

Abstract
By screening a chemical library for the compounds protecting cells from adriamycin (Adr), a series of small molecules was isolated that interfered with the accumulation of Adr in mouse fibroblasts by enhancing efflux of the drug. Isolated compounds also stimulated efflux of Rhodamine 123 (Rho-123), another substrate of multidrug transporters. Stimulation of drug efflux was detectable in the cells expressing P-glycoprotein (P-gp), but not in their P-gp-negative variants, and was completely reversible by the P-gp inhibitors. A dramatic stimulation of P-gp activity against Adr and Rho-123 by the identified compounds was accompanied by suppression of P-gp-mediated efflux of other substrates, such as Taxol (paclitaxel) or Hoechst 33342, indicating that they act as modulators of substrate specificity of P-gp. Consistently, P-gp modulators dramatically altered the pattern of cross-resistance of P-gp-expressing cells to different P-gp substrates: an increase in resistance to Adr, daunorubicin, and etoposide was accompanied by cell sensitization to Vinca alkaloids, gramicidin D, and Taxol with no effect on cell sensitivity to colchicine, actinomycin D, puromycin, and colcemid, as well as to several non-P-gp substrates. The relative effect of P-gp modulators against different substrates varied among the isolated compounds that can be used as fine tools for analyzing mechanisms of drug selectivity of P-gp. These results raise the possibility of a rational control over cell sensitivity to drugs and toxins through modulation of P-gp activity by small molecules.
Keywords: chemical library‖efflux
P-glycoprotein (P-gp) belongs to a superfamily of ABC transporters found both in pro- and eukaryotes that act as energy-dependent efflux pumps, transporting out of cells a wide variety of low molecular weight compounds (for review see ref. 1). In mammals, this superfamily includes P-gp transporters (MDR1 and MDR3 genes in human), the MRP subfamily (already composed of six members), and several other proteins (BCRP, etc.; refs. 2 and 3). These proteins can recognize and efflux numerous substrates with diverged chemical structure, including many anticancer drugs; the molecular mechanisms underlying broad substrate specificity of ABC transporters are generally unknown (3). Expression of such ABC transporters results in cross-resistance of the cells to many toxic compounds, known as multidrug resistance (MDR) (1, 3, 4). Increased expression of ABC transporters in tumor cells is one of the major mechanisms of cancer resistance to chemotherapy (1–3). Although the true clinical relevance of multidrug resistance is debated, P-gp and other ABC transporters are viewed as targets for therapeutic suppression to increase the susceptibility of multidrug-resistant cancers to chemotherapy (5).
Understanding of the normal physiological role of some of the ABC transporters (P-gp and MRP1) comes from the analysis of phenotypes of mice genetically deficient in the genes encoding these proteins. Mice lacking both mdr1a and mdr1b genes [two homologs of human P-gp-encoding MDR1 gene with different tissue expression (6)] developed normally, but were found to be extremely sensitive to certain xenobiotics and to exhibit strong alterations in pharmacokinetics of drugs known to be P-gp substrates (7, 8). Moreover, the deficiency in P-gp was associated with the ability of P-gp substrates to pass through the blood–brain barrier (6–8). In mammals, and particularly in humans, the expression of P-gp is restricted to specific organs, including intestine, kidney, liver, and endothelia of brain, testis, and placenta, consistent with their role in general detoxification and in establishment of blood–brain, blood–testis, and placental barriers (6). Although no physiological abnormalities were observed in the mice deficient in mrp1 gene, they are more sensitive to certain toxins, show changes in glutathione metabolism (9), and have decreased inflammatory response (10). In a recent publication, Robbiani et al. (11) reported possible involvement of MRP1 in dendritic cell migration to lymph nodes. Some members of the superfamily of ABC transporters seem to have a more narrow and specific spectrum of substrates: targeted disruption of mdr2 gene in mice results in a deviation in phosphatidylcholine and other phospholipid excretion in bile (12). Thus, in addition to their role in cancer resistance to chemotherapy, ABC transporters are involved in numerous physiological processes, and their value as therapeutic targets is likely to be far broader than just in cancer treatment.
Screening of chemical libraries for biologically active compounds aimed at certain cellular targets becomes a powerful approach to the isolation of small molecules regulating cellular function that can be useful as both research tools and therapeutic agents (13). Because ABC transporters are traditionally viewed as targets for therapeutic suppression, a number of chemical and protein inhibitors of these proteins were developed during the last decade, some of which have already been used in clinical trials to overcome MDR (5). We explored another aspect of the potential therapeutic value of ABS transporters: as targets for activation to increase their protective role against a variety of toxins. It was previously shown that P-gp activity against certain substrates could be significantly modulated by specific mutations presumably affecting conformation of drug-recognizing domains of the protein (14–17). These findings suggested that small molecules that modify conformation of the transporter might have a similar effect on its activity and can be isolated by screening compounds protecting P-gp-expressing cells from the toxicity of certain P-gp substrates. Here we report successful application of this approach to isolation of a series of small molecules that protect P-gp-expressing cells from adriamycin (Adr) by modulating substrate specificity of P-gp associated with dramatic changes in the spectrum of cross-resistance.
Materials and Methods
Drugs.
Rhodamine 123 (Rho-123), Adr, daunorubicin, Taxol, etoposide, vinblastine, vincristine, cytochalasin B, colcemid, colchicine, actinomycin D, gramicidin D, verapamil hydrochloride, prazosin, progesterone, puromycin, and Hoechst 33342 were all purchased from Sigma and dissolved in DMSO (except progesterone) in a 1-mM concentration. Progesterone was dissolved in PBS at a 1-mM concentration.
Cells.
Mouse fibroblast cell line ConA, human cell lines KB3-1, and its drug-resistance derivative KB8-5-11 were maintained in DMEM with 10% FBS, 2 mM glutamine, 100 units/ml penicillin G, and 100 μg/ml streptomycin sulfate (all from GIBCO/BRL). At every fourth passage, KB8-5-11 cells were cultivated in the presence of 25 ng/ml of Adr to eliminate cell variants with decreased expression of P-gp.
Screening of Chemical Library.
A Chemical Diverset library was purchased form Chembridge (San Diego). Primary screening was done on Con A cells for the compounds that interfere with Adr-induced activation of p53-responsive reporter LacZ gene as described (13). Individual library compounds were added to the cells in a 10–20-μM concentration. To identify P-gp modulators, all of the “hits” isolated after primary screening were tested for their ability to modulate cellular accumulation of Rho-123. Con A cells grown in 96-well plates were incubated for 4 h with 0.5 μM Rho-123 in combination with individual library compounds, and cell fluorescence was analyzed by using fluorescent microscopy.
Drug Sensitivity Assays.
To estimate drug-mediated growth inhibition, 103 cells were plated per well of 96-well plates and incubated with a range of drug concentrations added 24 h after plating. After 5–7 days of drug treatment, plates were fixed and stained with 2% methylene blue solution on 50% methanol. After elution with 1% SDS solution, optical density was analyzed on a Multiscan Ascent reader (Lybsystems, Helsinki) at 650 nm.
Drug Accumulation and Efflux Assay.
To determine drug accumulation, cells were incubated for 2 h with P-gp substrates Rho-123, Hoechst 33342, daunorubicin, or [3H]Taxol (Sigma), harvested by trypsinization, and suspended in ice-cold PBS for immediate flow cytometric analysis (for fluorescent substrates) or scintillation counting of incorporated radioactivity (for [3H]Taxol). For the drug efflux assay, the cells were washed thoroughly with PBS after incubation with the drugs, incubated in drug-free media with or without compounds from chemical library, and harvested at different time points for flow cytometric analysis (18). Library compound concentration was 10 μM in all of the experiments unless otherwise indicated.
Western Blot Analysis.
KB8-5-11 and KB3-1 cells were incubated for 4 h with selected compounds and the amounts of P-gp were estimated by using Western blotting with monoclonal antibody C219 (Signet Laboratories, Dedham, MA), as described (18).
Determination of cell surface expression of P-gp and the UIC2 shift assay were performed by using monoclonal antibodies C219 and UIC2, as described (19).
Analysis of Chemical Structures.
Flexible alignment of three-dimensional structures was performed by using MOE-FLEXALIGN, MOE 2001.0 software available from Chemical Computing Group (http://www.chemcomp.com). Similarity comparisons are based on the subdivision of two-dimensional representations of molecules into certain fragments (also called fingerprints). The measure of fingerprint overlap between two molecules is given by the Tanimoto coefficient, a numerical measure of molecular similarity; molecules showing high similarity coefficients would be expected to also show similar biological effects (20). A similarity correlation matrix was created by using the “Structure similarity” function of CHEMFINDER V. 6.0 (CambridgeSoft, Cambridge, MA) for EXCEL (Microsoft). All structures in Table 1 were placed in a column of the CHEMFINDER for EXCEL worksheet and for each the similarity coefficient related to all others was calculated by activating the above-mentioned function. Thus, a matrix displaying the similarity values for each molecule relative to all others in the set was obtained.
Table 1.
Names and structures of P-gp modulators
| QB no. | Name | Structure |
|---|---|---|
| 2 | 1-Carbazol-9-yl-3-(3,5-dimethylpyrazol-1-yl)-propan-2-ol | 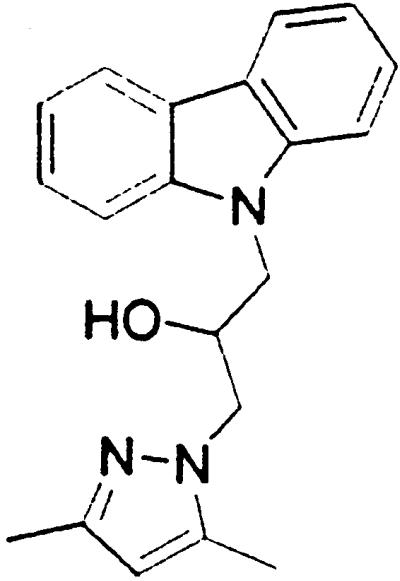 |
| 7 | 2-(4-Chloro-3,5-dimethylphenoxy)-N-(2-phenyl-2H-benzotriazol-5-yl)-acetamide |  |
| 8 | N-[2-(4-Chloro-phenyl)-acetyl]-N′-(4,7-dimethyl-quinazolin-2-yl)-guanidine |  |
| 9 | 1-Benzyl-7,8-dimethoxy-3-phenyl-3H-pyrazolo[3,4-c]isoquinoline |  |
| 10 | N-(3-Benzooxazol-2-yl-4-hydroxy-phenyl)-2-p-tolyloxyacetamide |  |
| 11 | 8-Allyl-2-phenyl-8H-1,3a,8-triaza-cyclopenta[a]indene |  |
| 12 | 3-(4-Chloro-benzyl)-5-(2-methoxy-phenyl)-[1,2,4]oxadiazole |  |
| 13 | 2-Phenethylsulfanyl-5,6,7,8-tetrahydro-benzo[4,5]thieno[2,3-d]pyrimidin-4-ylamine |  |
| 14 | (5,12,13-Triaza-indeno[1,2-b]anthracen-13-yl)-acetic acid ethyl ester |  |
| 15 | 2,2′-(1-phenyl-1H-1,2,4-triazole-3,5-diyl)bis-phenol |  |
| 16 | 2-(2-Chloro-phenyl)-5-(5-methylthiophen-2-yl)-[1,3,4]oxadiazole |  |
| 102 | 2-p-Tolyl-5,6,7,8-tetrahydrobenzo[d]imidazo[2,1-b]thiazole |  |
Results
Compounds That Suppress Cell Sensitivity to Adr in a p53-Independent Manner.
Treatment of cells with Adr results in the activation of the p53 pathway, followed by modulated expression of p53-responsive genes. Cells expressing p53-responsive reporter can, therefore, be used as a readout system for monitoring Adr activity and screening of chemicals that interfere with p53-mediated activation of the reporter. We have successfully used such a readout system for screening a chemical library for the compounds suppressing p53 (13). To prove that the isolated chemicals are in fact acting through p53 suppression, the initial “hits” were subjected to additional filtering for their p53 dependence. However, in addition to p53 inhibitors, we also identified a group of chemicals that prevented activation of p53-responsive reporter by Adr in a p53-independent manner. Because Adr is a known substrate for ABC-type transporters, we explored the potential involvement of active efflux in the biological effect of the isolated chemicals by introducing additional screening filters. First, we found that these compounds interfered with the accumulation of Adr in the cells as judged by fluorescent microscopy. Similarly, they reduced accumulation of another substrate of P-gp, Rho-123 (see Fig. 1a for example). Both effects were completely reversed by verapamil and reserpine, known inhibitors of P-gp. These results strongly indicated that the isolated compounds acted by an unusual mechanism: through activation of P-gp or other ABC transporters sensitive to P-gp inhibitors. The structural formulas of 12 compounds possessing the described properties are shown in Table 1.
Figure 1.

Stimulation of Rho-123 efflux from the cells by QB102 is suppressed by the P-gp inhibitor verapamil. (a) QB102-mediated reduction of Rho-123 accumulation by Con A fibroblasts is suppressed by verapamil. Cells were incubated for 2 h with the indicated concentrations of Rho-123, QB102 (10 μM), and verapamil (1 μM) and cellular fluorescence was analyzed by flow cytometry. (b) Cells were loaded with Rho-123 by incubation in the presence of 1 μM of the dye and quickly washed with Rho-123-free medium, and the intensity of cellular fluorescence was determined 1.5 h later in the presence or absence of QB102 and verapamil by fluorescent microscopy. (c) The dynamics of Rho-123 fluorescence reduction were monitored by flow cytometry.
Isolated Compounds Can Activate Efflux of Adr and Rho-123 by P-gp.
One of the most active compounds, QB102, was chosen for detailed characterization. First, QB102 did not have an effect on the excitation and emission spectra of Rho-123 and Adr fluorescence in solution, thus eliminating the possibility of its direct effect on the physico-chemical characteristics of the drugs (data not shown). We then tested whether QB102 affects the accumulation of fluorescent dyes by stimulating efflux or by interfering with their penetration through the cell membrane. Cells were loaded with Rho-123 or Adr by incubation in the medium with high concentration of the dye and quickly washed with the drug-free medium. The dynamics of reduction of cellular fluorescence were monitored at different time points in the presence or absence of QB102. The obtained results demonstrate that QB102 strongly increases efflux of Rho-123 and Adr from cells preloaded with the dye. This effect was completely reversed by verapamil and reserpine: in the presence of these P-gp inhibitors efflux of both Rho-123 and Adr was blocked regardless of the presence of QB102 (results obtained with Rho-123 are shown in Fig. 1 b and c). These observations confirmed the hypothesis that QB102 acts by activating some ABC transporter(s), presumably P-gp, that is known to be expressed in mouse fibroblasts (21).
To test the effect of QB102 specifically on P-gp, we used a pair of human cell lines that have been used as a conventional model for studying P-gp: P-gp-negative KB3-1 and its multidrug-resistant derivative KB8-5-11, overexpressing P-gp as a result of MDR1 gene amplification (22). KB3-1 and KB8-5-11 both have very low levels of MRP1 gene expression, as judged by reverse transcription (RT)-PCR assay (data not shown). Treatment with QB102 strongly decreased Rho-123 accumulation in KB8-5-11 cells and had no influence on KB3-1. Again, the decrease in accumulation correlated with stimulation of Rho-123 efflux and both effects were completely eliminated by verapamil (data not shown). Thus, QB102 definitely targets P-gp function, although we cannot exclude the possibility that activity of other ABC transporters could also be modulated by this compound.
The effect of QB102 on Rho-123 efflux can be detected as little as 5 min after addition of the compound (see Fig. 1c), leaving no time for gene expression to cause an effect. Four-hour incubation of 8-5-11 cells with QB102 does not change the amount of P-gp on the cell surface, as judged by the results of fluorescence-activated cell sorter (FACS) analysis with anti-P-gp antibodies UIC2 (data not shown). Hence, stimulation of P-gp function by QB102 cannot be explained by changes in gene expression or protein concentration on cell membrane.
QB102 Alters Cellular Cross-Resistance to Different Drugs by Modulating P-gp Function.
We tested whether activation of P-gp-mediated efflux of Adr by QB102 would affect cell sensitivity to this drug by using different types of drug assays. The results of representative growth inhibition assays are shown in Fig. 2. Treatment with QB102 increased resistance to Adr in both Con A and KB8-5-11, causing a pronounced shift in growth inhibition curves (see Fig. 2a for KB8-5-11); no effect on drug sensitivity was detected in P-gp-negative cell line KB3-1 (Fig. 2a). Interestingly, QB102 caused the opposite effect on the sensitivity of P-gp-expressing cells to Taxol (Fig. 2 a Right and b). Different effects of the compound on two different P-gp substrates suggested that QB102 is not a general activator of P-gp function, but rather a P-gp modulator.
Figure 2.

Different effects of QB102 on cell accumulation and sensitivity to different P-gp substrates. (a) Effect of QB102 on cell sensitivity to Adr and Taxol is P-gp-dependent. The results of colony assays carried out on P-gp positive (KB8-5-11) and P-gp negative (KB3-1) cells in constant to the presence of the indicated concentrations of the drugs. The effect of QB102 is dose-dependent. (b) QB102 (10 μM) has the opposite effect on cytotoxicity of Adr and Taxol and does not affect cell sensitivity to colcemide. Con A cells were growing 72 h in the presence of different concentrations of the indicated drugs with or without QB102, fixed with methanol, and stained with methylene blue. (c) QB102 has the opposite effect on accumulation of daunorubicin and Taxol. Con A cells were incubated 2 h with the indicated concentration of daunorubicin (Left) and Taxol (Right) and accumulation of the drugs was determined by flow cytometry (daunorubicin) or measurement of intracellular radioactivity ([3H]Taxol).
We tested the effects of QB102 on cell sensitivity to various P-gp substrates that, according to this criterion, fell into three groups (Table 2, Fig. 3). The first group includes drugs that become less toxic in the presence of QB102. Besides Adr and Rho-123, this group also contains daunorubicin and etoposide, although the effect of QB102 on cytotoxicity of the latter drug was much less pronounced. The second group consists of the drugs that become more potent in combination with QB102: Taxol, vincristine, vinblastine, gramicidin D, and Hoechst 33342. Finally, the third group is formed by those P-gp substrates that do not change their cytotoxicity in the presence of QB102 (actinomycin D, colcemid, cytochalasin D, and puromycin). Fig. 2b illustrates the influence of QB102 on cell sensitivity to the representatives of each of these three groups. Importantly, QB102 did not affect cell sensitivity to any of the non-P-gp substrates tested (see Table 2 and Fig. 3).
Table 2.
Influence of QB102 on cellular effects of different drugs
| Increased resistance/efflux | Decreased resistance/efflux | No effect |
|---|---|---|
| Adriamycin | Taxol | Colcemid |
| Daunorubicin | Vinblastine* | Colchicine |
| Etoposide* | Vincristine* | Actinomycin D |
| Rhodamine 123† | Gramicidin D* | Puromycin |
| Hoechst 33342† | Cytochalasin B* | |
| Camptothecin‡ | ||
| Carboplatin‡ | ||
| 5-Fluorouracil‡ |
Only resistance was tested.
Only efflux was tested.
Non-P-gp substrates.
Figure 3.

Effect of QB102 on cell sensitivity to different P-gp substrates. KB8-5-11 were grown 3 days in the presence of increasing concentrations of the indicated drugs with or without 10 μM of QB102. Fifty percent growth-inhibiting dose (IC50) was determined. Results of a representative experiment are shown.
Differential effect of QB102 on cell sensitivity to different P-gp substrates suggested that it might act by modulating P-gp activity, making it more effective against some and less effective against other substrates. To test this possibility, we analyzed how QB102 would affect the P-gp-mediated efflux of Taxol, the P-gp substrate belonging to the second group of drugs (i.e., those that become more potent in the presence of QB102). Consistent with the drug sensitivity data, QB102 suppressed P-gp-mediated efflux of Taxol by KB8-5-11 cells, acting as a P-gp inhibitor for this drug (Fig. 2c). These observations indicate that QB102 should be defined as a modulator rather than a general activator of P-gp that acts by changing relative substrate specificity of the transporter.
P-gp Modulators Vary in Their Relative Effect on Different P-gp Substrates.
We tested the effect of other compounds isolated for their stimulation of Adr accumulation on cellular uptake of two fluorescent P-gp substrates: Rho-123 and daunorubicin (daunorubicin was used in these experiments instead of Adr because of its brighter fluorescence, facilitating quantitative determination of drug accumulation). All of the compounds stimulated a decrease in accumulation of Rho-123 (see Fig. 4a). In all cases, the decreased accumulation correlated with stimulation of P-gp-dependent verapamil-sensitive drug efflux (data not shown). Interestingly, the isolated compounds fell into different categories according to their effect on daunorubicin accumulation. Some of the compounds behaved similarly to QB102 (QB11 and QB13), had a strong effect on Rho-123, and had 2–4 times less pronounced effect on daunorubicin. Compounds of another group (QB2, QB3, QB7, QB8, QB9, QB10, QB15, and QB16) were relatively effective in stimulating Rho-123 efflux but had a weak influence on daunorubicin accumulation; two compounds from this group (QB4 and QB14) had no effect on daunorubicin accumulation at all. Finally, one compound (QB12) was more effective in stimulating P-gp-mediated efflux of daunorubicin than Rho-123. Subsequently, we checked the effect of the compounds on cell resistance to daunorubicin.
Figure 4.

Differences in the relative effect of isolated P-gp modulators on different P-gp substrates correlate with their structures. (a) Comparison of the effect of each P-gp modulator on efflux of daunorubicin and Rho-123. Dependence of modulating effect on the dose of the compounds was determined for each modulator. In all cases, the modulating effect determined as stimulation of Rho-123 and daunorubicin efflux reached a plateau at a concentration of 5–10 μM. There was no difference in the dose dependence of modulating effect estimated for Rho-123 and daunorubicin. Con A cells were incubated for 2 h with Rho-123 (1 μM) or daunorubicin (500 ng/ml) in the presence of the indicated compounds (10 μM), and accumulation of the drugs was quantitated by flow cytometry. The compounds are shown in the order of their relative strength in stimulating Rho-123 efflux in comparison to the accumulation of the drugs in the absence of the P-gp modulators (c) or in the presence of verapamil (ver). (b) Flexible alignments of three-dimensional models of the indicated compounds. Light blue regions symbolize hydrogen bond acceptors.
As in the case of QB102, drug accumulation data correlated with the results of drug sensitivity assays. It is noteworthy that the compounds that demonstrated strong stimulatory effect on Rho-123 efflux increased cell sensitivity to Taxol and vinblastine (tested with all of the compounds at 10 μM concentration with the exception of QB15, which displayed growth inhibitory effect in long-term assays; data not shown).
Structural relationships among the different compounds were examined both by the evaluation of two-dimensional similarity and by molecular modeling. In general, the set of active compounds was found to be structurally diverse as evidenced by a relatively low structural likeness: in terms of Tanimoto metrics, only QB15 and QB9 had over 75% similarity. The latter compound also showed a 61% similarity quotient with QB7, whereas all other two-dimensional structure comparisons gave even lower similarity rates.
Alignment of the three-dimensional models of QB7, QB9, and QB15 showed two shared structural motifs (similarly placed hydrogen bond acceptors and six-member aromatic rings), suggesting that these particular compounds may be addressing some common P-gp-binding regions (Fig. 4b). Interestingly, the above molecules are indeed members of the group that stimulated Rho-123 efflux with a relatively weak influence on daunorubicin accumulation. Similarly, the three-dimensional alignment of QB102 with QB11 and QB13 showed good superposition (Fig. 3b), again in accordance with their biological activity.
Discussion
Isolation of P-gp modulators was a surprising and unexpected outcome of chemical library screening for p53 inhibitors, in which Adr was used as a DNA-damaging agent inducing p53. Many small molecules that target the activity of P-gp have been identified, the vast majority of which act as P-gp inhibitors (5), sensitizing cells to a variety of P-gp substrates. We, however, isolated a previously uncharacterized functional class of structurally diverged compounds that are capable of protecting cells from the drug by stimulating the P-gp-mediated efflux. They acted not as general stimulators of P-gp activity, but rather as P-gp modulators, making this transporter much more active against some and inactive against other substrates. “Retargeting” P-gp by the isolated compounds causes a dramatic change of the MDR phenotype, making P-gp-expressing cells more resistant to some drugs at the cost of losing resistance to the others. These findings indicate that cell resistance to toxic compounds can be dramatically increased by modulating the substrate specificity of multidrug transporters, broadening the approaches to rational control over the MDR phenotype by small molecules.
How do P-gp modulators work? The mechanism of P-gp modulation by the isolated compounds may imitate changes in substrate specificity of some P-gp mutants in which single amino acid substitutions greatly affect patterns of cross-resistance (14–17). Alternatively, they might alter the physico-chemical properties of the drug recognition pockets in the protein without changing conformation of the transporter. There is also a possibility that modulators do not interact with P-gp directly but modulate its function by altering plasma membrane properties or factors contributing to the activity of the transporter. In any event, they seem to be useful tools for the analysis of the mechanism of drug recognition function of P-gp.
There are several molecular models of ABC transporter function, all of which involve from two to three drug-binding sites (23–26). Thus, Shapiro and Ling (26) suggested a functional model of P-glycoprotein that contains at least two positive cooperative sites (the H and R sites) for drug binding and transport (the H site characterized as having high affinity to Hoechst and low affinity to Rho-123, and the R site as having opposite properties). Within this model, the activity of QB102 could be explained through its binding to the H site. P-gp substrates were classified as belonging to the R or H class, according to their effect on the transport of these two dyes. We sorted P-gp substrates according to the effect of QB102 on their efflux by P-gp (or cytotoxicity) and ended up with results resembling R–H classification. Thus, anthracyclines fell into the same category as Rho-123, whereas Hoechst formed the same group with Taxol. However, Vinca alkaloids and colchicines were found to belong to different categories in these two classifications, raising the possibility that P-gp modulators act through another mechanism.
Later Shapiro and Ling (27) found that P-gp substrates, prazosin and progesterone, could modulate efflux of other substrates in an in vitro system, suggesting that there is a third drug-binding site in P-gp and that these compounds might be modulators of the MDR phenotype. However, P-gp modulators described in the present study are clearly different from prazosin and progesterone because (i) they do not stimulate efflux of all P-gp substrates and (ii) they dramatically alter the cellular cross-resistance pattern of P-gp-expressing cells, whereas prazosin and progesterone do not. The use of thiol-reactive analogs of QB compounds for cysteine-scanning mutagenesis of P-gp would be a useful tool for identification of modulator-interacting sites within the transporter (28).
As far as other putative P-gp modulators are concerned, several previously published observations suggest that P-gp substrate specificity can be modulated by natural flavonoid polyphenols. Thus, quercetin was reported to act as a stimulator P-gp-mediated efflux of 7,12-dimethylbenz(a)anthracene (29) and Adr (30). However, others described quite the opposite effect of quercetin, at least on Adr efflux and resistance (31), and systematic study of flavonoid derivatives favored the latter results (32).
Comparison of biological effects of the identified P-gp modulators showed that they differ in their relative effect on the substrate specificity of P-gp, modifying the MDR phenotype in a manner specific for each compound. If they act by changing P-gp conformation, this would mean high conformational plasticity of this protein that could accept multiple different conformations and still retain its basic drug efflux function. Another assumption that follows from this observation is that P-gp activity can be finely “tuned” by the appropriate modulators to make it more effective against the desired toxins.
We did not test the effect of the small molecules modulating P-gp on other ABC transporters. Based on the frequency of P-gp modulators in our chemical library, we feel confident that similar selection performed on another cell-based readout system expressing non-P-gp ABC transporter (MRP, BCRP, etc.) would probably result in the isolation of the appropriate modulators. It would also be interesting to check the possibility of isolation of compounds that would target the transporter to efflux those toxins that have not originally been its substrates.
Considering their role in multidrug resistance of cancer, P-gp and other ABC transporters have thus far been viewed mainly as targets for suppression. Our observations offer another practical application of ABC transporters that stem from their natural function: their involvement in the protection of cells and tissues from a broad variety of cytotoxic compounds. The modulators of transporter substrate specificity make them more active against certain classes of drugs and can be used to facilitate chemoprotection under the conditions of acute or chronic poisoning with toxins or drugs. Selection of targeted modulators raises the possibility of developing detoxifying agents specific against certain classes of toxins. ABC transporters are known to be involved in the blood–brain placental barriers, thus defending brain tissue and fetuses against toxicity of a variety of toxic factors. Low efficiency of these defense systems against particular substrates may limit the application of high doses of otherwise useful drugs [i.e., cyclosporin A or ivermectin (7)]. P-gp modulators and other transporters could increase the tolerable doses of such drugs by increasing the effectiveness of these natural barriers. Moreover, the use of highly specific targeted modulators of ABC transporters can be beneficial for selective delivery of useful drugs through the blood–brain and placental barriers without generally affecting these defense systems against other factors.
Acknowledgments
We thank Alex Neyfakh for help and clever advise, Eugene Mechetner for UIC2 antibody, and Karen Toil for assistance in manuscript preparation. This work was supported by a grant from Quark Biotech, Inc. (to A.V.G.).
Abbreviations
- P-gp
P-glycoprotein
- MDR
multidrug resistance
- Adr
adriamycin
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Borst P. Semin Cancer Biol. 1997;8:131–134. doi: 10.1006/scbi.1997.0072. [DOI] [PubMed] [Google Scholar]
- 2.Konig J, Nies A T, Cui Y, Leier I, Keppler D. Biochim Biophys Acta. 1999;1461:377–394. doi: 10.1016/s0005-2736(99)00169-8. [DOI] [PubMed] [Google Scholar]
- 3.Gottesman M M, Pastan I. Annu Rev Biochem. 1993;62:385–427. doi: 10.1146/annurev.bi.62.070193.002125. [DOI] [PubMed] [Google Scholar]
- 4.Tan B, Piwnica-Worms D, Ratner L. Curr Opin Oncol. 2000;125:50–58. doi: 10.1097/00001622-200009000-00011. [DOI] [PubMed] [Google Scholar]
- 5.Sarkadi B, Muller M. Semin Cancer Biol. 1997;8:171–182. doi: 10.1006/scbi.1997.0069. [DOI] [PubMed] [Google Scholar]
- 6.Schinkel A. Semin Cancer Biol. 1997;8:161–170. doi: 10.1006/scbi.1997.0068. [DOI] [PubMed] [Google Scholar]
- 7.Schinkel A H, Smit J J, van Tellingen O, Beijnen J H, Wagenaar E, van Deemter L, Mol C A, van der Valk M A, Robanus-Maandag E C, te Riele H P, et al. Cell. 1994;77:491–502. doi: 10.1016/0092-8674(94)90212-7. [DOI] [PubMed] [Google Scholar]
- 8.Schinkel A H, Mayer U, Wagenaar E, Mol C A, van Deemter L, Smit J J, van der Valk M A, Voordouw A C, Spits H, van Tellingen O, et al. Proc Natl Acad Sci USA. 1997;94:4028–4033. doi: 10.1073/pnas.94.8.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lorico A, Rappa G, Finch R A, Yang D, Flavell R A, Sartorelli A C. Cancer Res. 1997;57:5238–5242. [PubMed] [Google Scholar]
- 10.Wijnholds J, Evers R, van Leusden M R, Mol C A, Zaman G J, Mayer U, Beijnen J H, van der Valk M, Krimpenfort P, Borst P. Nat Med. 1997;3:1275–1279. doi: 10.1038/nm1197-1275. [DOI] [PubMed] [Google Scholar]
- 11.Robbiani D F, Finch R A, Jager D, Muller W A, Sartorelli A C, Randolph G J. Cell. 2000;103:757–768. doi: 10.1016/s0092-8674(00)00179-3. [DOI] [PubMed] [Google Scholar]
- 12.Smit J J, Schinkel A H, Oude Elferink R P, Groen A K, Wagenaar E, van Deemter L, Mol C A, Ottenhoff R, van der Lugt N M, van Roon M A, et al. Cell. 1993;75:451–462. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- 13.Komarov P G, Komarova E A, Kondratov R V, Christov-Tselkov K, Coon J S, Chernov M V, Gudkov A V. Science. 1999;285:1733–1737. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 14.Choi K H, Chen C J, Kriegler M, Roninson I B. Cell. 1988;53:519–529. doi: 10.1016/0092-8674(88)90568-5. [DOI] [PubMed] [Google Scholar]
- 15.Gros P, Dhir R, Croop J, Talbot F. Proc Natl Acad Sci USA. 1991;88:7289–7293. doi: 10.1073/pnas.88.16.7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kajiji S, Talbot F, Grizzuti K, Van Dyke-Phillips V, Agresti M, Safa A R, Gros P. Biochemistry. 1993;32:4185–4194. doi: 10.1021/bi00067a005. [DOI] [PubMed] [Google Scholar]
- 17.Safa A R, Stern R K, Choi K, Agresti M, Tamai I, Metha N D, Roninson I B. Proc Natl Acad Sci USA. 1990;87:7225–7229. doi: 10.1073/pnas.87.18.7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hrycyna C A, Ramachandra M, Pastan I, Gottesman M M. Methods Enzymol. 1998;292:456–473. doi: 10.1016/s0076-6879(98)92035-3. [DOI] [PubMed] [Google Scholar]
- 19.Mechetner E B, Schott B, Morse B S, Stein W D, Druley T, Davis K A, Tsuruo T, Roninson I B. Proc Natl Acad Sci USA. 1997;94:12908–12913. doi: 10.1073/pnas.94.24.12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bajorath J. J Chem Inf Comput Sci. 2001;41:233–245. doi: 10.1021/ci0001482. [DOI] [PubMed] [Google Scholar]
- 21.Gupta R S. Biochem Biophys Res Commun. 1988;153:598–605. doi: 10.1016/s0006-291x(88)81137-9. [DOI] [PubMed] [Google Scholar]
- 22.Akiyama S, Fojo A, Hanover J A, Pastan I, Gottesman M M. Somat Cell Mol Genet. 1985;11:117–126. doi: 10.1007/BF01534700. [DOI] [PubMed] [Google Scholar]
- 23.Dey S, Ramachandra M, Pastan I, Gottesman M M, Ambudkar S V. Proc Natl Acad Sci USA. 1997;94:10594–10599. doi: 10.1073/pnas.94.20.10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Veen H W, Margolles A, Muller M, Higgins C F, Konings W N. EMBO J. 2000;19:2503–2514. doi: 10.1093/emboj/19.11.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharom F J, Liu R, Qu Q, Romsicki Y. Semin Cell Dev Biol. 2001;12:257–265. doi: 10.1006/scdb.2000.0251. [DOI] [PubMed] [Google Scholar]
- 26.Shapiro A B, Ling V. Eur J Biochem. 1997;250:130–137. doi: 10.1111/j.1432-1033.1997.00130.x. [DOI] [PubMed] [Google Scholar]
- 27.Shapiro A B, Fox K, Lam P, Ling V. Eur J Biochem. 1999;259:841–850. doi: 10.1046/j.1432-1327.1999.00098.x. [DOI] [PubMed] [Google Scholar]
- 28.Loo T W, Clarke D M. Biochim Biophys Acta. 1999;1461:315–325. doi: 10.1016/s0005-2736(99)00165-0. [DOI] [PubMed] [Google Scholar]
- 29.Phang J M, Poore C M, Lopaczynska J, Yeh G C. Cancer Res. 1993;53:5977–5981. [PubMed] [Google Scholar]
- 30.Critchfieod J M, Welsh C J, Phang J M, Yeh G C. Biochem Pharmacol. 1994;48:1437–1445. doi: 10.1016/0006-2952(94)90568-1. [DOI] [PubMed] [Google Scholar]
- 31.Scambia G, Ranelletti F O, Panici P B, De Vincenzo R, Bonanno G, Ferrandina G, Piantelli M, Bussa S, Rumi C, Cianfriglia M, et al. Cancer Chemother Pharmacol. 1994;34:459–464. doi: 10.1007/BF00685655. [DOI] [PubMed] [Google Scholar]
- 32.Ferte J, Kuhnel J-M, Chapuis G, Rolland Y, Lewin G, Schwaller M A. J Med Chem. 1999;42:478–489. doi: 10.1021/jm981064b. [DOI] [PubMed] [Google Scholar]