

Abstract
Copy number variation (CNV) is a common form of structural variation detected in human genomes, occurring as both constitutional and somatic events. Cytogenetic techniques like chromosomal microarray (CMA) are widely used in analyzing CNVs. However, CMA techniques cannot resolve the full nature of these structural variations (i.e. the orientation and location of associated breakpoint junctions) and must be combined with other cytogenetic techniques, such as karyotyping or FISH, to do so. This makes the development of a next-generation sequencing (NGS) approach capable of resolving both CNVs and breakpoint junctions desirable. Mate-pair sequencing (MPseq) is a NGS technology designed to find large structural rearrangements across the entire genome. Here we present an algorithm capable of performing copy number analysis from mate-pair sequencing data. The algorithm uses a step-wise procedure involving normalization, segmentation, and classification of the sequencing data. The segmentation technique combines both read depth and discordant mate-pair reads to increase the sensitivity and resolution of CNV calls. The method is particularly suited to MPseq, which is designed to detect breakpoint junctions at high resolution. This allows for the classification step to accurately calculate copy number levels at the relatively low read depth of MPseq. Here we compare results for a series of hematological cancer samples that were tested with CMA and MPseq. We demonstrate comparable sensitivity to the state-of-the-art CMA technology, with the benefit of improved breakpoint resolution. The algorithm provides a powerful analytical tool for the analysis of MPseq results in cancer.
Introduction
With the rapid advances in whole-genome sequencing (WGS) technology, genetic alterations like single nucleotide variations (SNVs) and structural variations (SVs) have been shown to play important roles in a variety of diseases. In particular, cancer is a disease that is characterized by genetic alterations that drive uncontrolled growth across all disease types.1–5 A major class of structural variant is copy number variation (CNV), defined as a loss or gain of at least 1kb genomic material.6 CNVs contribute to the natural variation in the healthy human genome7 and abnormal constitutional variants. They also represent a significant contributor to the pathogenic etiology of both constitutional and oncological disease. Because CNVs play a major role in cancer in modulating the expression levels of genes involved in cell growth and regulation8 it is important to precisely map which genes are gained or lost in the region and at what copy state. Traditionally, karyotyping and fluorescence in situ hybridization (FISH)9 have been used to identify CNVs. However, both methods have significant drawbacks in CNV detection. Karyotyping is low resolution and will only accurately determine large copy number variations (3–10 Mb) and without precise breakpoint location. FISH is limited in scope and only applicable in regions where a CNV may be suspected to occur. More recently, the most comprehensive and accurate way for CNVs to be detected is through an array-based comparative genomic hybridization (aCGH) or SNP-based microarrays, collectively described as chromosomal microarray (CMA).10,11
In brief, CMA provides an assessment of copy number in a sample genome by hybridization parameters compared to a reference, either by competitive hybridization of differentially labeled genome (aCGH) or by comparison to an in silico reference (SNP-based microarray). This allows for the detection of alterations in copy number throughout the genome at ~20–50kb resolution using the highest density arrays, and even greater resolution with high-density tiling of probes.10,12–15 While CMA offers state-of-the-art CNV detection at high resolution, there are also inherent advantages that NGS solutions offer.
NGS encompasses the broad advances in sequencing technology that allow for fast sequencing on the whole-genome scale. Mate-pair sequencing (MPseq) technology is one such whole-genome sequencing (WGS) method. MPseq is designed to allow for paired-end sequencing of large DNA fragments (2–5Kb) through the use of a modified library preparation.16 The longer fragments allow for the accurate determination of breakpoint junction locations at low base coverage. These junctions are created by the union of breakpoints in a genome and indicate where there has been rearrangement of chromosomal material. Because many CNVs involve the rearrangement of chromosomal material, the knowledge of a junction’s breakpoint locations afforded by MPseq greatly aids subsequent CNV detection and classification. With the development of such a CNV detection algorithm, MPseq would be a technology capable of providing a comprehensive assessment of SVs, while improving the breakpoint resolution of CNV calls.
In this study we present our CNV detection algorithm, called CNVDetect, which consists of normalization, segmentation, classification, and visualization steps. The segmentation of the genomic data into copy number regions, also referred to as step detection, is ubiquitous in CNV detection algorithms.17 Most NGS methods employ either a sliding window18–20 or a global approach like a Hidden Markov Model (HMM).21 Here we employ a sliding window approach in the interest of speed. Typically such approaches have the drawback of being low-resolution, being limited by the window size used and the ability to effectively capture the necessary statistics. However, MPseq provides breakpoint locations at ~200bp resolution for large structural rearrangements (gains or losses >30Kb).22 By integrating the discordant mate-pair reads into the algorithm we are able to effectively increase breakpoint resolution. In this way our method is unique in that it is optimized to take advantage of the high-resolution breakpoint information naturally provided by the MPseq NGS technology at low read depth coverage. Such methods have previously been developed for use with high read-depth paired-end sequencing data.23
This accurate breakpoint location information is especially important in analyzing cancer genome samples where normal cell contamination and minor clones often make precise CNV determination difficult. We present results from the CNV algorithm compared to 26 samples run using the CMA technique. We assess how the inclusion of breakpoint junction information reported by the BioMarker Discovery Structurel Variant Pipeline (BMD SV Pipeline), a collection of algorithms designed to report structural variants from NGS sequencing data (see Materials and Methods), can aid the CNVDetect algorithm in CNV edge detection. Further we present a series of examples ranging from simple to complex, which demonstrate the power of the CNVDetect in assisting in the characterization of complex cancer samples.
Materials and Methods
Study population
A series of de-identified clinical samples sent to the Mayo Cytogenetics Laboratory for karyotype and or FISH testing were chosen based on the presence of recurrent cytogenetic anomalies seen in acute myeloid leukemia, acute lymphocytic leukemia and chronic lymphocytic leukemia. This test set is intended to represent a wide variety of abnormalities to provide an initial assessment of MPseq suitability for clinical cytogenetic testing.
Cytogenetics chromosomal microarray analysis
Chromosomal microarray was performed on all samples using the CytoScan HD array (Affymetrix) according to the manufacturer’s protocol. Data were analyzed using the manufacturer-provided software (ChAS) with the following laboratory defined parameters: Default smooth and join settings were turned off. All copy number segments were flagged for review when involving at least 25 markers for deletions and 50 markers for duplications. Additionally, all microarray data were manually reviewed to dismiss artifactual calls, refine CNV breakpoints called by the software, as well as to identify additional subtle copy number variation that was not flagged by the algorithm. Such manual calling practices are widely used in clinical microarray review.24 Chromosomal microarray data were reviewed according to standard clinical protocols, and only those CNV segments meeting laboratory clinical reporting criteria were evaluated against MPseq in this study.
DNA library preparation and sequencing
DNA was isolated using the Qiagen Puregene extraction protocol. The DNA was processed using the Illumina Nextera Mate Pair library protocol and sequenced on the Illumina HiSeq 2000. Pooled libraries were hybridized per flow cell and sequenced using 101-basepair reads and paired-end sequencing.
BMD structural variant pipeline
The analysis pipeline for MPseq sequencing data was developed to find breakpoint junction locations and CNVs. The pipeline is termed the BMD Structural Variant Pipeline (BMD SV Pipeline) and is depicted in Figure 1A. It takes as input the MPseq sequencing data detailed in the DNA library preparation protocol and performs a two-step process: alignment and structural variant analysis. Alignment is performed using the BIMA alignment method.25 The mapped sequences are passed to the SVAtools module, which consists of a breakpoint junction detection step22 and a CNV detection step. This CNV detection step, termed CNVDetect, requires both the alignment and breakpoint junction detection steps to be performed before analysis can be completed.
Figure 1. Flow diagram for CNVDetect detection algorithm.
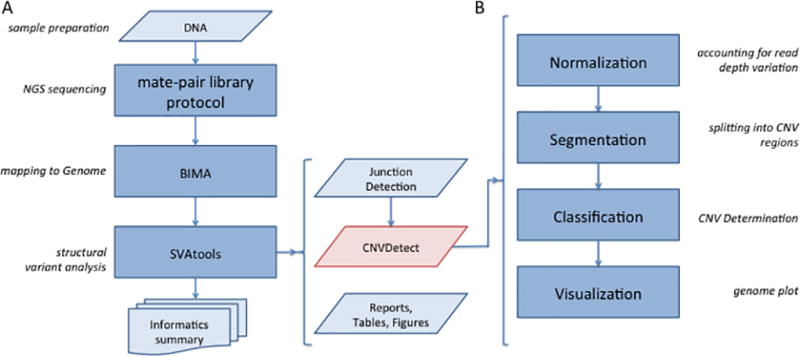
1A) The BMD SV Pipeline is presented in four stages: Library Protocol, NGS Sequencing, Mapping, and SV Analysis. CNVDetect’s place in the overall pipeline is highlighted in red. 1B) The CNVDetect algorithm proceeds in four stages: Normalization, Segmentation, Classification, and Visualization. It depends on output from the previous stages in the BMD SV Pipeline as well as from the Junction Detection algorithm within SVAtools.
The CNVDetect algorithm for determining CNVs in MPseq data was designed to proceed through four steps, normalization, segmentation, classification, and visualization (Figure 1B) to account for the following factors: i) the variation in copy number level due to structural or sequence biases, such as GC content, ii) the presence of normal cell contamination and iii) the presence of heterogeneous subclones with differing CNVs. Factor i) has the potential to increase the false positive rate of the algorithm, while factors ii) and iii) have the potential to increase the false negative rate.
CNVs are detected using the read count of concordant mapping fragments from MPseq sequencing data aligned using the BIMA alignment method.25 The first step of the algorithm aims to normalize the sample read count data using a previously sequenced normal genome sample that closely resembles the sample of interest. This step is designed to take into account all sequence, structural, or DNA processing biases that may contribute to variations in read count that do not reflect the signal of interest. The method does not rely on sequence data, such as GC content, to account for bias, but rather leverages existing genomic knowledge.
The second step of the algorithm segments the genomic data into copy number regions. It uses a sliding window algorithm for step detection, repeated for bin sizes ranging from 100Kb–1Mb. All positions with statistically significant changes in read depth are considered possible edges of CNV regions. We increase the edge detection resolution by incorporating breakpoint junctions detected in the SVAtools22 to supplement the statistically determined edges.
The third step of the algorithm takes the potential copy number regions and classifies them as loss, gain, or normal copy state. It is assumed that through segmentation the data within a copy number region belong to the same probability distribution. This probability distribution for the region is estimated and the peak of the distribution is taken as the expected read depth for the region. This value is then compared to the calculated read depth level for a normal 2N copy state to determine if it deviates significantly enough to be classified as a copy number loss or a copy number gain. The method can account for copy number variants present in at least 20% of cells. Each CNV is reported with a Normalized Read Depth (NRD) score that provides a quantitative measure of how far the CNV deviates from the calculated normal read depth level (normal NRD=2.0).
The final step of the algorithm visualizes the results using a novel genome visualization scheme called the genome plot.26 The results can also be visualized using other common techniques like a linear layout or a circos plot. Each of these steps is provided in more detail in the Supplementary Methods section. Full quality information for the MPseq data is provided in Supplementary Files (QCData_CNV.xlsx). In addition, a filtering was applied to the raw output from the pipeline for these cases to show how the reported CNV number can be reduced to a more reasonable number for manual analysis. This filter marked which called CNVs had >20% of the region covered by masked positions. These masked positions provide some indication as to whether the CNV called may be incorrect due to overlap with a polymorphic or highly homologous region. Filtered CNVs were occasionally incorporated into the final analysis for regions, such as the Y chromosome, where it was known that large areas are masked due to homology but the CNV was deemed likely real. The full BMD SV Pipeline is available for use by the wider scientific community by contacting the authors.
Results
A set of 26 bone marrow samples representing various hematologic neoplasms with CNVs previously characterized by CMA was selected for analysis using the BMD SV Pipeline. Following sequencing, mapping, and breakpoint junction detection, CNV analysis was performed in the CNVDetect module. These cases ranged from simple (1 small reported variant) to complex (>10 reported variants). In order to compare the CMA gold standard to the CNVDetect output a filtering was performed on the raw output, similar to filtering performed in the clinical CMA analysis. Both the CMA filtering technique and the CNVDetect filtering technique are described in the Methods section. The raw number of CNV calls and the number of CNV calls that pass the respective filters are presented in Table 1 and a full set of CMA and CNVDetect output files for the 26 sample cases are provided as Supplementary Files (CMA_Unfiltered.xlsx, CMA_Filtered.xlsx, and CNVDetect_Output.xlsx). The number of CMA and CNVDetect CNV calls can be compared to the final set of CNV calls reported through standard clinical analysis of the CMA data. This analysis includes a manual review process that further excludes CNV calls after filtering. The reasons a CNV call may be manually excluded during CMA analysis is described in more detail in the Methods section. The final number of reported CNV calls is considerably smaller than either the filtered CMA analysis or filtered CNVDetect analysis produces. Our method often produces a higher number of filtered CNV calls than the CMA filter, but on the same order, and the reasons for this are further described in the Discussion section along with strategies that could be employed to reduce the number. The CMA analysis reports a higher raw number of CNVs than CNVDetect, as there is no restriction on the size of a CNV call in the raw CMA analysis.
Table 1. Comparison of CMA and CNVDetect filtering.
The raw number of CNV calls output by both the CMA method and CNVDetect for each of the 26 patient samples are reported. These raw sets of CNVs are then filtered and the resulting number of CNVs is provided under the CMA Filter and CNV Filter heading. The final number of CNV calls reported using the clinical CMA analysis with manual review is provided for comparison.
| SV Folder Number |
CMA Raw | CMA Filter | CNV Raw | CNV Filter | REPORTED |
|---|---|---|---|---|---|
| 88044 | 183 | 8 | 56 | 29 | 3 |
| 88048 | 148 | 4 | 53 | 24 | 1 |
| 88059 | 125 | 9 | 68 | 42 | 5 |
| 88060 | 158 | 6 | 51 | 28 | 2 |
| 88061 | 101 | 11 | 72 | 47 | 8 |
| 88063 | 126 | 5 | 111 | 89 | 3 |
| 88064 | 177 | 7 | 72 | 42 | 2 |
| 88065 | 207 | 4 | 52 | 25 | 1 |
| 88067* | 834 | 415 | 110 | 73 | 10 |
| 88070 | 300 | 5 | 57 | 37 | 1 |
| 88073 | 329 | 15 | 51 | 28 | 2 |
| 88074 | 202 | 8 | 48 | 25 | 3 |
| 88076 | 391 | 26 | 36 | 19 | 1 |
| 88081 | 250 | 14 | 121 | 68 | 11 |
| 88086 | 152 | 17 | 146 | 89 | 7 |
| 88088 | 153 | 6 | 43 | 20 | 2 |
| 88089 | 193 | 6 | 55 | 22 | 1 |
| 88090 | 283 | 16 | 54 | 24 | 2 |
| 88091 | 135 | 5 | 47 | 25 | 2 |
| 88094 | 160 | 13 | 49 | 29 | 7 |
| 88096 | 221 | 16 | 28 | 10 | 2 |
| 88099 | 261 | 26 | 51 | 36 | 3 |
| 88100 | 120 | 6 | 74 | 47 | 3 |
| 88101 | 327 | 20 | 117 | 72 | 7 |
| 88102 | 144 | 15 | 58 | 34 | 8 |
| 88103 | 163 | 45 | 93 | 63 | 10 |
This sample failed QC in CMA processing, but was still analyzed for CNV reporting
Since our gold standard used a manual review to produce the final set of reported CNVs, the final analysis of our method focused only on these reported CNV locations. We aimed to determine whether these same CNV locations were called using MPseq+CNVDetect and thus determine whether MPseq+CNVDetect results could similarly be manually reviewed to recapitulate the gold standard results. Table 2 provides relevant sample data for each of the cases and a comparison to results found using CNVDetect.
Table 2. Comparison of CMA and CNVDetect results.
Both CMA and CNVDetect were run on 26 patient samples containing 107 reportable CNVs. In CMA, each CNV is classified as gain or loss (Type) with the log2 ration of probe intensities reported as the copy number level (CN) along with the chromosome location (Chr) and CNV boundaries (min and max). These results are provided under the CMA Analysis header. For comparison, the CNVDetect results were analyzed for CNV calls with similar bounds (MinCNV and MaxCNV). If such a call was found it was considered a true positive (Yes) and marked in the “Found?” column. If it was not found it was considered a false negative (No). The NRD value for the CNV was provided for comparison to the CN value from CMA. Additionally, the extent to which the called region overlaps with the results from CMA was calculated (Overlap). The CNVDetect results are provided under the CNVDetect Analysis header.
| CMA Analysis | CNVDetect Analysis | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Case | Type | CN | Chr | min | max | Found? | MinCNV | MaxCNV | NRD | Overlap |
| EV88044 | Gain | 2.15 | 15 | 75655926 | 101848096 | NO | ||||
| Loss | 1.85 | 17 | 150732 | 19941963 | NO | |||||
| Gain | 2.15 | 17 | 22266396 | 39610173 | NO | |||||
| EV88048 | Gain | 3 | 2 | 181249078 | 183436911 | YES | 181240000 | 183438000 | 3.107 | 1.000 |
| EV88059 | Loss | 1.6 | 6 | 85398516 | 116240537 | YES | 85379000 | 116251000 | 1.573 | 1.000 |
| Loss | 1.6 | 6 | 154631808 | 157214436 | YES | 154636000 | 157310000 | 1.560 | 0.998 | |
| Loss | 1.5 | 13 | 47898938 | 49957631 | YES | 47897000 | 49958000 | 1.500 | 1.000 | |
| Loss | 1.8 | 13 | 49971104 | 50097568 | YES | 49958000 | 50092000 | 0.999 | 0.956 | |
| Loss | 1.5 | 13 | 50108920 | 50922184 | YES | 50092000 | 50919000 | 1.444 | 0.996 | |
| EV88060 | Loss | 0.3 | 13 | 49925862 | 50516172 | YES | 49927000 | 50516000 | 0.253 | 0.998 |
| Loss | 1.5 | 13 | 50519349 | 51037765 | YES | 50516000 | 51027000 | 1.324 | 0.979 | |
| EV88061 | Loss | 1.7 | 2 | 31802162 | 32393289 | YES | 31728000 | 32385000 | 1.690 | 0.986 |
| Loss | 1.6 | 2 | 73555116 | 74697219 | YES | 73501000 | 74676000 | 1.799 | 0.981 | |
| Loss | 1.6 | 7 | 7398184 | 8546126 | YES | 7477000 | 8499000 | 1.696 | 0.890 | |
| Loss | 1.75 | 13 | 40904307 | 43361934 | NO | |||||
| Loss | 1.5 | 13 | 43368704 | 48382057 | YES | 43362000 | 48283000 | 1.514 | 0.980 | |
| Loss | 1 | 13 | 48386316 | 51084046 | YES | 48283000 | 51082000 | 1.253 | 0.999 | |
| Loss | 1.5 | 13 | 51085062 | 56092156 | YES | 51082000 | 56121000 | 1.541 | 1.000 | |
| Loss | 1.75 | 13 | 56104664 | 66805369 | YES | 56121000 | 66827000 | 1.676 | 0.998 | |
| EV88063 | Loss | 1.7 | 7 | 43360 | 57588468 | YES | 1 | 58028000 | 1.671 | 1.000 |
| Gain | 2.3 | 7 | 64208698 | 159327017 | YES | 62100000 | 159345973 | 2.270 | 1.000 | |
| Loss | 0.5 | 9 | 21178271 | 22202761 | YES | 21173000 | 22201000 | 0.414 | 0.998 | |
| EV88064 | Gain | 3.25 | 6 | 53161778 | 53731152 | YES | 53168000 | 53728000 | 2.943 | 0.984 |
| Gain | 2.75 | 6 | 57965290 | 58306189 | YES | 57968000 | 58311000 | 2.902 | 0.992 | |
| EV88065 | Loss | 1 | 18 | 1194375 | 1640812 | YES | 1190000 | 1645000 | 1.044 | 1.000 |
| EV88067 | Loss | 1.6 | 6 | 87159823 | 90471096 | YES | 87148000 | 90503000 | 1.658 | 1.000 |
| Loss | 1.6 | 9 | 21661728 | 22341057 | YES | 21682000 | 22330000 | 1.599 | 0.954 | |
| Loss | 1 | 11 | 23818009 | 39846329 | YES | 23808000 | 39860000 | 1.684 | 1.000 | |
| Loss | 1.6 | 13 | 49617252 | 51770641 | YES | 49597000 | 51785000 | 1.709 | 1.000 | |
| Loss | 1.6 | 16 | 35880 | 5538528 | YES | 1 | 5526000 | 1.646 | 0.998 | |
| Loss | 1.6 | 16 | 76126373 | 77157943 | YES | 76198000 | 77176000 | 1.690 | 0.931 | |
| Loss | 1.6 | 17 | 150732 | 5152903 | YES | 1 | 5210000 | 1.640 | 1.000 | |
| Loss | 1.6 | 17 | 7505268 | 20900684 | YES | 7492000 | 20832000 | 1.658 | 0.995 | |
| Loss | 1.3 | 17 | 20907120 | 21313672 | YES | 20832000 | 21324000 | 1.354 | 1.000 | |
| Loss | 1.6 | 17 | 47767250 | 48849786 | YES | 47770000 | 48859000 | 1.731 | 0.997 | |
| EV88070 | Loss | 1.4 | 4 | 53427547 | 54274695 | YES | 53418000 | 54275000 | 1.634 | 1.000 |
| EV88073 | Loss | 1.6 | 12 | 118516990 | 118588890 | YES | 118520000 | 118586000 | 1.101 | 0.918 |
| Gain | 3.2 | 17 | 73839502 | 74684979 | YES | 73832000 | 74695000 | 3.292 | 1.000 | |
| EV88074 | Loss | 1.3 | 11 | 119067603 | 121113378 | YES | 119066000 | 121121000 | 1.464 | 1.000 |
| Loss | 1.3 | 13 | 47767418 | 51606370 | YES | 47744000 | 51607000 | 1.358 | 1.000 | |
| Loss | 1.3 | 18 | 55080639 | 56205472 | YES | 55068000 | 56209000 | 1.342 | 1.000 | |
| EV88076 | Gain | 2.3 | 1 | 146149859 | 184080669 | YES | 143539000 | 183798000 | 2.272 | 0.993 |
| EV88081 | Loss | 1.6 | 1 | 1959099 | 7949703 | YES | 1971000 | 7949000 | 1.556 | 0.998 |
| Loss | 1.5 | 1 | 115806740 | 116739470 | YES | 115823000 | 116730000 | 1.656 | 0.972 | |
| Loss | 1.5 | 6 | 80892556 | 114303640 | YES | 80835000 | 114307000 | 1.659 | 1.000 | |
| Loss | 1.5 | 9 | 203861 | 21572243 | YES | 1 | 21577000 | 1.652 | 1.000 | |
| Loss | 1.2 | 9 | 21579259 | 22075597 | YES | 21577000 | 22079000 | 1.247 | 1.000 | |
| Loss | 1.5 | 9 | 22081850 | 26050211 | YES | 22079000 | 26047000 | 1.518 | 0.999 | |
| Gain | 3 | 12 | 52294886 | 52388067 | YES | 52257000 | 52394000 | 2.658 | 1.000 | |
| Gain | 3 | 15 | 61453278 | 61592715 | YES | 61454000 | 61591000 | 2.742 | 0.983 | |
| Loss | 1.5 | 17 | 150732 | 2931676 | YES | 1 | 2933000 | 1.578 | 1.000 | |
| Loss | 1.2 | 17 | 2936307 | 3391011 | YES | 2933000 | 3397000 | 1.396 | 1.000 | |
| Loss | 1.5 | 17 | 3397639 | 21542019 | YES | 3397000 | 21794000 | 1.608 | 1.000 | |
| EV88086 | Gain | 2.2 | 8 | 208048 | 145070385 | YES | 1 | 145138000 | 2.209 | 1.000 |
| Loss | 1.5 | 9 | 21817336 | 21968663 | YES | 21803000 | 21975000 | 1.409 | 1.000 | |
| Loss | 1.25 | 9 | 21976746 | 22005383 | YES | 21975000 | 22010000 | 1.029 | 1.000 | |
| Loss | 1.5 | 9 | 22009308 | 22104160 | YES | 22010000 | 22120000 | 1.520 | 0.993 | |
| Gain | 2.5 | 11 | 99772315 | 100264159 | YES | 99812000 | 100254000 | 2.524 | 0.899 | |
| Loss | 1.5 | 11 | 103229656 | 115453069 | YES | 103075000 | 115455000 | 1.522 | 1.000 | |
| Loss | 0.5 | Y | 2782099 | 26653790 | YES | 1 | 57227000 | 0.517 | 1.000 | |
| EV88088 | Gain | 3 | 8 | 208048 | 145070385 | YES | 1 | 145138000 | 2.927 | 1.000 |
| Gain | 1.5 | Y | 23257721 | 26277778 | YES | 22622000 | 26338000 | 1.434 | 1.000 | |
| EV88089 | Loss | 0.8 | Y | 2782099 | 26653790 | NO | ||||
| EV88090 | Loss | 1.25 | 9 | 130419555 | 130717717 | YES | 130412000 | 130728000 | 0.999 | 1.000 |
| Loss | 1.25 | 22 | 23293899 | 23414891 | YES | 23290000 | 23415000 | 1.001 | 1.000 | |
| EV88091 | Loss | 1.25 | 9 | 21828043 | 21996864 | YES | 21825000 | 21994000 | 1.262 | 0.983 |
| Loss | 1.5 | 9 | 71857107 | 107192688 | YES | 71594000 | 107005000 | 1.584 | 0.995 | |
| EV88094 | Gain | 2.3 | 1 | 171641445 | 248930485 | YES | 171635000 | 248956000 | 2.288 | 1.000 |
| Loss | 1 | 3 | 12582218 | 12675079 | YES | 12579000 | 12693000 | 1.514 | 1.000 | |
| Loss | 1.5 | 7 | 142251693 | 143399068 | YES | 142249000 | 143401000 | 1.554 | 1.000 | |
| Gain | 2.3 | 11 | 100524067 | 135068576 | YES | 100530000 | 135086000 | 2.327 | 1.000 | |
| Gain | 2.3 | 13 | 18862146 | 114342258 | YES | 18900000 | 114364328 | 2.305 | 1.000 | |
| Loss | 1.7 | 18 | 69603144 | 80256240 | YES | 69568000 | 80373000 | 1.715 | 1.000 | |
| Gain | 2.3 | 20 | 80927 | 64284202 | YES | 1 | 64444167 | 2.328 | 1.000 | |
| EV88096 | Loss | 1.7 | 2 | 120599518 | 130388693 | YES | 120383000 | 130359000 | 1.746 | 0.997 |
| Loss | 1.7 | 8 | 125355953 | 129686186 | NO | |||||
| EV88099 | Gain | 2.6 | 1 | 968121 | 248930485 | YES | 1 | 248956000 | 2.538 | 1.000 |
| Gain | 2.6 | 4 | 68453 | 190036318 | YES | 1 | 160254000 | 2.515 | 0.843 | |
| Loss | 1.4 | 16 | 66990865 | 67774694 | YES | 67038000 | 67779000 | 1.493 | 0.940 | |
| EV88100 | Loss | 1.1 | 5 | 82326551 | 153894589 | YES | 82327000 | 153900000 | 1.058 | 1.000 |
| Gain | 2.9 | 11 | 112186582 | 135068576 | YES | 112186000 | 135086000 | 2.862 | 1.000 | |
| Loss | 1.2 | 17 | 150732 | 8158478 | YES | 1 | 8157000 | 1.161 | 1.000 | |
| EV88101 | Loss | 1.7 | 9 | 19589909 | 21800760 | YES | 19592000 | 21803000 | 1.867 | 0.999 |
| Loss | 1.5 | 9 | 21807994 | 22492877 | YES | 21803000 | 22491000 | 1.619 | 0.997 | |
| Loss | 1 | 12 | 11618063 | 13133213 | YES | 11605000 | 13142000 | 1.024 | 1.000 | |
| Loss | 1.4 | 17 | 30233516 | 37700251 | YES | 30243000 | 37701000 | 1.358 | 0.999 | |
| Gain | 2.5 | 17 | 37710931 | 62716038 | YES | 37701000 | 62718000 | 2.556 | 1.000 | |
| Loss | 1.4 | 17 | 62718712 | 66002488 | YES | 62718000 | 66009000 | 1.407 | 1.000 | |
| Gain | 2.2 | 21 | 13634136 | 47701623 | YES | 13301000 | 46709983 | 2.171 | 0.971 | |
| EV88102 | Loss | 1.2 | 2 | 64635266 | 88825975 | YES | 64637000 | 88993000 | 1.337 | 1.000 |
| Loss | 1 | 6 | 78827961 | 115307126 | YES | 78835000 | 115310000 | 1.247 | 1.000 | |
| Loss | 1 | 6 | 143704644 | 149826658 | YES | 143700000 | 149829000 | 1.272 | 1.000 | |
| Loss | 1.8 | 9 | 19625174 | 21767405 | NO | |||||
| Loss | 1.6 | 9 | 21772930 | 22215463 | NO | |||||
| Loss | 1 | 12 | 11639155 | 11747039 | YES | 11640000 | 11748000 | 1.394 | 0.992 | |
| Loss | 1.3 | 17 | 159683 | 8094096 | YES | 1 | 8098000 | 1.206 | 1.000 | |
| Loss | 1.3 | X | 251879 | 156004066 | YES | 1 | 156040895 | 1.296 | 1.000 | |
| EV88103 | Gain | 2.75 | 1 | 144009402 | 168910115 | YES | 143672000 | 168901000 | 2.817 | 1.000 |
| Loss | 1.75 | 1 | 168915824 | 169888200 | NO | |||||
| Gain | 2.75 | 1 | 169892108 | 222802457 | YES | 169888000 | 222802000 | 2.820 | 1.000 | |
| Gain | 2.9 | 2 | 59929961 | 63403720 | YES | 59924000 | 63403000 | 2.907 | 1.000 | |
| Loss | 0.4 | 9 | 21604467 | 22258903 | YES | 21597000 | 22371000 | 0.353 | 1.000 | |
| Loss | 1.25 | 12 | 121574607 | 122813944 | YES | 121555000 | 122817000 | 1.271 | 1.000 | |
| Loss | 1.25 | 16 | 78701427 | 78967038 | YES | 78699000 | 78968000 | 1.155 | 1.000 | |
| Loss | 1.8 | 18 | 5594352 | 7718922 | NO | |||||
| Loss | 1.2 | 22 | 39447775 | 40832804 | YES | 39445000 | 40833000 | 1.237 | 1.000 | |
| Loss | 1.2 | X | 251879 | 156000606 | YES | 1 | 156040895 | 1.131 | 1.000 | |
For each case the reported variants were collected and CNVDetect output was analyzed to see if there was a copy number region available for comparison. If an equivalent region in the genome was segmented in our method, it was counted as a true call and the normalized read depth (NRD) and edge locations were compared to the copy number (termed CN in CMA) and edge locations from the CMA output. If CNVDetect segmented a region called by CMA into more than one region, but with similar NRD values and variant type, these regions were combined and a weighted average NRD reported for comparison. If no such region in the genome was found, a false negative was reported.
Overall we found that there were 107 CNVs reported by the cytogenetics laboratory in the 26 relevant patient cases. All but 10 were reported with high accuracy by our NGS method (91% true positive calls). Of the events that were not found by CNVDetect, eight were not called due to low calculated tumor percentage in the sample analyzed. Three of these eight were calculated by CMA to have a tumor percentage at or below the threshold of detection by the CNVDetect algorithm (20% tumor). Two of the eight were found to be >20% tumor by CMA and had mate-pair supporting fragments reported by the breakpoint junction detection algorithm, but the reported NRD value was below the 20% tumor threshold in CNVDetect and were not reported by the algorithm. The final three of the eight were found to be >20% tumor by CMA but had no read depth or MP support to suggest that the event occurred in our data. Additionally, one event was only partially called by CNVDetect and had to be considered a false negative (EV88089) and one event had MP and CNVDetect support for being a normal copy number region rather than the loss reported in the CMA data (EV88103). When analyzing the boundaries of the CNV regions it was found that our method covered >95% of the copy number calls in all but six of the calls. In these cases the discrepancy was due to regions with high homology or low probe density for CMA (centromere, telomere, or segmental duplication regions) where the CNV boundary location is difficult to determine in both methods.
All comparisons to CMA are limited to the CNV calls reported after manual cytogenetic analysis, as far more regions in any given genome will be predicted as a loss or gain by the CMA analysis software (ChAS) than will ultimately be reported (Table 1). However, we also allow for CNVDetect to calculate a variant cutoff value based on the noise detected in the sample and automatically make loss and gain calls, much like how filters are used in reporting raw CMA results. These results would still need to be manually curated to account for false positives due to noise, homology, etc., but provide a basis for visualization. An example of a visual representation of these CNVDetect results is presented as a genome plot Figure 2A for sample EV88086. Areas that have been calculated to be a loss of genetic material in comparison to the neutral 2N level are colored red, while areas that have been calculated to be a gain of genetic material in comparison to neutral 2N are colored blue. All regions that do not deviate from the calculated normal level in a statistically significant way are colored as grey. A comparison to the linear visualization provided in ChAS is provided in Figure 2B. This is also a good example of where the fuller context of breakpoint junctions and read-depth analysis allow for the reporting of CNVs that may not be considered significant enough for reporting through CMA alone. Note that in Figure 2A there is a small deletion in chromosome 2 connected to the complex event on chromosome 11 (connecting magenta lines in Figure 2A). While the gain and loss on chromosome 11 are reported in CMA (Table 2), this connecting loss is not. With the full context on how this complex event occurs, the deletion on chromosome 2 can be reported by MPseq.
Figure 2. Visualization of BMD SV Pipeline results for MPseq data.
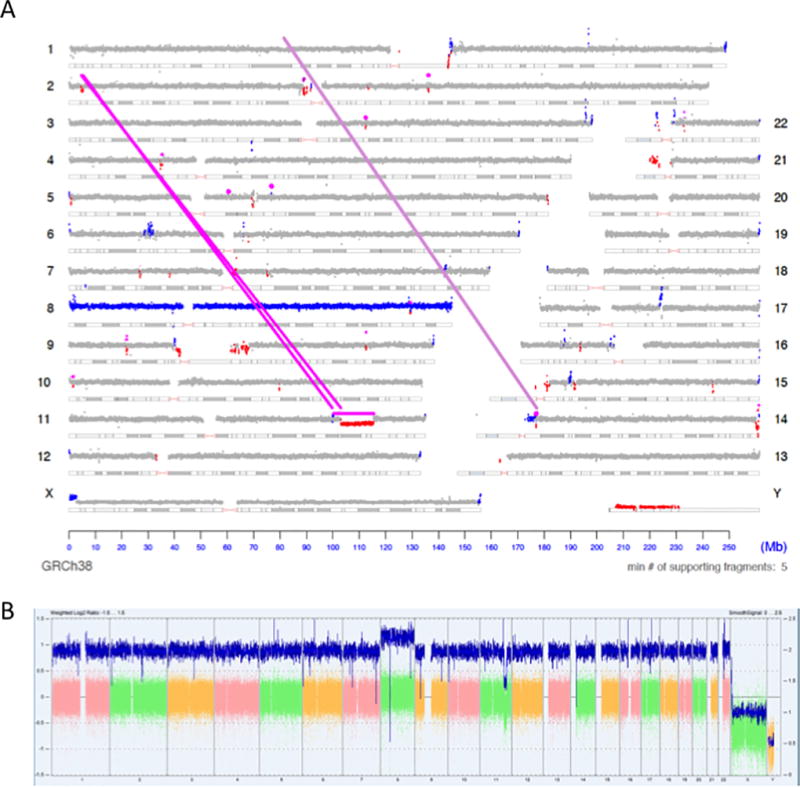
2A) The output of the BMD SV Pipeline for case EV88086 is presented as a genome plot. This plot depicts the read depth of the sequencing data as dots along a chromosome’s length. A magenta line is drawn where there is evidence of a chromosomal junction between two disparate locations in the genome. Regions where CNVs have been detected by the CNVDetect algorithm are colored red to indicate a loss in genomic material and blue to indicate a gain in genetic material. 2B) For comparison the CMA data for EV88086 is provided as visualized by ChAS.
This visualization of the CNV results highlights the major benefit of CNVDetect compared to other CNV detection methods, including the CMA method. SVAtools calculates and reports the location of breakpoint junctions within a sample (magenta lines in Figure 2A). CNVDetect has been designed to use the breakpoint locations from these junctions to segment the genome into potential CNV regions. The SVAtools algorithm will provide breakpoint locations at ~200bp resolution for >90% of the breakpoint junctions in a sample.22 CNVDetect combines this information with edges determined through a statistical edge detection method (see Methods). Statistical methods, like sliding window, top-down, or bottom-down methods, are what are commonly used for segmenting copy number regions. The combination of statistical information and breakpoint junction information allows for more accurate reporting of the edges of a CNV region compared to a statistical method alone. An example shown here from case EV88090 is a classic Philadelphia chromosome in a Chronic Myeloid Leukemia (CML) case where small deletions result from the translocation between chromosomes 9 and 22 (Figure 3A). From MPseq we have both a copy number loss illustrated (small red regions which indicate loss in read depth) and the breakpoint junction reported by SVAtools (magenta line). Using both pieces of information we are able to put the edges of the CNV region at positions 130728000 on chromosome 9 and 23290000 on chromosome 22 at 1kb resolution. Both these positions are ~11Kb and ~4Kb away from what is reported by CMA (positions 130717717 on chromosome 9 and 23293899 on chromosome 22).
Figure 3. CNV edge location comparison.
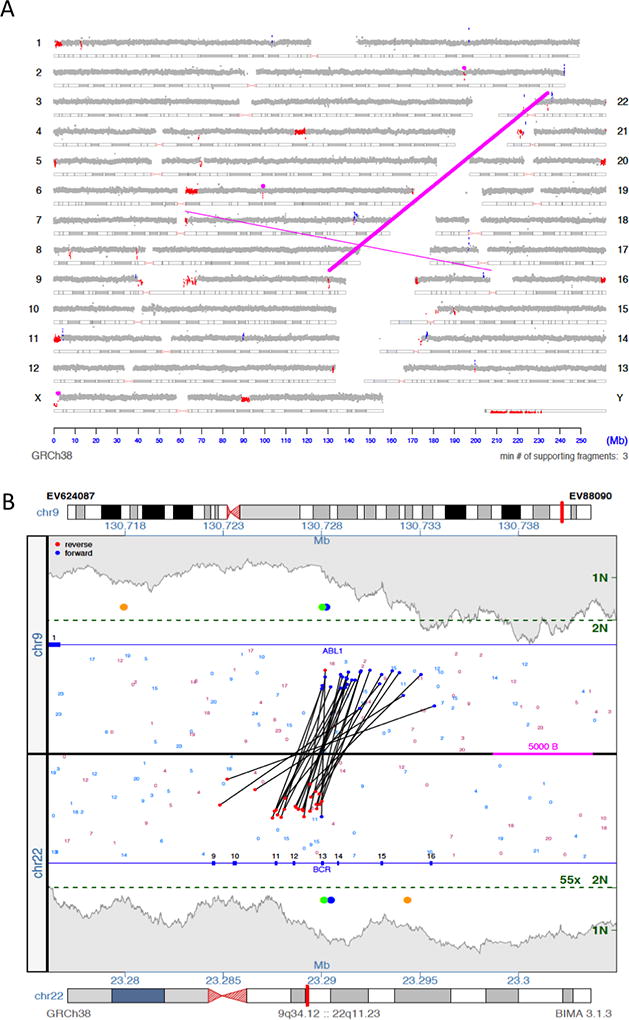
3A) The genome plot for EV88090 is depicted, which contains a Philadelphia chromosome t(9;22) (q34;q11) (magenta line connecting chromosomes 9 and 22). This event is balanced, but results in small regions of loss in both chromosome 9 and chromosome 22 (colored red in both locations). 3B) A junction plot for one of the junctions is provided. The orange dots indicate where CMA determined the CNV edges resulting from this translocation to be. The green dots indicate where CNVDetect determined the CNV edges to be. The blue dots indicate where a split read found sequence evidence for the exact location of the junction locations.
Figure 3B shows a close-up on this breakpoint junction visualizing the MPseq reads connecting 9q34.12 to 22q11.23. The two green dots indicate the CNV edges output by our algorithm for each loci, the orange dots are the copy number edges provided in CMA, and the blue dots are the edge locations as found in a sequenced split read that sequenced through the breakpoint junction. Unsurprisingly, our method outputs CNV edges close to the middle of the junction plot where the SVAtools junction detection algorithm estimates the breakpoints to be. CMA on the other hand does not have that supplemental information to help in edge location and must depend on individual probe performance and spacing to estimate the breakpoint. As expected, MPseq with SVAtools and CNVDetect provides more precise breakpoint prediction than CMA. Most importantly, we can see that the CNVDetect estimate, even on 1kb resolution, is close to the edge location found via the split read, which determined the Philadelphia translocation to occur in the common e13a2 form. CMA, without supplemental information, cannot provide edge locations with the necessary resolution to determine the fusion location on an exon resolution. This shows how the breakpoint locations determined in SVAtools can be used to supplement the otherwise low-resolution statistical method used in CNVDetect to call CNV edges at ~1000 bp resolution.
This ability to combine breakpoint and CNV information also allows for the ability to correctly call CNVs that are connected by a complex event that is not apparent by CMA or karyotyping alone. Shown here in Figure 4A is a complex event that was initially characterized by karyotyping as 46,XY,t(5;6)(q13;q23)[10]/47,XY,+12[2]/46,XY[8], with half of the cells demonstrating a t(5;6) translocation. From CMA, the deletions on chromosome 6 were called (EV88059 in Table 2), but little can be done from the information to fully elucidate the translocation and how the aberrant genome comes together.
Figure 4. Complex case elucidated by SVAtools.
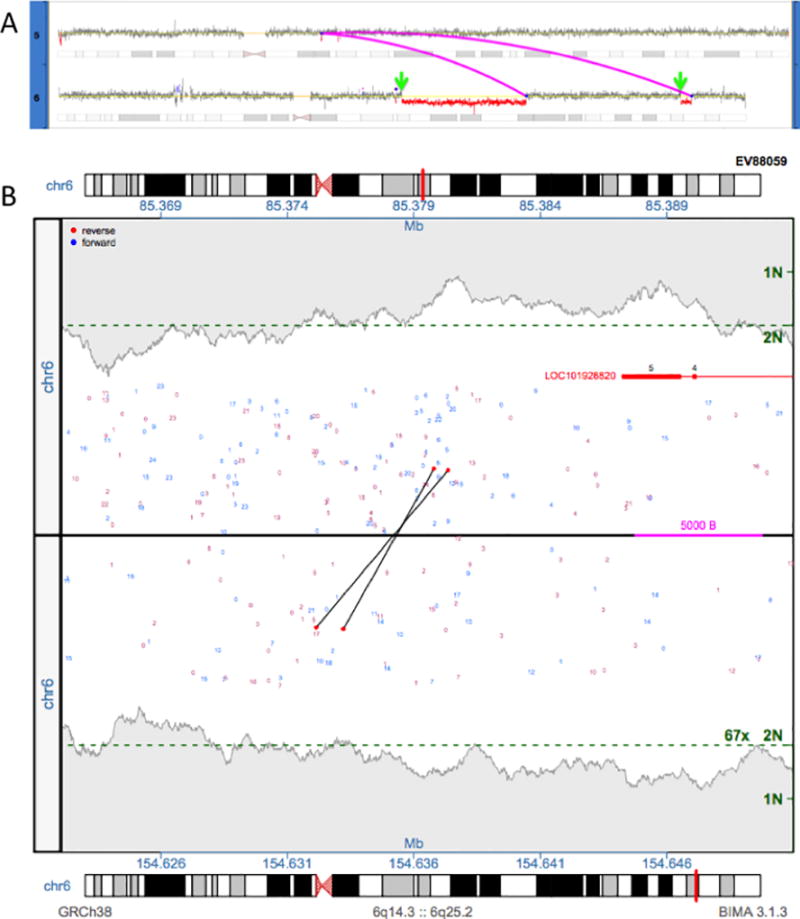
4A) A complex rearrangement between chromosomes 5 and 6 is depicted. Two junctions connecting the chromosomes are shown as magenta lines. We identify two breakpoint locations without junctions in CNVDetect (green arrows) using traditional statistical CNV segmentation techniques. 4B) A junction plot depicting the two regions around the green arrows is provided. Supporting fragments for the missing junction are shown as two black lines that span the two regions.
With the combination of the SVAtools breakpoint locations CNVDetect is able to correctly call a small deletion in chromosome 5, which is not called by CMA, and elucidate where each breakpoint of the t(5;6) translocation resides (magenta lines in Figure 4A). With the additional breakpoint junction information, this small deletion on chromosome 5, which was not significant enough to be reported through CMA analysis, was connected to the larger, more complex event. This context makes reporting through MPseq analysis straightforward. Note also that this is a case where the breakpoint junction detection module in SVAtools did not report a breakpoint junction that connects the proximal deletion in chromosome 6 to the distal deletion (green arrows in figure 4A). Despite the lack of breakpoint information in those two locations, CNVDetect was still able to call the CNVs by relying on the more traditional statistical method of CNV segmentation (see Methods). The proximal boundary of the first event was called at 85379000 of chromosome 6 and the proximal boundary of the second event was called at 154636000 of chromosome 6. Interrogation of the predicted edges revealed two discordant fragments in support of a breakpoint junction (Figure 4B). With this breakpoint junction the BMD SV Pipeline is able to fully elucidate the rearrangement, which can be characterized more accurately as 46,XY, der(5)t(5;6)(q12.3;q25.3),der(6)del(6)(q14.3;q22.1)inv(6)(q22.1;q25.2)del(6)(q25.2;q25.3)t(5;6)(q12.3;q22.1).
From these example we can see how traditional CNV detection methods can be supplemented with newer NGS breakpoint location information to not only increase the sensitivity of the method, but also the resolution of the calls made. For a method whose primary goal is to report which genes are gained, lost, deleted in homozygous state, or amplified, increasing the resolution and certainty on the bounds of these calls is important. A difference of 10kb in the call may be the difference between a gene being predicted to be included in an amplified region or not.
Discussion
Aberrant breakpoint junctions in tumor DNA can now be found by WGS. One such technique is MPseq, which is inexpensive, uses larger fragments capable of jumping over small repetitive regions, and can effectively bridge breakpoint junctions. Here we present a CNV detection algorithm, CNVDetect, which is designed for use with MPseq data. Most CNV detection algorithms rely solely on a statistical method to find boundaries between CNV regions and then to classify each region as either a loss or gain of genetic material. CNVDetect employs a similar statistical method, but supplements the edge detection algorithm with DNA breakpoint information to increase sensitivity and resolution of the reported CNV regions. This combination of information for genome segmentation is important as it allows the method to report CNVs with increased boundary resolution, while also maintaining the ability to call CNVs that are not expected to have breakpoint junction support. This is particularly important in cancer as terminal, whole-arm, and whole-chromosome rearrangements are widely observed, but may not have breakpoint support on one or both sides of the CNV region. In this way we are able to report CNVs with higher accuracy than state-of-the-art CMA technology, directly from NGS data in a single test. A single NGS-based test capable of detecting all classes of structural variation, balanced and unbalanced, throughout the genome, such as the one we have developed for MPseq is the natural next step in cytogenetics.
Here we show a comparison between CMA data and CNVDetect output to show the effectiveness of MPseq. Along with the accurate determination of breakpoint junctions shown in Johnson et al.,22 these data demonstrate how a single test may be a capable substitute for cytogenetic tests like CMA in the near future. We currently have the ability to detect over 90% of the copy number variants reported through CMA on 26 cases. The cases where CNVDetect did not report similar copy number variants were largely instances where tumor percentage dropped to near or below 20% in the sample analyzed by CNVDetect. At that level, the variation in the MPseq data can make it difficult for accurate CNV detection. Future research will aim to improve the normalization step to reduce noise in the data. With reduced noise, this detection limit could likely be lowered to <15%. Further, if the method were to be used to detect copy number variants on a large region, whole-arm, or whole-chromosome level, the statistical methods could be tailored to the region size and the lower limit could be further reduced. At the moment, the method has been developed to detect copy number variants down to ~100kb size, without consideration of how detection could be improved for known variant locations. Along with continued improvement of the resolution and sensitivity of the algorithm future avenues of research will aim to produce a head-to-head comparison of MPseq/CNVDetect and CMA, where the method’s sensitivity will be assessed without prior knowledge of CMA results.
As MPseq becomes more widely used, the sample preparation and sequencing will be further optimized to improve the quality and read-depth of the data used in the CNVDetect algorithm. This improvement in data quality could also reduce the lower limit of detection. Theoretically, the underlying statistical distribution for the read depth data is a Poisson distribution. Any increase or decrease in read depth should not affect the performance of CNV detection given this distribution as the mean and standard deviation both increase and decrease linearly at the same rate. However, particularly for low read depth cases like we have using MPseq technology, higher read depth will improve the sensitivity of the method due to a reduction in detection size. As read depth increases, smaller windows can be used in the copy number variant detection, and the increased available data will allow for more sensitive CNV detection and the detection of smaller CNVs. Additionally, increased read depth will increase the bridged-coverage for a sample and increase the sensitivity for breakpoint junction detection. This would allow for increased CNV edge resolution for a sample, and help increase the performance of the method, particularly in cases with low tumor cellularity. Such read depth dependent performance is typical for any read-based CNV detection method, however the use of both breakpoint junctions and read depth in the CNV calculation helps mitigate this effect compared to many other methods.
Much can be learned from comparing the CNVDetect results to a manually curated CMA output. Both the CMA method without manual review and our algorithmic CNV detection method are prone to reporting false positives, as demonstrated by the considerably higher number of CNV calls made by both CMA and CNVDetect as compared to the final number of Reported CNV calls (Table 1). In CMA analysis these excluded CNV calls often result from sequencing and hybridization artifacts and polymorphisms that may make reported results difficult to analyze. Alternately, CNVDetect reports a higher number of CNV calls after filtering for two reasons. The first is that since we are dealing with NGS data there are sequencing biases that must be dealt with prior to CNV calling. We use a normalization step to reduce this bias (see Methods) but for lower quality samples with abnormal levels of bias, normalization can be difficult and incorrect CNVs calls can result. Further sequencing of normals will make such failures in normalization more rare by better covering the sample landscape. Additionally, the incorporation of bias correction, such as GC bias correction, into the method could aid in reducing the effect of sequencing bias rather than relying only on matching normal samples.
The second reason CNVDetect reports a higher number of CNV calls is that our method often segments a single reportable CNV region into two or more regions. There are two ways we found that CNV regions were incorrectly split. The first is through the erroneous statistical identification of a CNV edge and the second is through the splitting of the region by a detected breakpoint junction that did not in fact result in a change in CNV level. If the regions were split without breakpoint support and through the statistical means alone, then adjacent regions with reported CNVs were merged in cases where the adjacent regions were within the expected detection limit from one another (limit of 20% deviation). If the regions were split due to a supporting breakpoint, then the supporting breakpoint junction was analyzed to determine whether it represented a junction type that would explain the lack or presence of a change in CNV. Balanced translocations, inversions, and transposons are all breakpoint junction types that would explain the presence of a junction without a resulting copy number change and the adjacent regions could be merged. Additionally, for cases where large copy number changes were detected (>40% deviation) without breakpoint junction support, the location of the change could be analyzed to locate a missing breakpoint junction that may have been filtered, masked, or have low mate-pair support, as was the case in EV88059 (Figure 4). Improved filtering criteria are valuable for the future development of MPseq with CNVDetection as a stand-alone tool for cytogenetic analysis of genomic samples.
As we look forward to the use of NGS technologies in individualized medicine, we can see how for a technology like MPseq, algorithmic methods like CNVDetect will become invaluable. This algorithm is able to resolve complex chromosomal rearrangements that involve numerous breakpoint junctions and CNVs. So while complex events like chromothripsis and chromoplexis, which have been shown to be pervasive in aggressive cancers, are typically difficult to analyze on a whole-genome level, our method can resolve them accurately. Additionally, we show how CNVDetect is capable of more accurately reporting the location of a CNV boundary. As individualized monitoring and therapeutics advance, the accurate determination of breakpoint junction locations will become increasingly valuable. Such breakpoint junctions can be used for the development of specific molecular assays for monitoring disease and tracking the effectiveness of targeted treatment.
Supplementary Material
Acknowledgments
This work was supported by the Mayo Clinic Center for Individualized Medicine (CIM) and by Award Number R01 CA177734 from the National Cancer Institute.
Bibliography
- 1.Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Killcoyne S, del Sol A. Identification of large-scale genomic variation in cancer genomes using in silico reference models. Nucleic Acids Research. 2016;44(1):e5–e5. doi: 10.1093/nar/gkv828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moncunill V, Gonzalez S, Bea S, et al. Comprehensive characterization of complex structural variations in cancer by directly comparing genome sequence reads. Nat Biotech. 2014;32(11):1106–1112. doi: 10.1038/nbt.3027. [DOI] [PubMed] [Google Scholar]
- 4.Tubio JMC. Somatic structural variation and cancer. Briefings in Functional Genomics. 2015;14(5):339–351. doi: 10.1093/bfgp/elv016. [DOI] [PubMed] [Google Scholar]
- 5.Yang L, Luquette LJ, Gehlenborg N, et al. Diverse mechanisms of somatic structural variations in human cancer genomes. Cell. 2013;153(4):919–929. doi: 10.1016/j.cell.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. nature. 2006;444(7118):444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sebat J, Lakshmi B, Troge J, et al. Large-Scale Copy Number Polymorphism in the Human Genome. Science. 2004;305(5683):525. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 8.Stranger BE, Forrest MS, Dunning M, et al. Relative Impact of Nucleotide and Copy Number Variation on Gene Expression Phenotypes. Science. 2007;315(5813):848. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buysse K, Delle Chiaie B, Van Coster R, et al. Challenges for CNV interpretation in clinical molecular karyotyping: Lessons learned from a 1001 sample experience. European Journal of Medical Genetics. 2009;52(6):398–403. doi: 10.1016/j.ejmg.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 10.Carter NP. Methods and strategies for analyzing copy number variation using DNA microarrays. Nat Genet. 2007 doi: 10.1038/ng2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snijders AM, Nowak N, Segraves R, et al. Assembly of microarrays for genome-wide measurement of DNA copy number. Nat Genet. 2001;29(3):263–264. doi: 10.1038/ng754. [DOI] [PubMed] [Google Scholar]
- 12.Haraksingh RR, Abyzov A, Urban AE. Comprehensive performance comparison of high-resolution array platforms for genome-wide Copy Number Variation (CNV) analysis in humans. BMC genomics. 2017;18(1):321. doi: 10.1186/s12864-017-3658-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wiszniewska J, Bi W, Shaw C, et al. Combined array CGH plus SNP genome analyses in a single assay for optimized clinical testing. European Journal of Human Genetics. 2014;22(1):79–87. doi: 10.1038/ejhg.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schaaf CP, Wiszniewska J, Beaudet AL. Copy number and SNP arrays in clinical diagnostics. Annual review of genomics and human genetics. 2011;12:25–51. doi: 10.1146/annurev-genom-092010-110715. [DOI] [PubMed] [Google Scholar]
- 15.Beaudet AL. The utility of chromosomal microarray analysis in developmental and behavioral pediatrics. Child development. 2012;84(1):121–132. doi: 10.1111/cdev.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murphy SJ, Cheville JC, Zarei S, et al. Mate pair sequencing of whole-genome-amplified DNA following laser capture microdissection of prostate cancer. DNA research. 2012;19(5):395–406. doi: 10.1093/dnares/dss021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang H, Nettleton D, Ying K. Copy number variation detection using next generation sequencing read counts. BMC Bioinformatics. 2014;15(1):109. doi: 10.1186/1471-2105-15-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chiang DY, Getz G, Jaffe DB, et al. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Meth. 2009;6(1):99–103. doi: 10.1038/nmeth.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoon S, Xuan Z, Makarov V, Ye K, Sebat J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Research. 2009;19(9):1586–1592. doi: 10.1101/gr.092981.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim T-M, Luquette LJ, Xi R, Park PJ. rSW-seq: Algorithm for detection of copy number alterations in deep sequencing data. BMC Bioinformatics. 2010;11(1):432. doi: 10.1186/1471-2105-11-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ivakhno S, Royce T, Cox AJ, Evers DJ, Cheetham RK, Tavare S. CNAseg - a novel framework for identification of copy number changes in cancer from second-generation sequencing data. Bioinformatics. 2010;26 doi: 10.1093/bioinformatics/btq587. [DOI] [PubMed] [Google Scholar]
- 22.Johnson SH, Smadbeck JB, Smoley SA, et al. Molecular karyotypes: SVAtools for junction detection of genome-wide chromosomal rearrangements by mate-pair sequencing (MPseq) Cancer Genetics. 2018;(221):1–18. doi: 10.1016/j.cancergen.2017.11.009. [DOI] [PubMed] [Google Scholar]
- 23.Medvedev P, Fiume M, Dzamba M, Smith T, Brudno M. Detecting copy number variation with mated short reads. Genome research. 2010;20(11):1613–1622. doi: 10.1101/gr.106344.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Genetics AWGotACoM, Genomics Laboratory Quality Assurance C. American College of Medical Genetics and Genomics technical standards and guidelines: microarray analysis for chromosome abnormalities in neoplastic disorders. Genet Med. 2013;15(6):484–494. doi: 10.1038/gim.2013.49. [DOI] [PubMed] [Google Scholar]
- 25.Drucker TM, Johnson SH, Murphy SJ, Cradic KW, Therneau TM, Vasmatzis G. BIMA V3: an aligner customized for mate pair library sequencing. Bioinformatics. 2014 doi: 10.1093/bioinformatics/btu078. btu078. [DOI] [PubMed] [Google Scholar]
- 26.Gaitatzes A, Johnson SH, Smadbeck JB, Vasmatzis G. Genome U-Plot: A Whole Genome Visualization. Bioinformatics. 2017 doi: 10.1093/bioinformatics/btx829. btx829. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.