

Abstract
Key points
A hypersensitive ventilatory control system or elevated “loop gain” during sleep is a primary phenotypic trait causing obstructive sleep apnoea (OSA).
Despite the multitude of methods available to assess the anatomical contributions to OSA during wakefulness in the clinical setting (e.g. neck circumference, pharyngometry, Mallampati score), it is currently not possible to recognize elevated loop gain in patients in this context.
Loop gain during sleep can now be recognized using simplified testing during wakefulness, specifically in the form of a reduced maximal breath‐hold duration, or a larger ventilatory response to voluntary 20‐second breath‐holds.
We consider that easy breath‐holding manoeuvres will enable daytime recognition of a high loop gain in OSA for more personalized intervention.
Abstract
Increased “loop gain” of the ventilatory control system promotes obstructive sleep apnoea (OSA) in some patients and offers an avenue for more personalized treatment, yet diagnostic tools for directly measuring loop gain in the clinical setting are lacking. Here we test the hypothesis that elevated loop gain during sleep can be recognized using voluntary breath‐hold manoeuvres during wakefulness. Twenty individuals (10 OSA, 10 controls) participated in a single overnight study with voluntary breath‐holding manoeuvres performed during wakefulness. We assessed (1) maximal breath‐hold duration, and (2) the ventilatory response to 20 s breath‐holds. For comparison, gold standard loop gain values were obtained during non‐rapid eye movement (non‐REM) sleep using the ventilatory response to 20 s pulses of hypoxic–hypercapnic gas (6% CO2–14% O2, mimicking apnoea). Continuous positive airway pressure (CPAP) was used to maintain airway patency during sleep. Additional measurements included gold standard loop gain measurement during wakefulness and steady‐state loop gain measurement during sleep using CPAP dial‐ups. Higher loop gain during sleep was associated with (1) a shorter maximal breath‐hold duration (r 2 = 0.49, P < 0.001), and (2) a larger ventilatory response to 20 s breath‐holds during wakefulness (second breath; r 2 = 0.50, P < 0.001); together these factors combine to predict high loop gain (receiver operating characteristic area‐under‐curve: 92%). Gold standard loop gain values were remarkably similar during wake and non‐REM sleep. The results show that elevated loop gain during sleep can be identified using simple breath‐holding manoeuvres performed during wakefulness. This may have implications for personalizing OSA treatment.
Keywords: OSA alternative treatments, OSA phenotyping, chemoreflex predictors
Key points
A hypersensitive ventilatory control system or elevated “loop gain” during sleep is a primary phenotypic trait causing obstructive sleep apnoea (OSA).
Despite the multitude of methods available to assess the anatomical contributions to OSA during wakefulness in the clinical setting (e.g. neck circumference, pharyngometry, Mallampati score), it is currently not possible to recognize elevated loop gain in patients in this context.
Loop gain during sleep can now be recognized using simplified testing during wakefulness, specifically in the form of a reduced maximal breath‐hold duration, or a larger ventilatory response to voluntary 20‐second breath‐holds.
We consider that easy breath‐holding manoeuvres will enable daytime recognition of a high loop gain in OSA for more personalized intervention.
Introduction
Obstructive sleep apnoea (OSA) is a common disorder with major health implications (Fu et al. 2017). Its treatment is currently limited to anatomical interventions, including continuous positive airway pressure (CPAP), oral appliances and surgery (Doff et al. 2013; Strollo et al. 2014; Furlow, 2016), without due consideration of the underlying pathogenesis. Aside from anatomical compromise, arguably the strongest determinant of OSA is a hypersensitive ventilatory control system or elevated “loop gain” (Younes et al. 2001; Wellman et al. 2008; Edwards et al. 2012, 2016a,b; Eckert et al. 2013; Xie et al. 2013; Joosten et al. 2017). For example, a high loop gain helps to predict a favourable response to non‐anatomical interventions (supplemental oxygen, acetazolamide, partial rebreathing) (Edwards et al. 2012, 2016b; Xie et al. 2013; Messineo et al. 2017; Sands et al. 2017a) and an unfavourable response to anatomical interventions (oral appliances, surgery) (Edwards et al. 2016a; Joosten et al. 2017). Thus, knowledge of a patient's loop gain may help determine what therapeutic options are of possible benefit. Currently, however, gold standard methods for assessing the loop gain contribution to OSA require a dedicated night of assessment in a specialized physiology laboratory, i.e. administration of hypoxic/hypercapnic gas (McClean et al. 1988; Ghazanshahi & Khoo, 1997; Hudgel et al. 1998; Younes et al. 2007; Loewen et al. 2009) or CPAP manipulation (Wellman et al. 2011, 2013) during sleep.
Previously, investigators have demonstrated that chemoreflex sensitivity (a key determinant of loop gain) during wakefulness – measured via CO2 rebreathing (Stanley et al. 1975) or dynamic CO2 administration (Trembach & Zabolotskikh, 2017) – can be estimated simply by measuring how long participants can voluntarily hold their breath for (“maximal breath‐hold duration”). Conceptually, during a breath‐hold, participants generate their own hypoxia–hypercapnia at a rate determined by the plant (e.g. lung volume), and a subsequent chemoreflex response, as determined by the controller (e.g. ventilatory response to hypercapnia). In principle, the loop gain (product of plant and controller factors) determines the rise in latent chemical drive during a breath‐hold. Thus, a higher loop gain should yield a shorter maximal breath‐hold duration, and a greater level of chemical drive manifest as elevated ventilation immediately after a breath‐hold of a prescribed duration.
There is also evidence that factors influencing loop gain might be conserved from wakefulness to sleep. For example, greater chemosensitivity during wakefulness is associated with central apnoeas during sleep (Javaheri, 1999; Solin et al. 2000; Giannoni et al. 2009). However, a tight relationship between loop gain during sleep and during wakefulness remains unproven.
Accordingly, the current study tested the hypothesis that loop gain during sleep could be predicted during wakefulness using breath‐holding manoeuvres. Our primary predictor was the maximal breath‐hold duration. Our secondary predictors were the ventilatory responses in the first two breaths following 20 s breath‐holds (a duration we expected all individuals could tolerate). As a gold standard, loop gain values during sleep were obtained by assessing the ventilatory response to 20 s pulses of hypoxic–hypercapnic gas during non‐REM sleep (6% CO2–14% O2, i.e. normal alveolar gas) (Sands et al. 2017b) while on CPAP to maintain airway patency. We also sought evidence to confirm (1) the association between wake and sleep loop gain values, by additionally performing similar pulses during wakefulness, and (2) that breath‐holding measures also predicted steady‐state loop gain measured using CPAP dial‐ups during sleep.
Methods
Ethical approval
This study conformed to the standards set by the latest revision of the Declaration of Helsinki and was approved by Partners Human Research Committee, Brigham and Women Hospital's Institutional Review Board (protocol number: 2016P002217), except for registration in a database. All subjects provided written informed consent prior to study enrolment.
Subjects
Twenty‐one participants (10 OSA, 11 controls) were studied overnight. OSA patients were otherwise healthy with an apnoea–hypopnoea index (AHI) of ≥10 events/h. Controls had an AHI < 5 events/h (n = 4 performed sleep studies) or were asymptomatic for snoring and daytime sleepiness (n = 6). We excluded patients using medications impacting control of respiration (e.g. opioids, antidepressants, anxiolytics, dopamine antagonists). We also excluded patients with claustrophobia, anaemia and pregnancy. One control was excluded from analysis due to CO2 analyser failure.
Equipment
In addition to standard polysomnographic setup (Flemons et al. 1999), subjects breathed through a sealed nasal mask attached to a pneumotachometer (Hans‐Rudolph, Kansas City, MO, USA; Validyne, Northridge, CA, USA). An intranasal catheter (Vacumetrics Inc., Ventura, CA, USA) measured inspired and end‐tidal partial pressures for oxygen () and carbon dioxide ().
To deliver hypoxic–hypercapnic pulses, the nasal mask was connected to a non‐rebreathing valve (22 mm diameter; Hans‐Rudolph) whose inspiratory line was switched from room air into a Douglas bag containing a gas mixture (6% CO2–14% O2). To deliver pulses during sleep on CPAP, both expiratory and inspiratory lines were connected to a positive/negative pressure source (Philips‐Respironics, Murrysville, PA, USA) whose input line could be switched into the Douglas bag; an exhalation valve in the expiratory line prevented rebreathing.
Protocol
Wakefulness measurements were performed in the evening prior to sleep; wakefulness was visually assessed and confirmed via EEG. All breath‐holding and gold‐standard loop gain measures were performed supine. Sleep measurements were performed in non‐REM.
Breath‐holding manoeuvres
Participants were instructed to perform breath‐holds starting from end‐expiratory lung volume (functional residual capacity) without prior hyperventilation. First, 20 s breath‐holds were performed every 3 min until six reliable manoeuvres were obtained (Fig. 1 A). Second, participants performed three maximal breath‐holds, separated by 3 min rest intervals; three additional maximal breath‐holds were then performed with verbal coaching to encourage a longer duration (Fig. 1 B). Coached breath‐holding involved the following. Prior to the breath‐hold, participants were told their previous breath‐hold time, and were encouraged to beat the previous time. During the coached breath‐hold, the investigator/technician stood at the bedside with a stopwatch and verbalized in real time the breath‐hold at least every 10 s. Participants were notified when they approached (within 5 s) their previous maximal breath‐hold duration. At this time enthusiastic verbal encouragement was provided, e.g. “keep going”, “go, go, go”, “try to resist”, along with feedback on the status of the breath‐hold duration every 5 s. Individuals were asked to exhale slightly at breath‐hold termination in order to document end‐tidal and for physiological interest (Fig. 1 A).
Figure 1. Example raw signals for gold standard loop gain measures and breath‐holding manoeuvres.
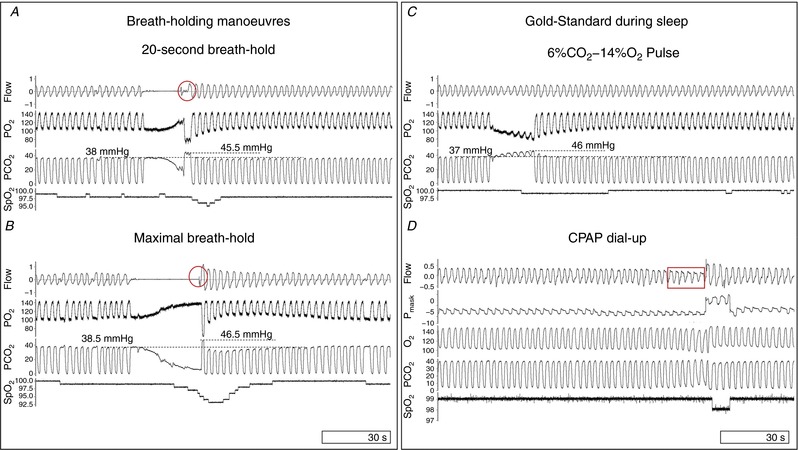
A, the ventilatory response on the 1st and 2nd breaths after 20 s breath‐holds were used as secondary predictors of loop gain. Individuals exhaled after breath‐holds to provide information on alveolar gas changes (circled). B, maximal breath‐holds at end‐expiratory lung volume (functional residual capacity) were timed to predict loop gain. C, hypoxic–hypercapnic pulses (6% CO2–14% O2) were administered for 20 s, every 3 min, to simulate apnoea and elicit a ventilatory response without mechanical or behavioural interference. Note the progressive increase in and the concurrent decrease in in the respective channels, and small ensuing rise in ventilation in this participant with low loop gain. D, CPAP dial‐ups were performed during sleep to assess steady‐state loop gain. CPAP is dialed‐up during a period of flow‐limited breathing (outlined by rectangle) to reveal the ventilatory drive response to the reduction in ventilation prior to the dial‐up. , intranasal oxygen partial pressure; , intranasal carbon dioxide partial pressure; P mask, CPAP delivered intramask pressure; , pulsed oxygen saturation. [Color figure can be viewed at http://wileyonlinelibrary.com]
Gold standard: hypoxic–hypercapnic pulses during wakefulness
To measure loop gain, we administered pulses of hypoxic–hypercapnic gas (Loewen et al. 2009), a modification of the pseudo‐random binary stimulation approach (Ghazanshahi & Khoo, 1997; Sands et al. 2017b). A gas mixture (6% CO2–14% O2) was chosen to mimic alveolar gases and thereby simulate apnoea without disturbing the participant. Gases were delivered in 20 s pulses, repeated every 3 min, to obtain 10 consecutive manoeuvres (Fig. 1 C).
Gold standard: hypoxic–hypercapnic pulses during sleep
CPAP was applied at the minimum level required to eliminate airflow limitation. Pulses were administered to assess loop gain as mentioned previously. If cortical arousals occurred, pulses were repeated.
Gold standard: steady‐state loop gain during sleep
While distinct from “dynamic” loop gain (determinant of central sleep apnoea; assessed above), the steady‐state loop gain – defined as the ventilatory response to a sustained disturbance – has also been shown to predict individual responses to therapies (Edwards et al. 2016a,b). Thus, we assessed steady‐state loop gain using CPAP dial‐ups (Wellman et al. 2013). Briefly, CPAP was lowered gradually (≤1 cmH2O/min) from optimal levels to reduce ventilation maximally without causing arousals and then rapidly returned to optimum to reveal the increase in ventilatory drive (Fig. 1 D).
Data analysis
Sleep stages and arousals were scored by a certified sleep technician using standard American Academy of Sleep Medicine criteria (Berry et al. 2012). Data were analysed with MATLAB (MathWorks, Natick, MA, USA). For each analysis, ventilation was assessed breath‐by‐breath as tidal volume × respiratory rate. End‐tidal gas values (,) were calculated after correcting for gas sampling delays; breaths without an end‐tidal plateau were manually excluded (linear interpolation between breaths). Breath‐by‐breath values were upsampled (4 Hz) to provide a continuous signal for analysis. Baseline ventilation was based on the 30 s prior to each manoeuvre. Loop gain values reported for the pulses describe the ventilatory response to a 1 cycle/min disturbance, i.e. the dynamic loop gain (Edwards et al. 2012; Terrill et al. 2015; Joosten et al. 2017; Sands et al. 2017b).
Breath‐hold manoeuvres
Ventilation data were overlaid (ensemble averaged) and aligned at end‐“apnoea”. Ventilatory responses were calculated from the first and the second breath after the 20 s breath‐hold. The mean of the maximal breath‐hold duration was recorded for each individual.
Gold standard: hypoxic–hypercapnic pulses during wakefulness and sleep
Pulses of hypoxic hypercapnia (20 s/3 min) stimulate the chemoreflex control system at several specific frequencies, including 1 cycle/min (Sands et al. 2017b). Fourier transform analysis of ventilation and changes at this frequency described controller (Δventilation/Δ) and plant gains (Δ/Δventilation*; *ventilation, corrected for inspired , yields a ventilatory disturbance (Ghazanshahi & Khoo, 1997; Sands et al. 2017b) as shown in Figs 2 and 3 (red dashed lines)). Overall loop gain equals controller × plant (Ghazanshahi & Khoo, 1997; Sands et al. 2017b). Plant and controller gain values were based on end‐tidal swings knowing that and vary closely in concert.
Figure 2. Example results in a participant with low loop gain.
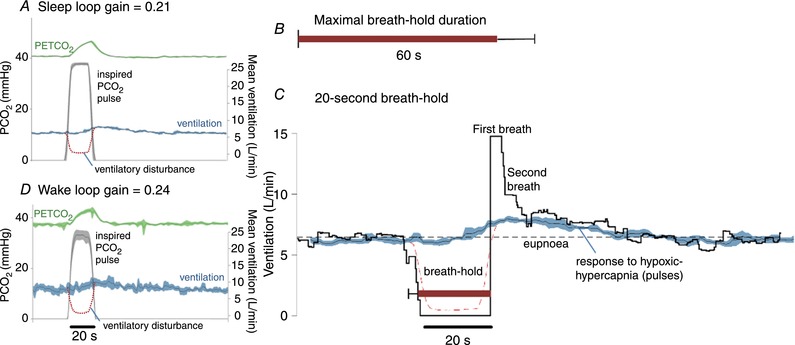
A, inspired pulses of hypoxic–hypercapnic gas (14% O2–6% CO2) cause an increase in end‐tidal () that then elicits a chemoreflex ventilatory response. In this example, the ventilatory response is small. Traces shown are the ensemble average of multiple pulses administered during sleep (black line represents mean, coloured shading represents 95% confidence interval). Dashed red line illustrates the calculated ventilatory disturbance (i.e. the calculated drop in ventilation that would cause the observed rise in , see Sands et al. 2017b); by design, the pulse is approximately equivalent to apnoea (ventilation = 0). Loop gain is taken as the ventilatory response to this disturbance (Fourier analysis, Sands et al. 2017b). B, maximal breath‐hold duration was long (60 ± 11 s, mean ± SD) in this participant (see A). C, ventilation during spontaneous 20 s breath‐holds. Ventilation falls to zero and, at breath‐hold termination (time = 0), there is a period of ventilatory recovery. Note the first recovery breath is much larger than subsequent breaths (15 L/min; 230% of eupnoea). The second breath is considerably smaller (10 L/min; 154% of eupnoea) and is more in line with the ventilatory response to hypoxic hypercapnia (pulses; 135% eupnoea, shown superimposed, from A). The first recovery breath was considered to be particularly influenced by factors beyond chemical stimuli. D, inspired pulses during wakefulness appear remarkably similar to those during sleep.
Figure 3. Example results in a participant with high loop gain.
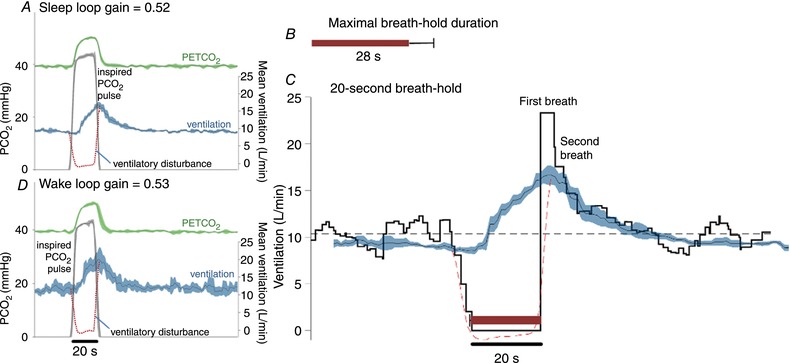
A, inspired pulses of hypoxic–hypercapnic gas elicit a large chemoreflex ventilatory response in this example, thus a greater value of loop gain is measured (response/disturbance). Traces shown are the ensemble average of multiple pulses administered during sleep (black line represents mean, shading represents 95% confidence interval). Dashed line illustrates the calculated ventilatory disturbance. B, maximal breath‐hold duration was short (28 ± 7 s, mean ± SEM) in this participant (see A). C, ventilation during spontaneous 20 s breath‐holds (ensemble average). Note the first recovery breath is larger than subsequent breaths (23.5 L/min; 229% of eupnoea). The second breath response is smaller but still high (17.5 L/min; 170% of eupnoea) and is more in line with the ventilatory response to hypoxic hypercapnia (pulses; 192% eupnoea, shown superimposed, from A). Just as seen in Fig. 2, the breaths after the first recovery breath appear consistent with the response to chemical stimuli. D, inspired pulses during wakefulness appear remarkably similar to those during sleep.
Steady‐state loop gain during sleep
Steady‐state loop gain was calculated during sleep as the ratio of the ventilatory drive response (Δventilation; breath 1 after dial‐up minus eupnoea (optimal CPAP)) to the steady‐state disturbance produced by the lower CPAP level (Δventilation; average of 5 breaths before dial‐up minus eupnoea) (Wellman et al. 2013). Median values for each participant were recorded. An average number of 7 ± 3 CPAP dial‐ups was recorded.
Statistics
Linear regression assessed the associations between predictors and gold‐standard loop gain. Multiple linear regression was used to confirm significant associations after adjusting for four clinical covariates (age, sex, body mass index (BMI), OSA diagnosis). Student's t tests and Wilcoxon/Mann–Whitney tests were used to compare values within and across groups.
Multiple linear regression was used to develop a predictive model for loop gain based on both primary and secondary predictors. Receiver operating characteristic analysis used the multiple regression model output to predict elevated loop gain (above median). Area under curve was used to quantify the predictive value (>0.8 was considered strong). We also assessed sensitivity and specificity to detect elevated loop gain (optimal cut‐off based on the Youden J statistic, i.e. maximizing sum of sensitivity and specificity), using (1) all data, and (2) leave‐one‐out cross‐validation (regression model and cut‐off were determined without each subject under investigation).
Unless specified otherwise, data are expressed as the mean ± SEM or median [75th minus 25th centile], and statistical significance was accepted if P < 0.05. Statistical analyses were performed using GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA, USA) and SPSS 23.00 (IBM, Armonk, NY, USA).
Results
Anthropometric and baseline data are summarized in Table 1. OSA patients were older and heavier than controls. OSA patients were placed on 9.0 ± 3.5 cmH2O, whereas controls were on 4.5 ± 3 cmH2O to maintain airway patency for loop gain measurements during sleep. While we did not perform separate sleep studies on the control group, we note that no control exhibited sleep apnoea when CPAP was lowered to atmospheric pressure during the dial‐downs. Specifically, steady‐state loop gain was measured (requiring stable breathing) at levels of −2 ± 3 cmH2O in controls and 2 ± 3 cmH2O in OSA. We are therefore highly confident that controls were OSA free.
Table 1.
Subject characteristics
| Characteristic | All participants (n = 20) | Controls (n = 10) | OSA patients (n = 10) |
|---|---|---|---|
| Age (years) | 49 [13] | 29 [22] | 54 [13] |
| Female sex (n (%)) | 8 (40) | 5 (50) | 3 (30) |
| Neck circumference (cm) | 39 [6] | 37 [4] | 43 [4] |
| Waist circumference (cm) | 108 [30] | 83 [8] | 113 [4] |
| Body mass index (kg/m2) | 29.7 [12.8] | 22.6 [3.6] | 33.8 [4.6] |
| Physical activity (h/week) | 3.5 [5] | 6.0 [5.5] | 2.5 [4] |
| Current smoking status (n (%)) | 4 (20) | 2 (20) | 2 (20) |
| Previous AHIa (events/h) | 21 [23] | 1.6 [3.1] | 27.6 [6] |
Data are represented as median [interquartile range]. AHI, apnoea–hypopnoea index; OSA, obstructive sleep apnoea. aNot available in 6/10 controls.
Example data from two participants, low and high loop gain, are illustrated in Figs 2 and 3. Compared with the participant with low loop gain, the participant with high loop gain had a shorter maximal breath‐hold duration, and a larger ventilation on the second breath after the 20 s breath‐holds (contrast Figs 3 and 2). Note how closely the ventilatory responses to hypoxic hypercapnia (pulses, sleep) overlie the ventilatory responses following 20 s breath‐holds (after the first breath; Figs 2 D and 3 D).
Primary predictor
Average breath‐hold measures are described in Table 2. As hypothesized, we found a strong and significant inverse univariate correlation between maximal breath‐hold duration (mean of all tests; primary predictor) and loop gain during sleep (pulses; coefficient of determination (r 2) = 0.49, P = 0.001; Fig. 4 A). This correlation was maintained after adjusting for age, sex, BMI and presence of OSA (P = 0.002 for breath‐hold duration, P = 0.01 for sex; predicted loop gain y = 0.86 ± 0.07 − (0.0087 ± 0.0017)[breath‐hold duration, s] + (0.14 ± 0.04)[male]; model r 2 = 0.68).
Table 2.
Average measures in all subjects
| Breath‐hold measurementa | Mean ± SD | Within‐subject SEM |
|---|---|---|
| Ventilation after 20 s breath‐holds | ||
| First breath response (%eupnoea)b | 229 ± 86 | 38 ± 31 |
| Second breath response (%eupnoea)b | 174 ± 48 | 34 ± 25 |
| Maximal breath‐hold duration | ||
| All datac (s) | 37.4 ± 12.8 | 3.7 ± 2.2 |
| Spontaneous maximal breath‐holding duration (s) | 34 ± 10 | 3.6 ± 3.0 |
| Coached maximal breath‐holding duration (s) | 47.2 ± 15.7 | 4.1 ± 4.4 |
| Maximal breath‐hold desaturation | ||
| All data (%) | 10.0 ± 6.7 | 1.0 ± 0.6 |
| Spontaneous maximal breath‐holding (%) | 8.7 ± 5.9 | 0.9 ± 0.7 |
| Coached maximal breath‐holding (%) | 12.3 ± 7.4 | 1.1 ± 0.8 |
aBreath‐holds were performed at end‐expiratory lung volume (functional residual capacity) while supine. bEupnoea is the baseline ventilation. cPrimary predictor.
Figure 4. Primary (A) and secondary (B) predictors of the respiratory system loop gain.
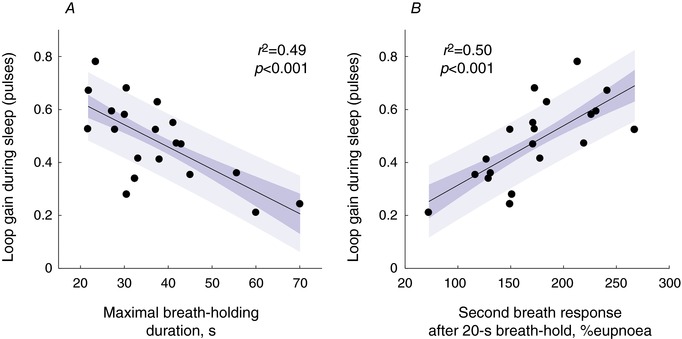
The dark and the light shaded areas represents SEM and SD of the regression line, respectively. [Color figure can be viewed at http://wileyonlinelibrary.com]
Secondary predictors
We did not find a significant relationship between the ventilatory response to 20 s breath‐holds at the first recovery breath and loop gain during sleep (pulses; r 2 = 0.19, P = 0.053). However, the ventilatory response at the second recovery breath was strongly and significantly correlated with loop gain during sleep (r 2 = 0.50, P < 0.001; Fig. 4 B; P value threshold = 0.025 after Bonferroni correction). This correlation was maintained after adjusting for age, sex, BMI and presence of OSA (P = 0.02; predicted loop gain y = 0.09 ± 0.10 + (0.0023 ± 0.0005)x).
Chemical drive physiology after breath‐holds
We considered that the ventilatory drive on the first breath following breath‐holds might be higher due to factors other than hypoxic/hypercapnic stimuli. Indeed, we found that, on average, the ventilation on the first recovery breath following breath‐holds (20 s) was larger than expected (+5 L/min, P = 0.001) based on the response to hypoxic hypercapnia (wake, pulses; Fig. 5 A). In contrast, the ventilation on the second recovery breath was similar in magnitude to the ventilatory response to hypoxic hypercapnia (Fig. 5 A); a tighter association was also evident on breath 2 (Fig. 5 A); see also Figs 2 D and 3 D. There is also clear concordance between the time course of ventilation during the breath‐hold recovery and the time course of chemical drive (response to hypoxic hypercapnia) at all times after the first breath (Fig. 5 B). Note that the magnitude of hypercapnic stimuli was similar (hypoxia was not identical) between 20 s breath‐holds and 20 s hypoxic–hypercapnic pulses (Table 3).
Figure 5. Comparison between chemical drive and recovery breaths after 20 s breath‐holding.
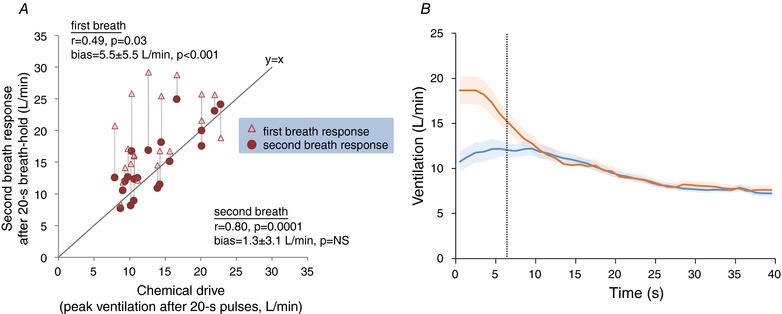
A, after 20 s breath‐holds, the ventilation on the second recovery breath reflects the underlying chemical drive based on hypoxic hypercapnia (pulses). The ventilation on the first recovery breath was substantially larger than the chemical drive, and only modestly correlated with it. The difference in ventilation between first and second recovery breaths may reflect a brief non‐chemical (e.g. behavioural) response to breath‐holding. Here, chemical drive was taken as the peak ventilation (ensemble average) observed following hypoxic–hypercapnic pulses during wakefulness (see Figs 2 and 3). NS, not significant. Bias reported is mean ± SD. B, ventilatory responses following 20 s breath‐holds (orange) superimposed on the ventilatory responses to hypoxic hypercapnia (blue). Continuous lines represent the mean ensemble‐averaged response (1 Hz). Shading represents SEM. The vertical black, dashed line represents the median time at which the second recovery breath ventilation was measured. Time zero for breath‐holds is the start of inspiration for the first recovery breath; time zero for the hypoxic hypercapnia is the time that ensemble‐averaged inspiratory falls back below 10 mmHg. Note that response to breath‐holding initially exceeds the responses to hypoxic hypercapnia but they are later concordant.
Table 3.
Changes in end‐tidal gases during 20 s breath‐holds versus pulses during wake
| Change from baseline | Pulses | Breath‐holds | P |
|---|---|---|---|
| ∆ (mmHg) | 7.0 ± 0.4 | 6.8 ± 0.5 | NS |
| ∆ (mmHg) | −18.4 ± 0.8 | −24.9 ± 1.4 | 0.0005 |
Data are expressed as means ± SEM. ∆ is the change in end‐tidal carbon dioxide partial pressure from baseline (mean); ∆ is the change in end‐tidal oxygen partial pressure. End‐tidal changes were similar and changes were considerably smaller with pulses versus breath‐holds. NS, not significant.
Regression model using primary and secondary predictors
Multiple regression analysis showed that loop gain during sleep was independently associated with both maximal breath‐hold duration (P = 0.016) and second recovery breath response (Δr 2 = 0.16, P = 0.013; y = 0.43 ± 0.15 − (0.0055 ± 0.0020)[breath‐hold duration, s] + (0.0015 ± 0.0005)[second breath response, %]). To assess the ability of this model to identify “high” loop gain (defined by above‐median values) (Terrill et al. 2015; Edwards et al. 2016b) we used receiver operator characteristic analysis and found an area under the curve of 0.92 ± 0.07 (P < 0.001). Sensitivity and specificity (all data) were 90% and 80% (cut‐off = 0.495). Leave‐one‐out cross‐validation yielded sensitivity and specificity of 60% and 80%.
Additional predictors
We observed an inverse association between loop gain during sleep and the magnitude of desaturation that occurred during maximal breath‐holds (mean desaturation from baseline, r 2 = 0.28, P = 0.019). However, this measure did not add significantly to the above regression model.
Impact of coaching
Coaching increased the maximal breath‐hold duration from 34.0 ± 3.1 to 47.2 ± 4.7 s (mean change = 13.3 ± 2.0 s, mean ± SEM, P < 0.0001). Both (mean) uncoached and (mean) coached maximal breath‐hold durations were significantly associated with loop gain during sleep (r 2 = 0.42, P = 0.007; r 2 = 0.45, P = 0.004).
Wake versus sleep loop gain (pulses)
Our hypothesis hinged on the assumption that wake and sleep loop gain values were similar or at least strongly associated. Indeed, we found that sleep loop gain was highly correlated with wake levels (Table 4). Sleep loop gain (pulses) was strongly associated with loop gain measured during wakefulness using the same technique (Table 4).
Table 4.
Sleep versus wake gold standard loop gain values (pulses)
| Gold standard variables | Wake | Sleep | Difference | Correlation | |
|---|---|---|---|---|---|
| r 2 | P | ||||
| Baseline ventilation (L/min) | 8.6 ± 0.5 | 7.2 ± 0.4 | −1.5 ± 0.5* | ||
| Baseline (mmHg) | 38.5 ± 0.6 | 39.5 ± 0.7 | 0.9 ± 0.5 | ||
| Loop gain | 0.42 ± 0.03 | 0.48 ± 0.34 | 0.06 ± 0.03** | 0.40 | 0.003 |
| Plant gain (mmHg/(L/min)) | 0.92 ± 0.07 | 1.07 ± 0.05 | 0.09 ± 0.06*** | 0.30 | 0.01 |
| Controller gain (L/min/mmHg) | 0.5 ± 0.05 | 0.48 ± 0.05 | −0.02 ± 0.04 | 0.38 | 0.004 |
Data are expressed as means ± SEM. Baseline ventilation fell significantly during sleep (* P = 0.004). Loop gain during sleep is slightly higher than loop gain during wakefulness (** P = 0.03), likely via increased plant gain, but the two are strongly correlated. Note that values during sleep were measured on CPAP to maintain airway patency; CPAP is expected to lower plant gain via increased lung volume, but the effect was considered slight, since plant gain was significantly higher during sleep (*** P = 0.01). Of note, sleep loop gain was measured on CPAP but wake loop gain was not (see Discussion).
Predicting steady‐state loop gain during sleep (CPAP dial‐up technique) using breath‐holding
We sought to separately test whether our breath‐holding measures predicted steady‐state loop gain, given its role in OSA pathogenesis (Eckert et al. 2013; Wellman et al. 2013) and predicting responses to treatment (Edwards et al. 2016a,b).
Maximal breath‐hold duration was significantly correlated with steady‐state loop gain during sleep (r 2 = 0.22, P = 0.036; y = 5.2 ± 1.1 − (0.064 ± 0.028)x); note that the correlation was less strong than for dynamic loop gain (pulses). The ventilatory response following 20 s breath‐holds was significantly associated with steady‐state loop gain during sleep at the second breath (r 2 = 0.31, P = 0.011; y = −0.67 ± 1.26 + (0.020 ± 0.007)x), but not at the first breath (r 2 = 0.24, P = 0.027; P threshold = 0.025). Multiple regression showed that the second breath response was the only significant independent contributor. Loop gain and breath‐holding measures for both controls and OSA patients are provided in Table 5.
Table 5.
Key measures of loop gain in OSA patients and controls
| Controls | OSA patients | |
|---|---|---|
| Gold standard loop gain during sleep | ||
| Dynamic loop gain (pulses) | 0.39 ± 0.04 | 0.57 ± 0.04 |
| Plant (mmHg/(L/min)) | 1.04 ± 0.08 | 1.10 ± 0.07 |
| Controller (L/min/mmHg) | 0.41 ± 0.06 | 0.55 ± 0.06 |
| Steady‐state loop gain (CPAP dial‐ups) | 2.1 ± 0.5 | 3.4 ± 0.6 |
| Breath‐hold predictors of loop gain | ||
| Ventilation after 20 s breath‐holds | ||
| First breath response (%eupnoea)a | 204 ± 20 | 254 ± 32 |
| Second breath response (%eupnoea)a | 150 ± 13 | 197 ± 58 |
| Maximal breath‐hold duration | ||
| All datab (s) | 43.1 ± 4.7 | 31.8 ± 2.4 |
| Spontaneous maximal breath‐holding duration (s) | 38.0 ± 3.4 | 28.8 ± 2.7 |
| Coached maximal breath‐holding duration (s) | 53.1 ± 5.8 | 39.7 ± 3.8 |
Data are expressed as means ± SEM. aEupnoea is the baseline ventilation. bPrimary predictor.
Relationship between dynamic and steady‐state loop gain
Loop gain during sleep (pulses; the dynamic test) was associated with the steady‐state loop gain during sleep (r 2 = 0.36, P = 0.005).
Discussion
The main finding of this pilot study was that the ventilatory control contribution to OSA (loop gain during sleep) can be estimated by performing voluntary breath‐holding manoeuvres during wakefulness. Specifically, a higher loop gain promotes a shorter maximal breath‐hold duration and a greater ventilatory response to voluntary apnoea. These variables predicted both an elevated dynamic loop gain (using gas pulses) and a greater steady‐state loop gain. Given that elevated loop gain has been found to determine the success/failure of a range of OSA therapies (Xie et al. 2013; Edwards et al. 2016a; Eckert et al. 2013; Edwards et al. 2012; Sands et al. 2017a), we envisage that breath‐holding measures of loop gain may play a role in personalized sleep medicine.
Novel physiological insights
Breath‐hold duration
To our knowledge, this is the first study to demonstrate a physiological link between breath‐holds during wakefulness and loop gain during sleep. We note, however, that breath‐hold duration has previously been linked with elevated chemosensitivity during wakefulness (Stanley et al. 1975; Trembach & Zabolotskikh, 2017). In 1975, Stanley and coworkers (Stanley et al. 1975) observed an inverse association (r 2 = 0.79) between maximal breath‐hold duration (while breathing CO2) and the hypercapnic ventilatory response (rebreathing test, i.e. a measure of controller gain) during wakefulness in healthy subjects. More recently, Trembach & Zabolotskikh found an inverse relationship (r 2 = 0.67) between maximal breath‐hold duration and peripheral chemosensitivity (single‐breath, 13% CO2). Additionally, patients with central congenital hypoventilation syndrome, who have substantially reduced chemosensitivity (Carroll et al. 2014), can hold their breath for prolonged durations and have little urge to breathe afterwards (Shea et al. 1993). To enable breath‐hold duration to become a relevant measure for OSA pathophysiology, we extended the above findings to predict the overall loop gain of the ventilatory control system (the parameter of interest, and arguably most responsible for the observable predictors) rather than chemosensitivity alone, and to predict this parameter during sleep.
Notably, the relationship between breath‐hold duration and loop gain hinges on the concept that breath‐holding is voluntarily terminated after a similar increase in chemical drive across patients. However, the strong associations observed, by us and others (Stanley et al. 1975; Trembach & Zabolotskikh, 2017), indicate that variability in maximal breath‐hold duration generally reflects variability in loop gain more than it reflects variability in the “breaking‐point” (i.e. the change in chemical drive that triggers voluntary breath‐hold termination). This finding is not trivial, since chemical drive is not the only determinant of breath‐hold termination. Conceptually, a feeling of “air hunger” builds up over time throughout a voluntary breath‐hold that somewhat exceeds the magnitude of hypoxic hypercapnia. Evidence includes (1) breath‐holds performed at lower lung volumes are terminated at greater levels of chemical drive (higher arterial and greater desaturation) (Parkes, 2006); (2) increasing initial leads to breath‐hold termination at higher levels of (Kelman & Wann, 1971; Parkes, 2006); subjects re‐breathing into a small bag can reach greater levels of hypoxia/hypercapnia than during a voluntary breath‐hold (Hill & Flack, 1908). The interpretation is that the absence of mechanical breathing movements is uncomfortable, and provides an additional stimulus to resume breathing independent of arterial gases. Likewise, motivation or coaching of individuals can increase breath‐hold duration (Table 2). Despite these potential confounding factors (differences in breaking point, motivation, non‐chemical i.e. mechanical contributions to ventilatory drive), breath‐hold duration is still strongly influenced by the sensitivity of the ventilatory chemoreflex control system.
Moreover, we also found that breath‐holding duration correlates more strongly with overall loop gain (r 2 = 0.48, P = 0.0007; wakefulness pulses) than with chemosensitivity (controller gain, r 2 = 0.20, P = 0.048) or plant gain (r 2 = 0.01, P = 0.76), which supports our view that overall loop gain contributes strongly to breath‐hold duration.
Ventilatory response to breath‐holding
We found that the magnitude of the ventilatory response to spontaneous breath‐holds during wakefulness closely tracks the ventilatory response to hypoxic hypercapnia (Figs 2, 3, 4 B and 5). However, this appears accurate only after excluding the first recovery breath, which may be influenced substantially by factors other than chemical drive to breathe (e.g. behavioural responses to breath‐holding). The magnitude of the first recovery breath exceeded the level expected based on the ventilatory response to hypoxic hypercapnia; we interpret that this difference reflects a transient non‐chemical (e.g. mechanical/behavioural) response to breath‐holding (see Fig. 5 and ‘Breath‐hold duration’ in Discussion, above). We interpret that this non‐chemical contribution to “air‐hunger” (based on the experience of not breathing per se) remains observable for several seconds after breath‐hold termination and contributes to the first large recovery breath following a breath‐hold. The overshoot is also reminiscent of the non‐chemical arousal‐related contribution to the ventilatory recovery from (non‐voluntary) obstructive respiratory events in sleep apnoea (Horner et al. 2001). Conversely, by the second recovery breath, ventilation is no greater than expected based on chemical drive responses to hypoxic hypercapnia (without the voluntary breath‐holding experience; comparison of the mean differences did not produce any significant difference, Fig. 5), thereby enabling ventilatory control assessment using the breath‐holding recovery period. Thus, we conclude that any behavioural contributions to ventilatory responses to breath‐holds are short‐lived. However, further studies that provide insight into the various sources of non‐chemical air‐hunger are warranted.
Oxygen desaturation and breath‐holding
We observed a 10.0 ± 6.7% desaturation after maximal breath‐holds. This magnitude of desaturation is substantial but is consistent with the literature. We emphasize that participants were studied at end‐expiratory lung volume/functional residual capacity (FRC) rather than total lung capacity, which accelerates desaturation. Subjects were also studied in the supine position rather than seated which further lowers FRC by ∼30% (Blair & Hickam, 1955). Previous reports are in line with our findings: Findley et al. showed that supine end‐expiratory breath‐holds led to an average minimum pulsed oxygen saturation () of 90.3 ± 6.3% after 30 s (Findley et al. 1983), a shorter duration than the average in our study. Series et al. observed a 12.8 ± 2.6% desaturation during 30 s obstructive apnoeas while supine in patients with OSA (Series et al. 1989).
Sleep versus wake
For the first time, loop gain during sleep was shown to directly correlate with loop gain during wakefulness with minimal bias (Table 4). This finding indicates that a high loop gain promoting OSA during sleep is, in fact, evident and observable during wakefulness. Our results also indicated that chemosensitivity (controller gain) was not different between wake and sleep (Table 4). On the surface, this similarity appears to contradict previous research indicating a reduced hypercapnic and hypoxic ventilatory responses from wake to non‐REM sleep (Douglas et al. 1982). However, these seminal studies did not consider the increased pharyngeal resistance in sleep. When increased pharyngeal resistance is accounted for, i.e. using the ventilatory effort response to , wake to sleep differences in chemosensitivity disappear (White, 1986). We found a small wake to non‐REM increase in loop gain due to a small rise in plant gain, as expected based on the known reduction in lung volume during sleep; of note, our use of CPAP to maintain airway patency likely offset this effect partially (Heinzer et al. 2005).
Clinical implications
A key limitation to progress in personalized medicine for OSA is the lack of a clinically available means to phenotype patients in the daytime and predict responses to treatment. Our study overcomes a major hurdle by demonstrating that daytime breath‐holding measurements can predict elevated loop gain during non‐REM sleep. The two key breath‐holding measures we used are easily obtained: (1) breath‐hold duration only requires a timer and is thus the easiest to measure; (2) the ventilatory response to breath‐holding requires an uncalibrated measure of changes in ventilation. The latter may be advantageous because it is, in principle, not affected by inter‐individual differences in chemical drive “breaking point”. Thus, it is highly feasible that such tests could be performed in the clinical setting to identify the approx. one‐third of patients with elevated loop gain (Eckert et al. 2013) who are at risk of failing anatomical therapies (Edwards et al. 2016a; Joosten et al. 2017) but might benefit from loop gain lowering treatments (Edwards et al. 2012, 2013). Phenotyping approaches are already needed in clinical trials to identify the subgroups of responders to emerging treatments. Further research is required to demonstrate the value of breath‐holding measures for predicting outcomes of interventions.
Methodological considerations
There are several limitations of our study. (1) CPAP was applied in order to maintain airway patency and measure sleep loop gain, as in virtually all previous studies on the topic (Edwards et al. 2012, 2014, 2016a; Taranto‐Montemurro et al. 2016). CPAP likely reduces loop gain slightly (via plant gain, through a lung volume increase). We note, however, that plant gain values during sleep were still not lower than wake values, thus the effect of CPAP was likely small. (2) It was unclear a priori whether coached or uncoached maximal breath‐hold durations would have greater predictive value, so we employed both approaches, repeated 3 times each, and used the average as our primary outcome variable. We recognize that testing breath‐hold 6 times may require more time than is available in some circumstances. We note, however, that both single approaches were highly reproducible (see intra‐subject SEM, Table 2) and both were adequate for predicting loop gain values (see Results). (3) We endeavoured to deliver a hypoxic–hypercapnic stimulus during the 20 s pulses that produced and values similar to that which occurred with the 20 s breath‐holds. However, the hypoxic stimulus turned out to be smaller during the pulses versus breath‐holding. Of note, the end‐tidal levels were similar, and this stimulus likely dominates the ventilatory response. The finding that the ventilatory responses to breath‐holds at the second breath have similar magnitude to those based on hypoxic hypercapnia suggests that the chemical drive was adequately matched.
Conclusions
Loop gain of the ventilatory control system during sleep can be estimated using breath‐hold testing in the daytime. Thus, physiological breath‐hold testing may have clinical utility for phenotyping patients with OSA.
Additional information
Competing interests
L.T.‐M. received consultancy fees from Novion Pharmaceuticals and Cambridge Sound Management. A.W. received consultancy fees from Cambridge Sound Management, Nox Medical, Bayer and Varnum. S.A.S. received consultancy fees from Cambridge Sound Management and Nox Medical. D.P.W. received consultancy fees from Philips‐Respironics. L.M., A.A., M.D.O.M. and N.C. have no conflicts of interest to disclose.
Author contributions
Study design: L.M., L.T.‐M., A.W., S.A.S. Data collection: L.M., N.C. Data analysis: L.M., A.A., S.A.S. Interpretation of results: all authors. Initial draft: L.M., S.A.S. Review of the manuscript for important intellectual content: all authors. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed. The experiments were carried out at Brigham and Women's Hospital ‐ Harvard Medical School.
Funding
This research project received generous funding from Fan Hongbing, president of OMPA Corporation, Kaifeng, China. This work was also supported by the National Institutes of Health (R01HL102321, R01HL128658, P01HL095491, P01HL094307, R35HL135818), as well as by Harvard Catalyst Clinical Research Center: UL1TR001102. L.M. is supported by the University of Brescia. L.T.‐M. is supported by the American Heart Association (15POST254800003). S.A.S. is supported by the American Heart Association (15SDG25890059). M.D.O.M. is supported by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) Foundation, Ministry of Education of Brazil.
Translational perspectives.
Oversensitive ventilatory control (increased “loop gain”) during sleep is a key determinant of sleep apnoea but there is no way to measure this trait in an office setting to guide therapy. We show for the first time that the increased loop gain during sleep is observable during wakefulness and can be measured simply by performing voluntary breath‐holds. Breath‐hold testing is feasible clinically and will enable novel pharmacological interventions to be tested specifically in patients with a high loop gain phenotype of sleep apnoea.
Acknowledgements
The authors are grateful to Lauren Hess for technical assistance.
Biography
Raised in the city of Brescia, Italy, Ludovico Messineo started his medical career studying at the University of Brescia. After getting his specialization in Sleep and Respiratory Medicine, he completed a postdoctoral research fellowship in the Sleep Medicine Department at Harvard Medical School, Boston, USA. Currently, he is working in clinical care and continuing his research in sleep medicine. He has also recently been accepted as a PhD candidate at University of New South Wales, Sydney, Australia. His current work focuses on obstructive sleep apnoea phenotyping and pharmacological treatment.

Edited by: Harold Schultz & Benedito Machado
Linked articles This article is highlighted by a Perspective by Pham et al. To read this Perspective, visit https://doi.org/10.1113/JP276590.
References
- Berry R, Budhiraja R, Gottlieb D, Gozal D, Iber C, Kapur V, Marcus CL, Mehra R, Parthasarathy S, Quan SF, Redline S, Strohl KP, Davidson Ward SL & Tangredi MM; American Academy of Sleep Medicine (2012). Rules for scoring respiratory events in sleep: update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med 8, 597–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair E & Hickam JB (1955). The effect of change in body position on lung volume and intrapulmonary gas mixing in normal subjects. J Clin Invest 34, 383–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll MS, Patwari PP, Kenny AS, Brogadir CD, Stewart TM & Weese‐Mayer DE (2014). Residual chemosensitivity to ventilatory challenges in genotyped congenital central hypoventilation syndrome. J Appl Physiol (1985) 116, 439–450. [DOI] [PubMed] [Google Scholar]
- Doff MH, Hoekema A, Wijkstra PJ, van der Hoeven JH, Huddleston Slater JJ, de Bont LG & Stegenga B (2013). Oral appliance versus continuous positive airway pressure in obstructive sleep apnea syndrome: a 2‐year follow‐up. Sleep 36, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas NJ, White DP, Weil JV, Pickett CK & Zwillich CW (1982). Hypercapnic ventilatory response in sleeping adults. Am Rev Respir Dis 126, 758–762. [DOI] [PubMed] [Google Scholar]
- Eckert DJ, White DP, Jordan AS, Malhotra A & Wellman A (2013). Defining phenotypic causes of obstructive sleep apnea. Identification of novel therapeutic targets. Am J Respir Crit Care Med 188, 996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards BA, Andara C, Landry S, Sands SA, Joosten SA, Owens RL, White DP, Hamilton GS & Wellman A (2016a). Upper‐airway collapsibility and loop gain predict the response to oral appliance therapy in patients with obstructive sleep apnea. Am J Respir Crit Care Med 194, 1413–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards BA, Connolly JG, Campana LM, Sands SA, Trinder JA, White DP, Wellman A & Malhotra A (2013). Acetazolamide attenuates the ventilatory response to arousal in patients with obstructive sleep apnea. Sleep 36, 281–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards BA, Sands SA, Eckert DJ, White DP, Butler JP, Owens RL, Malhotra A & Wellman A (2012). Acetazolamide improves loop gain but not the other physiological traits causing obstructive sleep apnoea. J Physiol 590, 1199–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards BA, Sands SA, Owens RL, Eckert DJ, Landry S, White DP, Malhotra A & Wellman A (2016b). The combination of supplemental oxygen and a hypnotic markedly improves obstructive sleep apnea in patients with a mild to moderate upper airway collapsibility. Sleep 39, 1973–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards BA, Sands SA, Owens RL, White DP, Genta PR, Butler JP, Malhotra A & Wellman A (2014). Effects of hyperoxia and hypoxia on the physiological traits responsible for obstructive sleep apnoea. J Physiol 592, 4523–4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findley LJ, Ries AL, Tisi GM & Wagner PD (1983). Hypoxemia during apnea in normal subjects: mechanisms and impact of lung volume. J Appl Physiol Respir Environ Exerc Physiol 55, 1777–1783. [DOI] [PubMed] [Google Scholar]
- Flemons WW, Buysse D, Redline S, Oack A, Strohl K, Wheatley J, Young T, Douglas N, Levy P, McNicolas W, Fleetham J, White D, Schmidt‐Nowarra W, Carley D & Romaniuk J (1999). Sleep‐related breathing disorders in adults: Recommendations for syndrome definition and measurement techniques in clinical research. Sleep 22, 667–689. [PubMed] [Google Scholar]
- Fu Y, Xia Y, Yi H, Xu H, Guan J & Yin S (2017). Meta‐analysis of all‐cause and cardiovascular mortality in obstructive sleep apnea with or without continuous positive airway pressure treatment. Sleep Breath 21, 181–189. [DOI] [PubMed] [Google Scholar]
- Furlow B (2016). SAVE trial: no cardiovascular benefits for CPAP in OSA. Lancet Respir Med 4, 860. [DOI] [PubMed] [Google Scholar]
- Ghazanshahi SD & Khoo MC (1997). Estimation of chemoreflex loop gain using pseudorandom binary CO2 stimulation. IEEE Trans Biomed Eng 44, 357–366. [DOI] [PubMed] [Google Scholar]
- Giannoni A, Emdin M, Bramanti F, Iudice G, Francis DP, Barsotti A, Piepoli M & Passino C (2009). Combined increased chemosensitivity to hypoxia and hypercapnia as a prognosticator in heart failure. J Am Coll Cardiol 53, 1975–1980. [DOI] [PubMed] [Google Scholar]
- Heinzer RC, Stanchina ML, Malhotra A, Fogel RB, Patel SR, Jordan AS, Schory K & White DP (2005). Lung volume and continuous positive airway pressure requirements in obstructive sleep apnea. Am J Respir Crit Care Med 172, 114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill L & Flack M (1908). The effect of excess of carbon dioxide and of want of oxygen upon the respiration and the circulation. J Physiol 37, 77–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner RL, Rivera MP, Kozar LF & Phillipson EA (2001). The ventilatory response to arousal from sleep is not fully explained by differences in CO2 levels between sleep and wakefulness. J Physiol 534, 881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudgel DW, Gordon EA, Thanakitcharu S & Bruce EN (1998). Instability of ventilatory control in patients with obstructive sleep apnea. Am J Respir Crit Care Med 158, 1142–1149. [DOI] [PubMed] [Google Scholar]
- Javaheri S (1999). A mechanism of central sleep apnea in patients with heart failure. N Engl J Med 341, 949–954. [DOI] [PubMed] [Google Scholar]
- Joosten SA, Leong P, Landry SA, Sands SA, Terrill PI, Mann D, Turton A, Rangaswamy J, Andara C, Burgess G, Mansfield D, Hamilton GS & Edwards BA (2017). Loop gain predicts the response to upper airway surgery in patients with obstructive sleep apnea. Sleep 40, zsx094. [DOI] [PubMed] [Google Scholar]
- Kelman GR & Wann KT (1971). Mechanical and chemical control of breath holding. Q J Exp Physiol Cogn Med Sci 56, 92–100. [DOI] [PubMed] [Google Scholar]
- Loewen A, Ostrowski M, Laprairie J, Atkar R, Gnitecki J, Hanly P & Younes M (2009). Determinants of ventilatory instability in obstructive sleep apnea: inherent or acquired? Sleep 32, 1355–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClean PA, Phillipson EA, Martinez D & Zamel N (1988). Single breath of CO2 as a clinical test of the peripheral chemoreflex. J Appl Physiol (1985) 64, 84–89. [DOI] [PubMed] [Google Scholar]
- Messineo L, Magri R, Corda L, Pini L, Taranto‐Montemurro L & Tantucci C (2017). Phenotyping‐based treatment improves obstructive sleep apnea symptoms and severity: a pilot study. Sleep Breath 21, 861–868. [DOI] [PubMed] [Google Scholar]
- Parkes MJ (2006). Breath‐holding and its breakpoint. Exp Physiol 91, 1–15. [DOI] [PubMed] [Google Scholar]
- Sands SA, Edwards BA, Terrill PI, Butler JP, Owens RL, Taranto‐Montemurro L, Azarbarzin A, Marques M, de Melo CM, White DP, Malhotra A & Wellman A (2017a). Phenotyping from polysomnography predicts obstructive sleep apnea responses to supplemental oxygen therapy [Abstract]. Am J Respir Crit Care Med 195 A2931. [Google Scholar]
- Sands SA, Mebrate Y, Edwards BA, Nemati S, Manisty CH, Desai AS, Wellman A, Willson K, Francis DP, Butler JP & Malhotra A (2017b). Resonance as the mechanism of daytime periodic breathing in patients with heart failure. Am J Respir Crit Care Med 195, 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Series F, Cormier Y & La Forge J (1989). Role of lung volumes in sleep apnoea‐related oxygen desaturation. Eur Respir J 2, 26–30. [PubMed] [Google Scholar]
- Shea SA, Andres LP, Shannon DC, Guz A & Banzett RB (1993). Respiratory sensations in subjects who lack a ventilatory response to CO2 . Respir Physiol 93, 203–219. [DOI] [PubMed] [Google Scholar]
- Solin P, Roebuck T, Johns DP, Walters EH & Naughton MT (2000). Peripheral and central ventilatory responses in central sleep apnea with and without congestive heart failure. Am J Respir Crit Care Med 162, 2194–2200. [DOI] [PubMed] [Google Scholar]
- Stanley NN, Cunningham EL, Altose MD, Kelsen SG, Levinson RS & Cherniack NS (1975). Evaluation of breath holding in hypercapnia as a simple clinical test of respiratory chemosensitivity. Thorax 30, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strollo PJ Jr, Soose RJ, Maurer JT, de Vries N, Cornelius J, Froymovich O, Hanson RD, Padhya TA, Steward DL, Gillespie MB, Woodson BT, Van de Heyning PH, Goetting MG, Vanderveken OM, Feldman N, Knaack L & Strohl KP (2014). Upper‐airway stimulation for obstructive sleep apnea. N Engl J Med 370, 139–149. [DOI] [PubMed] [Google Scholar]
- Taranto‐Montemurro L, Sands SA, Edwards BA, Azarbarzin A, Marques M, de Melo C, Eckert DJ, White DP & Wellman A (2016). Desipramine improves upper airway collapsibility and reduces OSA severity in patients with minimal muscle compensation. Eur Respir J 48, 1340–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrill PI, Edwards BA, Nemati S, Butler JP, Owens RL, Eckert DJ, White DP, Malhotra A, Wellman A & Sands SA (2015). Quantifying the ventilatory control contribution to sleep apnoea using polysomnography. Eur Respir J 45, 408–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trembach N & Zabolotskikh I (2017). Breath‐holding test in evaluation of peripheral chemoreflex sensitivity in healthy subjects. Respir Physiol Neurobiol 235, 79–82. [DOI] [PubMed] [Google Scholar]
- Wellman A, Eckert DJ, Jordan AS, Edwards BA, Passaglia CL, Jackson AC, Gautam S, Owens RL, Malhotra A & White DP (2011). A method for measuring and modeling the physiological traits causing obstructive sleep apnea. J Appl Physiol (1985) 110, 1627–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellman A, Edwards BA, Sands SA, Owens RL, Nemati S, Butler J, Passaglia CL, Jackson AC, Malhotra A & White DP (2013). A simplified method for determining phenotypic traits in patients with obstructive sleep apnea. J Appl Physiol (1985) 114, 911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellman A, Malhotra A, Jordan AS, Stevenson KE, Gautam S & White DP (2008). Effect of oxygen in obstructive sleep apnea: role of loop gain. Respir Physiol Neurobiol 162, 144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DP (1986). Occlusion pressure and ventilation during sleep in normal humans. J Appl Physiol (1985) 61, 1279–1287. [DOI] [PubMed] [Google Scholar]
- Xie A, Teodorescu M, Pegelow DF, Teodorescu MC, Gong Y, Fedie JE & Dempsey JA (2013). Effects of stabilizing or increasing respiratory motor outputs on obstructive sleep apnea. J Appl Physiol (1985) 115, 22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes M, Ostrowski M, Atkar R, Laprairie J, Siemens A & Hanly P (2007). Mechanisms of breathing instability in patients with obstructive sleep apnea. J Appl Physiol (1985) 103, 1929–1941. [DOI] [PubMed] [Google Scholar]
- Younes M, Ostrowski M, Thompson W, Leslie C & Shewchuk W (2001). Chemical control stability in patients with obstructive sleep apnea. Am J Respir Crit Care Med 163, 1181–1190. [DOI] [PubMed] [Google Scholar]