic formazanate radical (S = 1/2). This description is supported by the Mulliken spin population analysis, where 3.6 spins are found on each Fe with significant spin of the opposing sign on the coordinated dianionic formazanate radical ligands (Figure 10). This value is lower than four spins for each high-spin iron(II) center as a result of the covalent interaction with the exocyclic nitrogen that distributes some spin (of the same sign) on to the formazanate ligand. Though the calculations predict stronger antiferromagnetic coupling than we observe in the magnetic data, we ascribe the difference to the limitations of the BS formalism in a four-spin system as described above.
Figure 10.
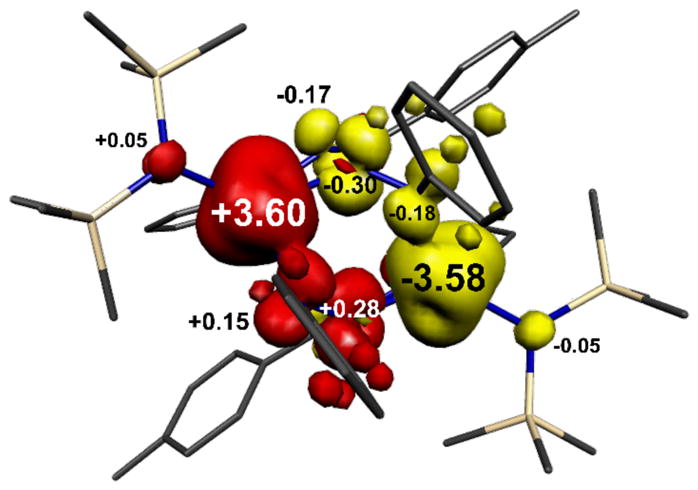
Mulliken spin population analysis for 3-Na (red: α-spin; yellow: β-spin). Counterions and solvents have been removed for clarity.
Overall, the resonance structures in Scheme 5 best describe the electronic structure of the rearranged formazanate radical dianions in 3 as they are in agreement with the presence two high-spin Fe centers, and reflect the intra- and interligand bond distances and computational data. The dimerization that is facilitated by the ligand rearrangement produces a pair of strongly coupled magnetic orbitals that are predominantly ligand in character though have a sizeable metal content by virtue of a bonding interaction between the exocyclic nitrogen and the other iron center. The net result is two high-spin iron(II) ions characterized by four highly localized magnetic orbitals that are weakly coupled on account of their large spatial separation. This general electronic structure description accurately reflects the physical characterization data despite the limitations of the BS-DFT calculations in reproducing the magnetic coupling. Moreover, it is unique among these iron-formazanate complexes, as the monometallic species 2 and 4 present a more common electronic configuration, that of a π-radical ligand (SL = 1/2) strongly antiferromagnetically coupled to a paramagnetic metal ion (SFe = 2) to give an S = 3/2 spin ground state.
Scheme 5.
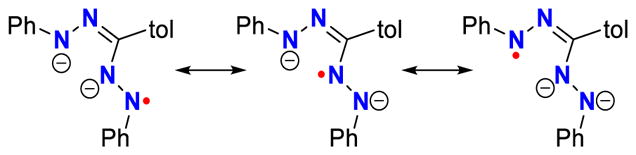
Limiting resonance forms for the rearranged formazanate radical dianion in dimers 3.
These studies show the significant effect that cation-π interactions can have on metal complexes with redox-active ligands. The observed rearranged formazanate binding mode is stabilized to different extent by the various alkali cations, which is reflected in the different solid state structures and observed decay rates for the dimers. The results show that cation-π interactions not necessarily only affect the electronic structure according to periodic trends but that the difference in size can also change result in different chemical structure.
Conclusion
In the absence of a crown ether, one-electron reduction of complex 1 by alkali metals results in the formation of dimeric complexes wherein the formazanate ligand has rearranged to form a five-membered metallacycle that is coordinated to the alkali cation. The electronic structure has two uncoupled high spin iron(II) centers and two ligand radicals as shown by Mössbauer, XAS, and DFT. Different alkali cations not only result in different orientations of the rearranged formazanate ligand but also affects how these dimers are converted to monomeric species in solution. Moreover, the difference in how the alkali cation is bound in the sodium dimer is shown to transcend periodic trends in terms of reactivity. This work demonstrates the considerable effect that Lewis-acidic cations can have on systems containing redox-active ligands, and shows the possibilities for tuning of structure and reactivity by choice of cation.
Supplementary Material
Acknowledgments
This work was supported by The Netherlands Organization for Scientific Research (Rubicon Postdoctoral Fellowship 680-50-1517 to D.L.J.B.) and the National Institutes of Health (Grant GM-065313 to P.L.H.). K.L.M. thanks the A. P. Sloan Foundation and the National Science Foundation (CHE-1454455) for funding.
Footnotes
Supporting information
The Supporting Information is available free of charge on the ACS Publications website at DOI: xxx
Synthetic procedures and characterization of all compounds, NMR, IR, Mössbauer, XAS and UV/vis spectral data, kinetic experiments, magnetic studies and details of computational and crystallographic studies (PDF)
References
- 1.a) Sigel A, Sigel H, Sigel RKO. The Alkali Metal Ions: Their Role for Life. Springer Cham; 2016. [PubMed] [Google Scholar]; b) Ma JC, Dougherty DA. The Cation–π Interaction. Chem Rev. 1997;97:1303–1324. doi: 10.1021/cr9603744. [DOI] [PubMed] [Google Scholar]; c) Dougherty DA. Cation-pi interactions in chemistry and biology: A new view of benzene, Phe, Tyr, and Trp. Science. 1996;271:163–168. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]; d) Riordan JF. The role of metals in enzyme activity. Ann Clin Lab Sci. 1977;7:119–129. [PubMed] [Google Scholar]
- 2.a) Connor GP, Holland PLH. Coordination chemistry insights into the role of alkali metal promoters in dinitrogen reduction. Catal Today. 2017;286:21–40. doi: 10.1016/j.cattod.2016.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Williams FJ, Palermo A, Tikhov MS, Lambert RM. Mechanism of alkali promotion in heterogeneous catalysis under realistic conditions: application of electron spectroscopy and electrochemical promotion to the reduction of NO by CO and by propene over rhodium. Surf Sci. 2001;482–485:177–182. [Google Scholar]; c) Oukaci R, Wu JCS, Goodwin JG., Jr Secondary Reactions during CO Hydrogenation on Zeolite-Supported Metal Catalysts: Influence of Alkali Cations. J Catal. 1987;107:471–481. [Google Scholar]; d) Mross WD. Alkali doping in heterogeneous catalysis. Catal Rev Sci Eng. 1983;25:591–637. [Google Scholar]
- 3.a) McWilliams SF, Rodgers KR, Lukat-Rodgers G, Mercado BQ, Grubel K, Holland PLH. Alkali Metal Variation and Twisting of the FeNNFe Core in Bridging Diiron Dinitrogen Complexes. Inorg Chem. 2016;55:2960–2968. doi: 10.1021/acs.inorgchem.5b02841. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) González-Riopedre G, Bermejo MR, Fernández-García MI, Gonzalez-Noya AM, Pedrido R, Rodríguez-Douton MJ, Maneiro M. Alkali-Metal-Ion-Directed Self-Assembly of Redox-Active Manganese(III) Supramolecular Boxes. Inorg Chem. 2015;54:2512–2521. doi: 10.1021/ic501842x. [DOI] [PubMed] [Google Scholar]
- 4.Reath AH, Ziller JW, Tsay C, Ryan AJ, Yang JY. Redox Potential and Electronic Structure Effects of Proximal Nonredox Active Cations in Cobalt Schiff Base Complexes. Inorg Chem. 2017;56:3713–3718. doi: 10.1021/acs.inorgchem.6b03098. [DOI] [PubMed] [Google Scholar]
- 5.Grubel K, Brennessel WW, Mercado BQ, Holland PL. Alkali Metal Control over N–N Cleavage in Iron Complexes. J Am Chem Soc. 2014;136:16807–16816. doi: 10.1021/ja507442b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Kita MR, Miller AJM. An Ion-Responsive Pincer-Crown Ether Catalyst System for Rapid and Switchable Olefin Isomerization. Angew Chem Int Ed. 2017;56:5498–5502. doi: 10.1002/anie.201701006. [DOI] [PubMed] [Google Scholar]; b) Kita MR, Miller AJM. Cation-Modulated Reactivity of Iridium Hydride Pincer-Crown Ether Complexes. J Am Chem Soc. 2014;136:14519–14529. doi: 10.1021/ja507324s. [DOI] [PubMed] [Google Scholar]; c) Moore CM, Bark B, Szymczak NK. Simple Ligand Modifications with Pendent OH Groups Dramatically Impact the Activity and Selectivity of Ruthenium Catalysts for Transfer Hydrogenation: The Importance of Alkali Metals. ACS Catal. 2016;6:1981–1990. [Google Scholar]; d) Bosch H, Van Ommen JG, Gellings PJ. On the role of alkali metals in ammonia synthesis. Appl Catal. 1985;18:405–408. [Google Scholar]
- 7.a) Rohling R, Hensen E, Pidko EA. Multi-site cooperativity in alkali-exchanged faujasites for the production of biomass derived aromatics. Chem Phys Chem. 2018;19:446–458. doi: 10.1002/cphc.201701058. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sivaguru J, Natarajan A, Kaanumalle LS, Shailaja J, Uppili S, Joy A, Ramamurthy V. Asymmetric Photoreactions within Zeolites: Role of Confinement and Alkali Metal Ions. Acc Chem Res. 2003;36:509–521. doi: 10.1021/ar020269i. [DOI] [PubMed] [Google Scholar]; c) O’Neill MA, Cozens FL. Influence of alkali metal cations on the photoheterolysis of 9-cyclopropyl-9-fluorenol and the reactivity of the 9-cyclopropyl-9-fluorenyl cation in non-acidic zeolites. Can J Chem. 2003;81:647–659. [Google Scholar]; d) Jackman LM, Lange BC. Structure and reactivity of alkali metal enolates. Tetrahedron. 1977;33:2737–2769. [Google Scholar]
- 8.a) Rodgers MT, Armentrout PB. Cationic noncovalent interactions: energetics and periodic trends. Chem Rev. 2016;116:5642–5687. doi: 10.1021/acs.chemrev.5b00688. [DOI] [PubMed] [Google Scholar]; b) Dougherty DA. The Cation-π Interaction. Acc Chem Res. 2013;46:885–893. doi: 10.1021/ar300265y. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mecozzi S, West AP, Jr, Dougherty DA. Cation-π interactions in simple aromatics: electrostatics provide a predictive tool. J Am Chem Soc. 1996;118:2307–2308. [Google Scholar]
- 9.a) Lambert C, von Raqué Schleyer P. Are Polar Organometallic Compounds “Carbanions”? The Gegenion Effect on Structure and Energies of Alkali-Metal Compounds. Angew Chem. 1994;106:1187–1199. [Google Scholar]; b) Shannon RD. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr, Sect A. 1976;A32:751–767. [Google Scholar]
- 10.Torvisco A, Ruhlandt-Senge K. s-Block Organometallics: Analysis of Ion-Association and Noncovalent Interactions on Structure and Function in Benzyl-Based Compounds. Inorg Chem. 2011;50:12223–12240. doi: 10.1021/ic201148d. [DOI] [PubMed] [Google Scholar]
- 11.Henderson KW, Dorigo AE, Liu Q-Y, Williard PG. Effect of Polydentate Donor Molecules on Lithium Hexamethyldisilazide Aggregation: An X-ray Crystallographic and a Combination Semiempirical PM3/Single Point ab Initio Theoretical Study. J Am Chem Soc. 1997;119:11855–11863. [Google Scholar]; Torvisco A, Decker K, Uhlig F, Ruhlandt-Senge K. Heavy Alkali Metal Amides: Role of Secondary Interactions in Metal Stabilization. Inorg Chem. 2009;48:11459–11465. doi: 10.1021/ic901767c. [DOI] [PubMed] [Google Scholar]; Bartlett RA, Hitchcock PB, Khvostov AV, Lappert MF, Protchenco AV. Ytterbium(II) amides and crown ethers: addition versus amide substitution. J Organomet Chem. 2002;647:198–204. [Google Scholar]; Power PP. A Silicon-Nitrogen Analogue of the [PPN]+ Cation: Synthesis and Structural Characterization of the [Ph3SiNSiPh3]-Ion. J Am Chem Soc. 1987;109:6509–6510. [Google Scholar]; DiBiase SA, Gokel GW. Crown-Cation Complex Effects. 8. Reactions of Crown Ether Activated tert-Butoxide Ion. J Org Chem. 1978;43:447–452. [Google Scholar]
- 12.Selected recent examples: Buscagan TM, Oyala PH, Peters JC. N2-to-NH3 Conversion by a triphos–Iron Catalyst and Enhanced Turnover under Photolysis. Angew Chem Int Ed. 2017;56:6921–692. doi: 10.1002/anie.201703244.MacLeod KC, Lewis RA, DeRosha DE, Mercado BQ, Holland PL. C-H and C-N Activation at Redox-Active Pyridine Complexes of Iron. Angew Chem Int Ed. 2017;56:1069–1072. doi: 10.1002/anie.201610679.Chakraborty U, Demeshko S, Meyers F, Rebreyend C, de Bruin B, Atanasov M, Neese F, Mühldorf B, Wolf R. Electronic Structure and Magnetic Anisotropy of an Unsaturated Cyclopentadienyl Iron(I) Complex with 15 Valence Electrons. Angew Chem Int Ed. 2017;56:7995–7999. doi: 10.1002/anie.201702454.McNeill E, Ritter T. 1,4-Functionalization of 1,3-Dienes With Low-Valent Iron Catalysts. Acc Chem Res. 2015;48:2330–2343. doi: 10.1021/acs.accounts.5b00050.Anderson JS, Rittle J, Peters JC. Catalytic conversion of nitrogen to ammonia by an iron model complex. Nature. 2013;501:84–87. doi: 10.1038/nature12435.
- 13.a) Hoyt JM, Schmidt VA, Tondreau AM, Chrik PJ. Iron-catalyzed intermolecular [2+2] cycloadditions of unactivated alkenes. Science. 2015;349:960–963. doi: 10.1126/science.aac7440. [DOI] [PubMed] [Google Scholar]; b) Wong L, Hernández Sánchez R, Logan JG, Zarkesh RA, Ziller JW, Heyduk AF. Disulfide reductive elimination from an iron(III) complex. Chem Sci. 2013;4:1906–1910. [Google Scholar]; c) Darmon JM, Stieber SCE, Sylvester KT, Fernández I, Lobkovsky E, Semproni SP, Bill E, Wieghardt K, DeBeer S, Chirik PJ. Oxidative Addition of Carbon–Carbon Bonds with a Redox-Active Bis(imino)pyridine Iron Complex. J Am Chem Soc. 2012;134:17125–17137. doi: 10.1021/ja306526d. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Smith AL, Hardcastle KI, Soper JD. Redox-Active Ligand-Mediated Oxidative Addition and Reductive Elimination at Square Planar Cobalt(III): Multielectron Reactions for Cross-Coupling. J Am Chem Soc. 2010;132:14358–14360. doi: 10.1021/ja106212w. [DOI] [PubMed] [Google Scholar]; d) Blackmore KJ, Ziller JW, Heyduk AF. “Oxidative Addition” to a Zirconium(IV) Redox-Active Ligand Complex. Inorg Chem. 2005;44:5559–5561. doi: 10.1021/ic050997c. [DOI] [PubMed] [Google Scholar]
- 14.a) Fujita D, Sugimoto H, Shiota Y, Morimoto Y, Yoshizawa K, Itoh S. Catalytic C–H amination driven by intramolecular ligand-to-nitrene one-electron transfer through a rhodium(III) centre. Chem Commun. 2017;53:4849–4852. doi: 10.1039/c7cc01840a. [DOI] [PubMed] [Google Scholar]; b) Jacquet J, Blanchard S, Derat E, Desage-El Murr M, Fensterbank L. Redox-ligand sustains controlled generation of CF3 radicals by well-defined copper complex. Chem Sci. 2016;7:2030–2036. doi: 10.1039/c5sc03636d. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Broere DLJ, van Leest NP, Siegler MA, van der Vlugt JI. Reversible Redox Chemistry and Catalytic C(sp3)–H Amination Reactivity of a Paramagnetic Pd Complex Bearing a Redox-Active o-Aminophenol-Derived NNO Pincer Ligand. Inorg Chem. 2016;55:8603–8611. doi: 10.1021/acs.inorgchem.6b01192. [DOI] [PubMed] [Google Scholar]; d) Broere DLJ, de Bruin B, Reek JNH, Lutz M, Dechert S, van der Vlugt JI. Intramolecular redox-active ligand-to-substrate single-electron transfer: radical reactivity with a palladium (II) complex. J Am Chem Soc. 2014;136:11574–11577. doi: 10.1021/ja502164f. [DOI] [PubMed] [Google Scholar]; e) Miyazato Y, Wada T, Muckerman JT, Fujita E, Tanaka K. Generation of a RuII–Semiquinone–Anilino-Radical Complex through the Deprotonation of a RuIII–Semiquinone–Anilido Complex. Angew Chem, Int Ed. 2007;46:5728–5730. doi: 10.1002/anie.200701600. [DOI] [PubMed] [Google Scholar]
- 15.a) Jacquet J, Desage-El Murr M, Fensterbank L. Metal-Promoted Coupling Reactions Implying Ligand-Based Redox Changes. Chem Cat Chem. 2016;8:3310–3316. [Google Scholar]; b) Broere DLJ, Plessius R, van der Vlugt JI. New avenues for ligand-mediated processes--expanding metal reactivity by the use of redox-active catechol. o-aminophenol and o-phenylenediamine ligands. Chem Soc Rev. 2015;44:6886–6915. doi: 10.1039/c5cs00161g. [DOI] [PubMed] [Google Scholar]; c) Luca OR, Crabtree RH. Redox-active ligands in catalysis. Chem Soc Rev. 2013;42:1440–1459. doi: 10.1039/c2cs35228a. [DOI] [PubMed] [Google Scholar]; d) Praneeth VKK, Ringenberg MR, Ward TR. Redox-Active Ligands in Catalysis. Angew Chem Int Ed. 2012;51:10228–10234. doi: 10.1002/anie.201204100. [DOI] [PubMed] [Google Scholar]; e) Lyaskovskyy V, de Bruin B. Redox Non-Innocent Ligands: Versatile New Tools to Control Catalytic Reactions. ACS Catal. 2012;2:270–279. [Google Scholar]
- 16.a) Chang H-C, Mochizuki K, Kitagawa S. Substituent-Directed Structural and Physicochemical Controls of Diruthenium Catecholate Complexes with Ligand-Unsupported Ru–Ru Bonds. Inorg Chem. 2005;44:3799–3809. doi: 10.1021/ic048251m. [DOI] [PubMed] [Google Scholar]; b) Osterloh F, Achim C, Holm RH. Molybdenum-Iron-Sulfur Clusters of Nuclearities Eight and Sixteen, Including a Topological Analogue of the P-Cluster of Nitrogenase. Inorg Chem. 2001;40:224–232. doi: 10.1021/ic000617h. [DOI] [PubMed] [Google Scholar]
- 17.a) Xu B, Ma A, Jia T, Hao Z, Gao, Mu Y. Synthesis and structural characterization of iron complexes bearing N-aryl-phenanthreno-iminoquinone ligands. Dalton Trans. 2016;45:17966–17973. doi: 10.1039/c6dt03572h. [DOI] [PubMed] [Google Scholar]; b) Piskunov AV, Ershova IV, Fukin GK, Shavyrin AS. Addition of allyl halides to the new bis-o-amidophenolate gallium(III) complex. Inorg Chem Commun. 2013;38:127–130. [Google Scholar]; c) Haneline MR, Heyduk AF. C-C Bond-Forming Reductive Elimination from a Zirconium(IV) Redox-Active Ligand Complex. J Am Chem Soc. 2006;128:8410–8411. doi: 10.1021/ja061107a. [DOI] [PubMed] [Google Scholar]
- 18.Hitchcock PB, Huang Q, Lappert MF, Wei X–H, Zhou M. Synthesis and structures of some heterometallic [(Li, Y)2, (M3, Ce) (M= Li or Na), (Li, Zr2) and (Li, Zr)4] oligomeric diamides derived from 1,2-bis(neopentylamino)benzene. Dalton Trans. 2006:2991–2997. doi: 10.1039/b515790k. [DOI] [PubMed] [Google Scholar]
- 19.Gallo E, Lolari E, Re N, Floriani C, Chiesi-Villa A, Rizzoli C. Carbon-Carbon Bonds Functioning as Electron Shuttles: The Generation of Electron-Rich Manganese(II)-Schiff Base Complexes and Their Redox Chemistry. J Am Chem Soc. 1997;119:5144–5154. [Google Scholar]
- 20.Letko CS, Panetier JA, Head-Gordon M, Tilley TD. Mechanism of the Electrocatalytic Reduction of Protons with Diaryldithiolene Cobalt Complexes. J Am Chem Soc. 2014;136:9364–9376. doi: 10.1021/ja5019755. [DOI] [PubMed] [Google Scholar]
- 21.a) Tondreau AM, Stieber SCE, Milsmann C, Lobkovsky E, Weyhermüller Semproni SP, Chirik PJ. Oxidation and Reduction of Bis(imino)pyridine Iron Dinitrogen Complexes: Evidence for Formation of a Chelate Trianion. Inorg Chem. 2013;52:635–646. doi: 10.1021/ic301675t. [DOI] [PubMed] [Google Scholar]; b) Scott J, Vidyaratne Korobkov I, Gambarotta S, Budzelaar PHM. Multiple Pathways for Dinitrogen Activation during the Reduction of an Fe Bis(iminepyridine) Complex. Inorg Chem. 2008;47:896–911. doi: 10.1021/ic701643d. [DOI] [PubMed] [Google Scholar]; c) Vidyaratne I, Scott J, Gambarotta S, Budzelaar PHM. Dinitrogen Activation, Partial Reduction, and Formation of Coordinated Imide Promoted by a Chromium Diiminepyridine Complex. Inorg Chem. 2007;46:7040–7049. doi: 10.1021/ic700810f. [DOI] [PubMed] [Google Scholar]; Bouwkamp MW, Lobkovsky E, Chirik PJ. Bis(imino)pyridine Ligand Deprotonation Promoted by a Transient Iron Amide. Inorg Chem. 2006;45:2–4. doi: 10.1021/ic051839o. [DOI] [PubMed] [Google Scholar]
- 22.Andrez J, Guidal V, Scopelliti R, Pécaut J, Gambarelli S, Mazzanti M. Ligand and Metal Based Multielectron Redox Chemistry of Cobalt Supported by Tetradentate Schiff Bases. J Am Chem Soc. 2017;139:8628–8638. doi: 10.1021/jacs.7b03604. [DOI] [PubMed] [Google Scholar]
- 23.a) Broere DLJ, Čorić I, Brosnahan A, Holland PLH. Quantitation of the THF Content in Fe[N(SiMe3)2]2·xTHF. Inorg Chem. 2017;56:3140–3143. doi: 10.1021/acs.inorgchem.7b00056. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Olmstead MM, Power PP, Shoner SC. Three-coordinate iron complexes: X-ray structural characterization of the amide-bridged dimers [Fe(NR2)2]2 (R = SiMe3, C6H5) and the adduct Fe[N(SiMe3)2]2(THF) and determination of the association energy of the monomer Fe(N(SiMe3)2]2 in solution. Inorg Chem. 1991;30:2547–2551. [Google Scholar]; c) Andersen RA, Faegri K, Green JC, Haaland A, Lappert MF, Leung W, Rypdal K. Synthesis of bis[bis(trimethylsilyl)-amido]iron(II). Structure and bonding in M[N(SiMe3)2]2 (M = Mn, Fe, Co): two-coordinate transition-metal amides. Inorg Chem. 1988;27:1782–1786. [Google Scholar]
- 24.Broere DLJ, Mercado BQ, Lukens JT, Vilbert AC, Banerjee G, Lant HMC, Lee SH, Bill E, Sproules S, Lancaster KM, Holland PLH. Redox-Active Formazanates in Low-Coordinate Iron Chemistry. doi: 10.1002/chem.201801298. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.a) Chang MC, Otten E. Intramolecular Hydride Transfer Reactions in Formazanate)Boron Dihydride Complexes. Organometallics. 2016;35:534–542. [Google Scholar]; b) Chang MC, Otten E. Synthesis and ligand-based reduction chemistry of boron difluoride complexes with redox-active formazanate ligands. Chem Commun. 2014;50:7431–7433. doi: 10.1039/c4cc03244f. [DOI] [PubMed] [Google Scholar]
- 26.Chang M-C, Roewen P, Travieso-Puente R, Lutz M, Otten E. Formazanate Ligands as Structurally Versatile, Redox-Active Analogues of β-Diketiminates in Zinc Chemistry. Inorg Chem. 2015;54:379–388. doi: 10.1021/ic5025873. [DOI] [PubMed] [Google Scholar]
- 27.Travieso-Puente R, Broekman JOP, Chang M-C, Demeshko S, Meyer F, Otten E. Spin-Crossover in a Pseudo-tetrahedral Bis(formazanate) Iron Complex. J Am Chem Soc. 2016;138:5503–5506. doi: 10.1021/jacs.6b01552. [DOI] [PubMed] [Google Scholar]
- 28.Mondol R, Snoeken DA, Chang M-C, Otten E. Stable, crystalline boron complexes with mono-, di- and trianionic formazanate ligands. Chem Commun. 2017;53:513–516. doi: 10.1039/c6cc08166e. [DOI] [PubMed] [Google Scholar]
- 29.Chang M-C, Dann T, Day DP, Lutz M, Wildgoose GG, Otten E. The formazanate ligand as an electron reservoir: bis(formazanate) zinc complexes isolated in three redox states. Angew Chem Int Ed. 2014;53:4118–4122. doi: 10.1002/anie.201309948. [DOI] [PubMed] [Google Scholar]
- 30.a) Ali A, Dhar D, Barman SK, Lloret F, Mukherjee R. Nickel(II) Complex of a Hexadentate Ligand with Two o-Iminosemiquinonato(1–) π-Radical Units and Its Monocation and Dication. Inorg Chem. 2016;55:5759–5771. doi: 10.1021/acs.inorgchem.5b02688. [DOI] [PubMed] [Google Scholar]; b) Broere DLJ, Plessius R, Tory J, Demeshko S, de Bruin B, Siegler MA, Hartl F, van der Vlugt JI. Localized Mixed Valence and Redox Activity within a Triazole-Bridged Dinucleating Ligand upon Coordination to Palladium. Chem Eur J. 2016;22:13965–13975. doi: 10.1002/chem.201601900. [DOI] [PubMed] [Google Scholar]; c) Broere DLJ, Metz LL, de Bruin B, Reek JNH, Siegler MA, van der Vlugt JI. Redox-Active Ligand-Induced Homolytic Bond Activation. Angew Chem, Int Ed. 2015;54:1516–1520. doi: 10.1002/anie.201410048. [DOI] [PubMed] [Google Scholar]
- 31.Blanchard S, Neese F, Bothe E, Bill E, Weyermüller T, Wieghardt K. Square Planar vs Tetrahedral Coordination in Diamagnetic Complexes of Nickel(II) Containing Two Bidentate π-Radical Monoanions. Inorg Chem. 2005;44:3636–3656. doi: 10.1021/ic040117e. [DOI] [PubMed] [Google Scholar]
- 32.Blanchard S, Bill E, Weyermüller T, Wieghardt K. N,N-Coordinated π Radical Anions of S-Methyl-1-phenyl-isothiosemicarbazide in Two Five-Coordinate Ferric Complexes [FeIII(LMe•)2X] (X = CH3S−, Cl−) Inorg Chem. 2004;43:2324–2329. doi: 10.1021/ic030285r. [DOI] [PubMed] [Google Scholar]
- 33.MacMillan SN, Lancaster KM. X-ray Spectroscopic Interrogation of Transition-Metal-Mediated Homogeneous Catalysis: Primer and Case Studies. ACS Catal. 2017;7:1776–1791. [Google Scholar]
- 34.Davidson MG, Garcia-Vivo D, Kennedy AR, Mulvey RE, Robertson SD. Exploiting σ/π Coordination Isomerism to Prepare Homologous Organoalkali Metal (Li, Na, K) Monomers with Identical Ligand Sets. Chem Eur J. 2011;17:3364–3369. doi: 10.1002/chem.201003493. [DOI] [PubMed] [Google Scholar]
- 35.a) Small D, Rosokha SV, Kochi JK, Head-Gordon M. Characterizing the Dimerizations of Phenalenyl Radicals by ab Initio Calculations and Spectroscopy: σ-Bond Formation versus Resonance π-Stabilization. J Phys Chem A. 2005;109:11261–11267. doi: 10.1021/jp054244n. [DOI] [PubMed] [Google Scholar]; b) Small D, Zaitsev V, Jung Y, Rosokha SV, Head-Gordon M, Kochi JK. Intermolecular π-to-π Bonding between Stacked Aromatic Dyads. Experimental and Theoretical Binding Energies and Near-IR Optical Transitions for Phenalenyl Radical/Radical versus Radical/Cation Dimerizations. J Am Chem Soc. 2004;126:13850–13858. doi: 10.1021/ja046770i. [DOI] [PubMed] [Google Scholar]; c) Pierpont CG, Buchanan RM. Transition metal complexes of o-benzoquinone, o-semiquinone, and catecholate ligands. Coord Chem Rev. 1981;38:45–87. [Google Scholar]; d) Pierpont CG, Buchanan RM. Distorted trigonal prismatic coordination in tris(9,10-phenanthrenequinone)molybdenum. J Am Chem Soc. 1975;97:4912–4917. [Google Scholar]
- 36.Ketterer NA, Fan H, Blackmore KJ, Yang X, Ziller JW, Baik M-H, Heyduk AF. π•-π• Bonding Interactions Generated by Halogen Oxidation of Zirconium(IV) Redox-Active Ligand Complexes. J Am Chem Soc. 2008;130:4364–4374. doi: 10.1021/ja077337m. [DOI] [PubMed] [Google Scholar]
- 37.Scarborough CC, Wieghardt K. Electronic Structure of 2,2′-Bipyridine Organotransition-Metal Complexes. Establishing the Ligand Oxidation Level by Density Functional Theoretical Calculations. Inorg Chem. 2011;50:9773–9793. doi: 10.1021/ic2005419. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.