

Abstract
Ischemic preconditioning is a process which serves to mitigate reperfusion injury. Preconditioning of the heart can be achieved through natural, pharmacological, and mechanical means. Mechanical preconditioning appears to have the greatest chance of good outcomes while methods employing pharmacologic preconditioning have been largely unsuccessful. Remote ischemic preconditioning achieves a cardioprotective effect by applying cycles of ischemia and reperfusion in a distal limb, stimulating the release of a neurohumoral cardioprotective factor incited by stimulation of afferent neurons. The cardioprotective factor stimulates the reperfusion injury salvage kinase (RISK) and survivor activator factor enhancement (SAFE) signaling cascades in cardiomyocytes which promote cell survival by the expression of anti-apoptotic genes and inhibition of the opening of mitochondrial permeability transition pores. Clinical application of ischemic preconditioning involving targets in the RISK and SAFE signaling appears promising in the treatment of acute myocardial infarction, however, clinical trials have yet to demonstrate additional benefit to current therapy.
Keywords: Ischemia, Ischemic conditioning, Cardiomyocytes, Reperfusion Injury, Cardioprotection, Reperfusion injury salvage kinase, Survival activator factor enhancement
Graphical abstract
INTRODUCTION
Ischemic heart disease is the leading cause of death globally, and death rates caused by ischemic heart disease are projected to increase1. Reperfusion of blood flow to the ischemic area is the current gold standard of treatment following a myocardial infarction or myocardial ischemia. Reperfusion has been shown to limit infarct size, improve long-term myocardial function and reduce mortality2. Additionally, restoration of blood flow has been shown to induce myocardial salvage with a significant decrease in morbidity and mortality3. Myocardial salvage is defined as the difference between actual and potential infarct size; inducing salvage means to save myocardial cells from ischemic damage. While reperfusion reduces mortality, a study by Jennings4,5 suggested reperfusion could facilitate the death of reversibly injured cardiomyocytes. Reperfusion injury significantly contributes to final infarct size, and infarct size directly relates to prognosis6–9. Reperfusion injury can be transient resulting in ventricular arrhythmias and myocardial stunning; or permanent resulting in lethal reperfusion injury10,11. Lethal reperfusion injury is a potentially preventable phenomenon and should be a target of therapy.
Reperfusion injury is characterized by rapid correction of acidic pH, calcium overload, and formation of reactive oxygen species (ROS). Those characterizations result in opening of the mitochondrial permeability transition pore (MPTP) which causes cell death by releasing cytochrome C into the cytoplasm and activating the caspase system9. Reperfusion injury is currently a neglected therapeutic target, but natural phenomena, mechanical interventions, and pharmacologic interventions mitigate reperfusion injury and limit infarct size. A review by Miura and Miki12 concluded infarct size had to be <20% of left ventricular size to confer any meaningful reduction of mortality13.
Reperfusion injury is mitigated through a process known as conditioning, where the myocardium is prepared for an ischemic insult through brief episodes of ischemia and reperfusion3,6. Conditioning can occur locally at the level of the heart, or remotely in distal organs. Conditioning the heart represents a form of cardioprotection, a term that refers to all strategies aimed at the attenuation of the injurious sequelae of myocardial ischemia and reperfusion. Sequelae include arrhythmias, decreased inotropy and coronary blood flow, and myocardial infarction7. Natural ischemic conditioning has been observed in patients who suffer from anginal pain immediately prior to an ischemic episode. These patients have smaller infarcts and a better prognosis than patients without pre-infarction angina14,15. Ischemic preconditioning is currently the best form of myocardial protection against infarction by attenuating ischemic damage.
Originally discovered in a study by Murray and colleagues4 in canine models, ischemic preconditioning significantly attenuated myocardial death by ischemic injury4,16,17. Murray demonstrated that 4–5 minutes of non-lethal coronary artery occlusion, followed by 5 minutes of reperfusion, is an adequate stimulus to produce a beneficial outcome. Ischemic preconditioning represents the most powerful action for attenuating myocardial infarction size in the ischemic heart6. Ischemic conditioning not only protects the myocardium, but also the coronary microcirculation13.
Infarct size is the preferred measurement to determine effects of conditioning since infarct size is an unambiguous endpoint with clinical significance7. The purpose of this review is to give an overview of cardiac ischemic conditioning by describing intracellular signaling cascades implicated in conditioning, discuss pharmacologic and mechanical modalities of ischemic conditioning, and provide a brief assessment of remote ischemic conditioning compared to other modalities.
SIGNALING CASCADES ASSOCIATED WITH CARDIAC CONDITIONING
Local ischemic conditioning can be obtained mechanically by clamping coronary vasculature, or pharmacologically by activation of cell surface receptors of adenosine, bradykinin, opioids, acetylcholine, and generation of ROS6. Pharmacological conditioning acts to prevent the opening of MPTP and thus prevent mitochondrial disruption without the induction of ischemia3,16. Preconditioning results in two windows of protection; the first window occurs due to protein modification and is transient in nature. It occurs 2–3 hours following the preconditioning stimulus7,16–20. The second window occurs 24 hours following the stimulus and lasts for approximately 48 hours due to the production of cytoprotective proteins7,16–20. The early phase of protection is induced through the mitochondria where a signaling ROS activates protein kinases such as Akt, Erk1/2, tyrosine kinase, and protein kinase C. Late protection is due to activation of transcription factors by these protein kinases, as well as recruitment of pro-survival signaling pathways to attenuate infarct size. The two major pro-survival signaling pathways are the reperfusion injury salvage kinase (RISK) pathway and the survivor activator factor enhancement pathway (SAFE). RISK acts through Akt and Erk1/2, and SAFE is potentiated by TNF-α to downstream Janus activated kinase (JAK) and signal transducer and activator of transcription −3 (STAT3) to reduce reperfusion injury6. Although the end effectors remain unknown, the proposed hypotheses suggest reperfusion injury is mitigated by the preservation of mitochondrial function through inhibition of the MPTP and opening of mitochondrial KATP channels6,10,18,17. This causes the mitochondrial membrane to depolarize, resulting in rapid loss of mitochondrial function and cell death through caspase activation7,16,17. Inhibiting the MPTP from opening improves cell survival. Molecules such as matrix metalloproteases (MMPs), gaseous messengers, and miRNA have also been implicated in pathways associated with cardiac conditioning20. However, the goal of this review article is to highlight the RISK and SAFE pathways to identify proposed therapies to interact with the key molecules in the signaling cascade.
RISK AND SAFE SIGNALING PATHWAYS
Preserving mitochondrial function in the presence of ischemia is the main mechanism of conditioning. Three parallel signaling pathways act to achieve the cardioprotective afforded by ischemic conditioning. One is the RISK pathway, which is activated by G-protein-coupled receptors (GPCRs) and potentiated through protein kinase B (Akt). Another is activation of protein kinase C which results in opening of mitochondrial KATP channels. A third is the SAFE pathway, which includes the JAK/STAT system, most prominently mitochondrial STAT37.
The RISK pathway was popularized by the studies of Yellon and Hausenloy21 and is composed of a group of kinases that confer cardioprotection when activated immediately prior to or at the time of reperfusion16,17. The RISK pathway can be activated by many agents which bind to GPCRs to include growth factors, opioids, adenosine, bradykinin and others16,22. Stimulation of GPCRs activates phosphoinositide 3 kinase (PI3K) and results in phosphorylation of its downstream targets Akt, glycogen synthase kinase-3 beta (GSK-3β), endothelial nitric oxide synthase(eNOS), protein kinase C, and anti-apoptotic factor Bcl-2 associated protein19,21. GSK-3β is proposed to be the convergence point of the RISK pathway. Activation of the RISK pathway results in the inhibition of GSK-3β following phosphorylation, and subsequent prevention of MPTP activation19,22.
Normally, the MPTP opens within the first moments of reperfusion due to the influx of ROS and calcium. Studies by Crompton in the 1980s first implicated MPTPs as critical to cardiomyocyte death23. Opening is triggered by ATP depletion, mitochondrial calcium overload, oxidative stress, and high phosphate levels. Cyclosporine-A prevents MPTP activation and prolongs cell survival22. Directly inhibiting the MPTP from opening before reperfusion has been shown to have successful clinical outcomes in ST segment elevation myocardial infarctions (STEMIs)16,24. However, the cardioprotective effects of MPTP inhibition are lost if the MPTP inhibitor is administered after the first 15 minutes of reperfusion22.
Like the RISK pathway, protein Kinase C (PKC) may be activated directly during ischemic preconditioning by binding of adenosine, opioids, or bradykinin to its GPCRs. The binding of GPCR and its ligand will result in activation of PI3K and alpha serine/threonine kinase (Akt) leading to nitric oxide synthase activation. Nitric oxide (NO) potentiates its signal through soluble guanylyl cyclase, activating protein kinase G leading to protein kinase C activation, which ultimately opens the mitochondrial KATP channel16,25. It has been reported that the MPTP and mitochondrial KATP channels are functionally linked, as opening of the MPTP counteracts the effects of an open KATP channel9,26. Since calcium overload is a stimulus to open the MPTP and an open KATP channel helps prevent calcium overload, opening the KATP channel helps to inhibit MPTP activation and thus prolong cell survival26. Recent studies have confirmed the role of PKC as a central mediator of ischemic conditioning during reperfusion by abrogating its cardioprotective effects with chlelerthyrine, a PKC inhibitor, during reperfusion26. Similarly, the PI3K/Akt pathway plays an important role against reperfusion injury, and cardioprotection could be abrogated following administration of the selective PI3K inhibitors wortmannin and LY29400226. Additionally, studies with overactivation of Akt in transgenic mice showed a reduced myocardial infarct size when compared to controls27. While the RISK pathway primarily activates or inhibits various proteins, the SAFE pathway stimulates anti-apoptotic factors and expression of genes to mitigate ischemic damage.
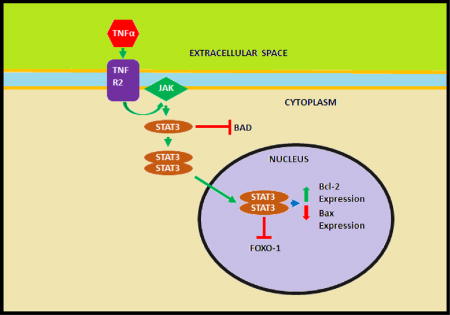
The SAFE pathway was originally described by Lecour and Opie28 in the in vitro murine models and is involved in both early and late phase ischemic preconditioning and postconditioning16,19. SAFE potentiates its effects though JAK, which is originally activated by various cytokines including interleukin-6, and Tumor necrosis factor alpha (TNF-α). JAK will subsequently activate STAT which will potentially affect mitochondrial respiration and swelling granting a protective response16. TNF-α may also stimulate the SAFE pathway after binding to TNF Receptor 2, which activates STAT-3, which will then inhibit the apoptotic factor forkhead box protein O1 (FOXO-1) in the nucleus. While TNF-α was originally thought to contribute to reperfusion injury, it may paradoxically contribute to the cardioprotection induced by pre- and postconditioning28,29. Increased concentrations of TNF-α are noted in myocardial infarctions, but attempts to neutralize TNF-α resulted in worsening heart failure29. STAT-3 has been shown to increase the anti-apoptotic gene Bcl-2 and decrease the apoptotic gene Bax in response to a conditioning stimulus. While STAT-3 is a transcription factor, many STAT-3 effects in a pre- or post-conditioning setting do not occur at transcription, but are resultant of phosphorylation of various components, such as the phosphorylation and inactivation of the pro-apoptotic factor Bad29. Interestingly, STAT 5 rather than STAT 3 activation was noted in patients undergoing remote ischemic conditioning before CABG, although the role of STAT 5 needs to be investigated further19.
PHARMACOLOGIC CONDITIONING IN RISK AND SAFE SIGNALING PATHWAYS
Targeting various levels of the RISK pathway to invoke a cardioprotective response has been a long-desired therapy in pharmacology following the discovery of ischemic conditioning. Thus far, pharmacologic therapy to protect against reperfusion injury has been largely disappointing5,10. This review describes several agents that interact with the molecules involved in the RISK and SAFE signaling pathways including: metoprolol, PPAR agonists, adenosine, nicorandil, volatile anesthetic agents, and cyclosporine-A17,18,20. Metoprolol administered prior to reperfusion in a porcine model reduced infarction size according to a study performed by Ibanez and colleagues6,31. Labne later supported this finding by undertaking a clinical trial (METOCARD-CNIC trial) that demonstrated administering metoprolol in the ambulance prior to angioplasty reduced infarct size and improved clinical outcomes in anterior STEMI patients10. However, the EARLY BAMI trials, in which 600 STEMI patients were randomized to IV metoprolol, did not show any reduction in infarct size31. The mixed results of the trials do not give credence to metoprolol’s efficacy as a conditioning agent. Further, the premise of preconditioning rests on the ability to administer agents before an ischemic event, making preconditioning an impossible therapy to apply outside of a scheduled surgical setting where the timing of an ischemic event is known. Patients with unstable angina or a recent MI have a higher risk of subsequent MI, so administering drugs that mimic ischemic preconditioning may have a beneficial effect on this group of patients18.
Like metoprolol, PPAR activating drugs were thought to derive a cardioprotective effect by activating PI3K-Akt proteins in the endothelial cells in the normal and diabetic heart32. The pleiotropic effects of PPAR activating agents could prove to be beneficial adjuncts to reperfusion therapy in the future. Activating those kinases were thought to lead to closure of the MPTP or opening of the mitochondrial KATP channel. An in vivo study by Ravingerova et al32 in hypertensive rats showed that PPARα agonists could serve as a mimetic of the delayed phase of cardioprotection, resulting in a reduction in infarct size. While metoprolol and PPAR activating drugs have been investigated, the most studied agents related to ischemic conditioning thus far are: adenosine/acadesine (adenosine regulatory agent), nicorandil (KATP channel opener), volatile anesthetics, and cyclosporine-A33. Unfortunately, in several studies neither adenosine nor nicorandil reduced infarct size or improved prognosis7. However, the PROTECTION AMI trial seems to demonstrate cyclosporine-A reduced infarct size by preventing the MPTP from opening, but patient outcome data are not currently available34. Figure 1 depicts the signaling cascades involved in ischemic conditioning.
Figure 1.
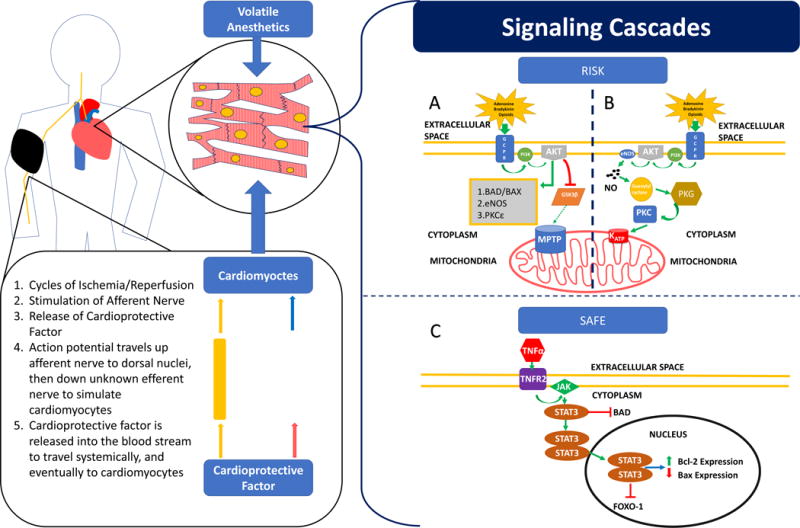
This figure depicts a patient undergoing remote ischemic preconditioning using a blood pressure cuff on his right arm. The cycles of ischemia and reperfusion stimulate release of the cardioprotective factor from the afferent neuron. The cardioprotective factor travels systemically via the blood interacting with various cells, including cardiomyocytes. The cardioprotective factor facilitates the RISK and SAFE signaling cascades in cardiomyocytes. At the same time, the afferent neuron potentiates an action potential to nuclei in the brainstem. An unknown efferent neuron potentiates a signal to the heart which triggers cardioprotective signaling pathways such as RISK and SAFE. Volatile anesthetic agents have been shown to adjust hemodynamic properties of the body and stimulate RISK and SAFE signaling cascades in cardiomyocytes to achieve a protective effect. (A) Depicts the RISK signaling cascade. Adenosine, bradykinin or opioids bind to its G-protein coupled receptor which results in phosphorylation and activation of PI3K. PI3K will phosphorylate and activate Akt which has several downstream effects, the most notable is the interaction with GSK3β. Akt can phosphorylate and hence inhibit GSK3β. GSK3β normally activates and opens the MPTP, but inhibition of GSK3β through phosphorylation prevents the MPTP from opening and promotes cell survival and resistance to ischemic damage. Akt can also activate other signaling kinases such as eNOS, and PKC and inhibit pro-apoptotic factors such as bad and bax; (B) Depicts a PKC signaling cascade. Adenosine, bradykinin or opioids bind to its G-protein coupled receptor which results in phosphorylation and activation of PI3K. PI3K will phosphorylate and activate Akt, which will activate eNOS to produce nitric oxide (NO). NO will activate soluble guanylyl cyclase, which will activate protein kinase G (PKG), leading to activation of PKC. PKC can open the mitochondrial KATP channel, which can prevent MPTP activation and promote cell survival and resistance to ischemic damage, and (C) Depicts the SAFE signaling cascade. TNF-α can bind to TNF Receptor 2 (TNFR2) which will activate the JAK kinase. JAK will phosphorylate and activate STAT-3. STAT-3 can inhibit the pro-apoptotic factor Bad, as well as form a homodimer which can translocate into the nucleus of the cell. The dimerized STAT-3 can induce transcription, increasing the expression of genes for Bcl-2, an anti-apoptotic protein, and decrease the expression of genes for Bax, a pro-apoptotic protein. STAT-3 may also inactivate FOXO-1, another transcription factor which has been shown to induce the expression of apoptotic genes.
ADENOSINE
Adenosine is widely distributed throughout the body and plays a major role in the cardiovascular system as a vasodilator, conduction blocker in the AV node and is thought to contribute to the reactive hyperemia observed in cardiac muscle16. Adenosine acts through GPCRs leading to activation of the RISK pathway25. Adenosine interacts with four receptors: A1, A 2A, A2B, and A3; A1, A2B, and A3 are expressed in cardiomyocytes, while A2A are more prevalent in coronary vessels1,16,19. A1 receptors are associated with slowing the atrial rate and conduction in nodal tissue, and adenosine released during ischemic conditioning binds to A1 and A3 receptors activating protein kinase C19. Ultimately adenosine acts to preserve mitochondrial function by preventing the MPTP from opening16,19. Use of adenosine in vivo for cardioprotection has been limited due to the short half-life of the molecule. Often the drug loses its efficacy before a cardioprotective effect can occur16.
Adenosine receptor agonists have been used with some success; however, adenosine use as adjunct therapy to reperfusion therapy in the Amistad I and II trials did not demonstrate any additional benefit compared to standard treatment16,30,35. Some experimental studies have shown varying effects of adenosine depending on the timing of administration. A beneficial effect was observed when adenosine was given before coronary occlusion with remarkable infarct reduction, a weaker response was noted when adenosine was given before reperfusion, and no effect is noted when given after reperfusion18,36. Thus, the lack of clinical effectiveness of adenosine observed in the AMISTAD trial may be due to delayed administration of adenosine (after thrombolytic administration) in 50% of the patients18,35.
The AMISTAD II trial appeared to demonstrate a reduction in infarct size associated with adenosine administration in patients in a 3-hour time-period with an evolving anterior MI receiving thrombolytic therapy or angioplasty18,30,37. The major limitation of this trial was sample size, the study population was too small to confirm the observed benefit of adenosine18,37. Opening mitochondrial KATP channels by nicorandil have been another target of pharmacologic conditioning to induce cardioprotection.
NICORANDIL
Nicorandil is the only KATP channel-opening agent currently available for cardiovascular use18. The first clinical trial to examine the cardioprotective effects of nicorandil was observed in patients with unstable angina in a study conducted by Patel and colleagues in the CESAR 2 trial18,38. The study suggested opening the KATP channel along with standard anginal therapy significantly reduced myocardial ischemic episodes and tachyarrhythmias18,38. Sakata and colleagues33 investigated the effects of an intravenous bolus of nicorandil following primary angioplasty or thrombolytic therapy one month after treatment. These investigators found improved myocardial blood flow and ventricular wall motion in patients given nicorandil compared to control. Another study by Ito and coworkers39 found that intravenous nicorandil in conjunction with coronary angioplasty was associated with improved with in-hospital outcomes compared to angioplasty alone. However, improved cardiac functioning following nicorandil in percutaneous coronary intervention (PCI) settings is most likely due to improvement in microvascular perfusion improvement opposed to myocardial conditioning18. Currently, exercise-based research appears to be the only viable and promising approach to uncovering KATP channel opening and cardioprotection40. Overall, the results of pharmacologic conditioning targeting signaling pathways with adenosine and nicorandil have been disappointing, as none have been able to demonstrate a prognostic benefit7. The only medications known to confer some cardioprotection are anesthetic agents.
VOLATILE ANAESTHETIC AGENTS
Volatile anesthetic agents could precondition myocardium, the kidney, and the brain17. Volatile anesthetics substantially alter the myocardial oxygen requirements through direct and indirect effects on systemic, pulmonary and coronary hemodynamics along with autonomic nervous activity in a dose dependent manner41. Those changes can give the appearance of attenuated infarct, but not confer clinical benefit41. Experiments conducted in the 1970s and 1980s using halothane, enflurane and isoflurane in canine models demonstrated a protective effect17,41,42. These studies were further supported by independent groups demonstrating cardioprotection induced by volatile anesthetics in 3 animal models17,43. These agents are hypothesized to trigger the RISK pathway by stimulation of GPCRs. Current evidence suggests isoflurane, sevoflurane and desflurane could precondition and protect the heart and vascular endothelium in vitro in human and animal myocardial tissue.17
Some experiments suggest that cardioprotective effects increase from isoflurane to sevoflurane to desflurane. Anesthetic agents other than volatile agents have been shown to influence preconditioning. Propofol, which is not a volatile anesthetic, has been shown to both block preconditioning by desflurane and remote ischemic preconditioning, but also act synergistically with other agents to confer cardioprotection7,17,44,45. Experimental trials conducted thus far demonstrate isoflurane, sevoflurane and desflurane could provide myocardial protection, and the combination of volatile anesthetics and high dose of propofol during bypass surgery and reperfusion might increase cardioprotection as demonstrated by reductions in troponin I17. Additionally, morphine has shown promise as an agent that enhances the pharmacologic preconditioning of isoflurane17.
One meta-analysis of patients in 27 trials showed reduced postoperative troponin I, higher cardiac indices, and lower inotropic support for those given volatile anesthetics in coronary artery bypass (CABG) surgery, but the authors noted low sample size to draw significant conclusions17. Laboratory experiments and proof of concept studies clearly suggest volatile anesthetics may provide a cardioprotective effect when larger areas of myocardium sustain ischemic injury41. Experimental proof-of-concept trials have been promising, but larger trials are necessary to demonstrate if anesthetic agents confer cardioprotection and if that effect can be translated into a clinical setting.
CYCLOSPORINE A
The most promising pharmacologic agent to confer a cardioprotective effect is cyclosporine-A, which inhibits cyclophilin D and thus prevents the opening of MPTP46,47. A study by Piot et al48 in human subjects demonstrated decreased creatinine kinase in patients who receive a bolus of cyclosporine-A immediately prior to PCI9,30,46. Cyclosporine has pleiotropic effects, and concerns over cardiac remodeling add skepticism to the effectiveness of the agent as potential therapy. However, of the patients that participated in a proof-of-concept trial, cardiac magnetic resonance imaging at 6 months’ follow-up showed that a single bolus of cyclosporine had no effect on left ventricular remodeling9. The placebo controlled CIRCUS (Cyclosporin and Prognosis in Acute Myocardial Infarction Patients) assessed total mortality, hospitalization for heart failure and left ventricular remodeling (increase of LV end-diastolic volume >15%) following administration of cyclosporine-A46,47. Cung and colleagues found that administration of cyclosporine just before PCI did not reduce the risk of death from any cause, worsening of heart failure, rehospitalization for heart failure, or adverse left ventricular remodeling when compared to placebo47. Unlike a previous smaller trial, the CIRCUS trial did not demonstrate a reduction in creatine kinase, which has been used as an approximate measure of infarction size47. While pharmacologic therapy has shown some promise, clinical application remains distant in the future. A more practical and less invasive approach to cardioprotection is mechanical conditioning.
NON-PHARMACOLOGICAL METHODS OF ISCHEMIC PRECONDITIONING
Mechanical modalities of ischemic condition have been far more successful than pharmacologic conditioning in achieving cardioprotective effects. Ischemic preconditioning, conditioning prior to an ischemic event, was first described in 1986 by Murray et al4 who demonstrated four 5-minute episodes of myocardial ischemia followed by 5 minutes of reperfusion led to a 75% reduction in infarct size in a canine model17,19,49. The first effective use of ischemic preconditioning in humans was observed by Yellon who found that intermittent clamping and declamping of the aorta preserved myocardial ATP levels of patients undergoing coronary artery bypass surgery16,24. Ischemic preconditioning mitigates infarct development opposed to mitigating reperfusion injury7,46. Several studies have confirmed the cardioprotective benefit of preconditioning by measuring serum cardiac enzymes, observing a reduction in ventricular arrhythmias, and shortened intensive care unit stay6. Ischemic preconditioning has become the archetype for cardioprotection, because it has demonstrated the greatest consistency and efficacy of any other intervention or drug7. The exact threshold or “maximal dose” of ischemic preconditioning remains unknown, but a recent meta-analysis described by Deng et al50 suggests ischemic episodes less than five minutes can be cardioprotective, but longer could induce deleterious effects. Natural ischemic preconditioning has been observed in the context of pre-infarction angina, and ischemia induced by exercise. Preconditioning has been studied in the setting of coronary angioplasty and cardiac surgery to develop clinical applications to the process. Pre-infarction angina mitigates infarction size by triggering ischemic pathways, such as RISK, that offer protection to the cardiomyocytes.
Exercise-induced ischemia, or warm up angina, describes attenuation or abolition of angina on a second effort when separated from a first exertion separated by a short period of rest51. Warm up angina is objectively defined by reduced ischemia (ST segment depression) or a raised ischemic threshold compared to a first exertion51. Studies by Okazaki and colleagues52 in human subjects with severe stenosis (>90%) in the left anterior descending artery stenosis, demonstrated that the warm-up phenomenon is not due to increased blood flow, but rather increased metabolic efficiency; a feature of ischemic preconditioning. Recent experimental evidence suggests that attenuation of ischemic/reperfusion by exercise is attributed to myocardial-specific biochemical alterations, and not on blood flow alteration40. Myocardial ischemia, rather than exercise, is the predominant stimulus required to initiate the maximal protective effect observed in warm-up angina51. This ischemia may activate intracellular mechanisms that confer a protective effect through the opening of KATP channels40,51.
Ischemic preconditioning in the context of coronary angioplasty was shown to reduce anginal pain, ST segment shift, and mean pulmonary pressure in studies originally published by Deutsch et al,53 then later confirmed by other angioplasty studies in human subjects18. These findings support ischemic preconditioning by demonstrating that prior ischemia lessens the malignant effects of subsequent ischemic episodes. ST-segment shift has been shown to correlate with metabolic, mechanical, and clinical parameters of myocardial ischemia, the greater the shift in the ST segment, the greater the derangement in metabolic, mechanic, and clinical behavior of the heart.
Intermittent ischemia has been achieved in cardiac surgery by cross clamping the aorta during coronary artery bypass graft surgery. Yellon and colleagues found significantly higher ATP content in the myocardium of patients exposed to aortic cross clamping than those who were not24. Increased ATP in a myocardial biopsy translates to increased function of those cells, as ATP depletion in ischemic injury usually translates to cell death. Additional therapies to facilitate preconditioning include the administration of hyperbaric oxygen prior to cardiac surgery. A study by Yogaratnam54 and others found a reduction in myocardial injury, intraoperative blood loss and postoperative complications following 10 minutes of pressurization, two 30-minute periods interrupted with a five-minute “air break” followed by a 25-minute depressurization period 24, 12, and 4 hours prior to CABG surgery54,55. However, the major disadvantage to ischemic preconditioning is the requirement to intervene prior to an ischemic episode, which is impossible in the case of a myocardial infarction6,7.
POSTCONDITIONING
The very initial study by Na et al56 showed that the heart could be protected against myocardial infarction by interrupting reperfusion with intermittent ischemia, thus demonstrating and coining the term “ischemic postconditioning”56–59. Ischemic post conditioning was originally performed by occluding then reperfusing the culprit lesion before allowing full reperfusion in PCI46. Blood flow is reintroduced to the ischemic myocardium in a stuttered or staccato-like manner before allowing complete restoration of blood flow3,49. Postconditioning is thought to limit reflow enough to maintain acidosis in the heart to prevent MPTP activation, and has been the best demonstration of therapy targeted against lethal reperfusion injury to date3,46. Ischemic postconditioning acts to decrease infarct size, while preconditioning attenuates ischemic damage although its cardioprotective effect does not seem to be as significant as preconditioning7. Since prognosis is related to infarct size, reducing infarct size is paramount to mitigate morbidity and mortality9,16. Postconditioning reduces infarct size by decreasing cardiomyocyte necrosis, reducing the generation of ROS, inhibiting inflammatory cell adherence and accumulation, and reducing calcium overload in the mitochondria16,46. Postconditioning potentiates its signals through the RISK pathway to inhibit opening of the MPTP.
The opportunity for therapeutic intervention in the context of STEMI for post-ischemic conditioning has helped facilitate its transition into the clinical setting6,60. Although no optimal protocol has been defined, postconditioning intervention must be performed within the first few minutes of reperfusion7. Like other modes of conditioning, the larger the area at risk during an infarct, the greater the amount of protection postconditioning can offer58. Staat and colleagues61 studied postconditioning in humans undergoing angioplasty via inflation and deflation of the angioplasty balloon. They found a reduction in creatinine kinase release, a 36% reduction in infarction size, and a 7% increase in left ventricular ejection fraction compared with control after 1 year7,18,58,61. However, other meta-analyses have failed to demonstrate any benefits of ischemic postconditioning in terms of ST-segment resolution, peak creatinine kinase, or major adverse cardiovascular events at 30 days6. The effects of postconditioning on other facets of reperfusion injury such as myocardial stunning, no-reflow phenomenon and arrhythmias remain largely unexplored; the only established benefit has been the attenuation of lethal reperfusion injury and reduction of infarct size58. While postconditioning has potential clinical application, the most promising application of ischemic conditioning in a clinical setting is remote ischemic conditioning.
REMOTE ISCHEMIC CONDITIONING
Remote ischemic conditioning (RIC) uses transient ischemia in one distal organ to confer ischemic conditioning to another. Unlike ischemic pre- and post-conditioning that involve manipulation of the culprit vessel, remote ischemic conditioning occurs at a site distal to the target organ and does not confer the same risk of microembolization7. The application of brief cycles of nonlethal ischemia followed by reperfusion to an organ or tissue protects the heart against acute reperfusion injury6,62. An in vivo study by Przyklenk et al63 in canine models showed that preconditioning the coronary bed opposite to the vasculature subjected to infarct still conferred a cardioprotective effect to the same extent as if the culprit vessel was conditioned16,43,63. The observation of remote protection was further evaluated in subsequent studies that suggest brief ischemia of noncardiac tissue such as the kidney, intestine or skeletal muscle can also protect the myocardium against a subsequent infarction7,18. Kharbanda et al64 discovered that cardioprotection could be achieved by inflating and deflating a blood pressure cuff in human subjects. This study established the protocol used in subsequent studies to evaluate remote ischemic preconditioning43.
A study by Hausenloy and coworkers compared the effects of remote ischemic preconditioning by measuring Troponin T release rates at 6,12, and 24 hours in patients undergoing CABG surgery24. Remote ischemic preconditioning (RICPre) was achieved through alternating cycles of right upper limb ischemia induced by an automated cuff inflator for 5 minutes followed by 5 minutes of reperfusion. Results showed an overall drop in troponin T release at 6, 12, 24 and 48 hours after surgery in CABG patients. Additional studies demonstrated reduced median troponin I release 24 hours after PCI in ST elevated myocardial infarction patients, along with decreased chest pain, ST segment deviation on the electrocardiogram, and decreased major cardiac and cerebral event rate 6 months after the intervention18. Zografos and colleagues demonstrated that even one five-minute cycle of transient upper limb ischemia reduced troponin I levels if administered before PCI65. The underlying mechanisms and pathways activated within the target organ are suggested to be the RISK and SAFE pathways6,62.
The RISK pathway appears to be the major signaling cascade potentiated by RICPre, as patients given various antagonists to effectors in the RISK cascade failed to demonstrate a reduction of infarct size following RIC administration44. Remote ischemic conditioning is the most attractive method of inducing cardioprotection because of its safety profile and feasibility7. An in vivo study by Theilmann66 described a reduction in all-cause mortality following administration of RICPre in human subjects. However, all-cause mortality included sepsis and when sepsis was excluded, all-cause mortality remained lower in the experimental group when compared to the control, but no longer significantly so66. This suggests RICPre may not have made any significant reduction in mortality or may protect against sepsis, but the latter notion remains neither tested nor currently supported. Additionally, RICPre elicits a systemic response opposed to the local one described in ischemic pre- and postconditioning and can therefore offer protection to other organs. A study by Manchurov et al67 demonstrated for the first time that remote ischemic preconditioning prior to primary PCI in patients with an acute myocardial infarction improves endothelial function assessed by flow mediated dilation test and thrombolysis in myocardial infarction (TIMI) flow grade. In fact, remote conditioning unlike local conditioning, might lead to persistent protection68. A meta-analysis conducted by Le Page and colleagues45 found that RICPre appears to be an effective method for reducing ischemic and reperfusion injury and possibly reduce long-term clinical events45.
The pathway that directs protection to the heart from a distal organ remains unclear, but the current paradigm describes a neurohormonal route6,7,43,69. A humoral factor and neural signal generated from an ischemic event is transported to the heart where it can induce protective effects6,62. The mechanism in which remote ischemia conveys its protective signal to the heart can be categorized into three interrelated events: generation of the cardioprotective signal in the conditioned organ or tissue, the pathway that conveys that signal to the heart, and the activation of signaling cascades within the heart that mediate the protective effect43. Similarly, three hierarchical levels of signal transduction are described in remote preconditioning: triggers such as adenosine, bradykinin or opioids that act on sarcolemmal membrane receptors, an intracellular signaling cascade composed of protein kinases, and the effectors such as mitochondria or the cytoskeleton7,19,62,70. Studies showed blood taken from a preconditioned rabbit and transfused into a naïve rabbit reduced myocardial infarction size in the naive rabbit, thus suggesting that humoral factors were transferred in the blood that carried a protective effect6,30,43,44. Furthermore, resection of the neural innervation of the limb, administration of a ganglion blocker (hexamethonium), inhibition of preganglionic vagal neurons in the brainstem, and resection of the vagal nerve supply to the heart have all been shown to abrogate the conditioning effects of limb RIC, suggesting intact neural pathways are essential to achieve the desired effect6,46. Figure 2 is a flow chart depciting the mechanism of remote ischemic preconditioning.
Figure 2.
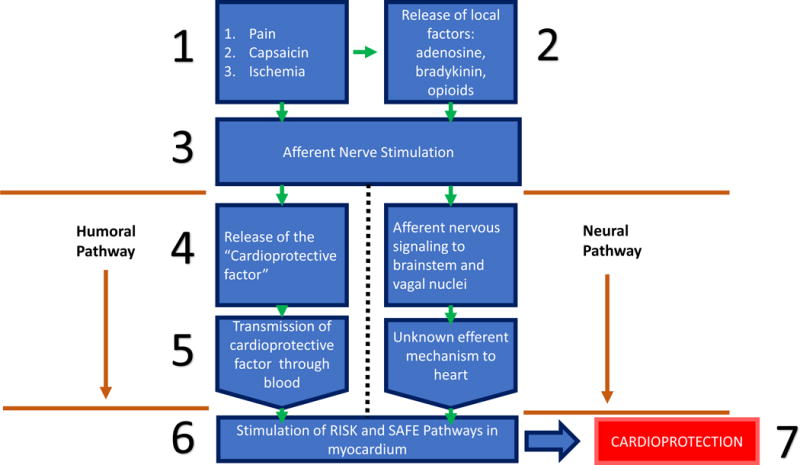
This flowchart depicts the current paradigm describing the mechanism in which remote ischemic conditioning provides cardioprotection. The initial stimulus to the limb may be from direct nervous stimulation with pain, capsaicin or ischemia induced by a blood-pressure cuff, or those modalities may result in the release of local factors such as adenosine, bradykinin or opioids to act on local afferent neurons. Stimulation or activation of local afferent neurons leads to the release of the “Cardioprotective factor” a humoral protein, or nervous signaling up though the central nervous system and synapse in the vagal nuclei in the brain stem. The cardioprotective factor released by the culprit afferent neurons will travel through the blood before reaching the heart and activating the RISK and SAFE pathways to confer a cardioprotective effect. The mechanism of the efferent limb of the neural pathway is unknown, but experiments have demonstrated vagal stimulation has induced activation of the RISK and SAFE pathways.
Studies by Jensen et al71 confirmed the need for an intact neural pathway to the limb by showing that the cardioprotective factor was not generated by limb RICPre in diabetic patients with sensory neuropathy of the limb. This indicates that transient ischemia or interruption of bloodflow is not a requisite trigger for remote protection44. This neural pathway appears to be stimulated by local factors, such as adenosine and bradykinin implicating afferent and autonomic neurons play a significant role in intra-organ protection46. Furthermore, the dialysate from diabetic subjects after RICPre provided protections to others if the donor did not have diabetic neuropathy, suggesting the nerves provide a humoral signal44.
Definitive evidence of the identity of the cardioprotective factor is lacking, but other agents still may participate in conferring a cardioprotective effect, but not to the same degree as the unknown “cardioprotective factor”6. Micro RNA-144 has been shown to increase the phosphorylation and activation of several kinases such as Akt, GSK-3β, which leads to increased activation of effectors in the RISK pathway72. Direct stimulation of the nerve leading to that limb, or other neurostimulation such as topical capsaicin resulted in the generation of the cardioprotective factor and reduced myocardial infarct size in animal models43,46,73. Non-traumatic peripheral nociceptive signal which instigated by topical capsaicin was found to have a >70% reduction in infarct size in mice 15 minutes prior to coronary artery occlusion44. It can be concluded from various animal studies as well as observations in diabetic patients that an intact neural pathway to the conditioned limb is required to generate the blood-borne cardioprotective factor43.
The final element of the pathway must concern an efferent neural limb to the target organ to activate downstream cardioprotective signal pathways43. The mechanism of this efferent pathway from the dorsal nuclei in the brainstem to the heart remains unknown43. Notable hypotheses suggest cardiac vagal and sympathetic efferent release cardioprotective substances44. The most exciting aspect of RIC is the ability to translate its use to a clinical setting. The most effective RIC stimulus is not currently known, but the timing of administration of RIC is better understood.
RIC can be delivered at various times with respect to ischemia and reperfusion, granting preconditioning (before ischemic event), perconditioning (after ischemic event but prior to reperfusion) and postconditioning (during reperfusion)43. The lack of significant temporal restraints makes RIC use as therapy more enticing. Initial studies focused on applying limb ischemia immediately prior to index myocardial ischemia in a preconditioning setting43. Limb ischemia to induce RIC was first investigated by Gunyadin et al74 in 2000, but Cheung et al75 were the first to describe a cardioprotective effect in children undergoing cardiac surgery for congenital heart disease.
RIC has been investigated in CABG surgery by observing a reduction in cardiac enzymes such as troponin T and I, in major non-cardiac vascular surgery by measuring reduction in the rise of cardiac enzymes such as creatinine kinase MB and troponin T and I, during elective PCI by measuring the reduction in the rise of serum cardiac enzymes, and STEMI treated by primary PCI by observing a reduction in infarct size and reduction in the rise of cardiac enzymes43. Yellon reported beneficial effects of RIC, describing a 43% reduction in perioperative myocardial injury in STEMI patients24. RIC has been shown to reduce the amount of cardiac enzyme release in patients undergoing CABG73. A clinical trial by Candilio and colleagues76 found similar results to D’Ascenzo with a 26% reduction of perioperative myocardial infarction, a 54% reduction in postoperative atrial fibrillation and a 47% decrease in acute kidney injury following a shortened remote ischemia preconditioning protocol in patients undergoing CABG. Zimmerman et al77 were the first to report a decrease in acute kidney injury (AKI) after administering RICPre to patients undergoing cardiac surgery, and others later confirmed those results14. Unfortunately, large prospective multicenter randomized clinical trials did not demonstrate RIC had any additional beneficial effect on major clinical outcomes in patients undergoing cardiac surgery6,78. Thielmann attributes these inconsistent to differences in study protocols, confounding comorbidities, anesthetic regimens, surgical procedures, techniques, and protection regimens66.
Limb RIC in the setting of PCI has also been examined to determine its cardioprotective effects. A recent meta-analysis described a benefit in patients undergoing elective PCI who were administered limb RIC6. The most promising application of limb RIC is in the setting STEMIs. Several proof-of-concept studies have reported cardioprotective effects with limb RIC in STEMI patients treated with primary coronary angioplasty79. Davies et al80 reports patients who received remote ischemic preconditioning prior to elective PCI resulted in lower troponin I release and decreased major adverse cardiac and cerebral events (MACCEs). The study by Davies and colleagues support the notion that RIC promotes lasting protection and maintained beneficial longitudinal outcomes, further indicating the potential of RIC as part of STEMI or cardiovascular surgery therapy. RIC can be effective in several settings such as when performed by paramedics in the ambulance, at the hospital prior to angioplasty, at the onset of reperfusion in the hospital, and even after reperfusion event6,43.
A study which applied 4 cycles of 5-minutes of upper arm ischemia and reperfusion in an ambulance in patients with anterior infarcts, demonstrated statistically significant infarct size reduction. Additionally, patients at highest risk were found to benefit more from RIC as an adjunctive therapy to primary PCI than lower risk patients44. Only certain STEMI patients may benefit from adjunctive therapy of RIC. Patients presenting with smaller infarcts often from occlusions of the right coronary artery or the left circumflex artery do not benefit as much as patients with larger infarcts such as those precipitated by occlusion of the left anterior descending artery13,44.
Remote ischemic postconditioning (RICPost) involves invoking limb ischemia at the time of reperfusion, and evidence suggest it involves activation of the SAFE pathway opposed to the RISK pathway normally activated in preconditioning43. In a randomized study of 100 patients, RICPost was determined to reduce infarct size by reducing creatine kinase-myocardial band (CK-MB) release44. A recent meta-analysis of cardiovascular interventions in STEMI patients suggests benefits from RICPost43. The greatest reduction in cardiac injury enzymes following RICPost was observed in cardiac surgery, while enzyme reductions in PCI were more disparate45. Confounders such as the use of propofol or volatile anesthetics may explain disparities14. Additionally, remote ischemic conditioning has been shown to reduce platelet activation and reactivity following coronary angioplasty in a study by Lanza and colleagues, and thus decrease risk for thrombotic events following coronary angioplasty81.
The ERICCA (Effect of Remote Ischemic Preconditioning on Clinical Outcomes in CABG surgery) and RIPHeart (Remote Ischemic Preconditioning in Heart Surgery) trials investigated RIC outcomes associated with cardiovascular death, non-fatal MI, coronary revascularization and stroke at one year82,83. ERICCA and RIPheart gave a neutral result, but was found that 90% of patients were given propofol anesthesia which has been demonstrated to abrogate remote ischemic conditioning13,82,83. The ongoing CONDI 2/ERIC-PPCI study (Effect of Remote Ischemic Conditioning on Clinical Outcome in STEMI Patients Undergoing Primary Percutaneous Coronary Intervention) on remote ischemic preconditioning in patients with STEMI may provide the answer of the efficacy of this potential new therapeutic intervention13,30.
All studies that gave a neutral result and provided the anesthetic regimen had used propofol; use of this drug appears to be the common denominator in all neutral or negative studies13. It has been difficult to adequately judge the effectiveness of RIC in large trial settings, in addition to prove its effectiveness in patients with comorbid conditions such as diabetes mellitus. Proof of concept trials and meta-analyses of postconditioning and remote ischemic conditioning have demonstrated the gold-standard outcome of decreased infarct size, but larger scale clinical trials have been unsuccessful in demonstrating any difference. The heterogeneity of the larger clinical trials includes methodological issues such as: primary determinants of infarct size, timing of the conditioning algorithms, ischemic durations, anesthetic regimen and others may account for the neutral results49,58. The lack of optimal postconditioning and remote conditioning protocol may result in hyperconditioning, where excessive stimulus leads to a deleterious effect49. Future applications of RICPost include solid organ transplantation, and trials such as REPAIR assess renal function in both the donor and recipient following limb RICPost14.
DIABETES AND HYPERLIPIDEMIA IN CARDIAC CONDITIONING
Of the morbidities with the potential to compromise the protective mechanisms of the heart, diabetes mellitus appears the most important to study36. Diabetes mellitus is associated with higher mortality following an acute myocardial infarction due to the atherosclerotic vessels and decreased function of the left ventricle36. A study by Lamblin et al84 showed that diabetic patients had increased rates of cardiovascular death and heart failure following their first acute myocardial infarction than patients who did not have diabetes. Haffner and colleagues showed that risk for a cardiovascular event in diabetic patients without a prior MI was the same as non-diabetic patients with a prior MI36,85. In fact, with few exceptions, most studies have shown that cardioprotection normally achieved by ischemic preconditioning or postconditioning is impaired in diabetes mellitus; Tsang and coworkers suggest that the threshold to induce cardioprotection is increased in diabetic hearts3,36,80,86. The most likely mechanism for cardioprotective-resistance is an impairment of intracellular signaling important for cardioprotection26,36. PI3K-Akt signaling, and STAT3 activity were found to be impaired in diabetic myocardium36. While the diabetic heart is at an increased risk for cardiovascular event, and is more resistant to cardioprotective signals, that resistance cannot be explained by hyperglycemia alone. Hyperglycemia has been shown to induce the expression of phosphatase and tensin homolog protein (PTEN), a negative regulator of PI3K/Akt and decreased ability of the cell to keep the MPTP closed26,36.
Studies by Tamareille et al87 using Langendorff-isolated-heart and in vivo ischemia-reperfusion rat models demonstrated the resistance to ischemic conditioning is related to the activation of GSK-3β,which impairs the RISK pathway. Activation, rather than the normal inhibition by phosphorylation, disrupts cardioprotection by opening the MPTP26. Expression and activation of GSK-3β in diabetic patients was found to be twice that of non-diabetic patients26. Blockade by cyclosporine-A may be a viable treatment. In fact, infarct size 5 days after acute myocardial infarction was limited by cyclosporine-A and beneficial effects continued 6 months later36. Similarly, closure of KATP channels has been shown in hyperlipidemic hearts, and subsequent resistance to ischemic conditioning8. Ravingerova and colleagues suggest administration of PPAR agonists may be a potential target for treating ischemic injury in pathologically altered myocardium, such as the myocardium in patients with diabetes mellitus32.
A study which investigated rabbits in New Zealand who were exposed to high cholesterol diets and cardiac ischemic and reperfusion injury, found that hyperlipidemia significantly exacerbated reperfusion injury by increasing infarct size and extent of cardiomyocyte death compared to rabbits with a normal diet8. Andreadou et al90 discussed the effects of a hyperlipidemic heart on cardiac conditioning in animal models in a recent review. The underlying mechanisms associated with hyperlipidemia is disruption of the endothelial nitrous oxide-cyclic guanosine monophosphate cascade, RISK pathway, MMP-2 cascade, and KATP and apoptotic pathways which serve to interrupt potential conditioning effects. Other small clinical studies reviewed by Ferdninandy et al88 showed hyperlipidemia attenuates cardioprotective effects and exasperates cardiac ischemic and reperfusion injury. The loss of cardioprotection in hyperlipidemic hearts has been associated with redistribution of sarcolemmal and mitochondrial Connexin 43, leading to reduced activation of Akt, and decreased opening of KATP channels8. Connexin 43 has been identified to be important in cardiac ischemia and reperfusion injuries89. Loss or redistribution of connexin 43 results in a loss of gap junction communication at the intercalated disks or inner mitochondrial membrane30,72. Conversely, RIC leads to increased phosphorylation of connexin 43 which preserves the function of connexin and offers a protective role72. Figure 3 depicts where disease states or other agents can affect ischemic preconditioning.
Figure 3.
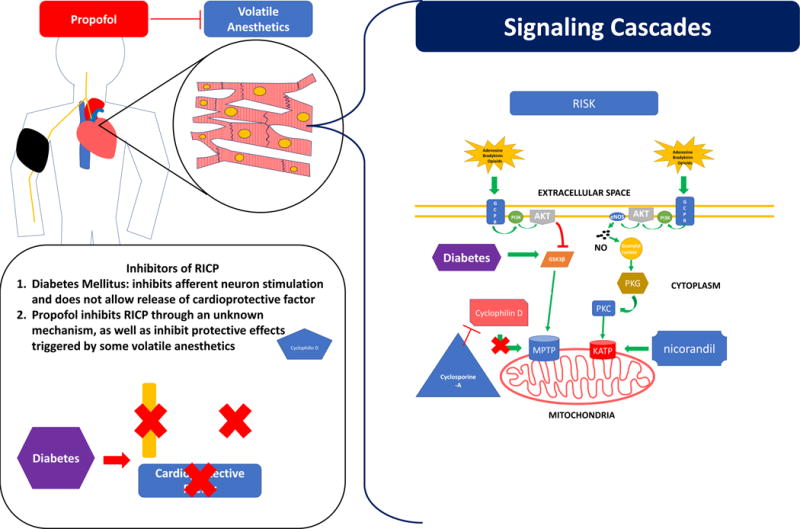
This figure depicts points along the remote ischemic preconditioning process where pharmacologic agents or disease states can disrupt or potentiate cardioprotective effects. Diabetes mellitus disrupts cardioprotective effects at various levels. Diabetic neuropathy leads to a failure to secrete the cardioprotective factor and potentiate action potentials along the by afferent neuron. RISK and SAFE cascades would not be activated by a remote stimulus and no cardioprotection would be achieved. Increased sugars due to diabetes increase activation of GSK3β, which increases activation of the MPTP and promotes cell death. Propofol is a sedative agent that acts at various levels to disrupt ischemic preconditioning. Propofol has been shown to disrupt remote ischemic preconditioning though an unknown mechanism, as well as disrupt the cardioprotective effects stimulated by volatile anesthetic agents. Cyclophilin D activates the MPTP and promotes cell death. Cyclosporine A inhibits cylcophilin D and was suggested to help promote a cardioprotective effect by its downstream inhibition of the MPTP. Current data does not suggest cyclosporine A achieves a significant cardioprotective effect. Nicorandil stimulates opening of the KATP channel and was suggested to give a cardioprotective effect. Recent studies show nicorandil does not give a significant cardioprotective effect.
LIMITATIONS AND CONTROVERSIES OF REMOTE CONDITIONING VERSUS PHARMACOLOGIC CONDITIONING
While many proof-of-concept studies demonstrate some promising results of RIC, incorporation of this evidence into treatment guidelines requires demonstration of benefit to clear clinical hard endpoints46. The most effective endpoint would be measurement of infarct size, which can be quantified by cardiac magnetic resonance (CMR), with evaluation of left ventricular ejection fraction as a surrogate functional parameter because it has been associated with long-term mortality and morbidity following STEMI46. Since RIC reduces final tissue necrosis, its effectiveness must be assessed by reduced post-infarction left ventricular dysfunction and heart failure combined with mortality reduction as the primary outcome70. Furthermore, a study by Pryds et al91 demonstrated that RIC in the setting of STEMI attenuates the detrimental effect of health system delay on myocardial salvage. This means that more at risk or threatened myocardium was salvaged during reperfusion because of remote ischemic preconditioning than what would have been salvaged with just reperfusion due to the inherent delay of administering primary PCI in a hospital. Pryds and colleagues suggest their findings indicate RIC as a potential adjunct to reperfusion therapy in patients with prolonged treatment delays to alleviate the detrimental effects of those delays91.
All the published meta-analyses confirm a significant biomarker reduction following RIC, but found no significant reductions in hard endpoints60,78,79. No pharmacologic study has been shown to effectively add benefit via either of those parameters while actions such as remote ischemic conditioning and postconditioning have been shown to reduce infarct size via CMR46. RIC has been shown to reduce myocardial reperfusion injury in the setting of STEMI, CABG, and open-heart surgery43,92. A randomized clinical trial conducted by Theilmann et al66 demonstrated that RICPre reduces perioperative myocardial injury during elective CABG surgery, and is a safe perioperative method that improves prognosis in patients. Several trials do not factor comorbidities, such as diabetes, which may modify responses to pre-and postconditioning and final infarct size36. If RIC proves to be beneficial following clinical trials, anesthetists may have the option of expanding ischemic protection to the heart, but also vulnerable organs such as the kidneys in the future76. Yang and colleagues confirmed kidney protection in meta-analysis studying patients in cardiac surgery79.
The only organ hypothesized not to benefit from remote ischemia is the bowel; mesenteric ischemia in the setting of cardiovascular surgery is usually the result of large atheroemboli, inadequate collateral circulation or systemic vasoconstriction, so relative resistance to ischemia-reperfusion is unlikely to protect anything except some mucosal ischemia78. Due to the lack of efficacy of pharmacologic treatments and the ability to easily perform RIC techniques and add to protocols, it is more reasonable to focus on mechanical means of cardioprotection to optimize protocols70. Although other therapies, such as hyperbaric oxygen may be used as conditioning stimulus prior to CABG surgery and has been shown to limit myocardial damage, improve myocardial function and exert a protective effect against ischemic reperfusion injury preoperatively and postoperatively when performed correctly55.
CONCLUSION
Infarct size is the great prognostic indicator in the setting of acute myocardial infarction, and reperfusion of the ischemic myocardium is the gold standard of treatment following coronary occlusion, but also results in expansion of the original infarct. Therapies that target reperfusion injury have been explored, and the most promising include ischemic conditioning of the heart. The heart can be conditioned naturally, mechanically, or pharmacologically in a local setting, or remotely via ischemic events on distant organs. A cardioprotective effect is propagated through several signaling cascades that include the RISK and SAFE pathways which ultimately result in inhibition of the MPTP, opening of KATP channels, or expression of prosurvival/protective genes. Pharmacologic conditioning that serves to close the MPTP, open KATP or potentiate RISK and SAFE pathways have been largely unsuccessful apart from some volatile anesthetic agents and cyclosporine-A. Postconditioning has been employed as another strategy to reduce infarct size and mitigate reperfusion injury and acts in a similar mechanistic manner to preconditioning. The greatest potential for clinical application is remote ischemic conditioning (RIC) that can confer a cardioprotective effect that can be achieved by inflating and deflating a blood pressure cuff on the upper limb. RIC induces its cardioprotective effect though a neurohumoral pathway that is incited by neural stimulation. Intact peripheral nerve pathways in the limb along with intact vagal pathways are necessary for limb RIC to be effective. Additionally, RIC can be applied in a flexible timeframe granting either pre- or post-conditioning effects. It should be noted that many scientists call for more clinical trials to confirm the perceived beneficial effects of RIC. While the common complaint of heterogeneity and confounders is valid, it may be possible that RIC fits such a specific niche that its addition to treatment guidelines or protocols may not add any benefits to patients. If drugs such as propofol or conditions such as diabetes abrogate cardioprotective effects of RIC, assessing the population of patients who suffer from STEMIs with comorbidities of diabetes mellitus or those given propofol is vital to determining if ischemic conditioning will benefit enough of the overall population to incite a change in current protocols or guidelines. Since propofol administration can be controlled, additional trials such as the CONDI/ERIC-PPCI trials, where propofol is not administered should be assessed to determine the benefits of RIC. Evaluation of RIC in both diabetic and non-diabetic patients would also be able to provide greater insight into the application of RIC in clinical settings.
Acknowledgments
Funding
This work was supported by research grants R01 HL112597, R01 HL116042, and R01 HL120659 to DK Agrawal from the National Heart, Lung and Blood Institute, National Institutes of Health, USA. The content of this review article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Compliance with Ethical Standard
Conflict of Interest
Author JHR declares no competing financial or non-financial interest. Author JHW declares no competing financial or non-financial interest. Author MJM declares no competing financial or non-financial interest. Author DKA declares no competing financial or non-financial interest. No writing assistance was utilized in the production of this manuscript.
Animals/Human Subjects
This article does not contain any studies with human participants or animals performed by any of the authors. Findings were gathered from the published articles in the literature.
References
- 1.Bell RM, Yellon DM. Conditioning the whole heart-not just the cardiomyocyte. J Mol Cell Cardiol. 2012;53(1):24–32. doi: 10.1016/j.yjmcc.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 2.Ibanez B, Heusch G, Ovize M, Van De Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol. 2015;65(14):1454–1471. doi: 10.1016/j.jacc.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 3.Iliodromitis EK, Cohen MV, Dagres N, Andreadou I, Kremastinos DT, Downey JM. What is Wrong With Cardiac Conditioning? We May be Shooting at Moving Targets. J Cardiovasc Pharmacol Ther. 2015;20(4):357–369. doi: 10.1177/1074248414566459. [DOI] [PubMed] [Google Scholar]
- 4.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74(5):1124–1136. doi: 10.1161/01.CIR.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 5.Jennings RB. Historical Perspective on the Pathology of Myocardial Ischemia/Reperfusion Injury. Circ Res. 2013;113(4):428–438. doi: 10.1161/CIRCRESAHA.113.300987. [DOI] [PubMed] [Google Scholar]
- 6.Ibanez B, Heusch G, García-Dorado D, Fuster VYD. In: MOLECULAR AND CELLULAR MECHANISMS OF MYOCARDIAL ISCHEMIA/REPERFUSION INJURY. 14th. Fuster V, Harrington RA, Narula JEZ, editors. New York, NY: McGraw-Hill; 2017. http://accessmedicine.mhmedical.com.cuhsl.creighton.edu/content.aspx?bookid=2046§ionid=155633823. [Google Scholar]
- 7.Heusch G. Cardioprotection: Chances and challenges of its translation to the clinic. Lancet. 2013;381(9861):166–175. doi: 10.1016/S0140-6736(12)60916-7. [DOI] [PubMed] [Google Scholar]
- 8.Balakumar P, Babbar L. Preconditioning the hyperlipidemic myocardium: Fact or fantasy? Cell Signal. 2012;24(3):589–595. doi: 10.1016/j.cellsig.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 9.Bernink FJP, Timmers L, Beek AM, et al. Progression in attenuating myocardial reperfusion injury: An overview. Int J Cardiol. 2014;170(3):261–269. doi: 10.1016/j.ijcard.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 10.Ibanez B, MacAya C, Sánchez-Brunete V, et al. Effect of early metoprolol on infarct size in ST-segment-elevation myocardial infarction patients undergoing primary percutaneous coronary intervention: The effect of Metoprolol in Cardioprotection during an Acute Myocardial Infarction (METOCARD-CNIC) Tri. Circulation. 2013;128(14):1495–1503. doi: 10.1161/CIRCULATIONAHA.113.003653. [DOI] [PubMed] [Google Scholar]
- 11.Lee SM, Hutchinson M, Saint DA. The role of Toll-like receptor 4 (TLR4) in cardiac ischaemic-reperfusion injury, cardioprotection and preconditioning. Clin Exp Pharmacol Physiol. 2016;43(9):864–871. doi: 10.1111/1440-1681.12602. [DOI] [PubMed] [Google Scholar]
- 12.Miki T, Yuda S, Kouzu H, Miura T. Diabetic cardiomyopathy: Pathophysiology and clinical features. Heart Fail Rev. 2013;18(2):149–166. doi: 10.1007/s10741-012-9313-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heusch G, Rassaf T. Time to Give Up on Cardioprotection?: A Critical Appraisal of Clinical Studies on Ischemic Pre-, Post-, and Remote Conditioning∗. Circ Res. 2016;119(5):676–695. doi: 10.1161/CIRCRESAHA.116.308736. [DOI] [PubMed] [Google Scholar]
- 14.Le Page S, Prunier F. Remote ischemic conditioning: Current clinical perspectives. J Cardiol. 2015;66(2):91–96. doi: 10.1016/j.jjcc.2015.01.009. [DOI] [PubMed] [Google Scholar]
- 15.Lorgis L, Gudjoncik A, Richard C, et al. Pre-Infarction Angina and Outcomes in Non-ST-Segment Elevation Myocardial Infarction: Data from the RICO Survey. Moretti C, ed. PLoS One. 2012;7(12):e48513. doi: 10.1371/journal.pone.0048513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lefer DJ, Bolli R. Cardioprotection. First. Vol. 1. Elsevier Inc; 2012. [DOI] [Google Scholar]
- 17.Kunst G, Klein AA. Peri-operative anaesthetic myocardial preconditioning and protection - Cellular mechanisms and clinical relevance in cardiac anaesthesia. Anaesthesia. 2015;70(4):467–482. doi: 10.1111/anae.12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morley CGD, Stadtman TC. Studies on the Fermentation of D-a-Lysine Purification and Properties of an Adenosine Triphospate Regulated B12-Coenzyme-Dependent D-a Lysine Mutase Complex from Clostridium Sticklandii. Second. Vol. 9. Elsevier Inc; 2011. [DOI] [PubMed] [Google Scholar]
- 19.Heusch G. Molecular basis of cardioprotection signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res. 2015;116(4):674–699. doi: 10.1161/CIRCRESAHA.116.305348. [DOI] [PubMed] [Google Scholar]
- 20.Hausenloy DJ, Garcia-Dorado D, Bøtker HE, et al. Novel targets and future strategies for acute cardioprotection: Position Paper of the European Society of Cardiology Working Group on Cellular Biology of the Heart. doi: 10.1093/cvr/cvx049. [DOI] [PubMed] [Google Scholar]
- 21.Hausenloy DJ, Tsang A, Yellon DM. The reperfusion injury salvage kinase pathway: A common target for both ischemic preconditioning and postconditioning. Trends Cardiovasc Med. 2005;15(2):69–75. doi: 10.1016/j.tcm.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 22.Ong SB, Dongworth RK, Cabrera-Fuentes HA, Hausenloy DJ. Role of the MPTP in conditioning the heart - Translatability and mechanism. Br J Pharmacol. 2015;172(8):2074–2084. doi: 10.1111/bph.13013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341:233–249. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1220352/pdf/10393078.pdf. Accessed April 22, 2018. [PMC free article] [PubMed] [Google Scholar]
- 24.Yellon DM, Ackbarkhan AK, Balgobin V, et al. Remote ischemic conditioning reduces myocardial infarct size in STEMI patients treated by thrombolysis. J Am Coll Cardiol. 2015;65(25):2764–2765. doi: 10.1016/j.jacc.2015.02.082. [DOI] [PubMed] [Google Scholar]
- 25.Swyers T, Redford D, Larson D. Volatile anesthetic-induced preconditioning. Perfusion. 2014;29(1):10–15. doi: 10.1177/0267659113503975. [DOI] [PubMed] [Google Scholar]
- 26.Rana A, Goyal N, Ahlawat A, Jamwal S, Reddy B, Sharma S. Mechanisms involved in attenuated cardio-protective role of ischemic preconditioning in metabolic disorders. Perfusion. 2015;30(2):94–105. doi: 10.1177/0267659114536760. [DOI] [PubMed] [Google Scholar]
- 27.Moreira JBN, Wohlwend M, Alves MNM, Wisløff U, Bye A. A small molecule activator of AKT does not reduce ischemic injury of the rat heart. J Transl Med. 2015;13:76. doi: 10.1186/s12967-015-0444-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Somers SJ, Frias M, Lacerda L, Opie LH, Lecour S. Interplay Between SAFE and RISK Pathways in Sphingosine-1-Phosphate – Induced Cardioprotection. 2012:227–237. doi: 10.1007/s10557-012-6376-2. [DOI] [PubMed] [Google Scholar]
- 29.Lecour S. Activation of the protective Survivor Activating Factor Enhancement (SAFE) pathway against reperfusion injury: Does it go beyond the RISK pathway? J Mol Cell Cardiol. 2009;47(1):32–40. doi: 10.1016/j.yjmcc.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 30.Schmidt MR, Redington A, B??tker HE. Remote conditioning the heart overview: Translatability and mechanism. Br J Pharmacol. 2015;172(8):1947–1960. doi: 10.1111/bph.12933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roolvink V, Ibáñez B, Ottervanger JP, et al. Early Intravenous Beta-Blockers in Patients With ST-Segment Elevation Myocardial Infarction Before Primary Percutaneous Coronary Intervention. J Am Coll Cardiol. 2016;67(23):2705–2715. doi: 10.1016/j.jacc.2016.03.522. [DOI] [PubMed] [Google Scholar]
- 32.Ravingerova T, Ledvenyiova-Farkasova V, Ferko M, et al. Pleiotropic preconditioning-like cardioprotective effects of hypolipidemic drugs in acute ischemia-reperfusion in normal and hypertensive rats. Can J Physiol Pharmacol. 2015;93(7):495–503. doi: 10.1139/cjpp-2014-0502. [DOI] [PubMed] [Google Scholar]
- 33.Sakata Y, Nakatani D, Shimizu M, et al. Oral treatment with nicorandil at discharge is associated with reduced mortality after acute myocardial infarction. J Cardiol. 2012;59(1):14–21. doi: 10.1016/j.jjcc.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 34.Lincoff AM, Roe M, Aylward P, et al. Inhibition of delta-protein kinase C by delcasertib as an adjunct to primary percutaneous coronary intervention for acute anterior ST-segment elevation myocardial infarction: Results of the PROTECTION AMI randomized controlled trial. Eur Heart J. 2014;35(37):2516–2523. doi: 10.1093/eurheartj/ehu177. [DOI] [PubMed] [Google Scholar]
- 35.Mahaffey KW, Puma JA, Barbagelata NA, et al. Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction - Results of a multicenter, randomized, placebo-controlled trial: The acute myocardial infarction STudy of ADenosine (AMISTAD) trial. J Am Coll Cardiol. 1999;34(6):1711–1720. doi: 10.1016/S0735-1097(99)00418-0. [DOI] [PubMed] [Google Scholar]
- 36.Miki T, Itoh T, Sunaga D, Miura T. Effects of diabetes on myocardial infarct size and cardioprotection by preconditioning and postconditioning. Cardiovasc Diabetol. 2012;11(1):67. doi: 10.1186/1475-2840-11-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ross AM, Gibbons RJ, Stone GW, Kloner RA, Alexander RW. A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II) J Am Coll Cardiol. 2005;45(11):1775–1780. doi: 10.1016/j.jacc.2005.02.061. [DOI] [PubMed] [Google Scholar]
- 38.Patel DJ, Purcell HJ, Fox KM. Cardioprotection by opening of the K ATP channel in unstable angina Is this a clinical manifestation of myocardial preconditioning? Results of a randomized study with nicorandil. 1999:51–57. doi: 10.1053/euhj.1998.1354. [DOI] [PubMed] [Google Scholar]
- 39.Ito H, Taniyama Y, Iwakura K, et al. Intravenous nicorandil can preserve microvascular integrity and myocardial viability in patients with reperfused anterior wall myocardial infarction. J Am Coll Cardiol. 1999;33(3):654–660. doi: 10.1016/S0735-1097(98)00604-4. [DOI] [PubMed] [Google Scholar]
- 40.Quindry JC, Hamilton KL. Exercise and Cardiac Preconditioning Against Ischemia Reperfusion. Curr Cardiol Rev. 2013;9(3):220–229. doi: 10.2174/1573403x113099990033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pagel PS. Myocardial protection by volatile anesthetics in patients undergoing cardiac surgery: A critical review of the laboratory and clinical evidence. J Cardiothorac Vasc Anesth. 2013;27(5):972–982. doi: 10.1053/j.jvca.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 42.Moscarelli M, Punjabi PP, Miroslav GI, Del Sarto P, Fiorentino F, Angelini GD. Myocardial conditioning techniques in off-pump coronary artery bypass grafting. J Cardiothorac Surg. 2015;10:7. doi: 10.1186/s13019-014-0204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sivaraman V, Pickard JMJ, Hausenloy DJ. Remote ischaemic conditioning: Cardiac protection from afar. Anaesthesia. 2015;70(6):732–748. doi: 10.1111/anae.12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heusch G, Bøtker HE, Przyklenk K, Redington A, Yellon D. Remote ischemic conditioning. J Am Coll Cardiol. 2015;65(2):177–195. doi: 10.1016/j.jacc.2014.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Le Page S, Bejan-Angoulvant T, Angoulvant D, Prunier F. Remote ischemic conditioning and cardioprotection: a systematic review and meta-analysis of randomized clinical trials. Basic Res Cardiol. 2015;110(2) doi: 10.1007/s00395-015-0467-8. [DOI] [PubMed] [Google Scholar]
- 46.Ibáñez B, Heusch G, Ovize M, Van De Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol. 2015;65(14):1454–1471. doi: 10.1016/j.jacc.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 47.Cung T-T, Morel O, Cayla G, et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N Engl J Med. 2015;373(11):1021–1031. doi: 10.1056/NEJMoa1505489. [DOI] [PubMed] [Google Scholar]
- 48.Piot C, Croisille P, Staat P, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359(5):473–481. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 49.Przyklenk K. Ischaemic conditioning: Pitfalls on the path to clinical translation. Br J Pharmacol. 2015;172(8):1961–1973. doi: 10.1111/bph.13064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deng Q-W, Xia Z-Q, Qiu Y-X, et al. Clinical benefits of aortic cross-clamping versus limb remote ischemic preconditioning in coronary artery bypass grafting with cardiopulmonary bypass: a meta-analysis of randomized controlled trials. J Surg Res. 2015;193(1):52–68. doi: 10.1016/j.jss.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 51.Williams RP, Manou-Stathopoulou V, Redwood SR, Marber MS. Republished: “Warm-up Angina”: harnessing the benefits of exercise and myocardial ischaemia. Postgrad Med J. 2014;90(1069):648–656. doi: 10.1136/postgradmedj-2013-304187rep. [DOI] [PubMed] [Google Scholar]
- 52.Okazaki Y, Kodama K, Sato H, et al. Attenuation of increased regional myocardial oxygen consumption during exercise as a major cause of warm-up phenomenon. J Am Coll Cardiol. 1993;21(7):1597–1604. doi: 10.1016/0735-1097(93)90374-A. [DOI] [PubMed] [Google Scholar]
- 53.Deutsch E, Berger M, Kussmaul WG, Hirshfeld JW, Herrmann HC, Laskey WK. Adaptation to ischemia during percutaneous transluminal coronary angioplasty. Clinical, hemodynamic, and metabolic features. Circulation. 1990;82(6):2044–2051. doi: 10.1161/01.CIR.82.6.2044. [DOI] [PubMed] [Google Scholar]
- 54.Yogaratnam JZ, Laden G, Guvendik L, Cowen M, Cale A, Griffin S. Hyperbaric oxygen preconditioning improves myocardial function, reduces length of intensive care stay, and limits complications post coronary artery bypass graft surgery. Cardiovasc Revascularization Med. 2010;11(1):8–19. doi: 10.1016/j.carrev.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 55.Allen MW, Golembe E, Gorenstein S, Butler GJ. Protective effects of hyperbaric oxygen therapy (HBO2) in cardiac care–A proposal to conduct a study into the effects of hyperbaric pre-conditioning in elective coronary artery bypass graft surgery (CABG) Undersea Hyperb Med. 2015;42(2):107–114. http://www.ncbi.nlm.nih.gov/pubmed/26094285. [PubMed] [Google Scholar]
- 56.Na HS, Kim YI, Yoon YW, Han HC, Nahm SH, Hong SK. Ventricular premature beat— driven intermittent restoration of coronary blood flow reduces the incidence of reperfusion-induced ventricular fibrillation in a cat model of regional ischemia. Am Heart J. 1996;132(1):78–83. doi: 10.1016/S0002-8703(96)90393-2. [DOI] [PubMed] [Google Scholar]
- 57.Lucas DN, Yentis SM. Unsettled weather and the end for thiopental? Obstetric general anaesthesia after the NAP5 and MBRRACE-UK reports. Anaesthesia. 2015;70(4):375–379. doi: 10.1111/anae.13034. [DOI] [PubMed] [Google Scholar]
- 58.Ovize M, Thibault H, Przyklenk K. Myocardial conditioning: Opportunities for clinical translation. Circ Res. 2013;113(4):439–450. doi: 10.1161/CIRCRESAHA.113.300764. [DOI] [PubMed] [Google Scholar]
- 59.Halabisky B, Strowbridge BW. Copyright (c) 2003 by the American Physiological Society. 2003;44106(216):1–35. [Google Scholar]
- 60.Elbadawi A, Ha LD, Abuzaid AS, Crimi G, Azzouz MS. Meta-Analysis of Randomized Trials on Remote Ischemic Conditioning during Primary Percutaneous Coronary Intervention in Patients with ST-Segment Elevation Myocardial Infarction. Am J Cardiol. 2016;119(6):832–838. doi: 10.1016/j.amjcard.2016.11.036. [DOI] [PubMed] [Google Scholar]
- 61.Staat P, Rioufol G, Piot C, et al. Postconditioning the human heart. Circulation. 2005;112(14):2143–2148. doi: 10.1161/CIRCULATIONAHA.105.558122. [DOI] [PubMed] [Google Scholar]
- 62.Ravingerova T, Farkasova V, Griecsova L, et al. Remote preconditioning as a novel “ conditioning” approach to repair the broken heart: Potential mechanisms and clinical applications. Physiol Res. 2016;65:S55–S64. doi: 10.33549/physiolres.933392. [DOI] [PubMed] [Google Scholar]
- 63.Przyklenk K, Bauer B, Ovize M, Kloner RA, Whittaker P. Regional ischemic “preconditioning” protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation. 1993;87(3):893–899. doi: 10.1161/01.cir.87.3.893. [DOI] [PubMed] [Google Scholar]
- 64.Kharbanda RK, Mortensen UM, White PA, et al. Transient limb ischemia induces remote ischemic preconditioning in vivo. Circulation. 2002;106(23):2881–2883. doi: 10.1161/01.CIR.0000043806.51912.9B. [DOI] [PubMed] [Google Scholar]
- 65.Zografos TA, Katritsis GD, Tsiafoutis I, Bourboulis N, Katsivas A, Katritsis DG. Effect of one-cycle remote ischemic preconditioning to reduce myocardial injury during percutaneous coronary intervention. Am J Cardiol. 2014;113(12):2013–2017. doi: 10.1016/j.amjcard.2014.03.043. [DOI] [PubMed] [Google Scholar]
- 66.Thielmann M, Kottenberg E, Kleinbongard P, et al. Cardioprotective and prognostic effects of remote ischaemic preconditioning in patients undergoing coronary artery bypass surgery: A single-centre randomised, double-blind, controlled trial. Lancet. 2013;382(9892):597–604. doi: 10.1016/S0140-6736(13)61450-6. [DOI] [PubMed] [Google Scholar]
- 67.Manchurov V, Ryazankina N, Khmara T, et al. Remote Ischemic Preconditioning and Endothelial Function in Patients with Acute Myocardial Infarction and Primary PCI. Am J Med. 2014;127(7):670–673. doi: 10.1016/j.amjmed.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 68.Mewton N, Ovize M. Remote preconditioning and all-cause mortality. Lancet. 2013;382(9892):579–580. doi: 10.1016/S0140-6736(13)61607-4. [DOI] [PubMed] [Google Scholar]
- 69.Yang H, Tracey KJ. HHS Public Access. 2015;1799(0):149–156. doi: 10.1016/j.bbagrm.2009.11.019.Targeting. [DOI] [Google Scholar]
- 70.Pickard JMJ, Bøtker HE, Crimi G, et al. Remote ischemic conditioning: from experimental observation to clinical application: report from the 8th Biennial Hatter Cardiovascular Institute Workshop. Basic Res Cardiol. 2015;110(1) doi: 10.1007/s00395-014-0453-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jensen RV, Støttrup NB, Kristiansen SB, Bøtker HE. Release of a humoral circulating cardioprotective factor by remote ischemic preconditioning is dependent on preserved neural pathways in diabetic patients. Basic Res Cardiol. 2012;107(5):1–9. doi: 10.1007/s00395-012-0285-1. [DOI] [PubMed] [Google Scholar]
- 72.Gill R, Kuriakose R, Gertz ZM, Salloum FN, Xi L, Kukreja RC. Remote ischemic preconditioning for myocardial protection: update on mechanisms and clinical relevance. Mol Cell Biochem. 2015;402(1-2):41–49. doi: 10.1007/s11010-014-2312-z. [DOI] [PubMed] [Google Scholar]
- 73.D’Ascenzo F, Cavallero E, Moretti C, et al. Remote ischaemic preconditioning in coronary artery bypass surgery: a meta-analysis. Heart. 2012;98(17):1267–1271. doi: 10.1136/heartjnl-2011-301551. [DOI] [PubMed] [Google Scholar]
- 74.Günaydin B, Cakici I, Soncul H, et al. Does remote organ ischaemia trigger cardiac preconditioning during coronary artery surgery? Pharmacol Res. 2000;41(4):493–496. doi: 10.1006/phrs.1999.0611. [DOI] [PubMed] [Google Scholar]
- 75.Cheung MMH, Kharbanda RK, Konstantinov IE, et al. Randomized Controlled Trial of the Effects of Remote Ischemic Preconditioning on Children Undergoing Cardiac Surgery. First Clinical Application in Humans. J Am Coll Cardiol. 2006;47(11):2277–2282. doi: 10.1016/j.jacc.2006.01.066. [DOI] [PubMed] [Google Scholar]
- 76.Candilio L, Malik A, Ariti C, et al. Effect of remote ischaemic preconditioning on clinical outcomes in patients undergoing cardiac bypass surgery: a randomised controlled clinical trial. Heart. 2015;101(3):185–192. doi: 10.1136/heartjnl-2014-306178. [DOI] [PubMed] [Google Scholar]
- 77.Zimmerman RF, Ezeanuna PU, Kane JC, et al. Ischemic preconditioning at a remote site prevents acute kidney injury in patients following cardiac surgery. Kidney Int. 2011;80(8):861–867. doi: 10.1038/ki.2011.156. [DOI] [PubMed] [Google Scholar]
- 78.Healy DA, Khan WA, Wong CS, et al. Remote preconditioning and major clinical complications following adult cardiovascular surgery: Systematic review and meta-analysis. Int J Cardiol. 2014;176(1):20–31. doi: 10.1016/j.ijcard.2014.06.018. [DOI] [PubMed] [Google Scholar]
- 79.Yang L, Wang G, Du Y, Ji B, Zheng Z. Remote ischemic preconditioning reduces cardiac troponin i release in cardiac surgery: A meta-analysis. J Cardiothorac Vasc Anesth. 2014;28(3):682–689. doi: 10.1053/j.jvca.2013.05.035. [DOI] [PubMed] [Google Scholar]
- 80.Davies WR, Brown AJ, Watson W, et al. Remote ischemic preconditioning improves outcome at 6 years after elective percutaneous coronary intervention: The CRISP stent trial long-term follow-up. Circ Cardiovasc Interv. 2013;6(3):246–251. doi: 10.1161/CIRCINTERVENTIONS.112.000184. [DOI] [PubMed] [Google Scholar]
- 81.Lanza GA, Stazi A, Villano A, et al. Effect of Remote Ischemic Preconditioning on Platelet Activation Induced by Coronary Procedures. Am J Cardiol. 2016;117(3):359–365. doi: 10.1016/j.amjcard.2015.10.056. [DOI] [PubMed] [Google Scholar]
- 82.Hausenloy DJ, Candilio L, Evans R, et al. Remote Ischemic Preconditioning and Outcomes of Cardiac Surgery. N Engl J Med. 2015;373(15):1408–1417. doi: 10.1056/NEJMoa1413534. [DOI] [PubMed] [Google Scholar]
- 83.Meybohm P, Bein B, Brosteanu O, et al. A Multicenter Trial of Remote Ischemic Preconditioning for Heart Surgery. N Engl J Med. 2015;373(15):1397–1407. doi: 10.1056/NEJMoa1413579. [DOI] [PubMed] [Google Scholar]
- 84.Bauters C, Lamblin N, Fadden EPM, Van Belle E, Millaire A, De Groote P. Influence of diabetes mellitus on heart failure risk and outcome. 2003;16:1–16. doi: 10.1186/1475-2840-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Haffner SM. Relationship of metabolic risk factors and development of cardiovascular disease and diabetes. Obesity. 2006 Jun;14(Suppl 3):121S–127S. doi: 10.1038/oby.2006.291. [DOI] [PubMed] [Google Scholar]
- 86.Tsang A, Hausenloy DJ, Mocanu MM, Carr RD, Yellon DM. Preconditioning the Diabetic Heart. Diabetes. 2005;54(8):2360–2364. doi: 10.2337/diabetes.54.8.2360. [DOI] [PubMed] [Google Scholar]
- 87.Tamareille S, Ghaboura N, Treguer F, et al. Myocardial reperfusion injury management: erythropoietin compared with postconditioning. Am J Physiol Heart Circ Physiol. 2009;297(6):H2035–43. doi: 10.1152/ajpheart.00472.2009. [DOI] [PubMed] [Google Scholar]
- 88.Ferdinandy P, Hausenloy DJ, Heusch G, Baxter GF, Schulz R. Interaction of Risk Factors, Comorbidities, and Comedications with Ischemia/Reperfusion Injury and Cardioprotection by Preconditioning, Postconditioning, and Remote Conditioning. Pharmacol Rev Pharmacol Rev. 2014;66:1142–1174. doi: 10.1124/pr.113.008300. [DOI] [PubMed] [Google Scholar]
- 89.Schulz R, Görge PM, Görbe A, Ferdinandy P, Lampe PD, Leybaert L. Connexin 43 is an emerging therapeutic target in ischemia/reperfusion injury, cardioprotection and neuroprotection. Pharmacol Ther. 2015;153:90–106. doi: 10.1016/j.pharmthera.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Andreadou I, Iliodromitis EK, Lazou A, et al. Effect of hypercholesterolaemia on myocardial function, ischaemia-reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning. Br J Pharmacol. 2017;174(12):1555–1569. doi: 10.1111/bph.13704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pryds K, Terkelsen CJ, Sloth AD, et al. Remote ischaemic conditioning and healthcare system delay in patients with ST-segment elevation myocardial infarction. Heart. 2016;102(13):1023–1028. doi: 10.1136/heartjnl-2015-308980. [DOI] [PubMed] [Google Scholar]
- 92.Payne RE, Aldwinckle J, Storrow J, Kong RS, Lewis ME. RIPC remains a promising technique for protection of the myocardium during open cardiac surgery: A meta-analysis and systematic review. Heart Surg Forum. 2015;18(2):77–84. doi: 10.1532/hsf.1251. [DOI] [PubMed] [Google Scholar]