

Abstract
The D1 dopamine receptor is linked to a variety of neuropsychiatric disorders and represents an attractive drug target for the enhancement of cognition in schizophrenia, Alzheimer disease, and other disorders. Positive allosteric modulators (PAMs), with their potential for greater selectivity and larger therapeutic windows, may represent a viable drug development strategy, as orthosteric D1 receptor agonists possess known clinical liabilities. We discovered two structurally distinct D1 receptor PAMs, MLS6585 and MLS1082, via a high-throughput screen of the NIH Molecular Libraries program small-molecule library. Both compounds potentiate dopamine-stimulated G protein- and β-arrestin-mediated signaling and increase the affinity of dopamine for the D1 receptor with low micromolar potencies. Neither compound displayed any intrinsic agonist activity. Both compounds were also found to potentiate the efficacy of partial agonists. We tested maximally effective concentrations of each PAM in combination to determine if the compounds might act at separate or similar sites. In combination, MLS1082 + MLS6585 produced an additive potentiation of dopamine potency beyond that caused by either PAM alone for both β-arrestin recruitment and cAMP accumulation, suggesting diverse sites of action. In addition, MLS6585, but not MLS1082, had additive activity with the previously described D1 receptor PAM “Compound B,” suggesting that MLS1082 and Compound B may share a common binding site. A point mutation (R130Q) in the D1 receptor was found to abrogate MLS1082 activity without affecting that of MLS6585, suggesting this residue may be involved in the binding/activity of MLS1082 but not that of MLS6585. Together, MLS1082 and MLS6585 may serve as important tool compounds for the characterization of diverse allosteric sites on the D1 receptor as well as the development of optimized lead compounds for therapeutic use.
Introduction
There is great interest in identifying small-molecule ligands for G protein-coupled receptors (GPCRs), as nearly 50% of all FDA-approved drugs target these important receptor proteins (Eglen, 2007). Unfortunately, many of these drugs are not very selective and exhibit problematic or limiting side effects owing to undesirable off-target signaling. A therapeutically important subclass of GPCRs is that activated by dopamine (DA), a crucial neurotransmitter in both the central nervous system and the periphery (Sibley and Monsma, 1992; Rankin et al., 2010). In mammals, five distinct DA receptor (DAR) subtypes exist and are divided into two subfamilies on the basis of their structure, pharmacology, and signaling properties (Sibley and Monsma, 1992; Beaulieu and Gainetdinov, 2011). The D1-like DARs (D1R and D5R) are coupled to Gαs/olf proteins and activate adenylyl cyclase, resulting in increased intracellular cAMP levels. In contrast, the D2-like DARs (D2R, D3R, and D4R) are coupled to Gαi/o proteins, which inhibit adenylyl cyclase activity and also modulate K+- and Ca2+-channel activities. Dysfunction of the central nervous system dopaminergic system is involved in the etiology and/or therapy of many neuropsychiatric disorders, which are treated with drugs that either stimulate or block DAR subtypes, making these receptors key therapeutic targets.
The most highly expressed DAR subtype is the D1R, which is found in high abundance in various regions of the mammalian forebrain, including the striatum, cerebral cortex, hippocampus, and the olfactory bulb. Physiologically, it plays a crucial role in regulating movement, cognition, learning and memory, as well as reward and reinforcement. As such, the D1R provides an attractive drug target for the treatment of several disorders including the decline of cognition and memory, both hallmarks of Alzheimer disease, schizophrenia, and Parkinson disease. Seminal work by Goldman-Rakic and others have shown that an optimal level of D1-like receptor activity in the prefrontal cortex (PFC) is required for ideal performance in learning and memory tasks (Cai and Arnsten, 1997; Castner and Goldman-Rakic, 2004; Goldman-Rakic et al., 2004; Nakako et al., 2013). Either too little or too much D1R stimulation (the latter can occur with high levels of DA release during stress) impairs PFC function (Arnsten and Dudley, 2005; Arnsten and Li, 2005; Vijayraghavan et al., 2007). These observations have led to the “inverted U” hypothesis of the relationship between D1R stimulation and normal physiologic functioning of the PFC (Arnsten et al., 2009), suggesting that “fine-tuning” D1R stimulation may be effective for enhancing cognition. Notably, the Measurement and Treatment Research to Improve Cognition in Schizophrenia (MATRICS) program evaluated a range of molecular targets for enhancing cognition and rated the D1R in the PFC as the most promising target (Tamminga, 2006). Additionally, animal tests have confirmed the positive effects of D1R stimulation on working memory and cognitive function (Goldman-Rakic et al., 2004).
One approach for optimizing D1R stimulation is the development of compounds that enhance dopamine activity, without directly activating the receptor itself. Such compounds, termed positive allosteric modulators (PAMs), may bind to sites on the receptor separate from the orthosteric binding site. PAMs may enhance endogenous ligand activity through augmenting agonist potency, efficacy, or both. PAMs may confer advantages over traditional orthosteric agonists; for instance, PAMs may have fewer off-target side effects, as they bind to less conserved regions of a receptor. Likewise, PAMs may also exhibit decreased or no receptor desensitization compared with orthosteric agonists (May et al., 2007; Gjoni and Urwyler, 2008). Importantly, allosteric compounds can also function as “stabilizers” of signaling pathways, as they exert their effects by enhancing the activity of endogenous neurotransmitters without overwhelming the underlying neuronal tone. Taken together, development of D1R PAMs may be an attractive approach for developing therapeutic compounds as well as tools to further advance our understanding of D1R signaling. Indeed, D1R PAMs have been described recently by other groups (Lewis et al., 2015; Svensson et al., 2017; Bruns et al., 2018).
To discover novel D1R PAM molecules, we employed a high-throughput screening paradigm to interrogate the NIH Molecular Libraries Screening Center Network small-molecule library. Here, we report the discovery and characterization of two structurally distinct positive allosteric modulators of D1R signaling, MLS1082 and MLS6585. These compounds have no inherent agonist activity but potentiate both G protein- and β-arrestin-mediated signaling stimulated by both the endogenous ligand dopamine and other D1R agonists. Further, using functional additivity as well as mutational approaches, we obtained evidence that MLS1082 and MLS6585 probably bind to diverse receptor sites. Overall, our current studies describe novel D1R PAM compounds and also provide evidence for multiple allosteric sites on the D1R.
Materials and Methods
Materials.
Original screening quantities of MLS1082 and MLS6585 were obtained from the Molecular Libraries Screening Center Network Library. Compounds were subsequently purchased from MolPort (Riga, Latvia) for follow up triage studies. All other chemicals were obtained from MilliporeSigma (St. Louis, MO) unless otherwise indicated within the Methods. All tissue culture media and components were obtained from Mediatech, Inc./Corning Inc. (Manassas, VA). Finally, MLS1082, MLS6585, and Compound B were synthesized in-house at the KU Specialized Chemistry Center, as described in the supplemental section (Supplemental Methods). Some batches of Compound B were also synthesized at Monash University. Identical results were obtained with all batches of compounds from all suppliers.
Calcium Mobilization Assay.
HEK293T cells were stably transfected with human D1R and Gα15 protein using the Flp-In T-REx expression system (Thermo Fisher Scientific, Waltham, MA). Cells were first stably transfected with the human D1R in pcDNA3.1+ and selected with G418. Colonies were validated by radioligand binding for D1R expression. Cells were then stably transfected with Gα15 protein in G15/pIREShygro (Clonetech, Mountain View, CA) vector that imparted hygromycin resistance and subsequently selected with hygromycin. G15 expression was validated from individual colonies using the Ca2+ mobilization assay. Cells lines giving the most robust calcium response were selected for screening assays. D1R-stimulated calcium mobilization was measured using methods similar to those previously published by our laboratory (Chun et al., 2013). For high-throughput screening, D1R-G15 cells (4000 cells/well and 3 μl/well) were added directly to the culture media and plated in 1536-well, optical, clear-bottomed, black-walled plates (Greiner Bio-one, Monroe, NC). The following day, cells were incubated for 60 minutes at room temperature in the dark with Fluo-8 NW calcium dye in the presence of an extracellular signal quencher (Screen Quest Fluo-8 NW Calcium Assay Kit; AAT Bioquest, Inc., Sunnyvale, CA), as recommended by the manufacturer. The plates were then treated with 40 μM test compound and read kinetically in real time (every 0.6 seconds) both before compound addition and for 2 minutes after compound addition. Compound additions were done in unison using an onboard 1536-pintool simultaneously with continuous reading at an excitation wavelength of 480 nm and an emission wavelength of 540 nm on an FDSS 7000 (Hamamatsu, Bridgewater, NJ). For potentiation assays, cells were first treated with test compound as described above followed by a second addition of an EC20 concentration of DA (∼300 nM) to give a small response that allows for measurement of the potentiation of the dopamine response. In this paradigm both agonist activity and potentiation could be examined in a single read. Data were recorded and quantified as maximum minus minimum (max-min) relative fluorescence units within the assay window using FDSS software. Hit compounds were defined as compounds that significantly (>3 SD) potentiated the EC20 response of DA and were chosen for further study.
cAMP Accumulation Assay.
Assays were performed on D1R-HEK293 cells stably expressing the human D1R (Codex Biosolutions, Gaithersburg, MD). HEK293 cell lines were maintained in Dulbecco’s modified Eagle’s medium, supplemented with 10% fetal bovine serum, 100 IU/ml penicillin, 100 μg/ml streptomycin, 1 mM sodium pyruvate, and 250 μg/ml G418, and incubated at 37°C, 5% CO2, and 90% humidity. For the assay, cells were seeded in 384-well black, clear-bottomed plates at a density of 5000 cells/well, 10 μl/well. After 18- to 24-hour incubation at 37°C, 5% CO2, and 90% humidity, the media was removed and replaced with 5 μl/well phosphate-buffered saline (PBS). Cells were then treated with 2.5 μl of varying concentrations of compound diluted in <3% dimethylsulfoxide (DMSO) in PBS containing 25 μM 4-(3-butoxy-4-methoxybenzyl)imidazolin-2-one (Ro 20-1724), 1 μM propranolol, 0.2 mM sodium metabisulfite, and incubated for 30 minutes at 37°C, 5% CO2, and 90% humidity. cAMP was measured using the DiscoverX HitHunter kit (DiscoverX, Fremont, CA) according to the manufacturer’s recommendations. Briefly, antibody and working solution were added to each well according to the manufacturer’s protocol and incubated in the dark at room temperature for 60 minutes. Following incubation, enzyme acceptor reagent was added to the plates and luminescence [as relative luminescence units (RLUs)] was measured (FDSS/μCell; Hamamatsu Photonics K. K., Hamamatsu, Japan) following a 3-hour incubation in the dark at room temperature. Data are represented as a percentage of the control maximum dopamine-stimulated cAMP signal.
β-Arrestin Recruitment Assay.
Agonist-mediated recruitment of β-arrestin-2 to all five dopamine receptor subtypes was determined using the DiscoverX PathHunter complementation assay (DiscoverX Inc.), as previously described by our laboratory (Free et al., 2014; Conroy et al., 2015). Briefly, CHO-K1 cells stably expressing either human D1R, D2R, D3R, D4R, or D5R, as indicated, were seeded in cell plating media (DiscoverX) at a density of 2625 cells/well and 7.5 μl/well in 384-well black, clear-bottomed plates. Following 18- to 24-hour of incubation, the cells were treated with the indicated concentrations of compound in PBS buffer containing <2% DMSO and 0.2 mM sodium metabisulfite and incubated at 37°C for 90 minutes. DiscoverX reagent was added to cells according to the manufacturer’s protocol, followed by a 60-minute incubation in the dark at room temperature. Luminescence was measured on a Hamamatsu FDSS/μCell reader (Hamamatsu) and data were collected using the FDSS software. Data were collected as RLUs and normalized to a percentage of the control luminescence seen with a maximum concentration of dopamine, with zero percent being RLUs produced in the absence of any compound.
Radioligand Binding Assays.
Radioligand binding competition assays were conducted with slight modifications as previously described by our laboratory (Chun et al., 2013). HEK293 cells stably transfected with human D1R (Codex Biosolutions, Inc.) were dissociated from plates using Earle’s balanced salt solution lacking calcium and magnesium, and intact cells were collected by centrifugation at 1000g for 10 minutes. Cells were resuspended and lysed using 5 mM Tris-HCl and 5 mM MgCl2 at pH 7.4 at 4°C. Cell lysate was pelleted by centrifugation at 30,000g for 30 minutes and re-suspended in 50 mM Trizma + 5 mM MgCl2 at pH 7.4. Cell membrane preparations (100 μl, containing ∼25 μg protein) were incubated for 90 minutes at room temperature with the indicated concentrations of MLS1082 or MLS6585 in the presence or absence of dopamine and 0.5 nM [3H]-SCH23390 in a final reaction volume of 250 μl. Non-specific binding was determined in the presence of 4 μM (+)-butaclamol. Bound ligand was separated from free by filtration through a PerkinElmer Unifilter-96 GF/C 96-well micro-plate using the PerkinElmer Unifilter-96 Harvester, washing three times, 1 ml per well in ice-cold assay buffer. After drying, 50 μl of liquid scintillation cocktail (MicroScint PS; Perkin Elmer, Waltham, MA) was added to each well, plates were sealed, and analyzed on a PerkinElmer Topcount NXT.
Data Analysis.
Binding-interaction studies with allosteric ligands were fitted to the following allosteric ternary complex model (May et al., 2007), eq. (1):
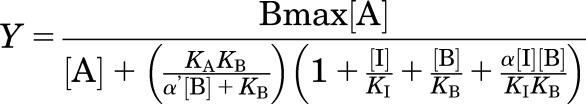 |
(1) |
Where Y is percentage (vehicle control) binding; Bmax is the total number of receptors; [A], [B], and [I] are the concentrations of radioligand, allosteric modulator, and the orthosteric ligand, respectively; KA, KB, and KI are the equilibrium dissociation constants of the radioligand, allosteric modulator, and orthosteric ligand, respectively. α′ and α are the binding cooperativities between the allosteric ligand and [3H]-SCH23390 and the allosteric modulator and the agonist dopamine, respectively. Saturation binding experiments were used to determine the value of pKA for [3H]-SCH23390 (pKA = 9.30, KA = 0.5 nM). Values of α (or α′) > 1 denote positive cooperativity; values <1 (but >0) denote negative cooperativity, and value = 1 denotes neutral cooperativity. For compound MLS6585, a near complete inhibition of [3H]-SCH23390 binding by the allosteric modulator was observed, which is consistent with a very high level of negative cooperativity. In this case to allow fitting of the data, logα’ was fixed to −3 to reflect this high negative cooperativity. For compound MLS1082, no displacement of [3H]-SCH23390 binding was observed, which is consistent with neutral cooperativity (logα′ = 0). The dissociation constant of dopamine (KI) was not fixed in these analyses but rather determined for each separate experiment.
Concentration-response curves for the interaction between the allosteric ligand and the orthosteric ligand in the β-arrestin recruitment assays were globally fitted to the following operational model of allosterism and agonism (Leach et al., 2007), eq. (2):
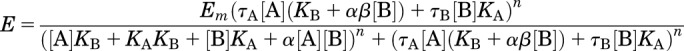 |
(2) |
Where Em is the maximum possible cellular response; [A] and [B] are the concentrations of orthosteric and allosteric ligands, respectively; KA and KB are the equilibrium dissociation constant of the orthosteric and allosteric ligands, respectively; τA and τB are operational measures of orthosteric and allosteric ligand efficacy, respectively; α is the binding cooperativity parameter between the orthosteric and allosteric ligand; β denotes the magnitude of the allosteric effect of the modulator on the efficacy of the orthosteric agonist and n denotes the transducer slope that describes the underlying stimulus-response coupling of the ligand-occupied receptor to the signal pathway. This parameter was constrained to be shared between all curves within a fitted dataset for each interaction study. In many instances, the individual model parameters of eq. (2) could not be directly estimated via the nonlinear regression algorithm by analysis of the functional data alone owing to parameter redundancy. To facilitate model convergence, therefore, we fixed the equilibrium dissociation constant of each ligand to that determined from the binding assays. For all compounds no agonism was observed so log τB was fixed to −3.
Bioluminescence Resonance Energy Transfer Assays.
Experiments were performed in HEK239 cells transiently transfected with D1R-RLuc8 and β-arrestin-mVenus [β-arrestin bioluminescence resonance energy transfer (BRET)] or Gαs-mVenus + β1 + γ2 (Gs BRET) using the polyethylenimine transfection method. Briefly, 4 × 106 cells/plate were seeded on 10-cm dishes and incubated overnight. Appropriate amounts of DNA were combined with 3 μg/μl polyethylenimine per microgram of DNA in nonsupplemented DMEM and incubated with the cells overnight. Experiments were performed 48 hours post-transfection. On experiment day, cells were collected and resuspended in Dulbecco’s phosphate-buffered saline with Ca2+ and Mg2+ + 0.2 mM sodium metabisulfite + 5.5 mM glucose. Cells were plated in 96-well white, solid-bottomed plates (Greiner Bio-One) and incubated at room temperature for 45 minutes. Coelenterazine H (5 μM; NanoLight Technology, Pinetop, AZ) was added to the cells and incubated for 5 minutes at room temperature protected from light, followed by incubation with the indicated concentration of compounds for 5 minutes in >3% DMSO. Luminescence and fluorescence signals were measured using a PHERAstar plate reader (BMG LABTECH, Cary, NC). BRET ratio was calculated by dividing the fluorescence signal by the luminescence signal for each well and normalized to a percentage of the control BRET ratio with a maximum concentration of dopamine, with zero percent being the BRET ratio produced in the absence of any compound.
Internalization Assay.
Agonist-mediated D1R internalization was assessed using the PathHunter Total GPCR Internalization Assay system (Eurofins DiscoverX, Inc., Fremont, CA), which utilizes an U2OS cell line stably expressing the D1R tagged with a Prolink tag, and an enzyme acceptor tag fused to an endosomal marker protein. Trafficking of the tagged receptor to the endosomes results in complementation of the two enzyme fragments and a subsequent chemiluminescent signal. The assay was conducted according to the manufacturer’s recommendation as described in Conroy et al. (2015).
Statistical Analysis.
Data were analyzed using GraphPad Prism 6.01 (GraphPad Software, Inc., La Jolla, CA). All results are normalized to dopamine control. Maximum efficacies are expressed as mean ± S.E.M. Affinities and potencies are expressed as geometric mean [95% confidence interval (CI)]. Statistical significance was determined using two-tailed Student’s t tests when two groups were compared and one-way analysis of variance (ANOVA) with Bonferroni post-test when multiple groups were compared, with P < 0.05 used as the cutoff for statistical significance.
Results
High-Throughput Screening.
To identify novel, positive allosteric modulators (PAMs) of the dopamine D1 receptor (D1R), we performed a high-throughput screen of the entire Molecular Libraries Probe Production Centers Network small-molecule library, comprising more than 400,000 compounds. For the primary screen, an HEK293 cell line that coexpresses the human D1R along with Gα15 was engineered. G15 has been documented to link many GPCRs, including the D1R, to phospholipase C-mediated Ca2+ signaling pathways (Offermanns and Simon, 1995). We found that DA stimulation of our D1R-G15 stable cell line robustly and reproducibly elevated intracellular Ca2+ levels as measured by a fluorescent readout (data not shown). Although cAMP is the primary signaling pathway for the D1R, we used a Ca2+ assay for the screen owing to robustness of the signal, ease of use in high-throughput screen, and cost value. Potentiation screens were conducted using a two-add, two-read protocol by which a single concentration of library compound was added, followed by the addition of an EC20 concentration of DA with continual reading of the fluorescence signal in real time. Primary hits were identified as compounds that resulted in a potentiation of the Ca2+ fluorescence signal produced by the EC20 concentration of DA without any intrinsic agonist activity. Approximately 1000 primary hits were selected on the basis of efficacy and chemical diversity and were subsequently validated by repetition of the primary Ca2+ assay using full concentration-response curves in D1R-G15-containing cells or parental cells lacking the D1R. Importantly, all hit compounds were validated in an orthogonal, D1R-mediated cAMP assay to confirm potentiation of an endogenous signaling pathway. This hit validation process winnowed the primary hit compounds from approximately 1000 down to 96 lead compounds. These lead compounds underwent extensive counter-screening and triage experiments resulting in the final selection of two structurally distinct lead PAMs, MLS1082 and MLS6585 (Fig. 1), for further study.
Fig. 1.

Structures of lead compounds MLS1082 and MLS6585.
MLS1082 and MLS6585 Potentiate β-Arrestin- and G Protein-Mediated D1R Signaling.
The two lead compounds were initially characterized in β-arrestin recruitment and cAMP accumulation assays to quantify D1R signal potentiation. β-arrestin recruitment to GPCRs is canonically associated with termination of receptor signaling; however, there is now appreciation that GPCRs, including the D1R, can activate downstream signaling pathways via β-arrestin recruitment (Urs et al., 2011). For these experiments, we used the DiscoverX β-arrestin-D1R complementation assay to measure β-arrestin recruitment in response to DA stimulation (Fig. 2). The ability of MLS1082 and MLS6585 to potentiate the DA response was determined by addition of a single, high concentration (50 μM) of each compound. Addition of MLS1082 in the β-arrestin assay increased the potency of DA by ∼7-fold and the efficacy by ∼20% (Fig. 2A). MLS6585 also increased DA’s potency and efficacy by ∼8-fold and ∼34%, respectively (Fig. 2B). Neither compound promoted recruitment of β-arrestin to the D1R in the absence of dopamine, indicating that these compounds have no intrinsic agonist efficacy (Fig. 2, A and B). These data demonstrate that both compounds are potentiators of DA-stimulated D1R mediated β-arrestin recruitment.
Fig. 2.

Stimulation of β-arrestin recruitment and cAMP accumulation by dopamine is potentiated by a single, high concentration of MLS1082 or MLS6585. β-arrestin recruitment and cAMP accumulation were measured following stimulation by the indicated concentrations of dopamine either alone (DA) or in the presence of 50 μM of either MLS1082 or MLS6585. Each PAM was also examined in the absence of DA, as indicated. (A) MLS1082 increased dopamine’s potency for recruiting β-arrestin (EC50 [95% CI]: DA = 1.5 μM [0.66–3.4]; DA + MLS1082 = 0.22 μM [0.09–0.58], P = 0.009), and efficacy (Emax ± S.E.M.: DA = 98.6% ± 2.6%; DA + MLS1082 = 118% ± 2.6%, P = 0.013). (B) MLS6585 increased dopamine’s potency for recruiting β-arrestin (EC50 [95% CI]: DA = 1.24 μM [0.78–2.0]; DA + MLS6585 = 0.15 μM [0.08–0.29], P = 0.0001), and efficacy (Emax ± S.E.M. DA = 100% ± 2.1%; DA + MLS6585 = 134.8% ± 2.6%, P = 0.024). Neither PAM displayed any measurable agonist activity in the β-arrestin recruitment assay. (C) MLS1082 increases dopamine’s potency for cAMP accumulation (EC50 [95% CI]: DA = 0.16 μM [0.08–0.29]; DA + MLS1082 = 0.04 μM [0.02–0.08], P = 0.0006) with no increase in efficacy (Emax ± S.E.M.: DA = 98.7% ± 1.5%; DA + MLS1082 = 109.4% ± 1.6%, P = 0.35). (D) MLS6585 increases dopamine’s potency for cAMP accumulation (EC50 [95% CI]: DA = 0.1 μM [0.05–0.21]; DA + MLS6585 = 0.03 μM [0.01–0.05], P = 0.0001) with no increase in efficacy (Emax ± S.E.M.: DA = 98.7% ± 1.5%; DA + MLS6585 = 99.4% ± 1.6%, P = 0.56). Neither MLS1082 nor MLS6585 demonstrated any agonist activity for cAMP accumulation. Data are displayed as a percentage of the maximum control stimulation seen with dopamine, mean ± S.E.M., n = 5 of experiments run in quadruplicate.
We next examined the ability of the PAMs to potentiate DA-stimulated cAMP accumulation, the primary G protein-mediated signaling mechanism of the D1R. In the presence of a single, high concentration (50 μM) of MLS1082, the potency of DA for stimulating cAMP accumulation was increased by ∼3-fold without a change in efficacy (Fig. 2C). Likewise, addition of MLS6585 (50 μM) to the assay increased the potency of DA by ∼6-fold with no change in efficacy (Fig. 2D). The lack of efficacy potentiation (compared with the β-arrestin recruitment assays) may be attributable to a ceiling effect in the cAMP assays. To determine if the PAMs have any intrinsic agonist activity in the cAMP assay, they were examined in the absence of DA, but no measurable agonist activity was observed with either compound (Fig. 2, C and D). These findings indicate that both compounds are PAMs at D1R-mediated G protein signaling as detected using cAMP accumulation.
Since both compounds showed potentiation of cAMP accumulation, we sought to ensure that this was occurring at the level of the D1R. To address this, we investigated the effects of the PAMs on activation of adenylyl cyclase by forskolin, which is a direct activator of this enzyme, in cells lacking the D1R (Supplemental Fig. 1). Stimulation via forskolin results in a robust cAMP accumulation; however, neither MLS1082 (50 μM) nor MLS6585 (50 μM) potentiated either the potency or efficacy of forskolin. Further, neither PAM demonstrated any measurable cAMP accumulation on its own. Together, these data indicate that, in addition to β-arrestin potentiation, MLS1082 and MLS6585 also potentiate G protein-mediated signaling, and this occurs at the level of the D1R.
PAMs Increase Dopamine’s Potency and Efficacy for D1R Signaling.
PAMS can act by altering the signaling potency or efficacy of an endogenous ligand, or by altering the affinity of the endogenous ligand to bind to its receptor, or both. To understand the mechanisms underlying the activity of these compounds, we used radioligand binding assays to measure DA’s ability to compete with a radiolabeled antagonist ([3H]-SCH23390) for binding to the orthosteric site of the D1R. Initially, however, we determined if either PAM had any direct effects on [3H]-SCH23390 binding. Figure 3A shows that MLS1082 had minimal effects on [3H]-SCH23390, decreasing binding by ∼17% at a high (50 μM) concentration. However, MLS6585 had a greater effect, decreasing [3H]-SCH23390 binding by ∼66% at a 50 μM concentration. Notably, both MLS1082 and MLS6585 lack a positively charged nitrogen at physiologic pH, a critical feature of all orthosteric monoaminergic receptor ligands (Michino et al., 2015), suggesting that they probably would not be orthosteric ligands of the D1R. Given these observations, and consideration of the allosteric ternary complex model (May et al., 2007), MLS6585 may function as a negative allosteric modulator (NAM) of [3H]-SCH23390 binding, perhaps by stabilizing the active state of the D1R, which favors agonist binding versus antagonist binding (Canals et al., 2012). Notably, allosteric modulators can exhibit “probe dependency,” whereby they may affect the binding and/or efficacy of diverse orthosteric ligands in different ways (Christopoulos, 2014).
Fig. 3.

MLS1082 and MLS6585 increase dopamine affinity as measured using [3H]-SCH23390 binding assays. Data are represented as a percentage of the control specific [3H]-SCH23390 binding in the absence of competitor, mean ± S.E.M., N = 3. (A) MLS1082 decreases [3H]-SCH23390 binding by 17.2% ± 3.4% at the highest concentration tested (50 μM); MLS6585 had a greater effect, decreasing [3H]-SCH23390 binding by 65.8% ± 4.4% at 50 μM. Unlabeled SCH23990 was used a positive control. (B) Increasing concentrations of MLS1082 significantly shifted the dopamine competition curve by ∼3-fold leftward indicating an increase in dopamine affinity in the presence of MLS1082 (Ki [95% CI]; DA Ki = 0.71 μM [0.49–1.2]; + 30 μM MLS1082 = 0.26 μM [0.08–0.84], P = 0.03). (C) Increasing concentrations of MLS6585 significantly shifted the dopamine competition curve ∼7-fold leftward (DA Ki + 30 μM MLS6585 = 0.13 μM [0.04–0.45], P = 0.03. Higher concentrations of MLS6585 reduce [3H]-SCH23390 binding as shown in (A).
We next measured DA’s ability to displace [3H]-SCH23390 binding in the presence of increasing concentrations of MLS1082 or MLS6585 (Fig. 3, B and C). DA alone fully competed with [3H]-SCH23390 with a Ki of 0.7 ± 0.04 μM. However, in the presence of increasing concentrations of MLS1082, DA’s affinity for the D1R increased by ∼3-fold with no effect on maximum [3H]-SCH23390 binding (Fig. 3B). MLS6585 showed a larger potentiation, increasing DA’s affinity ∼7-fold. As discussed above, we also observed a decrease in [3H]-SCH23390 binding in the DA + MLS6585 binding conditions owing to the negative interaction of MLS6585 with the SCH23390 scaffold (see Fig. 3A). Curve-shift data were fit to the allosteric ternary complex model (May et al., 2007) to estimate the affinity of each PAM for the D1R in the absence of the endogenous ligand (Kb) as well as the binding cooperativity between the PAM and dopamine (α), with α < 1 indicating negative cooperativity, α = 1 consistent with neutral cooperativity, and α > 1 indicating positive cooperativity. For MLS1082, the model estimated an affinity for the D1R in the submicromolar range (pKb = 6.27 ± 0.19, Kb = 0.54 μM) and an α of 3.09 (logα = 0.49 ± 0.07) that equates to the maximal fold shift in DA affinity that we observed. MLS6585 had a lower affinity compared with MLS1082 but still in the low micromolar range (pKb = 5.27 ± 0.03, Kb = 5.37 μM) but a greater α of 6.61 (logα = 0.82 ± 0.11). These data suggest that MLS6585 may have a slightly greater effect on DA affinity for the D1R than MLS1082, suggesting that the two PAMs may be acting via separate mechanisms of potentiation.
The β-arrestin recruitment assay was employed to determine the effect of the PAMs on the efficacy of dopamine. Here, β-arrestin recruitment to the D1R was determined in the presence of an increasing concentration of each PAM, ranging from 0.1 to 100 μM (Fig. 4). Increasing concentrations of MLS1082 or MLS6585 progressively shifted the DA dose-response curve to the left and increased maximum efficacy in a concentration-dependent manner. These data were used in conjunction with the outputs from the allosteric ternary complex model described above to assess the effect of the PAMs on dopamine’s efficacy using an operational model of allosterism (Leach et al., 2007). In addition to estimates of modulator affinity (Kb) and cooperativity with dopamine affinity (α), this model also allows estimation of a factor, β, as a measure of the modulatory effect of a compound upon the efficacy of the orthosteric agonist, with β values <1 indicating negative modulation, β = 1 indicating neutral modulation, and β values >1 indicating positive modulation. The model reported very similar affinities of the two PAMs for the D1R. For MLS1082, Kb was determined to be 0.46 μM (pKb = 6.34 ± 0.08) and for MLS6585, the analysis reported a Kb of 5.4 μM (pKb = 5.27 ± 0.24). By fixing values of cooperativity with dopamine affinity (α) to those determined in the binding studies, we determined that both PAMs had β factors greater than one, indicating a positive modulatory effect on dopamine efficacy [(logβ (β)): MLS1082 = 0.41 ± 0.06 (2.57); MLS6585 = 0.32 ± 0.1 (2.09)]. Taken together, this series of experiments indicate the PAMs potentiate the affinity and efficacy of dopamine at the D1R, with MLS6585 having a greater effect on dopamine’s affinity than MLS1082. Both compounds have affinities for the D1R in the low micromolar range, with MLS1082 having a slightly higher affinity than MLS6585. Neither compound displayed appreciable allosteric agonism in this assay.
Fig. 4.

MLS1082 and MLS6585 increase dopamine’s potency and efficacy as measured in β-arrestin recruitment assays. Increasing concentrations of MLS1082 (A) or MLS6585 (B) were used to potentiate dopamine-stimulated β-arrestin recruitment. The maximum shift potentiated by MLS1082 was a 4.96 ± 0.29-fold increase in the EC50 for dopamine (P < 0.001 vs. DA control, paired Student’s t test) and an Emax value of 121.7% ± 12.9% (P = 0.017 vs. DA control, paired Student’s t test). MLS6585 potentiated the EC50 for dopamine by 9.7 ± 3.4-fold (P = 0.045 vs. DA control, paired Student’s t test) and showed an Emax increase of 122.4% ± 11.4% (P = 0.0093 vs. DA control, paired Student’s t test). Data are displayed as a percentage of the maximum control stimulation seen with dopamine, mean ± S.E.M., n = 6–8 of experiments run in quadruplicate. Insets illustrate the concentration-dependence effects of each PAM on the DA EC50 and Emax values, showing that the PAMs appear more potent at increasing DA efficacy than potency for this signaling output.
MLS1082 and MLS6585 Potentiate DA-Induced D1R Internalization.
Given that the PAM compounds potentiated DA-induced recruitment of β-arrestin to the D1R, we thought that it would be informative to examine receptor internalization, the natural sequela of β-arrestin-GPCR interactions. For this series of experiments, we used U2OS cells that are stably transfected with both the D1R fused to a small fragment of β-galactosidase and a complementing fragment of β-galactosidase that is fused to an endosomal marker protein. When the receptor is internalized into endosomes, β-galactosidase is complemented and provides a luminescent signal upon addition of substrate (Conroy et al., 2015). Figure 5 shows that DA produces a robust internalization of the D1R when added to the cells. Cotreatment with MLS1082 results in a 6-fold increase in the potency of DA for promoting D1R internalization as well as a 20% increase in the maximum response (Fig. 5). Cotreatment with MLS6585 produced similar results, increasing the potency of DA by 6-fold, although no increase in Emax was observed (Fig. 5). These results are largely in agreement with those observed for β-arrestin recruitment and cAMP accumulation and further support the notion that MLS1082 and MLS6585 are PAMs of the D1R.
Fig. 5.

MLS1082 and MLS6585 potentiate dopamine-induced D1R internalization. Receptor internalization was measured using the DiscoverX GPCR internalization assay as described in Materials and Methods. Cells were treated with the indicated concentrations of dopamine for 3 hours in the absence or presence of 50 μM MLS1082 (+ 50 μM 1082), or 50 μM MLS6585 (+ 50 μM 6585). Both MLS1082 and MLS6585 potentiated DA’s potency for inducing receptor internalization (EC50 [95% CI]: DA = 2.79 μM [1.4–5.6], DA + MLS1082 = 0.53 μM [0.16–1.8], P = 0.004), DA + MLS6585 = 0.46 μM [0.15–1.45], P = 0.04). Further, MLS1082 increased DA’s efficacy for internalization (P = 0.04), but MLS6585 showed no potentiation of efficacy (Emax ± S.E.M.: DA = 97.4% ± 4.7%, DA + MLS1082 = 113.5% ± 2.8%, DA + MLS6585 = 97.2% ± 3.0%). Statistical comparisons via Student’s t test, n = 5.
MLS1082 and MLS6585 Potentiate the Activity of Synthetic Agonists.
We next wanted to investigate the ability of the PAM compounds to potentiate the activity of D1R agonists other than DA. Initially, we tested dihydrexidine (DHX), a well characterized agonist with high efficacy at the D1R (Lovenberg et al., 1989; Mottola et al., 1992). Figure 6 shows that DHX stimulates robust recruitment of β-arrestin to the D1R. Further, MLS1082 promotes a 3-fold shift in the EC50 for DHX with a 30% increase in DHX’s efficacy for this response. Likewise, the addition of MLS6585 to the assay increased the potency of DHX by 4-fold with a 30% increase in the maximum response. These results indicate that, in addition to exhibiting positive allostery with the endogenous agonist dopamine, MLS1082 and MLS6585 can also potentiate the activity of nonendogenous synthetic agonists.
Fig. 6.

MLS1082 and MLS6585 increase the efficacy and potency of the D1R agonist dihydrexidine. β-arrestin recruitment was measured following stimulation by the indicated concentrations of dihydrexidine in the absence or presence of 50 μM MLS1082 (+ 50 μM 1082) or MLS6585 (+ 50 μM 6585). DA was run as a control in every experiment and the data were plotted as the percentage of the maximum DA response observed. Both MLS1082 and MLS6585 increased the efficacy and potency of dihydrexidine: (EC50 [95% CI]) dihydrexidine = 73.3 nM [42.8–125.2], dihydrexidine + MLS1082 = 20.9 nM [9.7–45.5], P < 0.0001, dihydrexidine + MLS6585 = 18.6 nM [11.3–30.6], P < 0.0001; (Emax ± S.E.M.) dihydrexidine = 70.5% ± 2.5%; dihydrexidine + MLS1082 = 95.3% ± 3.8%, P < 0.001; dihydrexidine + MLS6585 = 96.9% ± 3.2%, P < 0.001. Data are displayed as a percentage of the maximum control stimulation seen with dopamine, mean ± S.E.M., and statistical comparisons are via paired two-tailed Student’s t test, n = 5.
When examining efficacy potentiation of a full agonist it may be possible to underestimate the degree of potentiation owing to ceiling effects from any given assay. However, potentiation of partial agonists may provide a larger window to examine efficacy potentiation as, by definition, partial agonists do not maximally activate the signaling output. To investigate this, we first used fenoldopam (SKF82526) and apomorphine, partial agonists with low to moderate efficacy to stimulate β-arrestin recruitment in the presence or absence of 50 μM MLS1082 or MLS6585 (Fig. 7, A and B). In the absence of any PAM, the Emax of fenoldopam was 48% of the DA Emax. MLS1082 increased the Emax of fenoldopam to 70%, whereas MLS6585 increased the fenoldopam Emax to 87% (Fig. 7A). Apomorphine alone exhibited an Emax of 29%, whereas in the presence of MLS1082 or MLS6585, the Emax values increased to 54% and 81%, respectively (Fig. 7B). These efficacy increases are significantly higher than the increases seen with dopamine (Figs. 2 and 4). Importantly, like their effects on dopamine, the PAMs caused an increase in the potencies of fenoldopam (MLS1082: 5-fold, MLS6585: 4-fold) (Fig. 7A) and of apomorphine (MLS1082: 7-fold, MLS6585: 4-fold) (Fig. 7B).
Fig. 7.

MLS1082 and MLS6585 potentiate the activity of D1R partial agonists. β-arrestin recruitment or cAMP assays were performed in concentration-response curve format using known partial agonists of the D1R in either the presence or absence of the indicated PAM compounds at 50 μM concentration. DA was run as a control in every experiment and the data were plotted as the percentage of the maximum DA response observed. (A) MLS1082 and MLS6585 increased both the efficacy and potency of the partial agonist fenoldopam. Emax ± S.E.M. (% DA response): fenoldopam = 47.7% ± 1.6%; fenoldopam + MLS1082 = 70.8% ± 2.1%, P < 0.05; fenoldopam + MLS6585 = 87.4% ± 2.7%, P < 0.0001. EC50 [95% CI]: fenoldopam = 37.9 nM [22.5–63.8]; fenoldopam + MLS1082 = 7.5 nM [4.7–11.9], P < 0.0001; fenoldopam ± MLS6585 = 8.6 nM [5.3–13.9], P = 0.0002. (B) MLS1082 and MLS6585 increased both the efficacy and potency of the partial agonist apomorphine. Emax ± S.E.M.: apomorphine = 28.6% ± 2.2%; apomorphine + MLS1082 = 54.3% ± 3.2%, P < 0.01; apomorphine + MLS6585 = 80.8% ± 2.7%, P < 0.0001. EC50 [95% CI]: apomorphine = 0.1 μM [0.04–0.26]; apomorphine + MLS1082 = 0.014 μM [0.007–0.027], P = 0.0006; apomorphine + 6585 = 0.024 μM [0.015–0.036], P = 0.001. (C) The G protein-biased agonist SKF38393 exhibited no measurable agonist activity for β-arrestin recruitment but gained efficacy upon concurrent treatment with the PAM compounds. Emax ± S.E.M.: SKF38393 + MLS1082 = 24.1% ± 1.3%; SKF38393 + MLS6585: 28.7% ± 1.3%. EC50 [95% CI]: SKF38393 + MLS1082 = 0.12 μM [0.05–0.29]; SKF38393 + MLS6585 = 0.14 μM [0.08–0.27]. (D) MLS1082 and MLS6585 potentiated the efficacy of SKF77434-stimulated cAMP accumulation. Emax ± S.E.M.: SKF77434 = 24.7% ± 2.0%; SKF77434 + MLS1082 = 56.8% ± 2.9%, P < 0.0001; SKF7743 + MLS6585 = 48.3% ± 2.6%, P < 0.01. Neither PAM affected SKF77434 potency, however. EC50 [95% CI]: SKF77437 = 0.06 μM [0.01–0.03], SKF77434 + MLS1082 = 0.03 μM [0.01–0.07], SKF77434 + MLS6585 = 0.02 μM [0.01–0.05]. Statistical comparisons were determined for Emax values using one-way ANOVA testing, and Student’s t test for potency values, n = 6–8.
Second, we examined the G protein-biased agonist SKF38393 (Conroy et al., 2015) (Fig. 7C). As we previously observed (Conroy et al., 2015), we were not able to detect significant β-arrestin recruitment by this compound. However, when cotreated with our PAM compounds, SKF38393 gained efficacy, stimulating β-arrestin recruitment as a weak partial agonist (Fig. 7C). We interpret these results as the PAMs promoting an increase in SKF38393 activity that is below the threshold of detection in our assay as opposed to the PAMs imparting efficacy to a compound completely lacking in agonist activity. Last, we examined SKF77434, a compound with low efficacy for stimulating D1R-mediated cAMP accumulation (Conroy et al., 2015) (Fig. 7D). In the absence of the PAMs, SKF77434 stimulated cAMP accumulation to about 25% of the maximum DA response. MLS1082 and MLS6585 potentiated the maximum efficacy for SKF77434 stimulation of cAMP accumulation to 56% and 48%, respectively. Although MLS1082 and MLS6585 tended to increase the EC50 for SKF77434, this did not reach statistical significance (Fig. 7D). Notably, the PAM-induced increase in efficacy for the partial agonists appears significantly larger than that for DA. Taken together, these results show that MLS1082 and MLS6585 are effective potentiators of agonist efficacy at the D1R.
Receptor Selectivity of MLS1082 and MLS6585.
We next evaluated MLS1082 and MLS6585 for potential activity at other dopamine receptor subtypes and one other catecholamine receptor. Interestingly, we found that both compounds exhibit PAM activity at the closely related D5R, which, along with the D1R, compose the D1-like receptor subfamily. Supplemental Fig. 2 and Supplemental Table 1 show that both MLS1082 and MLS6585 potentiate the potency and efficacy of DA-induced recruitment of β-arrestin to the D5R. Neither compound, however, exhibits intrinsic agonist efficacy. In contrast, using the β-arrestin recruitment assay, we found that MLS1082 and MLS6585 were completely devoid of PAM activity at the D2R, D3R, D4R, or the β2-adrenergic receptor (Supplemental Fig. 3; Supplemental Table 1). These data suggest that MLS1082 and MLS6585 have selectivity for the D1-like dopamine receptor family.
MLS1082 and MLS6585 Act at Different Allosteric Sites on the D1R.
Because the two PAMS described in this study have structural and activity differences, we asked if the compounds might act at different binding sites on the D1R. As an initial approach to this question, we repeated the β-arrestin recruitment and cAMP accumulation assays described above and characterized the ability of the PAMs to potentiate the DA response alone or in combination. We hypothesized that additive results from using the PAMs in combination support the notion that separate binding sites may exist, whereas nonadditive results could suggest that the compounds bind to the same site or that there is a ceiling effect such that maximum potentiation is achieved by either compound. β-arrestin recruitment (Fig. 8A) or cAMP accumulation (Fig. 8B) were stimulated with increasing concentrations of DA with or without maximally effective concentrations (50 μM) of MLS1082, MLS6585, or MLS1082 and MLS6585 in combination. In the β-arrestin recruitment experiments, MLS1082 or MLS6585 increased the potency for DA by 4- and 7-fold respectively (Fig. 8A), whereas the combination of MLS1082 and MLS6585 increased the potency for DA by 43-fold, which was 11- and 6-fold greater than the EC50 shifts seen with either PAM alone. Likewise, in the cAMP accumulation assays, MLS1082 or MLS6585 increased the potency for DA by 3- and 4-fold respectively (Fig. 8A), whereas the combination of MLS1082 and MLS6585 increased the potency for DA by 14-fold, which was 5- and 4-fold greater than the EC50 shifts seen with MLS1082 or MLS6585 alone, respectively. Taken together, these data suggest that the MLS1082 and MLS6585 probably act at two separate allosteric sites on the D1R to potentiate signaling activity by dopamine.
Fig. 8.

MLS1082 and MLS6585 show additive potentiation of β-arrestin recruitment and cAMP accumulation. β-arrestin and cAMP assays were performed as described in Materials and Methods. Potentiation of the DA response with either 50 μM MLS1082 or 50 μM MLS6585, or both, was performed as described in Figs. 2 and 4. (A) MLS1082 or MLS6585 potentiated the potency (EC50) for DA-stimulated β-arrestin recruitment by 4- and 7-fold, respectively (EC50 [95% CI]: DA = 2.6 μM [1.3–5.0]; DA + MLS1082 = 0.68 μM [0.35–1.3], P = 0.011; DA + MLS6585 = 0.38 μM [0.19–0.74], P = 0.003; fold shift EC50 vs. DA control, MLS1082: P = 0.029, MLS658: P = 0.016). The two PAM combination (50 μM MLS1082 + 50 μM MLS6585) increased the potency by 43-fold vs. DA alone and by 11- and 6-fold vs. DA+MLS1082 or DA+MLS6585, respectively (EC50 [95% CI]: DA + MLS1082 + MLS6585 = 0.05 μM [0.02–0.15], P = 0.0022; MLS1082 alone vs. combo P = 0.005; MLS6585 alone vs. combo P = 0.008). (B) Both MLS1082 and MLS6585 potentiated the potency for DA-stimulated cAMP accumulation by 3- and 4-fold, respectively (EC50 [95% CI]: DA = 0.13 μM [0.07–0.25], DA + MLS1082 = 0.05 μM [0.01–0.2], P = 0.035, DA + MLS6585 = 0.031 μM [0.017–0.06], P = 0.005). Combination of the two PAMS potentiated the potency of dopamine by 14-fold with no effect on efficacy (EC50 [95% CI]: DA + MLS1082 + MLS6585 = 0.0092 μM [0.0043–0.02], P = 0.0002 vs. control, 1082 vs. Combo P = 0.008; 6585 vs. Combo P = 0.006). Statistical comparisons via Student’s t test, n = 3–5.
Similar experiments were performed using a compound (“Compound B”) previously described as exhibiting D1R PAM activity (Lewis et al., 2015) (Fig. 9A). For these experiments, DA-stimulated β-arrestin recruitment was measured with and without maximally effective concentrations of MLS1082, MLS6585, Compound B, or in combination. When tested alone, MLS1082 or Compound B increased the potency for DA by 6-fold, whereas the combination of MLS1082 and Compound B increased the potency for DA by 8-fold, which was not significantly different from the potentiation observed using each compound alone (Fig. 9B). In contrast, MLS6585 or Compound B increased the potency for DA by 11-fold and 6-fold, respectively, whereas the combination of MLS6585 and Compound B increased DA’s potency by 30-fold, which was 3- and 5-fold greater than the EC50 shifts seen with MLS6585 or Compound B alone, respectively (Fig. 9B). These results suggest that MLS1082 and Compound B share a common allosteric site on the D1R, which is different from the site that is modulated by MLS6585.
Fig. 9.

Combination experiments suggest that MLS1082 and Compound B act at the same site on D1R, which is separate from that of MLS6585. β-arrestin recruitment was measured following stimulation with dopamine in the absence (DA) or in the presence of 50 μM MLS1082 (+ 50 μM 1082), 50 μM MLS6585 (+ 50 μM 6585), 100 μM Compound B (+ 100 μM Cmpd B), or a combination of the three compounds. (A) Structure of Compound B. (B) MLS1082 and Compound B both potentiate DA’s potency (EC50 [95% CI]: DA = 4.19 μM [2.4–7.5]; DA + MLS1082 = 0.68 μM [0.36–1.3], P = 0.004; DA + Compound B = 0.56 μM [0.22–1.4], P = 0.01). Addition of MLS1082 and Compound B together caused the same level of potentiation as either compound alone (EC50 [95% CI]: DA + MLS1082 + Compound B = 0.5 μM [0.29–0.86]). (C) MLS6585 and Compound B both potentiated DA’s potency for β-arrestin recruitment (EC50 [95% CI]: DA = 4.19 μM [2.4–7.5]; DA + MLS6585 = 0.39 μM [0.27–0.54], P = 0.0002); DA + Compound B = 0.56 μM [0.22–1.4], P = 0.01). Addition of MLS6585 and Compound B together resulted in a greater potentiation of DA’s potency than either compound alone (EC50 ± S.E.M.: DA + MLS6585 + Compound B = 0.09 μM [0.02–0.46], P = 0.004). Statistical comparisons via paired two-tailed Student’s t test, n = 5.
R130Q D1R Point Mutation Selectively Abolishes MLS1082 Activity.
Compound B was initially described as exhibiting PAM activity for the human but not rat D1R (Lewis et al., 2015). In that the study, the authors noted that there was a sequence difference at position 130 of the D1R—arginine in the human but glutamine in the rat. Interestingly, mutating the rat D1R sequence to match that of the human (Q130R) resulted in a gain of PAM activity of Compound B for the rat D1R. This further lead to the hypothesis that residue 130 is involved in Compound B binding (Lewis et al., 2015). We used this information to determine if residue R130 is involved in the activity of either of our PAMs. Using mutagenesis, we changed R130 in the human D1R to Q and measured the ability of our PAMs to potentiate DA-stimulated responses. For these experiments, we used BRET assays to measure DA-stimulated β-arrestin recruitment or Gαs engagement with the D1R. No differences between wild-type and R130Q D1R were observed for the DA control responses in either assay (Fig. 10). As in previous results, both MLS1082 and MLS6585 potentiated DA’s potency for stimulating β-arrestin recruitment to the wild-type D1R by 4-fold and 5-fold, respectively (Fig. 10A). MLS1082 and MLS6585 also increased DA’s efficacy, although this effect was not as pronounced as that seen with the DiscoverX β-arrestin assay, suggesting that the BRET assay may be more efficiently coupled. In contrast to the wild-type D1R, MLS1082 was without effect using the R130Q D1R, whereas MLS6585 still potentiated DA’s potency for this mutant receptor by 5-fold—identical to that of the wild-type D1R (Fig. 10B). With respect to the D1R-Gs BRET assays, both MLS1082 and MLS6585 potentiated the potency of DA for stimulating this response by 3- and 5-fold, respectively using the wild-type D1R (Fig. 10C). In contrast, using the R130Q mutant D1R, MLS1082 was inactive at potentiating DA-stimulation of D1R-Gs interactions, whereas MLS6585 enhanced DA’s potency by 6-fold (Fig. 10D). These results suggest that MLS1082 and MLS6585 are potentiating the D1R through two separate sites of action and that amino acid residue R130 may be involved the PAM activity of MLS1082 but not MLS6585.
Fig. 10.

R130Q mutation abolishes MLS1082 but not MLS6585 PAM activity. Dopamine-stimulated β-arrestin recruitment and G protein (Gαs) engagement were measured using BRET assays as described in Materials and Methods. Briefly, cells were transfected with either the wild-type D1R or the R130Q mutant along with the indicated biosensor. Cells were then stimulated with the indicated concentrations of dopamine alone (DA) or in the presence of 50 μM MLS1082 (+ 50 μM 1082) or 50 μM MLS6585 (+ 50 μM 6585). (A) Both MLS1082 and MLS6585 potentiated DA’s potency for β-arrestin recruitment to the wild-type D1R (EC50 [95% CI]: DA = 1.76 μM [0.69–4.5]; DA + MLS1082 = 0.45 μM [0.2–1.0], P < 0.001; DA + MLS6585 = 0.43 μM [0.21–0.86], P < 0.001). Further, both MLS1082 and MLS6585 increased DA’s efficacy (Emax ± S.E.M.: DA = 99.9% ± 1.2%; DA + MLS1082 = 132.7% ± 2.2%; DA + MLS6585 = 114.5% ± 1.6%, P < 0.01). (B) With the mutant R130Q receptor, MLS6585, but not MLS1082, potentiated DA’s potency (EC50 [95% CI]: DA = 1.67 μM [0.79–3.5]; DA + MLS1082 = 1.23 μM [0.44–3.5]; DA + MLS6585 = 0.38 μM [0.19–0.75], P < 0.0001). Further, MLS1082 did not potentiate DA’s efficacy for activating the R130Q mutant (Emax ± S.E.M.: DA = 98.4% ± 5.2%; DA + MLS1082 = 105.4% ± 2.9%); however, MLS6585 did potentiate the Emax (DA + MLS6585 = 111.3 ± 3.1, P < 0.03). (C) With the wild-type receptor, both MLS1082 and MLS6585 enhanced DA’s potency for stimulating D1R-Gs interactions (EC50 [95% CI]: DA = 0.37 μM [0.24–0.57]; DA + MLS1082 = 0.12 μM [0.09–0.16], P = 0.0001; DA + MLS6585 (0.07 μM [0.04–0.12], P = 0.001). MLS1082 also promoted a measurable increase in DA efficacy, whereas MLS6585 did not (Emax ± S.E.M.: DA = 100.3% ± 2.1%; DA+MLS1082 = 109.4% ± 2.2%; DA+MLS6585 = 101.7% ± 3.4%). (D) With the mutant R130Q receptor, MLS6585, but not MLS1082, enhanced DA’s potency (EC50 [95% CI]: DA = 0.39 μM [0.25–0.6]; DA + MLS1082 = 0.23 μM [0.17–0.31]; DA + MLS6585 = 0.069 μM [0.05–0.11], P < 0.0001). Further, neither compound increased DA’s efficacy at the R130Q receptor (Emax ± S.E.M.: DA = 100.3% ± 49%; DA+MLS1082 = 98.2% ± 4.8%; DA+MLS6585 = 107.3% ± 5.3%). Statistical comparisons via paired two-tailed Student’s t test, and one-way ANOVA; n = 5 or 6.
Discussion
Using high-throughput screening, we have identified two positive allosteric modulators of the D1 dopamine receptor. These PAMs have dissimilar chemical structures, although neither possess a nitrogen atom that is predicted to be protonated at physiologic pH. This is a hallmark of all positively charged dopaminergic ligands, which interact with a highly conserved Asp residue [Asp3.32 in the Ballesteros-Weinstein numbering system (Ballesteros and Weinstein, 1995)] present in the orthosteric binding sites of biogenic amine receptors (Michino et al., 2015). Thus, these PAMs probably will not bind to the orthosteric site of the D1R. Both PAMs were found to potentiate agonist stimulation of two signaling arms of the D1R, namely cAMP accumulation and β-arrestin recruitment. Although no signaling bias, at least qualitatively, was observed using the PAM compounds, other D1R signaling pathways (p-ERK, etc.) remain to the be examined. Both PAMs were also found to potentiate the activity of dihydrexidine, a well characterized synthetic D1R agonist with high functional efficacy.
We found that both PAMs also potentiated DA-induced D1R internalization, which might be expected given the observed enhancement of β-arrestin recruitment to the D1R. Although both PAMs increased the potency for DA-stimulation of cAMP accumulation and β-arrestin recruitment, each PAM also appeared to increase the efficacy for DA in the β-arrestin assays but not in the cAMP or Gs BRET assays. This may be an inherent property of the PAMs or, more probably, is owing to a ceiling effect in the G protein-coupled assays, which are more amplified than the β-arrestin assays. Notably, neither PAM exhibited agonist efficacy at either cAMP accumulation or β-arrestin recruitment, appearing instead to exhibit pure PAM properties for these two signaling outputs.
To investigate the mechanism(s) of the PAM compounds, we first examined their ability to enhance DA binding to the D1R using radioligand binding competition assays. Interestingly, at high concentrations MLS6585, but not MLS1082, partially inhibited the binding of the orthosteric antagonist [3H]-SCH23390, suggesting weak negative cooperativity with this radioligand (Fig. 3A). As previously described, allosteric modulators can exhibit probe-dependency for their modulatory effects on ligand-receptor interactions (Christopoulos, 2014; Gentry et al., 2015). Using curve-shift analyses, MLS1082 was found to promote a dose-dependent 3-fold potentiation in the ability of DA to compete for [3H]-SCH23390 binding, whereas MLS6585 promoted a 7-fold increase in affinity (Fig. 3, B and C). Fitting the curve-shift data to the allosteric ternary complex model (May et al., 2007) revealed a Kb of 0.54 μM for MLS1082 and an α factor of 3.09, whereas MLS6585 exhibited a Kb of 5.37 μM but a greater α factor of 6.61. Thus, MLS1082 appeared to possess greater potency but less efficacy than MLS6585 for increasing DA affinity for the D1R.
Functional curve-shift analyses were also performed with the β-arrestin recruitment assay and analyzed using the operational model of allosterism and agonism (Leach et al., 2007). In these experiments, both PAMs produced a dose-dependent increase in DA potency (4- and 7-fold) and efficacy (20%–25%) (Fig. 4). These experiments yielded very similar D1R affinities of the two PAMs as was determined in the binding assays. For MLS1082, the Kb was 0.46 μM, and for MLS6585, the Kb was 5.4 μM. Both PAMs exhibited β factors greater than one, indicating a positive modulatory effect upon dopamine efficacy. For MLS1082 the β factor was 2.57, whereas for MLS6585 the β factor was 2.09. Notably, although the ability of these PAMs to potentiate DA activity at the D1R is not extraordinarily high (potency shift <10-fold), their PAM activity may actually be close to that desired for a clinical therapeutic. As discussed above, cognitive impairment in disease states has been associated with low levels of D1R activity in the prefrontal cortex; however, too much D1R stimulation (e.g., associated with stress) can lead to decreased cognition (an inverted U relationship) (Goldman-Rakic et al., 2004; Arnsten and Dudley, 2005; Arnsten and Li, 2005; Vijayraghavan et al., 2007). Thus, the desired attribute for a procognitive therapeutic might be one that will moderately potentiate D1R activity without producing an overshoot that could result in decreased cognition. Further experiments in animals, and eventually in man, will be required to fully test this hypothesis.
Interestingly, when we examined the ability of the PAM compounds to potentiate the activity of D1R partial agonists for stimulating β-arrestin recruitment or cAMP accumulation, we observed a similar shift in agonist potency compared with DA; however the increase in efficacy was much larger. Similar results were observed by Livingston and Traynor (2014), in which PAMs of the μ-opioid receptor increased the efficacy of agonists in a way that was correlated with their intrinsic activity. Overall, these results are in agreement with a two-state model of receptor activation, in which the degree of positive cooperativity exhibited by an allosteric modulator is correlated with the coupling efficiency of the orthosteric ligand and signaling output (Canals et al., 2012).
Given the diverse structures of MLS1082 and MLS6585, we wondered whether they might be binding to the same or different allosteric sites on the D1R. As an initial assessment, we performed additivity experiments using maximally effective concentrations of each PAM. Strikingly, we found that the potentiating effects of both PAMs were additive in nature, which was true for both cAMP accumulation and β-arrestin recruitment assays. Such results are difficult to explain without invoking the existence of two allosteric sites on the D1R that independently mediate the actions of these diverse PAMS. In such a model, simultaneous occupancy of the two allosteric sites on the receptor can promote an even greater stabilization of the active signaling state(s) of the D1R than that achievable by either PAM alone.
As discussed above, a D1R PAM, Compound B, was recently described that is active in potentiating agonist stimulation of primate D1Rs but not rodent D1Rs (Lewis et al., 2015). Using a human/rat chimeric receptor approach coupled with mutagenesis, these authors identified residues within the second intracellular loop (ICL2) region that were necessary for the activity of Compound B and responsible for the species differences identified. Specifically, residue R130 was delineated in the human receptor that is a glutamine residue in the rodent receptor. Notably, changing the rodent sequence to human (Q130R) in the rat D1R imparted PAM activity to Compound B (Lewis et al., 2015). Interestingly, we also found that MLS1082 lacked PAM activity at the rat D1R, whereas MLS6585 was equally effective as a PAM in both the human and rat D1Rs (data not shown). These results suggested that MLS1082 and Compound B might share a similar binding site or require similar residues for their activity. Perhaps not surprisingly, we found that the PAM activities of MLS1082 and Compound B were nonadditive, whereas those of MLS6585 and Compound B were completely additive. Further, analysis of a human R130Q mutant D1R showed that this mutation rendered MLS1082 inactive, whereas the PAM activity of MLS6585 was not affected. Taken together, these results support the hypothesis that MLS1082 and Compound B function through the same allosteric site on the D1R, which probably involves R130 within the ICL2 of the receptor, and that this differs from the site mediating the effects of MLS6585.
Notably, we found that both MLS1082 and MLS6585 exerted PAM activity at the closely related D5R. Although the site mediating the effects of MLS6585 remains unclear, it is interesting to note that the D5R possesses an arginine residue (R147) in the ICL2 region that is aligned with the R130 residue in the D1R that may compose the binding site for MLS1082. Thus, the D1R and the D5R may possess a conserved allosteric site within the ILC2 region, at least for the MLS1082 and Compound B scaffolds. Such a finding is not without precedent, as Livingston et al. (2018) have recently provided evidence for a conserved allosteric site across all three traditional opioid receptor subtypes (μ, δ, and κ). From a therapeutic standpoint, potentiation of both the D1R and D5R may prove advantageous, as both subtypes may contribute to the enhanced cognition seen through stimulation of D1-like receptors in the prefrontal cortex (Cai and Arnsten, 1997; Castner and Goldman-Rakic, 2004; Goldman-Rakic et al., 2004; Nakako et al., 2013).
Interestingly, another structurally distinct PAM for the D1R has recently been reported, referred to as DETQ (Svensson et al., 2017; Bruns et al., 2018). Like Compound B and MLS1082, DETQ is inactive as a PAM at rodent D1Rs but is active in potentiating agonist stimulation of the human D1R. Although this group has not reported additivity or mutational experiments, it would not be surprising to find that MLS1082, Compound B, and DETQ all function through the same allosteric site on the D1R. Interestingly, although Compound B and DETQ each possess a dichloro-substituted phenyl ring in their structure (Lewis et al., 2015; Svensson et al., 2017), this moiety is absent in MLS1082. Obviously, further structure-activity-relationship information will be needed as well as a clear delineation of the allosteric binding site for the MLS1082/Compound B/DETQ series of scaffolds to understand how these compounds interact with the D1R. Notably, a human D1R knock-in mouse was created to evaluate DETQ in vivo (Svensson et al., 2017; Bruns et al., 2018). One potential limitation of this mouse model is that the human D1R was found to express at only 50% of the normal D1R levels in wild-type mice. Nonetheless, these authors have generated encouraging data showing that DETQ can potentiate D1R-mediated behaviors (particularly motor activity) in the “humanized” mouse, although effects on learning, memory, or cognition have not yet been reported (Bruns et al., 2018).
In summary, we have identified two novel PAMs of the D1R and provided evidence that they bind to diverse sites on the D1R. It is probable that the MLS1082 scaffold binds to an intracellular allosteric site, potentially involving the ICL2 region, that mediates the effects of previously identified D1R PAM compounds DETQ and Compound B. At present, the receptor site mediating the allosteric effects of the MLS6585 scaffold is unknown. The current identification of multiple allosteric sites on the D1R may provide opportunities for developing a diverse allosteric pharmacology (PAMs, negative allosteric modulators, and silent allosteric modulators) for this receptor subtype as well the prospect for moving novel lead compounds into the clinic.
Acknowledgments
The authors thank Benjamin Neuenswander (KU Specialized Chemistry Center) for HPLC purification of MLS1082 and MLS6585 and compound purity analysis. They also thank Dr. Peter J. Scammells and Dr. Ben Capuano for synthesis of initial batches of Compound B.
Abbreviations
- BRET
bioluminescence resonance energy transfer
- Compound B
Cmpd B, rel-(9R,10R,12S)-N-(2,6-dichloro-3-methylphenyl)-12-methyl-9,10-dihydro-9,10-ethanoanthracene-12-carboxamide
- DA
dopamine
- DAR
dopamine receptor
- DETQ
2-(2,6-dichlorophenyl)-1-((1S,3R)-3-(hydroxymethyl)-5-(2-hydroxypropan-2-yl)-1-methyl-3,4-dihydroisoquinolin-2(1H)-yl)ethan-1-one
- DHX
dihydrexidine
- DMSO
dimethyl sulfoxide
- GPCR
G protein-coupled receptor
- ICL2
second intracellular loop
- PAM
positive allosteric modulator
- PBS
phosphate-buffered saline
- PFC
prefrontal cortex
- RLUs
relative luminescence units
Authorship Contributions
Participated in research design: Luderman, Conroy, Free, Jain, Hazelwood, Aubé, Frankowski, Sibley.
Conducted experiments: Luderman, Conroy, Sanchez-Soto, Moritz, Willette, Titus.
Contributed new reagents or analytic tools: Southall, Ferrer, Fyfe, Jain, Hazelwood, Aubé, Frankowski.
Performed data analysis: Luderman, Conroy, Free, Southall, Ferrer, Sanchez-Soto, Moritz, Willette, Lane.
Wrote or contributed to the writing of the manuscript: Luderman, Free, Aubé, Lane, Frankowski, Sibley.
Footnotes
This project was funded by the Intramural Research Program of the National Institute of Neurologic Disorders and Stroke (NINDS), in the National Institutes of Health (NIH), and Molecular Libraries Initiative funding to the KU Specialized Chemistry Center [U54HG005031], to J.A.
Primary Laboratory of Origin: Molecular Neuropharmacology Section, National Institute of Neurologic Disorders and Stroke, Intramural Research Program, National Institutes of Health, 35 Convent Drive, MSC-3723, Bethesda, MD 20892-3723.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Arnsten AF, Dudley AG. (2005) Methylphenidate improves prefrontal cortical cognitive function through alpha2 adrenoceptor and dopamine D1 receptor actions: relevance to therapeutic effects in attention deficit hyperactivity disorder. Behav Brain Funct 1:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AF, Li BM. (2005) Neurobiology of executive functions: catecholamine influences on prefrontal cortical functions. Biol Psychiatry 57:1377–1384. [DOI] [PubMed] [Google Scholar]
- Arnsten AFT, Vijayragavan S, Wang M, Gamo NJ, Paspalas CD. (2009) Dopamine’s influence on prefrontal cortical cognition: actions and circuits in behaving primates, in Dopamine Handbook (Iversen LL, Iversen SD, Dunnett SB, Bjorklund A. eds) pp 230–248, Oxford University Press, New York. [Google Scholar]
- Ballesteros JA, Weinstein H. (1995) [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors, in Methods in Neurosciences (Sealfon SC ed) pp 366–428, Academic Press, New York. [Google Scholar]
- Beaulieu JM, Gainetdinov RR. (2011) The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev 63:182–217. [DOI] [PubMed] [Google Scholar]
- Bruns RF, Mitchell SN, Wafford KA, Harper AJ, Shanks EA, Carter G, O’Neill MJ, Murray TK, Eastwood BJ, Schaus JM, et al. (2018) Preclinical profile of a dopamine D1 potentiator suggests therapeutic utility in neurological and psychiatric disorders. Neuropharmacology 128:351–365. [DOI] [PubMed] [Google Scholar]
- Cai JX, Arnsten AF. (1997) Dose-dependent effects of the dopamine D1 receptor agonists A77636 or SKF81297 on spatial working memory in aged monkeys. J Pharmacol Exp Ther 283:183–189. [PubMed] [Google Scholar]
- Canals M, Lane JR, Wen A, Scammells PJ, Sexton PM, Christopoulos A. (2012) A Monod-Wyman-Changeux mechanism can explain G protein-coupled receptor (GPCR) allosteric modulation. J Biol Chem 287:650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castner SA, Goldman-Rakic PS. (2004) Enhancement of working memory in aged monkeys by a sensitizing regimen of dopamine D1 receptor stimulation. J Neurosci 24:1446–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A. (2014) Advances in G protein-coupled receptor allostery: from function to structure. Mol Pharmacol 86:463–478. [DOI] [PubMed] [Google Scholar]
- Chun LS, Free RB, Doyle TB, Huang XP, Rankin ML, Sibley DR. (2013) D1-D2 dopamine receptor synergy promotes calcium signaling via multiple mechanisms. Mol Pharmacol 84:190–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conroy JL, Free RB, Sibley DR. (2015) Identification of G protein-biased agonists that fail to recruit β-arrestin or promote internalization of the D1 dopamine receptor. ACS Chem Neurosci 6:681–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eglen RM. (2007) Assessing GPCR activation using protein complementation: a novel technique for HTS. Biochem Soc Trans 35:746–748. [DOI] [PubMed] [Google Scholar]
- Free RB, Chun LS, Moritz AE, Miller BN, Doyle TB, Conroy JL, Padron A, Meade JA, Xiao J, Hu X, et al. (2014) Discovery and characterization of a G protein-biased agonist that inhibits β-arrestin recruitment to the D2 dopamine receptor. Mol Pharmacol 86:96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry PR, Sexton PM, Christopoulos A. (2015) Novel Allosteric Modulators of G Protein-coupled Receptors. J Biol Chem 290:19478–19488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjoni T, Urwyler S. (2008) Receptor activation involving positive allosteric modulation, unlike full agonism, does not result in GABAB receptor desensitization. Neuropharmacology 55:1293–1299. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, Castner SA, Svensson TH, Siever LJ, Williams GV. (2004) Targeting the dopamine D1 receptor in schizophrenia: insights for cognitive dysfunction. Psychopharmacology (Berl) 174:3–16. [DOI] [PubMed] [Google Scholar]
- Leach K, Sexton PM, Christopoulos A. (2007) Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci 28:382–389. [DOI] [PubMed] [Google Scholar]
- Lewis MA, Hunihan L, Watson J, Gentles RG, Hu S, Huang Y, Bronson J, Macor JE, Beno BR, Ferrante M, et al. (2015) Discovery of D1 dopamine receptor positive allosteric modulators: characterization of pharmacology and identification of residues that regulate species selectivity. J Pharmacol Exp Ther 354:340–349. [DOI] [PubMed] [Google Scholar]
- Livingston KE, Traynor JR. (2014) Disruption of the Na+ ion binding site as a mechanism for positive allosteric modulation of the mu-opioid receptor. Proc Natl Acad Sci USA 111:18369–18374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston KE, Stanczyk MA, Burford NT, Alt A, Canals M, Traynor JR. (2018) Pharmacologic evidence for a putative conserved allosteric site on opioid receptors. Mol Pharmacol 93:157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovenberg TW, Brewster WK, Mottola DM, Lee RC, Riggs RM, Nichols DE, Lewis MH, Mailman RB. (1989) Dihydrexidine, a novel selective high potency full dopamine D-1 receptor agonist. Eur J Pharmacol 166:111–113. [DOI] [PubMed] [Google Scholar]
- May LT, Leach K, Sexton PM, Christopoulos A. (2007) Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol 47:1–51. [DOI] [PubMed] [Google Scholar]
- Michino M, Beuming T, Donthamsetti P, Newman AH, Javitch JA, Shi L. (2015) What can crystal structures of aminergic receptors tell us about designing subtype-selective ligands? Pharmacol Rev 67:198–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottola DM, Brewster WK, Cook LL, Nichols DE, Mailman RB. (1992) Dihydrexidine, a novel full efficacy D1 dopamine receptor agonist. J Pharmacol Exp Ther 262:383–393. [PubMed] [Google Scholar]
- Nakako T, Murai T, Ikejiri M, Ishiyama T, Taiji M, Ikeda K. (2013) Effects of a dopamine D1 agonist on ketamine-induced spatial working memory dysfunction in common marmosets. Behav Brain Res 249:109–115. [DOI] [PubMed] [Google Scholar]
- Offermanns S, Simon MI. (1995) G alpha 15 and G alpha 16 couple a wide variety of receptors to phospholipase C. J Biol Chem 270:15175–15180. [DOI] [PubMed] [Google Scholar]
- Rankin ML, Hazelwood LA, Free RB, Namkung Y, Rex EB, Roof RA, Sibley DR. (2010) Molecular pharmacology of the dopamine receptors, in Dopamine Handbook (Iversen LL, Iversen SD, Dunnett SB, Bjorklund A. eds) pp 63–87, Oxford University Press, New York. [Google Scholar]
- Sibley DR, Monsma FJ., Jr (1992) Molecular biology of dopamine receptors. Trends Pharmacol Sci 13:61–69. [DOI] [PubMed] [Google Scholar]
- Svensson KA, Heinz BA, Schaus JM, Beck JP, Hao J, Krushinski JH, Reinhard MR, Cohen MP, Hellman SL, Getman BG, et al. (2017) An allosteric potentiator of the dopamine D1 receptor increases locomotor activity in human D1 knock-in mice without causing stereotypy or tachyphylaxis. J Pharmacol Exp Ther 360:117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamminga CA. (2006) The neurobiology of cognition in schizophrenia. J Clin Psychiatry 67:e11. [PubMed] [Google Scholar]
- Urs NM, Daigle TL, Caron MG. (2011) A dopamine D1 receptor-dependent β-arrestin signaling complex potentially regulates morphine-induced psychomotor activation but not reward in mice. Neuropsychopharmacology 36(3):551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayraghavan S, Wang M, Birnbaum SG, Williams GV, Arnsten AF. (2007) Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci 10:376–384. [DOI] [PubMed] [Google Scholar]