

Abstract
Hepatocellular carcinoma (HCC) is the third leading cause of cancer-related death worldwide; however, the mutational properties of HCC-associated carcinogens remain largely uncharacterized. We hypothesized that mechanisms underlying chemical-induced HCC can be characterized by evaluating the mutational spectra of these tumors. To test this hypothesis, we performed exome sequencing of B6C3F1/N HCCs that arose either spontaneously in vehicle controls (n=3) or due to chronic exposure to gingko biloba extract (GBE; n=4) or methyleugenol (MEG; n=3). Most archived tumor samples are available as formalin-fixed paraffin-embedded (FFPE) blocks, rather than fresh-frozen (FF) samples, hence, exome sequencing from paired FF and FFPE samples was compared. FF and FFPE samples showed 63-70% mutation concordance. Multiple known (e.g., Ctnnb1T41A, Braf V637E) and novel (e.g., Erbb4C559S, Card10A700V, Klf11P358L) mutations in cancer-related genes were identified. The overall mutational burden was greater for MEG than for GBE or spontaneous HCC samples. To characterize the mutagenic mechanisms, we analyzed the mutational spectra in the HCCs according to their trinucleotide motifs. The MEG tumors clustered closest to COSMIC Signatures 4 and 24, which are respectively associated with benzo(a)pyrene and aflatoxin induced HCCs in humans. These results establish a novel approach for classifying liver carcinogens and understanding the mechanisms of hepatocellular carcinogenesis.
Keywords: Exome sequencing, Sanger sequencing, methyleugenol (MEG), gingko biloba extract (GBE), mutational spectra, formalin fixed paraffin embedded tissues
Introduction
Hepatocellular carcinoma (HCC) is a leading cause of cancer-related deaths worldwide (Global Burden of Disease Cancer et al., 2017), and its incidence is increasing (Balogh et al., 2016). Efforts to understand the molecular basis of HCC and other cancers have motivated researchers to establish databases to classify the spectra of genomic changes involved in subtypes of human cancers (https://www.genome.gov/17516564/the-cancer-genome-atlas/; http://icgc.org/; http://cancer.sanger.ac.uk/cosmic/signatures). Multiple large-scale analyses suggest that tumors carry “mutational signatures”, which are unique combinations of mutations that are a consequence of genetic alterations influenced by various environmental exposures (Helleday et al., 2014, Alexandrov et al., 2013). The mutational signature is determined by the frequency of the unique combination of the base substitutions within the trinucleotide context (5′ and 3′ to each mutated base) (Alexandrov et al., 2013). Because the extent and types of chemical exposure in humans are not always quantifiable, efforts have been made to develop model systems for assessing the effects of chemical exposure in immortalized rodent or human cells (Olivier et al., 2014, Nik-Zainal et al., 2015, Severson et al., 2014) and in rodent tumors (Westcott et al., 2015). On the basis of these analyses, more than 30 mutational signatures have been identified, some which have been attributed to specific environmental agents. The signatures are conserved in human and rodent cancers and, as a result, are expected to have direct translational relevance (Schulze et al., 2015).
B6C3F1/N mouse, which is the F1 hybrid of C57BL6/N and C3H/HeN mice, has been used to evaluate toxicity and carcinogenicity by the National Toxicology Program (NTP) due to its ability to correctly identify several known carcinogens (King-Herbert and Thayer, 2006, King-Herbert et al., 2010). Though the B6C3F1/N mouse genome has not been fully assembled, HCCs from these mice have been characterized for the GTPase-encoding oncogene Hras, which is preferentially mutated in spontaneous tumors, and the Wnt signaling pathway gene Ctnnb1, which is preferentially mutated in some chemically-induced tumors (Devereux et al., 1999, Hoenerhoff et al., 2013). Because of their well-characterized mutation patterns, these genes can be used to validate the viability, power and accuracy of the exome sequencing approach for identifying additional mutations in B6C3F1/N mouse HCCs. Direct comparison of the mutational spectra for aflatoxin B1 from human and mouse HCCs show that they are remarkably similar (Chawanthayatham et al., 2017). Therefore, the B6C3F1/N mouse provides a relevant model system for comprehensive investigation of the mutational responses to environmental carcinogens.
In previous NTP studies, B6C3F1/N mice were used to identify the carcinogenic potential of ginkgo biloba extract (GBE) (National Toxicology, 2013) and methyleugenol (MEG) (National Toxicology, 2000). The selection of GBE, a widely used herbal supplement in traditional Chinese medicine for a variety of central neurodegenerative disorders (Nash and Shah, 2015), was based on concerns about its potential human health risks and its wide-spread use as a neutraceutical. Additionally, MEG, a plant-derived phenylpropanoid chemical that is commonly used as a flavoring agent in food products, was selected because it bears structural resemblance to safrole, which is a known carcinogen (Bode and Dong, 2015). Our results and those of others verified that both of these compounds have carcinogenic activity, but indicated that their mechanisms of carcinogenesis may differ fundamentally in terms of their levels of genotoxicity (non-genotoxic for GBE; genotoxic for MEG) (Li et al., 2009, Umegaki et al., 2007, Hoenerhoff et al., 2013, Maeda et al., 2015, Maeda et al., 2014, Burkey et al., 2000, Miller et al., 1983, Zhou et al., 2007). Genotoxic or non-genotoxic classification of chemical compounds is based on various in vitro and in vivo genotoxicity assays (Smith et al., 2016), where genotoxic carcinogens induce DNA damage or mutations and non-genotoxic carcinogens do not directly alter DNA, but instead influence cellular transformation secondarily, in response to proliferative or metabolic effects. Despite the circumstantial and mechanistic evidence linking GBE and MEG to cancer, the mutational signatures for these compounds have not been determined.
To explore the potential utility of mutational spectra in characterizing the mechanisms of GBE and MEG, we performed exome sequencing on HCCs from B6C3F1/N mice. Three HCCs that arose in untreated animals (spontaneous), four HCCs from animals treated with high dose levels of GBE, and three HCCs from animals treated with MEG were sequenced. As a control, exome sequencing was also performed on DNA extracted from non-tumor normal liver tissues from untreated age-matched vehicle control animals (n=3) from the GBE and MEG studies. Most tumor samples from NTP studies are not archived as fresh frozen (FF) samples, but are instead stored as formalin fixed paraffin embedded (FFPE) blocks following pathology evaluation. However, FFPE sample preparation is notorious for introducing DNA degradation and base modification (including cytosine deamination) (Do and Dobrovic, 2015). Therefore, to determine the feasibility of using NTP FFPE samples, we prepared paired FF and FFPE samples from each HCC and subjected them to exome sequencing, in parallel.
Materials and Methods
Mouse strains, tumor induction, and preparation of samples
Hepatocellular carcinomas from male and female B6C3F1/N mice (F1 hybrid mice from a C57BL6/N dam and a C3H/HeN sire) were procured from archival tissue from the NTP chronic GBE and MEG bioassays (National Toxicology, 2000, National Toxicology, 2013). The mice were exposed daily by oral gavage for 2 years to 2000 mg/kg GBE (n=4); 37, 75, or 150 mg/kg MEG (n=3); or a vehicle control (n=3). HCCs that developed after 2-year exposures were collected and one half was flash frozen (FF) in liquid nitrogen and the other half was collected in 10% neutral buffered formalin and routinely processed and embedded in paraffin blocks (FFPE) within 24-48 hours. Representative slides were stained with hematoxylin and eosin for microscopic examination. As a technical reference and also as a control for germline mutations, non-tumor normal liver tissues from untreated age-matched vehicle control animals were also collected from the GBE and MEG bioassays (n=3). Experiments were performed in compliance with the requirements set forth by the Guide for the Care and Use of Laboratory Animals.
DNA isolation and exome sequencing
DNA was extracted from FF and FFPE normal liver and GBE-treated, MEG-treated and spontaneous HCCs with DNeasy Tissue kits (Qiagen, Valencia, CA). For genomic library preparation, paired-end libraries were prepared using the Paired-End DNA Sample Preparation Kit (Illumina, San Diego, CA). Exome sequences were enriched from the genomic libraries according to the manufacturer’s protocol with the SureSelect Mouse Exome kit (G7550A-001: Agilent, CA). Sequencing was performed on the Illumina HiSeq2000 according to the manufacturer’s protocol. The 26 libraries were indexed and run on a total of 13 lanes (2 samples per lane) to generate an average of ≥50x depth of coverage required for the analyses.
Identification of germline variants in the B6C3F1 genome
Due to the lack of an assembled genome for the B6C3F1/N mouse, we used the assemblies of the C3H/HeN and C57BL6/N genomes from the Mouse Methylome Project (NIH/NIEHS grant #ZI-ES103146-01) to identify the differences between the C3H/HeN and C57BL6/N genomes using MUMmer (version 3.23). Differences (single nucleotide variation, small insertions/deletions events and multi-nucleotide variations) between the C3H/HeN and C57BL6/N genomes were identified, and those that were on exons (97,962 genomic locations; 0.15 % of the exonic bases) were compiled as a “truth set” of known germline variants in the B6C3F1/N mouse.
Identification of variants in HCC tumors and filtering against B6C3F1/N germline variants
Before analyzing the exome sequences, FASTQ files were subjected to quality control with the FastQC tool (www.bioinformatics.babraham.ac.uk/projects/fastqc/). The low-quality bases at the ends of the reads were removed. The exome sequencing reads were then aligned to the C3H/HeN genome using the Burrows-Wheeler aligner (BWA) mem algorithm (version 0.7.12). Aligned reads were pre-processed using a GATK best practices workflow consisting of marking duplicates, realigning around indels and recalibrating the base quality scores (Van der Auwera et al., 2013, McKenna et al., 2010). The pre-processed exome sequencing reads from each individual tumor sample were used to make tumor vs normal variant calls using the MuTect tool (version 1.1.4) (Cibulskis et al., 2013). In the initial preprocessing step, the MuTect tool ignores reads with too many mismatches or very low-quality scores since these represent noisy reads. The MuTect tool used default cutoffs of at least 14 reads in the tumor and at least 8 in the normal. For the normal variant calls, pools of either the three FF or the three FFPE samples were used. The “truth set” of known variants between the C3H/HeN and C57BL6/N genomes were used to classify the variants as somatic or germ-line. MuTect was used for variant calling in the C3H exonic regions.
Classification of variants with predicted oncogenic function
The variants were functionally annotated using the SnpEff tool (Cingolani et al., 2012) to predict their effects. The following functional information on each variant was obtained: type of the variant (synonymous/non-synonymous), functional class (Silent, Missense, Nonsense), and amino acid and codon change. Subsequently, the non-synonymous mutations were subjected to SIFT analysis (http://sift.bii.a-star.edu.sg/downloads/SIFTBLink.tar.gz), which considers the degree of conservation of amino acid residues of closely related sequences identified through PSI-BLAST, to predict whether the amino acid change would be tolerated (score ≥0.05) or would affect protein function (score <0.05). The variants were then compared with the known cancer driver mutations from: NIEHS Gene alteration database (GACDB) (Jackson et al., 2006, Lea et al., 2007), IntOGen (Integrative Oncogenomics database) (Gonzalez-Perez et al., 2013) and Catalogue of Somatic Mutations in Cancer (COSMIC database) (Forbes et al., 2017). Sanger sequencing was used to confirm some of these variants.
Analysis of mutational spectra
Mutational spectra were obtained by examining the trinucleotide context (one nucleotide before and one nucleotide after the mutation). The frequency of occurrence of each such “motif” was computed using the R package “Somatic Signatures”. Six substitution subtypes were included, based on the pyrimidine of the mutated Watson-Crick base pair: C>A, C>G, C>T, T>A, T>C, and T>G. In addition, the substitutions were examined by incorporating information on the nucleotides immediately 5′ and 3′ to each mutated nucleotide generating 96 possible mutation types (6 types of substitutions * 4 types of nucleotides at the 5′ position * 4 types of nucleotides at the 3′ position).
To classify the mutational signatures obtained in our study, the known signatures for mutational processes in human cancers were downloaded from COSMIC (http://cancer.sanger.ac.uk/cosmic/signatures.txt). Hierarchical clustering based on the Kullback-Leibler distance method was used to compare the mutational signatures in human cancers to the mutational spectra from the mouse HCC FF/FFPE exome sequencing data after accounting for the differences in the trinucleotide composition of the human and mouse exomes.
Results
Identification of germline variants in the B6C3F1/N mouse
The genome of the standard NTP mouse model B6C3F1/N is not assembled. Therefore, we performed a series of initial analyses to identify differences between the C3H/HeN and C57BL6/N genomes using MUMmer (version 3.23) and the genome assemblies from the NIH/NIEHS B6C3F1/N Mouse Methylome Project (grant #ZI-ES103146-01). Differences (single nucleotide variation, small insertions/deletions events and multi-nucleotide variations) between the C3H/HeN and C57BL6/N genomes were identified at 7,087,821 genomic locations. Among these, 97,962 locations were on exons (0.15 % of the exonic bases; data not shown). This set of differences between the parental genomes was used as a “truth set” of known germline variants in the B6C3F1/N mice for comparison to the tumors.
To obtain a reliable set of tumor-specific variants from the exome sequencing data, we first filtered out the low-quality reads and aligned them to the C3H/HeN genome using the BWA mem algorithm (version 0.7.12). Analysis of the number of total and aligned reads from each of the tumor and normal tissue samples verified that a high percentage (>90%) of both the FF and FFPE samples aligned to the C3H/HeN genome (Supplementary Table 1). Furthermore, 60-90% of the bases in coding exons had a depth coverage of 50 reads or more for the majority of samples (Supplementary Figure 1). The exception was for the normal samples 2 and 3 that were processed by the FFPE method and had a much lower number of target bases (27-45%, versus 90-95% for the other samples). These findings verify the uniformity and high quality of the FF sample set, and suggest that two of the FFPE reference samples may have been compromised in their quality.
Next, we pre-processed the aligned reads using a GATK best practices workflow consisting of marking duplicates, realigning the sequences around indels and recalibrating the base quality scores. These pre-processed exome sequencing reads were used to make tumor vs normal variant calls using the MuTect tool (version 1.1.4). The “truth set” of known variants between the C3H/HeN and C57BL6/N genomes were used to classify the tumor variants as somatic or germline, and the final sets of tumor-specific variants for each sample were compiled for further functional analyses (Figure 1).
Figure 1.
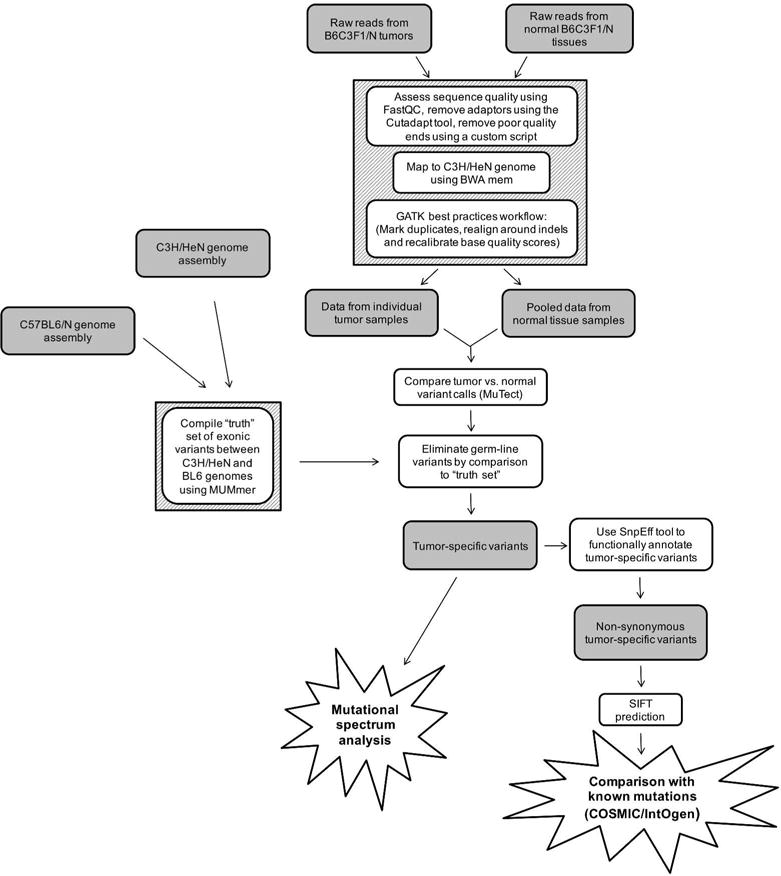
Steps used to obtain and analyze sets of tumor-specific variants. Raw reads from FF and FFPE normal tissues and individual tumor samples were filtered to remove low quality reads, mapped to the C3H genome, and subjected to a GATK best practices workflow protocol. Then, the resultant data sets for each tumor sample were compared to the pooled normal variant data set for the corresponding FF or FFPE samples. Germ-line variants were eliminated by removing the C3H versus BL6 variants in the “truth set”. The tumor-specific variants were then subject to mutational spectra analysis or filtered for non-synonymous variants and low SIFT scores prior to comparison to known mutations.
Identification of known and novel mutations in different tumor samples
To assess the accuracy and sensitivity of exome sequencing, we first evaluated the oncogenic mutations in Ctnnb1 and Hras that were identified by exome sequencing of the FF and FFPE samples and compared them to data obtained by Sanger sequencing of the FF samples from each tumor (Table 1). As expected (Devereux et al., 1999, Hoenerhoff et al., 2013), mutations in Ctnnb1 were identified only in chemically induced HCCs, whereas mutations in Hras were identified in both spontaneous and chemically-induced HCCs. A variety of different Ctnnb1 variants was identified by the Sanger method in GBE and MEG-induced HCCs, whereas only a subset of these mutations (T41A in a single GBE tumor and D32N and D32Y in two different MEG tumors) were identified by exome sequencing. Furthermore, these three Ctnnb1 mutations (T41A, D32N and D32Y) were detected only in the FF samples, whereas most of the variants of the Hras gene that were identified by Sanger sequencing were also detected by exome sequencing of both the FF and FFPE samples. Detailed analysis of the individual HCCs demonstrated that each had a unique pattern of Ctnnb1 and Hras variants (Supplementary Table 2), suggesting that heterogeneity exists among the variant-positive HCCs. This result was verified by Sanger sequencing of an independent set of spontaneous, GBE-induced and MEG-induced HCCs (n=10 each), which also showed a non-uniform distribution of the variants and a propensity for Ctnnb1 variants in chemically-induced HCCs and Hras in spontaneous HCCs (Table 1, last three columns).
Table 1.
Variants of Ctnnb1 and Hras identified by Sanger sequencing and exome sequencing*
| Gene | mutation | Tumors from mice that were used for exome sequencing | Tumors from other mice | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Spontaneous | GBE | MEG | Spontaneous | GBE | MEG | ||||||||
| Sanger (n=3) |
Exome-Seq, FF (n=3) |
Exome-Seq, PE (n=3) |
Sanger (n=4) |
Exome-Seq, FF (n=4) |
Exome-Seq, PE (n=4) |
Sanger (n=3) |
Exome-Seq, FF (n=3) |
Exome-Seq, PE (n=3) |
Sanger (n=10) |
Sanger (n=10) |
Sanger (n=7) |
||
| Ctnnb1 | Q28L | 1 | |||||||||||
| D32N | 1 | 1 | 1 | ||||||||||
| D32H | 1 | ||||||||||||
| D32Y | 1 | 1 | |||||||||||
| G34R | 1 | ||||||||||||
| T41A | 1 | 1 | |||||||||||
| T41L | 2 | ||||||||||||
| T42P | 1 | ||||||||||||
| S45F | 1 | ||||||||||||
| Δ5-13 | 1 | ||||||||||||
| Δ5-26 | 1 | ||||||||||||
| Hras | Q61L | 1 | 1 | 1 | |||||||||
| Q61K | 2 | 2 | 2 | 6 | 2 | ||||||||
| Q61R | 1 | 1 | 1 | 1 | 1 | ||||||||
Abbreviations: GBE, ginkgo biloba extract; MEG, methyleugenol; FF, exome sequencing for fresh frozen tissue; PE, exome sequencing for formalin-fixed paraffin-embedded tissue
To assess the composition of additional variant genes in each tumor tissue, we evaluated the matches to known variants in cancer-related genes from COSMIC (Forbes et al., 2017), IntOGen (Gonzalez-Perez et al., 2013) and NIEHS Gene alteration database (GACDB) (Jackson et al., 2006, Lea et al., 2007). Several mutations that align with known human mutations in cancer-related genes were identified in the MEG tumors, many of which were calculated to have SIFT scores of <0.05, indicating a greater potential for altered biological effects (Table 2). Among these known mutations, variants were identified in Braf (Shiraha et al., 2013), Elmo1 (Jiang et al., 2011), and Gnas (Nault et al., 2012), each of which have been established to play a role in the pathogenesis of HCC.
Table 2.
Variants from exome sequencing that correspond to known mutations in human cancer-related genes from publicly available databases*
| Gene | mutation | Spontaneous | GBE | MEG | Database match | SIFT score | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | ||||||||||||||||
| FF | PE | FF | PE | FF | PE | FF | PE | FF | PE | FF | PE | FF | PE | FF | PE | FF | PE | FF | PE | COSMIC | IntOGen | NIEHS | |||
| Acss3 | G672V | X | X | yes | 0.08 | ||||||||||||||||||||
| Bcl11a | A189T | X | X | yes | 0.06 | ||||||||||||||||||||
| Braf | V637E | X | X | yes | yes | yes | 0 | ||||||||||||||||||
| Clta | N88S | X | X | X | X | yes | 0.28 | ||||||||||||||||||
| Ctnnb1 | D32N | X | yes | yes | yes | 0 | |||||||||||||||||||
| D32Y | X | yes | 0 | ||||||||||||||||||||||
| T41A | X | yes | 0 | ||||||||||||||||||||||
| Dnahc5 | E3279K | X | X | yes | 0.38 | ||||||||||||||||||||
| Elmo1 | G125A | X | yes | 0 | |||||||||||||||||||||
| Elk3 | P88L | X | yes | 0.06 | |||||||||||||||||||||
| Gnas | R926C | X | X | yes | 0 | ||||||||||||||||||||
| Hras | Q61K | X | X | X | X | yes | yes | yes | 0.1 | ||||||||||||||||
| Q61R | X | X | yes | yes | 0 | ||||||||||||||||||||
| Kif3c | E76K | X | yes | 0.09 | |||||||||||||||||||||
| Lrp1b | R1646K | X | X | yes | 0.85 | ||||||||||||||||||||
| Lyst | Y2952C | X | yes | 0 | |||||||||||||||||||||
| Rbbp5 | L399V | X | yes | 0.08 | |||||||||||||||||||||
| Slc2a13 | R601W | X | yes | 0.01 | |||||||||||||||||||||
Abbreviations: GBE, ginkgo biloba extract; MEG, methyleugenol; FF, fresh frozen tissue; PE, formalin-fixed paraffin-embedded tissue
To further extend the array of cancer-related gene variants identified in HCCs from spontaneous, GBE and MEG-treated mice, we performed a global search of uncharacterized mutations within known cancer-related genes. Several previously uncharacterized mutations had SIFT scores <0.05, the majority of which were in tumors from mice treated with the higher MEG doses (Supplementary Table 3). Some notable examples include a C559S mutation in Erbb4 (Soung et al., 2006); an A700V mutation in Card10 (Wang et al., 2001); and a P358L mutation in Klf11 (Fluhr et al., 2017), all of which have not been previously characterized, though the genes have established roles in HCC or other cancers. Several variants were also identified exclusively in spontaneous or GBE-induced HCCs, but not in MEG-induced HCCs, such as a G97C mutation in Ulk1 (Lee and Jang, 2015, Xu et al., 2013) in a spontaneous HCC sample and a D226E mutation in Actn1 (Feng et al., 2013) in a GBE-induced HCC sample. These results further support the validity of our model and suggest the potential utility of the exome sequencing approach for new gene mutation discovery.
MEG displays a dose-dependent and higher mutational burden than GBE or spontaneous HCCs
To achieve a global perspective on the effects of the two chemicals on mutagenicity, we evaluated the number of non-synonymous variant calls for each treatment group. The mutation spectra and total mutation counts from FF and FFPE tissues for each of the exposures are presented in supplementary figure 2 and supplementary tables 6, respectively. The overall mutation burden was higher in HCCs from MEG-treated mice than from GBE-treated mice or HCCs arising spontaneously. Furthermore, a dose-related increase in mutation burden was observed for the MEG-treated mice (Figure 2A). This pattern was replicated for both FF and FFPE samples, though the overall number of variant calls was uniformly greater for the FFPE samples. The dose-dependent increase in mutation burden was also observed for combined non-synonymous and synonymous variants (Supplementary Table 4).
Figure 2.
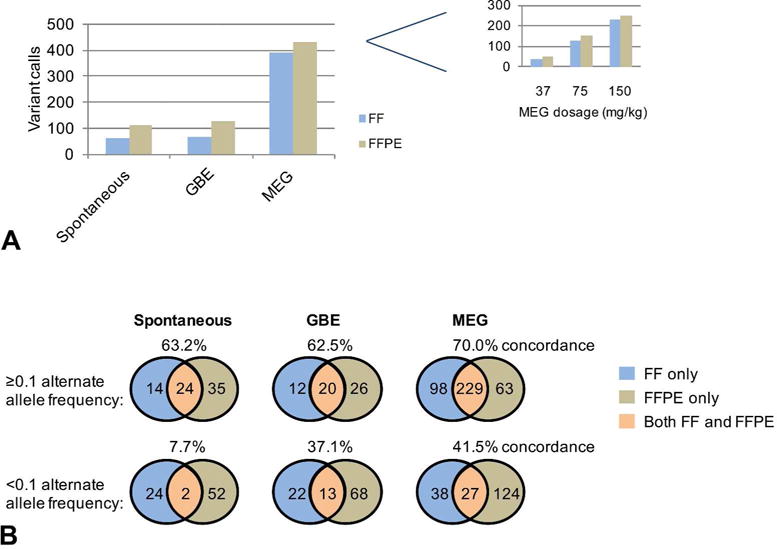
Non-synonymous mutational burdens in FF and FFPE samples. (A) The total numbers of non-synonymous mutations are shown for sets of Spontaneous (n=3), GBE (n=4) and MEG (n=3) tumors that were processed as FF and FFPE samples. The inset shows the numbers of non-synonymous mutations identified in individual MEG samples from mice treated at the specified dosages. (B) Venn diagram of the total numbers of non-synonymous mutations identified uniquely in the FF or FFPE samples and in both FF and FFPE samples for Spontaneous, GBE and MEG sets of tumors at higher (≥0.1) or lower (<0.1) alternate allele frequency. The concordance was calculated as a percentage of FF mutations that were also identified in the FFPE samples.
MEG tumors display distinct mutational spectra
To determine whether MEG and GBE might preferentially induce mutations in specific nucleotides, we generated mutational spectra of the FF and FFPE exome sequencing samples based on the trinucleotide context as described in the method section (Supplementary table 7). The spectrum plots (Figure 3A) show some unique differences in the spectral patterns for the MEG samples compared to the GBE and spontaneous HCCs, which were similar to each other.
Figure 3.
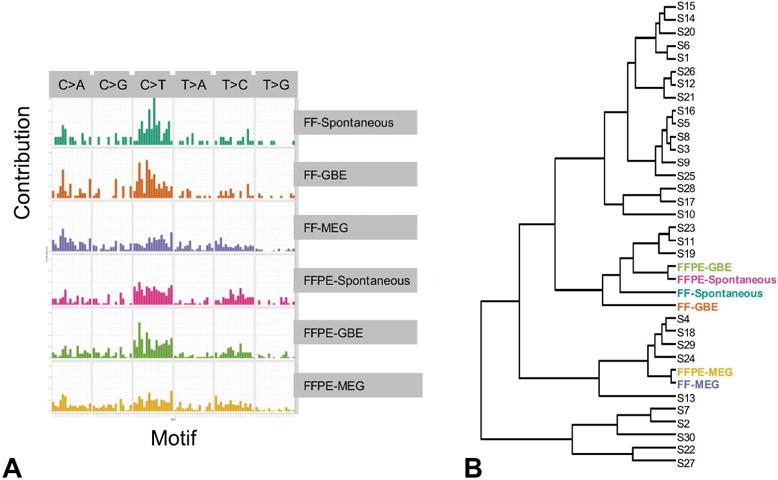
Analysis of mutational spectra of spontaneous tumors and tumors from mice treated with GBE or MEG. (A) The spectra of mutations based on the trinucleotide content (one nucleotide before to one nucleotide after the mutation). (B) Hierarchical clustering based on the Kullback-Leibler distance method after accounting for the differences in the trinucleotide composition of the human and mouse genomes. The six sets of samples were classified together with 30 “signatures” from COSMIC database
To further examine these differences, we performed clustering using the Kullback-Leiber distance measure for the mutational spectra of FF and FFPE samples and 30 publicly available COSMIC signatures that have been previously characterized (Forbes et al., 2017). The FF and FFPE MEG tumors clustered closest to a cluster containing Signatures 4 and 24, which has been observed in a subset of human liver cancers, including cancers with known exposure to benzo(a)pyrene and aflatoxin B1 (Figure 3B) (Chawanthayatham et al., 2017). Furthermore, the mutational spectra for both the FF and FFPE GBE samples clustered with those of the spontaneous HCCs near Signatures 2, 7, 11, 19 and 23.
Single nucleotide variants (SNV) Concordance between FF and FFPE samples
To assess the concordance between the FF and FFPE samples, we examined the overlap in the exonic non-synonymous variants identified in paired samples. Approximately 63-70% of the variants in the FF exome sequencing data set were also represented in the FFPE exome sequencing data set (Figure 2A). The primary reason for the discordance between the FF and the FFPE mutations is the poor signal in 2 out of the 3 FFPE normal samples. This could be due to the degradation, library prep, sample handling, etc. of the FFPE normal samples. Also, we noted that at a very low alternate allele frequency (<0.1), the concordance between FF and FFPE was reduced (8-40%) (Figure 2B). These results suggest that variant calls in FFPE samples preserve the majority of the mutations from FF samples, but that FFPE sample variants that are of higher alternate allele frequency are likely to be most reliable. However, when we used a more stringent cutoff of alternate allele frequency >=0.2 and it produced a slightly higher rate of concordance (~73-77%) between FF and FFPE (data not presented).
To gain further insight into potential sources of discordance in the exome sequencing data, we performed Sanger sequencing of a concordant gene (Braf) and a discordant gene (Bves). The Braf V637E mutation, which was identified by exome sequencing in both FF and FFPE samples, could be verified by Sanger sequencing. However, the three variants of Bves (M166K, R180S and P195L), which showed discordance in the FF and FFPE samples for mice from different groups, were not verifiable by Sanger sequencing (Supplementary Table 5). We also noted that the alternate allele frequency for Bves (average 0.12; range 0.059-0.32), was lower than the alternate allele frequency for Braf (0.368).These results suggest that the discordance in our exome sequencing data could potentially be explained, in part, by a level of false positivity, which has been shown to be inherent to the exome sequencing method (Mu et al., 2016), and which may be more prominent for mutations that have low alternate allele frequency.
We also re-examined the discordant Ctnnb1 variants from Table 1 to determine whether low alternate allele frequency may explain their observed discordance in the FF and FFPE samples. However, the alternate allele frequencies of the discordant Ctnnb1 variants (D32N: 0.25; D32Y: 0.375; T41A: 0.375) were relatively high, suggesting that other factors may contribute to discordance. Further investigation demonstrated that Ctnnb1 had an unusually low number of supporting reads (8-10 reads), which could provide an alternate potential explanation for discordance between FF and FFPE samples; We took a very careful look at all the reads in the FF and FFPE samples for the Ctnnb1 variants. From this investigation, we found that the low number of supporting reads and noisy signal in the FFPE normal samples may be the cause of the discordance between the FF and FFPE samples for the Ctnnb1 gene.
To determine whether the inflated number of variant calls in the FFPE samples might be related to an increase in the incidence to C to T mutations, which is a common artifact of FFPE processing (Do and Dobrovic, 2015), we examined the percentage of C:T transitions among the variants. However, the number of C:T variants in FFPE samples (Spontaneous, 15%; GBE, 21%; MEG, 9.4%) did not follow a consistent trend relative to the number of C:T variants in FF samples (Spontaneous, 21.4%; GBE, 12.8%; MEG, 11.0%) (data not shown).
Discussion
Similar to other cancers, HCCs are characterized by diverse networks of molecular events, which include the introduction of genomic mutations, the modulation of epigenetic regulation (histone modification and miRNAs/lncRNAs), the dysregulation of gene expression profiles, and the modulation of post-translational modification. Emerging research studies have demonstrated that external factors, such as chemical carcinogen exposure, also leave a “signature” on the HCCs at the genomic, epigenetic and transcriptional levels (Westcott et al., 2015, Lin et al., 2017, Shi et al., 2016). In this study, we sought to characterize the genomic alterations caused by exposure of B6C3F1/N mice to GBE and MEG for two years. We also sought to determine whether a mutational signature for these carcinogens could be identified for both FF and FFPE tumor sample preparation methods. Our results defined a mutational signature for MEG that was similar in both FF and FFPE samples. Furthermore, we uncovered an array of mutations in cancer-related genes in spontaneous, GBE and MEG HCCs, including several that have not been previously identified. These findings suggest an approach for mechanistic characterization of suspected carcinogens, as well as a strategy for identifying novel molecular biomarkers of HCC.
Though GBE and MEG have each been shown to contribute to HCC development in NTP carcinogenicity bioassays (National Toxicology, 2000, National Toxicology, 2013), evidence suggests that they act through disparate mechanisms. GBE has traditionally been categorized to be non-genotoxic due to early evidence that it activates several nuclear receptors and induces xenobiotic metabolizing enzymes (Li et al., 2009, Umegaki et al., 2007). Furthermore, GBE induces hepatocellular hypertrophy in B6C3F1/N mice and modulates the expression of genes that control oxidative stress and proliferation (Hoenerhoff et al., 2013). These findings are supported by mechanistic evaluation of GBE in constitutive androstane receptor (CAR) knockout mice, which confirmed a predominant non-genotoxic mode of carcinogenicity (Maeda et al., 2015, Maeda et al., 2014). However, results from in vitro bacterial gene mutation assays (Ames assays) that were performed using the same lot of GBE suggest that GBE is mutagenic to different bacterial strains (Hoenerhoff et al., 2013). Thus, though the mechanism of GBE is likely to be primarily non-genotoxic in mice, it is conceivable that high doses of GBE in the Ames assay may contribute to some level of genotoxicity as evidenced by bacterial assays.
The mechanism of carcinogenesis induced by MEG, on the other hand, is likely to be genotoxic. Though MEG is not mutagenic in bacterial assays, evidence suggests that it can be metabolically converted in vivo to a DNA reactive electrophile, 1-hydroxymethyleugenol, which induces DNA adduct formation and hepatocellular carcinomas in male B6C3F1 mice (Burkey et al., 2000, Miller et al., 1983). MEG and other structurally similar compounds (e.g., safrole and estragole) can produce reactive intermediates that are capable of forming DNA adducts that result in genotoxicity (Zhou et al., 2007).
The B6C3F1/N is the standard mouse model for NTP bioassays and the hepatocellular carcinomas examined in this study were derived from bioassays using this mouse model (King-Herbert and Thayer, 2006, King-Herbert et al., 2010). However, because their genome is not assembled, we had to incorporate a series of additional analyses after exome sequencing to rule out single nucleotide polymorphisms and germ-line mutations and to ensure the reliability of the single nucleotide variant calls. The resultant data sets displayed >90% alignment with the C3H/HeN genome. To further eliminate non-somatic mutations, we used non-tumor liver tissues from vehicle control age-matched mice as reference samples. Because the mutagenic properties of chemicals typically involve specific structural elements that reside in DNA from any species, these findings are expected to have translational relevance in terms of the mechanistic action, though it is likely that the repertoire of genetic mutations that leads to HCC may have some features of species specificity, especially for chemicals with a non-genotoxic mode of action.
Though FFPE samples are notoriously compromised in their genomic quality (Do and Dobrovic, 2015), they constitute a vast resource of patient and experimental animal tumor samples for biomedical research throughout the world. Therefore, an important component of our work was the comparison of data resulting from FF and FFPE samples obtained from the same tumor mass. Although the FFPE variants aligned well to the C3H genome, the variants for two of the FFPE non-tumor control samples displayed a reduced depth of coverage, which could be due to reduced DNA quality from FFPE samples. Furthermore, the FF and FFPE samples displayed mutations in sets of genes that overlapped by approximately 60-70%. By comparison, approximately 70-80% concordance was obtained in a recent study using human tumors that were prepared by FF and FFPE methods (Hedegaard et al., 2014). Given that heterogeneity of HCCs is common, even within adjacent parts of the same tumor mass, incomplete concordance between variant sets might be expected for any study that compares adjacent samples (Lin et al., 2017). In this study, every HCC mass was dissected into 2 halves with each of them either FF or FFPE. DNA was isolated from the entire HCC section from the FFPE blocks. For FF HCCs, a part of the frozen tumor was used for DNA extraction. Hence, there is a chance that sample heterogeneity may have contributed to the discordance to some extent. Notably, the concordance between the FF and FFPE samples in our study was reduced for variants in genes with lower alternate allele frequency, for which several Sanger sequencing–verified mutations were incompletely detected by exome sequencing. These results suggest that there were qualitative differences in the content of the FF and FFPE variant sets that could be explained, in large part, by variable representation of low alternate allele frequency genes. Additional Sanger sequencing of a concordant gene (Braf) and a discordant gene (Bves) demonstrated that the discordance might also be explained, in part, by an inherent level of false positivity in the experimental assay. A low level of false positivity in next-generation sequencing (approximately 1.3%) has been shown to occur primarily in complex genomic regions; however, raising the quality-score threshold to a predicted zero false positive rate has led to a significant (2.2%) loss of detection for Sanger-confirmed variants (Mu et al., 2016). Therefore, though raising the threshold could improve the quality of the data, it could also lead to loss of information.
In addition to the qualitative differences in the FF and FFPE samples indicated by the incomplete concordance, the samples in our study also differed quantitatively in that more variant calls were uniformly observed in FFPE samples as compared to FF samples. This finding could be contributed in part by a slightly greater number of sequence reads that were performed for the FFPE samples than for the FF samples (Supplementary Table 1) or by the reduced quality of two of the FFPE normal reference tissue samples (Supplementary Figure 1), which may have resulted in inflation of the tumor:normal variant ratio. Alternatively, it is possible that the FFPE preparation method introduced more artifactual mutations. We did not, however, observe a consistent pattern of increase in C:G>T:A mutations, that are a common artifact due to spontaneous deamination reactions secondary to prolonged formalin fixation (Do and Dobrovic, 2015). The absence of increased abundance of C:G>T:A mutations in the FFPE samples compared to the FF samples may be related to the relatively short time (24-48 hours) of formalin fixation in these samples that have a frozen tissue counterpart (Prentice et al., 2018). The artifactual increase in C:G>T:A mutations may be remedied by pretreating with uracil N-glycosylase (UNG). In contrast to the FFPE samples in this study, the majority of the FFPE archival samples in NTP rodent carcinogenicity bioassays are fixed in formalin for weeks due to the large number of animals (~800) at study termination and NGS studies on these samples may benefit UNG pretreatment. We are currently conducting a prospective study, in order to identify the time that leads to increased C:G>T:A mutations during formalin fixation.
Despite the differences in the FF and FFPE data sets, the most obvious trend in the overall mutational burden was that it was much higher for MEG HCCs than for GBE or spontaneous HCCs and displayed a dose-dependent pattern of increase for MEG HCCs, regardless of the sample preparation method. The latter findings suggest that the potent genotoxic effects of MEG treatment superseded the subtler effects of the FFPE versus FF preparation method on the data sets. The most important consideration in favor of this possibility is that the hierarchical clustering revealed a similar mutational spectra signature for the FF and FFPE samples for each of the paired samples. Based on these findings, the approach used in this study should be applicable to both FF and FFPE samples, given that the effects of the chemical carcinogen are sufficiently potent, though additional filters, such as the application of alternate allele number restriction and the use of additional replicates, may help to ensure the quality of the data for FFPE samples.
Our results also suggest the possibility of using this approach to verify the roles of mutations that have been described in the literature and to identify new roles for previously uncharacterized mutations, genes, and pathways, which could help to establish potential novel biomarkers and therapeutic targets. By comparison with databases that catalogue known mutations in cancer-related genes, we revealed variants that align with mutations in the Wnt-signaling activator Ctnnb1 for chemically-induced tumors and in the MAPK/ERK pathway activator Hras for spontaneous tumors, which is consistent with the induction of HCC by distinct carcinogenic processes (Hoenerhoff et al., 2013). Other mutations that are predicted to affect protein function were identified in several genes that are known to participate in the development of HCC, including Braf (Shiraha et al., 2013), Elmo1 (Jiang et al., 2011), and Gnas (Nault et al., 2012). We also identified numerous variants in genes that are known to be associated with HCC and other cancers. These include a C559S mutation in Erbb4, a receptor tyrosine kinase that harbors at least 8 other exonic mutations in HCC and various other types of cancers (Soung et al., 2006); an A700V mutation in Card10, a member of the caspase recruitment domain/guanylate kinase class of signaling proteins that interacts with BCL10 and NF-kappa B (Wang et al., 2001); and a P358L mutation in Klf11, a Krüppel-like transcription factor that is dysregulated at the epigenetic level in cancers of the blood. A G97C mutation in the autophagy regulator Ulk1, which has an established role in HCC and in a wide variety of other cancers (Lee and Jang, 2015, Xu et al., 2013), was identified in a spontaneous tumor sample in our study, while a D226E mutation in the focal adhesion gene Actn1 (Feng et al., 2013) was identified in a GBE sample. Thus, our analysis may also contribute to the understanding of functional roles for previously uncharacterized mutations.
The clustering of MEG induced HCCs very close to other liver cancer signatures such as signature 4 and 24 based on its mutational spectra is a particularly compelling finding of this study, given the extreme potency of the prototypical signature 4 carcinogen benzo(a)pyrene and tobacco carcinogens, and 24 carcinogen aflatoxin B1, which is a common contaminant of a variety of food products (Gross-Steinmeyer and Eaton, 2012) suggesting these chemicals may have potentially similar mutagenic mechanisms. As a genotoxin, aflatoxin has been proposed to induce lipid peroxidation and generate aldehydes that inhibit DNA repair, leading to p53 codon 249 mutations in more than 60% of aflatoxin B1-associated HCCs (Weng et al., 2017). However, we did not identify p53 mutations in our analysis of MEG. The extent to which MEG overlaps in its mechanism of toxicity and carcinogenicity with benzo(a)pyrene and aflatoxin B1 will be important to assess in future studies. Though the mutational spectra for the MEG-induced HCCs had distinctly unique clustering patterns, the mutational spectra for the GBE-induced and spontaneous HCCs clustered together near Signature 2, 7, 11, 19 and 23. The underlying etiologies for signature 2 resembles cytosine mutations caused by APOBEC enzymes, signature 7 resembles mutations in pyrimidine dimers due to UV light, signature 11 resembles that of alkylating agents and that Signature 19 and 23 has not been reported; however, further studies may help determine its significance to the establishment of spontaneous and GBE-induced HCCs. Based on the genes that are mutated, it appears that GBE may accelerate intrinsic biochemical mutagenic processes leading to dose-related increases in HCC with similar mutation spectra to the spontaneous tumors. In addition, accumulation of mutations in additional genes such as Ctnnb1 might confer greater proliferative advantage to the GBE-exposed HCCs. Thus, these observations highlight the need for both mutational spectra and analysis of specific genes and pathways as complementary approaches to a more complete understanding of the underlying mechanisms of carcinogenesis.
In summary, the findings of this study are significant in terms of providing mechanistic insight into the mutagenic outcome of MEG exposure in mice, which is likely to be applicable to its outcome in humans. These results suggest that our approach for characterizing suspected carcinogens could be applied to tumor samples that are prepared by the FFPE method, the primary method of tissue fixation at the NTP archives and those of other worldwide research organizations. This study would be benefitted by expansion to a larger investigation with more samples and a greater variety of chemical compounds. Nevertheless, we have verified the utility of this approach for classifying chemical carcinogens and identifying molecular biomarkers of HCC, which could help in understanding the mechanisms of carcinogenesis and eventually prove instructive in the development of novel biomarkers of exposures and disease as well as personalized therapy.
Supplementary Material
Acknowledgments
We would like to acknowledge the assistance provided by Ms. Leslie Couch at the NTP archives in retrieving the fresh frozen tissues as well as the formalin fixed paraffin embedded tissues for this project as well as the NTP Pathology support core laboratory for providing tissue sections for DNA isolation. The efforts by the NTP staff that generated the cancer data for the GBE and MEG chronic bioassays are also acknowledged. We appreciate the help provided by Ms. Beth Mahler with the images. We would like to thank the reviewers for their constructive criticism of this manuscript.
Footnotes
Declaration of Conflicting Interests Statement
The authors declared no potential, real, or perceived conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjord JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jager N, Jones DT, Jones D, Knappskog S, Kool M, Lakhani SR, Lopez-Otin C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt AN, Valdes-Mas R, van Buuren MM, van‘t Veer L, Vincent-Salomon A, Waddell N, Yates LR, Australian Pancreatic Cancer Genome, I., Consortium, I.B.C., Consortium, I.M.-S. PedBrain I, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balogh J, Victor D, 3rd, Asham EH, Burroughs SG, Boktour M, Saharia A, Li X, Ghobrial RM, Monsour HP., Jr Hepatocellular carcinoma: a review. Journal of hepatocellular carcinoma. 2016;3:41–53. doi: 10.2147/JHC.S61146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode AM, Dong Z. Toxic phytochemicals and their potential risks for human cancer. Cancer prevention research. 2015;8:1–8. doi: 10.1158/1940-6207.CAPR-14-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkey JL, Sauer JM, McQueen CA, Sipes IG. Cytotoxicity and genotoxicity of methyleugenol and related congeners– a mechanism of activation for methyleugenol. Mutation research. 2000;453:25–33. doi: 10.1016/s0027-5107(00)00070-1. [DOI] [PubMed] [Google Scholar]
- Chawanthayatham S, Valentine CC, 3rd, Fedeles BI, Fox EJ, Loeb LA, Levine SS, Slocum SL, Wogan GN, Croy RG, Essigmann JM. Mutational spectra of aflatoxin B1 in vivo establish biomarkers of exposure for human hepatocellular carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:E3101–E3109. doi: 10.1073/pnas.1700759114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devereux TR, Anna CH, Foley JF, White CM, Sills RC, Barrett JC. Mutation of beta-catenin is an early event in chemically induced mouse hepatocellular carcinogenesis. Oncogene. 1999;18:4726–4733. doi: 10.1038/sj.onc.1202858. [DOI] [PubMed] [Google Scholar]
- Do H, Dobrovic A. Sequence artifacts in DNA from formalin-fixed tissues: causes and strategies for minimization. Clinical chemistry. 2015;61:64–71. doi: 10.1373/clinchem.2014.223040. [DOI] [PubMed] [Google Scholar]
- Feng Y, Ngu H, Alford SK, Ward M, Yin F, Longmore GD. alpha-actinin1 and 4 tyrosine phosphorylation is critical for stress fiber establishment, maintenance and focal adhesion maturation. Experimental cell research. 2013;319:1124–1135. doi: 10.1016/j.yexcr.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluhr S, Krombholz CF, Meier A, Epting T, Mucke O, Plass C, Niemeyer CM, Flotho C. Epigenetic dysregulation of the erythropoietic transcription factor KLF1 and the beta-like globin locus in juvenile myelomonocytic leukemia. Epigenetics. 2017;12:715–723. doi: 10.1080/15592294.2017.1356959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, Stefancsik R, Harsha B, Kok CY, Jia M, Jubb H, Sondka Z, Thompson S, De T, Campbell PJ. COSMIC: somatic cancer genetics at high-resolution. Nucleic acids research. 2017;45:D777–D783. doi: 10.1093/nar/gkw1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Global Burden of Disease Cancer, C. Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, Brenner H, Dicker DJ, Chimed-Orchir O, Dandona R, Dandona L, Fleming T, Forouzanfar MH, Hancock J, Hay RJ, Hunter-Merrill R, Huynh C, Hosgood HD, Johnson CO, Jonas JB, Khubchandani J, Kumar GA, Kutz M, Lan Q, Larson HJ, Liang X, Lim SS, Lopez AD, MacIntyre MF, Marczak L, Marquez N, Mokdad AH, Pinho C, Pourmalek F, Salomon JA, Sanabria JR, Sandar L, Sartorius B, Schwartz SM, Shackelford KA, Shibuya K, Stanaway J, Steiner C, Sun J, Takahashi K, Vollset SE, Vos T, Wagner JA, Wang H, Westerman R, Zeeb H, Zoeckler L, Abd-Allah F, Ahmed MB, Alabed S, Alam NK, Aldhahri SF, Alem G, Alemayohu MA, Ali R, Al-Raddadi R, Amare A, Amoako Y, Artaman A, Asayesh H, Atnafu N, Awasthi A, Saleem HB, Barac A, Bedi N, Bensenor I, Berhane A, Bernabe E, Betsu B, Binagwaho A, Boneya D, Campos-Nonato I, Castaneda-Orjuela C, Catala-Lopez F, Chiang P, Chibueze C, Chitheer A, Choi JY, Cowie B, Damtew S, das Neves J, Dey S, Dharmaratne S, Dhillon P, Ding E, Driscoll T, Ekwueme D, Endries AY, Farvid M, Farzadfar F, Fernandes J, Fischer F, TT GH, Gebru A, Gopalani S, et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA oncology. 2017;3:524–548. doi: 10.1001/jamaoncol.2016.5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Tamborero D, Schroeder MP, Jene-Sanz A, Santos A, Lopez-Bigas N. IntOGen-mutations identifies cancer drivers across tumor types. Nature methods. 2013;10:1081–1082. doi: 10.1038/nmeth.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross-Steinmeyer K, Eaton DL. Dietary modulation of the biotransformation and genotoxicity of aflatoxin B(1) Toxicology. 2012;299:69–79. doi: 10.1016/j.tox.2012.05.016. [DOI] [PubMed] [Google Scholar]
- Hedegaard J, Thorsen K, Lund MK, Hein AM, Hamilton-Dutoit SJ, Vang S, Nordentoft I, Birkenkamp-Demtroder K, Kruhoffer M, Hager H, Knudsen B, Andersen CL, Sorensen KD, Pedersen JS, Orntoft TF, Dyrskjot L. Next-generation sequencing of RNA and DNA isolated from paired fresh-frozen and formalin-fixed paraffin-embedded samples of human cancer and normal tissue. PloS one. 2014;9:e98187. doi: 10.1371/journal.pone.0098187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nature reviews. Genetics. 2014;15:585–598. doi: 10.1038/nrg3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenerhoff MJ, Pandiri AR, Snyder SA, Hong HH, Ton TV, Peddada S, Shockley K, Witt K, Chan P, Rider C, Kooistra L, Nyska A, Sills RC. Hepatocellular carcinomas in B6C3F1 mice treated with Ginkgo biloba extract for two years differ from spontaneous liver tumors in cancer gene mutations and genomic pathways. Toxicologic pathology. 2013;41:826–841. doi: 10.1177/0192623312467520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MA, Lea I, Rashid A, Peddada SD, Dunnick JK. Genetic alterations in cancer knowledge system: analysis of gene mutations in mouse and human liver and lung tumors. Toxicological sciences: an official journal of the Society of Toxicology. 2006;90:400–418. doi: 10.1093/toxsci/kfj101. [DOI] [PubMed] [Google Scholar]
- Jiang J, Liu G, Miao X, Hua S, Zhong D. Overexpression of engulfment and cell motility 1 promotes cell invasion and migration of hepatocellular carcinoma. Experimental and therapeutic medicine. 2011;2:505–511. doi: 10.3892/etm.2011.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King-Herbert A, Thayer K. NTP workshop: animal models for the NTP rodent cancer bioassay: stocks and strains–should we switch? Toxicologic pathology. 2006;34:802–805. doi: 10.1080/01926230600935938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King-Herbert AP, Sills RC, Bucher JR. Commentary: update on animal models for NTP studies. Toxicologic pathology. 2010;38:180–181. doi: 10.1177/0192623309356450. [DOI] [PubMed] [Google Scholar]
- Lea IA, Jackson MA, Li X, Bailey S, Peddada SD, Dunnick JK. Genetic pathways and mutation profiles of human cancers: site- and exposure-specific patterns. Carcinogenesis. 2007;28:1851–1858. doi: 10.1093/carcin/bgm176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YJ, Jang BK. The Role of Autophagy in Hepatocellular Carcinoma. International journal of molecular sciences. 2015;16:26629–26643. doi: 10.3390/ijms161125984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stanton JD, Tolson AH, Luo Y, Wang H. Bioactive terpenoids and flavonoids from Ginkgo biloba extract induce the expression of hepatic drug-metabolizing enzymes through pregnane X receptor, constitutive androstane receptor, and aryl hydrocarbon receptor-mediated pathways. Pharmaceutical research. 2009;26:872–882. doi: 10.1007/s11095-008-9788-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DC, Mayakonda A, Dinh HQ, Huang P, Lin L, Liu X, Ding LW, Wang J, Berman BP, Song EW, Yin D, Koeffler HP. Genomic and Epigenomic Heterogeneity of Hepatocellular Carcinoma. Cancer research. 2017;77:2255–2265. doi: 10.1158/0008-5472.CAN-16-2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda J, Inoue K, Ichimura R, Takahashi M, Kodama Y, Saito N, Yoshida M. Essential role of constitutive androstane receptor in Ginkgo biloba extract induced liver hypertrophy and hepatocarcinogenesis. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association. 2015;83:201–209. doi: 10.1016/j.fct.2015.06.010. [DOI] [PubMed] [Google Scholar]
- Maeda J, Kijima A, Inoue K, Ishii Y, Ichimura R, Takasu S, Kuroda K, Matsushita K, Kodama Y, Saito N, Umemura T, Yoshida M. In vivo genotoxicity of Ginkgo biloba extract in gpt delta mice and constitutive androstane receptor knockout mice. Toxicological sciences: an official journal of the Society of Toxicology. 2014;140:298–306. doi: 10.1093/toxsci/kfu090. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EC, Swanson AB, Phillips DH, Fletcher TL, Liem A, Miller JA. Structure-activity studies of the carcinogenicities in the mouse and rat of some naturally occurring and synthetic alkenylbenzene derivatives related to safrole and estragole. Cancer research. 1983;43:1124–1134. [PubMed] [Google Scholar]
- Mu W, Lu HM, Chen J, Li S, Elliott AM. Sanger Confirmation Is Required to Achieve Optimal Sensitivity and Specificity in Next-Generation Sequencing Panel Testing. The Journal of molecular diagnostics: JMD. 2016;18:923–932. doi: 10.1016/j.jmoldx.2016.07.006. [DOI] [PubMed] [Google Scholar]
- Nash KM, Shah ZA. Current Perspectives on the Beneficial Role of Ginkgo biloba in Neurological and Cerebrovascular Disorders. Integrative medicine insights. 2015;10:1–9. doi: 10.4137/IMI.S25054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Toxicology, P. NTP Toxicology and Carcinogenesis Studies of Methyleugenol (CAS NO. 93-15-2) in F344/N Rats and B6C3F1 Mice (Gavage Studies) National Toxicology Program technical report series. 2000;491:1–412. [PubMed] [Google Scholar]
- National Toxicology, P. Toxicology and carcinogenesis studies of Ginkgo biloba extract (CAS No. 90045-36-6) in F344/N rats and B6C3F1/N mice (Gavage studies) National Toxicology Program technical report series. 2013:1–183. [PubMed] [Google Scholar]
- Nault JC, Fabre M, Couchy G, Pilati C, Jeannot E, Tran Van Nhieu J, Saint-Paul MC, De Muret A, Redon MJ, Buffet C, Salenave S, Balabaud C, Prevot S, Labrune P, Bioulac-Sage P, Scoazec JY, Chanson P, Zucman-Rossi J. GNAS-activating mutations define a rare subgroup of inflammatory liver tumors characterized by STAT3 activation. Journal of hepatology. 2012;56:184–191. doi: 10.1016/j.jhep.2011.07.018. [DOI] [PubMed] [Google Scholar]
- Nik-Zainal S, Kucab JE, Morganella S, Glodzik D, Alexandrov LB, Arlt VM, Weninger A, Hollstein M, Stratton MR, Phillips DH. The genome as a record of environmental exposure. Mutagenesis. 2015;30:763–770. doi: 10.1093/mutage/gev073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier M, Weninger A, Ardin M, Huskova H, Castells X, Vallee MP, McKay J, Nedelko T, Muehlbauer KR, Marusawa H, Alexander J, Hazelwood L, Byrnes G, Hollstein M, Zavadil J. Modelling mutational landscapes of human cancers in vitro. Scientific reports. 2014;4:4482. doi: 10.1038/srep04482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentice LM, Miller RR, Knaggs J, Mazloomian A, Aguirre Hernandez R, Franchini P, Parsa K, Tessier-Cloutier B, Lapuk A, Huntsman D, Schaeffer DF, Sheffield BS. Formalin fixation increases deamination mutation signature but should not lead to false positive mutations in clinical practice. PLoS One. 2018;13:e0196434. doi: 10.1371/journal.pone.0196434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, Calatayud AL, Pinyol R, Pelletier L, Balabaud C, Laurent A, Blanc JF, Mazzaferro V, Calvo F, Villanueva A, Nault JC, Bioulac-Sage P, Stratton MR, Llovet JM, Zucman-Rossi J. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nature genetics. 2015;47:505–511. doi: 10.1038/ng.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severson PL, Vrba L, Stampfer MR, Futscher BW. Exome-wide mutation profile in benzo[a]pyrene-derived post-stasis and immortal human mammary epithelial cells. Mutation research Genetic toxicology and environmental mutagenesis. 2014;775-776:48–54. doi: 10.1016/j.mrgentox.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, He J, Lin J, Sun X, Sun F, Ou C, Jiang C. Distinct response of the hepatic transcriptome to Aflatoxin B1 induced hepatocellular carcinogenesis and resistance in rats. Scientific reports. 2016;6:31898. doi: 10.1038/srep31898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraha H, Yamamoto K, Namba M. Human hepatocyte carcinogenesis (review) International journal of oncology. 2013;42:1133–1138. doi: 10.3892/ijo.2013.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MT, Guyton KZ, Gibbons CF, Fritz JM, Portier CJ, Rusyn I, DeMarini DM, Caldwell JC, Kavlock RJ, Lambert PF, Hecht SS, Bucher JR, Stewart BW, Baan RA, Cogliano VJ, Straif K. Key Characteristics of Carcinogens as a Basis for Organizing Data on Mechanisms of Carcinogenesis. Environmental health perspectives. 2016;124:713–721. doi: 10.1289/ehp.1509912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soung YH, Lee JW, Kim SY, Wang YP, Jo KH, Moon SW, Park WS, Nam SW, Lee JY, Yoo NJ, Lee SH. Somatic mutations of the ERBB4 kinase domain in human cancers. International journal of cancer. 2006;118:1426–1429. doi: 10.1002/ijc.21507. [DOI] [PubMed] [Google Scholar]
- Umegaki K, Taki Y, Endoh K, Taku K, Tanabe H, Shinozuka K, Sugiyama T. Bilobalide in Ginkgo biloba extract is a major substance inducing hepatic CYPs. The Journal of pharmacy and pharmacology. 2007;59:871–877. doi: 10.1211/jpp.59.6.0014. [DOI] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Current protocols in bioinformatics. 2013;43:11-10–11-33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Guo Y, Huang WJ, Ke X, Poyet JL, Manji GA, Merriam S, Glucksmann MA, DiStefano PS, Alnemri ES, Bertin J. Card10 is a novel caspase recruitment domain/membrane-associated guanylate kinase family member that interacts with BCL10 and activates NF-kappa B. The Journal of biological chemistry. 2001;276:21405–21409. doi: 10.1074/jbc.M102488200. [DOI] [PubMed] [Google Scholar]
- Weng MW, Lee HW, Choi B, Wang HT, Hu Y, Mehta M, Desai D, Amin S, Zheng Y, Tang MS. AFB1 hepatocarcinogenesis is via lipid peroxidation that inhibits DNA repair, sensitizes mutation susceptibility and induces aldehyde-DNA adducts at p53 mutational hotspot codon 249. Oncotarget. 2017;8:18213–18226. doi: 10.18632/oncotarget.15313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westcott PM, Halliwill KD, To MD, Rashid M, Rust AG, Keane TM, Delrosario R, Jen KY, Gurley KE, Kemp CJ, Fredlund E, Quigley DA, Adams DJ, Balmain A. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature. 2015;517:489–492. doi: 10.1038/nature13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Yu H, Zhang X, Shen X, Zhang K, Sheng H, Dai S, Gao H. UNC51-like kinase 1 as a potential prognostic biomarker for hepatocellular carcinoma. International journal of clinical and experimental pathology. 2013;6:711–717. [PMC free article] [PubMed] [Google Scholar]
- Zhou SF, Xue CC, Yu XQ, Wang G. Metabolic activation of herbal and dietary constituents and its clinical and toxicological implications: an update. Current drug metabolism. 2007;8:526–553. doi: 10.2174/138920007781368863. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.