

Abstract
The development of intestinal‐type gastric cancer is preceded by the emergence of metaplastic cell lineages in the gastric mucosa. In particular, intestinal metaplasia and spasmolytic polypeptide‐expressing metaplasia (SPEM) have been associated with the pathological progression to intestinal‐type gastric cancer. The development of SPEM represents a physiological response to damage that recruits reparative cells to sites of mucosal injury. Metaplastic cell lineages are characterized by mucus secretion, adding a protective barrier to the epithelium. Increasing evidence indicates that the influence of alarmins and cytokines is required to initiate the process of metaplasia development. In particular, IL‐33 derived from epithelial cells stimulates IL‐13 production by specialized innate immune cells to induce chief cell transdifferentiation into SPEM following the loss of parietal cells from the corpus of the stomach. While SPEM represents a physiological healing response to acute injury, persistent injury and chronic inflammation can perpetuate a recurring pattern of reprogramming and metaplasia that is a risk factor for gastric cancer development. The transdifferentiation of zymogen secreting cells into mucous cell metaplasia may represent both a general repair mechanism in response to mucosal injury in many epithelia as well as a common pre‐neoplastic pathway associated with chronic injury and inflammation.
Keywords: SPEM, spasmolytic polypeptide‐expressing metaplasia, macrophage polarization, IL‐13, IL‐33, ST2, transdifferentiation
Introduction
Globally, gastric cancer is a major health concern and remains one of the leading causes of cancer‐related death (de Martel et al. 2012; Burkitt et al. 2017). This health burden has put a spotlight on determining the underlying mechanisms that are responsible for the pathological progression to gastric cancer. The most common type of gastric cancer, intestinal‐type adenocarcinoma, almost always evolves in a field of pre‐existing parietal cell loss (oxyntic atrophy) and metaplasia (Correa, 1988; Schmidt et al. 1999). Metaplasia in the stomach arises from the reprogramming of differentiated epithelial cells (Nam et al. 2010; Leushacke et al. 2017). There is increasing interest in understanding the cross‐talk between inflammatory signals and the epithelium; however, we are just beginning to comprehend how inflammation contributes to the pathological progression to gastric cancer. Here we will review the recent insights into the role of inflammation in the emergence and progression of metaplasia in the stomach.
Context for metaplasia development: injury and repair
Metaplasia refers to the presence of a normal cell lineage in a tissue where it is not normally found. In most cases, metaplastic lineages are characterized by mucus secretion (Petersen et al. 2017b). Pelayo Correa was the first to describe the association of intestinal metaplasia (the presence of Muc2/TFF3 expressing intestinal‐type goblet cells in the stomach) with intestinal‐type gastric cancer (Correa, 1988; Correa et al. 2010). In 1999, we later described a second metaplastic process associated with intestinal‐type gastric cancer designated spasmolytic polypeptide‐expressing metaplasia or SPEM (Schmidt et al. 1999). SPEM refers to the presence of Muc6 and TFF2 expressing cells at the base of corpus glands with a morphology more characteristic of mucus‐producing deep antral glands (thus SPEM has also been designated ‘pseudopyloric metaplasia’) (Weis et al. 2013). Similar to the case for mucous cell metaplasia present in Barrett's oesophagus, metaplasia in the stomach corpus is associated with damage to the gastric mucosa (Wang, 2015). Indeed, recent investigations have noted that SPEM lineages are present at the edges of healing ulcers in the corpus of the stomach, and these lineages directly contribute to the mucosal response to injury (Engevik et al. 2016). Thus, in the stomach where injury may be compounded by the caustic nature of acidic gastric secretions, the presence of increased mucus secretion at the site of damage would represent a critical physiological response to local injury (Demitrack et al. 2012; Hoffmann, 2015). Nevertheless, increasing evidence indicates that maintenance of metaplasia in the presence of chronic inflammation, and perhaps progression of SPEM to intestinal metaplasia, can predispose an individual to develop dysplasia in the gastric mucosa (Schmidt et al. 1999; Halldorsdottir et al. 2003; Zhang et al. 2017).
Transdifferentiation of chief cells
The normal mammalian stomach corpus has a constitutively active stem cell compartment located near the top of the gland in a region known as the isthmus (Fig. 1; Karam & Leblond, 1993). Injury to the epithelial layer and inflammation in the stomach, including Helicobacter pylori‐induced oxyntic atrophy (Schmidt et al. 1999) or ulceration (Engevik et al. 2016), can cause increased proliferation in these stem cells, but it also initiates the reprogramming and proliferation of a differentiated zymogen secretory cell type known as the chief cell (Nam et al. 2010). Chief cells are mature, post‐mitotic cells located at the base of the gland that are responsible for secreting pepsin and other digestive enzymes. Following acute or chronic injury, chief cells are capable of transdifferentiating into SPEM (Fig. 1). SPEM is a common metaplastic phenotype observed in the atrophic human (Yamaguchi et al. 2002; Halldorsdottir et al. 2003) and rodent (Goldenring et al. 2000; Weis et al. 2013) stomach that is highly correlated with the development of intestinal‐type gastric cancer (Zhang et al. 2017).
Figure 1. Schematic diagram of the normal mammalian stomach corpus gland and epithelial reprogramming into SPEM following damage.
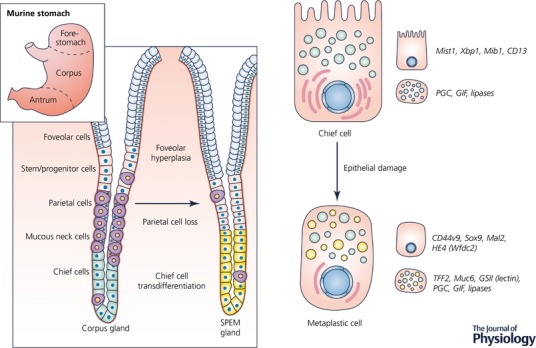
Epithelial damage in the acid‐secreting region of the stomach known as the corpus results in the replacement of normal cell lineages with a metaplastic cell lineage designated SPEM. Damage, such as parietal cell loss, is also associated with expansion of mucus‐secreting foveolar cells. SPEM (yellow cells in the metaplastic gland at right) develops via the transdifferentiation of chief cells (green cells in the normal corpus gland at left) and is characterized by a distinct gene expression profile and protein signature, as well as the production of mucus granules containing Muc6 and TFF2. The SPEM lineage is reminiscent of mucus‐producing cells found in deep antral glands. Mucus‐secreting metaplastic cells develop to protect and fuel the repair of the stomach.
The transdifferentiation of chief cells into SPEM occurs through a highly regulated process that involves dismantling the chief cell secretory architecture and distinct transcriptional changes that include the upregulation of TFF2, Muc6 and CD44 variant 9 (CD44v9) (Fig. 1; Mills & Sansom, 2015). Although currently it cannot be ruled out that some phenotypically metaplastic cells are derived from mucous neck cells or isthmal stem cells, there is convincing evidence that chief cells reprogramme following injury to fuel repair of the stomach (Nam et al. 2010; Hayakawa et al. 2015; Leushacke et al. 2017; Radyk et al. 2017). When this repair process is combined with chronic injury and inflammation, it can put the individual at risk for developing progressive metaplasia and dysplasia. Interestingly, a similar process occurs in the pancreas following injury and inflammation, where digestive enzyme‐secreting acinar cells transdifferentiate into a metaplastic cell lineage analogous to SPEM, a process described as acinar to ductal metaplasia or ADM (Jensen et al. 2005; De La et al. 2008; Shi et al. 2013; Mills & Sansom, 2015).
Utilization of acute damage models
To elucidate the role of the inflammatory infiltrate and cytokines in the transdifferentiation of chief cells into SPEM, our group has turned to acute damage models of metaplasia. In particular, we have focused on the administration of two parietal cell toxic drugs, DMP‐777 and L‐635, which function as parietal cell‐specific protonophores and cause parietal cell necrosis. In addition to acting as a parietal cell‐specific protonophore, DMP‐777 functions as a neutrophil elastase inhibitor and prevents inflammatory infiltration. On the other hand, L‐635 (a chiral cousin of DMP‐777) lacks elastase inhibitor activity and causes parietal cell necrosis with a prominent inflammatory infiltration in only 3 days (Goldenring et al. 2000). The parietal cell loss induced by both drugs is reversible within 10–14 days after the cessation of drug treatment. Similar induction of reversible acute parietal cell has also been observed following administration of high doses of tamoxifen in mice (Huh et al. 2012).
In humans, the loss of parietal cells is thought to occur as a result of chronic Helicobacter pylori infection (Roth et al. 1999; Burkitt et al. 2017). H. pylori infection can take months to cause parietal cell loss in mice, and years in humans. Utilization of drug‐induced models of parietal cell loss allows us to bypass the extended period required for H. pylori‐associated pathology. While treatment with either DMP‐777 or L‐635 results in the emergence of SPEM, the inflammatory infiltrate associated with L‐635 administration or H. pylori infection is required to promote increased proliferation and intestinalization of metaplasia (Petersen et al. 2014). Furthermore, recent work suggests that a coordinated cytokine signalling cascade is required for the emergence and progression of SPEM (Petersen et al. 2017a). By studying the administration of either DMP‐777 or L‐635 to mice lacking components of the immune system, we have gained insights into how this coordinated response influences the development of metaplasia and its pathological progression.
Epithelial alarmins: IL‐33 signalling
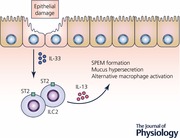
Previous investigations have suggested that interleukin 33 (IL‐33) functions as a secreted signal released from cells at sites of mucosal damage. We have recently reported that mice lacking IL‐33 or a subunit of the IL‐33 receptor complex (ST2) fail to develop metaplasia following acute parietal cell loss (Petersen et al. 2017a). IL‐33 belongs to the IL‐1 family of cytokines. Under healthy conditions, IL‐33 translocates to the nucleus where it participates in regulating gene expression (Carriere et al. 2007). IL‐33 also functions as a stored alarmin that is released when there is cell injury or necrosis that damages the epithelial cell barrier (Buzzelli et al. 2015; Schwartz et al. 2016). Although not well understood, there are also regulated mechanisms of IL‐33 release from stimulated living cells (Kouzaki et al. 2011; Kakkar et al. 2012; Byers et al. 2013). Extracellular IL‐33 works to coordinate immune defenses and repair mechanisms, while also triggering adaptive immune responses. IL‐33 signals through the IL‐33 receptor complex that consists of ST2 and IL1RAcP (Schmitz et al. 2005). In the normal mammalian stomach corpus, IL‐33 is expressed in a subset of surface mucous foveolar epithelial cells (Buzzelli et al. 2015; Petersen et al. 2017a). Following the administration of L‐635 and subsequent parietal cell loss, there is an expansion of IL‐33 expressing foveolar epithelial cells as well as an inflammatory infiltrate, especially macrophages, that produce IL‐33. IL‐33 release and signalling after epithelial injury results in the upregulation of type II cytokines, including interleukin 13 (IL‐13) (Fig. 2; Petersen et al. 2017a). The tissue resident cells in the gastrointestinal tract that express the IL‐33 receptor include mast cells, macrophages, dendritic cells and a cell type that has recently emerged, known as type II innate lymphoid cells or ILC2s (Liew et al. 2010; Molofsky et al. 2015). ILC2s are involved in tissue remodelling and mucous metaplasia through the production of type II cytokines in many tissues (Bernink et al. 2014). ILC2s represent the most likely source of IL‐13 in the stomach downstream of acute parietal cell loss and IL‐33 signalling, although future studies are required to determine this definitively.
Figure 2. The coordinated regulation of metaplasia development.
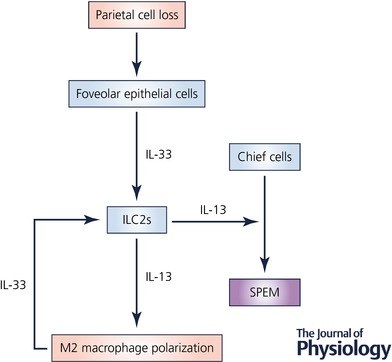
IL‐33 functions as an alarmin released from foveolar epithelial cells following damage to the mucosa, such as parietal cell loss. The IL‐33 receptor (ST2) is expressed on the surface of a variety of tissue‐resident immune cell populations. However, in this case, we hypothesize that type II innate lymphoid cells (ILC2s) respond to IL‐33 by producing type II cytokines including IL‐13. IL‐13 signalling downstream of IL‐33 drives SPEM formation and alternative activation of macrophages. Alternatively activated macrophages subsequently promote the advancement of SPEM to a more proliferative metaplasia and produce IL‐33 which may further promote IL‐13 release.
Type II cytokines and IL‐13 signalling
Robust upregulation of type II cytokines including: IL‐4, IL‐5, IL‐9 and IL‐13 occurs following drug‐induced acute parietal cell loss. The requirement for each of the type II cytokines in the development of metaplasia has been interrogated using knockout models or neutralizing antibodies (Petersen et al. 2017a). Mice lacking IL‐13, but not other type II cytokines, fail to develop metaplasia following acute parietal cell loss. Although not contributing directly to the development of SPEM, the other type II cytokines may work to promote inflammatory infiltration, type II polarization, and maintenance of the response. Interestingly, the addition of recombinant IL‐13 to ST2 knockout mice restores SPEM development following L635‐treatment (Petersen et al. 2017a). IL‐13 is an immunoregulatory cytokine that shares many functional properties with IL‐4. However, IL‐13 appears to be more important for mucus hypersecretion (Grunig et al. 1998; Wills‐Karp et al. 1998), an essential characteristic of SPEM (Weis et al. 2013; Engevik et al. 2016). IL‐13 signals through a heterodimeric receptor system comprising IL4Rα complexed with IL13Rα1 or IL13Rα2 (Seyfizadeh et al. 2015). In the proximal mammalian glandular stomach, IL13Rα1 is localized to the apical membrane of chief cells, but further investigations are required to determine how IL‐13 signalling, downstream of IL‐33, is promoting the transdifferentiation of chief cells (Petersen et al. 2017a).
Macrophage infiltration and polarization
Epithelial damage signals and cytokine release result in substantial inflammatory infiltration into the stomach. This inflammatory infiltration is predominantly made up of neutrophils, eosinophils, T and B cells, and macrophages. The role each of these immune cell populations plays in the development and progression of metaplasia has been studied using genetic mouse models or depletion studies. Previous studies have found that induction of parietal cell loss by Helicobacter infection requires T and B cells (Roth et al. 1999). However, following acute parietal cell loss, metaplasia develops and progresses normally in mice lacking T and B cells, neutrophils and eosinophils. On the other hand, the progression of metaplasia is hindered in clodronate‐treated (macrophage‐depleted) mice (Petersen et al. 2014, 2017a). Depending on the cytokines that macrophages are exposed to, these cells are either classically (type I) activated or alternatively (type II) activated (Labonte et al. 2014; Liu et al. 2014). In this case, IL‐4 and IL‐13 work to alternatively activate macrophages following acute parietal cell loss (Petersen et al. 2014). Transcriptional profiling of macrophages after acute metaplasia development has revealed that IL‐33 is one of the top upregulated genes in macrophages associated with SPEM (Petersen et al. 2017a). Alternatively activated macrophages generally promote the resolution of inflammation and wound repair (Van Dyken & Locksley, 2013). With regard to the advancement of metaplasia, alternatively activated macrophages are required for the upregulation of intestinalizing genes including TFF3, CFTR and DMBT1, as well as SPEM cell proliferation (Petersen et al. 2014). Interestingly, alternatively activated macrophages and IL‐13 signalling are also required for the progression of ADM in the pancreas (Liou et al. 2013, 2017).
Implications of metaplasia regulation by the immune system
Helicobacter‐induced parietal cell loss requires the action of specific lymphocytic immune cell populations. However, recent work suggests that the emergence of SPEM, as a physiological response to damage, does not require the action of inflammatory infiltration into the stomach. Instead, alarmins released from epithelial cells, particularly IL‐33, work to coordinate a cytokine signalling cascade by tissue resident cells that promote the transdifferentiation of chief cells into SPEM. In the setting of chronic injury, inflammatory infiltration, especially alternatively activated macrophages, subsequently drives the progression of metaplasia to become more proliferative with increased intestinal characteristics. Furthermore, profiling studies have revealed that Ras activity is elevated in at least 40% of gastric cancers (Deng et al. 2012; Cancer Genome Atlas Research, 1988). Expression of constitutively active Kras (G12D) in chief cells leads to development of SPEM within 1 month, which progresses to intestinal metaplasia after 3–4 months (Choi et al. 2016). Induction of KRAS in chief cells also results in the infiltration of alternatively activated macrophages into the gastric mucosa (Choi et al. 2016). Thus, activated Kras signalling in the epithelial cells and SPEM may promote pro‐inflammatory signalling that could in turn drive progression of SPEM towards more intestinalized metaplasia.
We hypothesize that the advancement of SPEM to a more proliferative and/or intestinalized metaplasia increases the risk for developing dysplasia in the gastric mucosa and intestinal‐type gastric cancer. Not only has this work connected the pathogenesis of metaplasia in the stomach to pancreatic ADM, but there are also parallels to other type II‐mediated diseases including fungal allergy and asthma, all of which involve robust tissue remodelling and increased mucus secretion. It seems likely that zymogenic cells in a number of mucosal contexts including stomach, pancreas, salivary glands and perhaps even Paneth cells in the small intestine, may respond to injury by transdifferentiating into mucus‐secreting lineages. While such processes are normally programmed to recede following resolution of injury, in the face of continued damage and chronic inflammation, these reparative metaplastic lineages may evolve into more proliferative and self‐renewing pre‐neoplastic or dysplastic lineages that can predispose to development of cancer.
Additional information
Competing interests
The authors have declared that no conflict of interest exists.
Funding
J.R.G. is supported by grants from a Department of Veterans Affairs Merit Review Award (I01BX000930), a Department of Defense IDEA grant (CA160479) and NIH RO1 DK071590. A.R.M. is supported by NIH T32 GM008554.
Biographies
Anne Meyer is in her third year of graduate school in the Cell Developmental Biology Department at Vanderbilt University. Anne is interested in the impact of inflammation on the development and progression of cancer. She joined the Goldenring laboratory in May of 2016 and is focused on elucidating the initiating mechanisms of metaplasia development in the stomach.

James Goldenring is a Professor of Surgery and Cell and Developmental Biology and Co‐Director of the Epithelial Biology Centre at Vanderbilt University School of Medicine. Dr Goldenring first described spasmolytic polypeptide‐expressing metaplasia (SPEM) two decades ago as a possible precancerous metaplasia. The mechanisms responsible for induction of SPEM and its links to intestinal metaplasia and intestinal‐type gastric cancer are the continuing research interests of the Goldenring lab.
Edited by: Kim Barrett & Ole Petersen
This review was presented at the symposium ‘Gastrointestinal Tract XVII: Current Biology of the GI Tract, Mucosa, Microbiota, and Beyond’, which took place at FASEB 2017, Steamboat Springs, Colorado, USA, 30 July–4 August 2017.
References
- Bernink JH, Germar K & Spits H (2014). The role of ILC2 in pathology of type 2 inflammatory diseases. Curr Opin Immunol 31, 115–120. [DOI] [PubMed] [Google Scholar]
- Burkitt MD, Duckworth CA, Williams JM & Pritchard DM (2017). Helicobacter pylori‐induced gastric pathology: insights from in vivo and ex vivo models. Dis Model Mech 10, 89–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzelli JN, Chalinor HV, Pavlic DI, Sutton P, Menheniott TR, Giraud AS & Judd LM (2015). IL33 is a stomach alarmin that initiates a skewed Th2 response to injury and infection. Cell Mol Gastroenterol Hepatol 1, 203–221 e203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers DE, Alexander‐Brett J, Patel AC, Agapov E, Dang‐Vu G, Jin X, Wu K, You Y, Alevy Y, Girard JP, Stappenbeck TS, Patterson GA, Pierce RA, Brody SL & Holtzman MJ (2013). Long‐term IL‐33‐producing epithelial progenitor cells in chronic obstructive lung disease. J Clin Invest 123, 3967–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2014). Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, Bouche G & Girard JP (2007). IL‐33, the IL‐1‐like cytokine ligand for ST2 receptor, is a chromatin‐associated nuclear factor in vivo. Proc Natl Acad Sci USA 104, 282–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi E, Hendley AM, Bailey JM, Leach SD & Goldenring JR (2016). Expression of activated ras in gastric chief cells of mice leads to the full spectrum of metaplastic lineage transitions. Gastroenterology 150, 918–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa P ( 1988). A human model of gastric carcinogenesis. Cancer Res 48, 3554–3560. [PubMed] [Google Scholar]
- Correa P, Piazuelo MB & Wilson KT (2010). Pathology of gastric intestinal metaplasia: clinical implications. Am J Gastroenterol 105, 493–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La OJ, Emerson LL, Goodman JL, Froebe SC, Illum BE, Curtis AB & Murtaugh LC (2008). Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci USA 105, 18907–18912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D & Plummer M (2012). Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol 13, 607–615. [DOI] [PubMed] [Google Scholar]
- Demitrack ES, Aihara E, Kenny S, Varro A & Montrose MH (2012). Inhibitors of acid secretion can benefit gastric wound repair independent of luminal pH effects on the site of damage. Gut 61, 804–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng N, Goh LK, Wang H, Das K, Tao J, Tan IB, Zhang S, Lee M, Wu J, Lim KH, Lei Z, Goh G, Lim QY, Tan AL, Sin Poh DY, Riahi S, Bell S, Shi MM, Linnartz R, Zhu F, Yeoh KG, Toh HC, Yong WP, Cheong HC, Rha SY, Boussioutas A, Grabsch H, Rozen S & Tan P (2012). A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co‐occurrence among distinct therapeutic targets. Gut 61, 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engevik AC, Feng R, Choi E, White S, Bertaux‐Skeirik N, Li J, Mahe MM, Aihara E, Yang L, DiPasquale B, Oh S, Engevik KA, Giraud AS, Montrose MH, Medvedovic M, Helmrath MA, Goldenring JR & Zavros Y (2016). The development of spasmolytic polypeptide/TFF2‐expressing metaplasia (SPEM) during gastric repair is absent in the aged stomach. Cell Mol Gastroenterol Hepatol 2, 605–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenring JR, Ray GS, Coffey RJ, Meunier PC, Haley PJ, Barnes TB & Car BD (2000). Reversible drug‐induced oxyntic atrophy in rats. Gastroenterology 118, 1080–1093. [DOI] [PubMed] [Google Scholar]
- Grunig G, Warnock M, Wakil AE, Venkayya R, Brombacher F, Rennick DM, Sheppard D, Mohrs M, Donaldson DD, Locksley RM & Corry DB (1998). Requirement for IL‐13 independently of IL‐4 in experimental asthma. Science 282, 2261–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halldorsdottir AM, Sigurdardottrir M, Jonasson JG, Oddsdottir M, Magnusson J, Lee JR & Goldenring JR (2003). Spasmolytic polypeptide‐expressing metaplasia (SPEM) associated with gastric cancer in Iceland. Dig Dis Sci 48, 431–441. [DOI] [PubMed] [Google Scholar]
- Hayakawa Y, Ariyama H, Stancikova J, Sakitani K, Asfaha S, Renz BW, Dubeykovskaya ZA, Shibata W, Wang H, Westphalen CB, Chen X, Takemoto Y, Kim W, Khurana SS, Tailor Y, Nagar K, Tomita H, Hara A, Sepulveda AR, Setlik W, Gershon MD, Saha S, Ding L, Shen Z, Fox JG, Friedman RA, Konieczny SF, Worthley DL, Korinek V & Wang TC (2015). Mist1 expressing gastric stem cells maintain the normal and neoplastic gastric epithelium and are supported by a perivascular stem cell niche. Cancer Cell 28, 800–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann W ( 2015). TFF2, a MUC6‐binding lectin stabilizing the gastric mucus barrier and more (Review). Int J Oncol 47, 806–816. [DOI] [PubMed] [Google Scholar]
- Huh WJ, Khurana SS, Geahlen JH, Kohli K, Waller RA & Mills JC (2012). Tamoxifen induces rapid, reversible atrophy, and metaplasia in mouse stomach. Gastroenterology 142, 21–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen JN, Cameron E, Garay MV, Starkey TW, Gianani R & Jensen J (2005). Recapitulation of elements of embryonic development in adult mouse pancreatic regeneration. Gastroenterology 128, 728–741. [DOI] [PubMed] [Google Scholar]
- Kakkar R, Hei H, Dobner S & Lee RT (2012). Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J Biol Chem 287, 6941–6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karam SM & Leblond CP (1993). Dynamics of epithelial cells in the corpus of the mouse stomach. I. Identification of proliferative cell types and pinpointing of the stem cell. Anat Rec 236, 259–279. [DOI] [PubMed] [Google Scholar]
- Kouzaki H, Iijima K, Kobayashi T, O'Grady SM & Kita H (2011). The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL‐33 release and innate Th2‐type responses. J Immunol 186, 4375–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labonte AC, Tosello‐Trampont AC & Hahn YS (2014). The role of macrophage polarization in infectious and inflammatory diseases. Mol Cells 37, 275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leushacke M, Tan SH, Wong A, Swathi Y, Hajamohideen A, Tan LT, Goh J, Wong E, Denil S, Murakami K & Barker N (2017). Lgr5‐expressing chief cells drive epithelial regeneration and cancer in the oxyntic stomach. Nat Cell Biol 19, 774–786. [DOI] [PubMed] [Google Scholar]
- Liew FY, Pitman NI & McInnes IB (2010). Disease‐associated functions of IL‐33: the new kid in the IL‐1 family. Nat Rev Immunol 10, 103–110. [DOI] [PubMed] [Google Scholar]
- Liou GY, Bastea L, Fleming A, Doppler H, Edenfield BH, Dawson DW, Zhang L, Bardeesy N & Storz P (2017). The presence of Interleukin‐13 at pancreatic ADM/PanIN lesions alters macrophage populations and mediates pancreatic tumorigenesis. Cell Reports 19, 1322–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou GY, Doppler H, Necela B, Krishna M, Crawford HC, Raimondo M & Storz P (2013). Macrophage‐secreted cytokines drive pancreatic acinar‐to‐ductal metaplasia through NF‐kappaB and MMPs. J Cell Biol 202, 563–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YC, Zou XB, Chai YF & Yao YM (2014). Macrophage polarization in inflammatory diseases. Int J Biol Sci 10, 520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills JC & Sansom OJ (2015). Reserve stem cells: Differentiated cells reprogram to fuel repair, metaplasia, and neoplasia in the adult gastrointestinal tract. Sci Signal 8, re8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AB, Savage AK & Locksley RM (2015). Interleukin‐33 in tissue homeostasis, injury, and inflammation. Immunity 42, 1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam KT, Lee HJ, Sousa JF, Weis VG, O'Neal RL, Finke PE, Romero‐Gallo J, Shi G, Mills JC, Peek RM Jr, Konieczny SF & Goldenring JR (2010). Mature chief cells are cryptic progenitors for metaplasia in the stomach. Gastroenterology 139, 2028–2037 e2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen CP, Meyer AR, De Salvo C, Choi E, Schlegel C, Petersen A, Engevik AC, Prasad N, Levy SE, Peebles RS, Pizarro TT & Goldenring JR (2017a). A signalling cascade of IL‐33 to IL‐13 regulates metaplasia in the mouse stomach. Gut (in press; 10.1136/gutjnl-2016-312779). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen CP, Mills JC & Goldenring JR (2017b). Murine Models of Gastric Corpus Preneoplasia. Cell Mol Gastroenterol Hepatol 3, 11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen CP, Weis VG, Nam KT, Sousa JF, Fingleton B & Goldenring JR (2014). Macrophages promote progression of spasmolytic polypeptide‐expressing metaplasia after acute loss of parietal cells. Gastroenterology 146, 1727–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radyk MD, Burclaff J, Willet SG & Mills JC (2017). Metaplastic cells in the stomach arise, independently of stem cells, via dedifferentiation or transdifferentiation of chief cells. Gastroenterology (in press; 10.1053/j.gastro.2017.11.278). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth KA, Kapadia SB, Martin SM & Lorenz RG (1999). Cellular immune responses are essential for the development of Helicobacter felis‐associated gastric pathology. J Immunol 163, 1490–1497. [PubMed] [Google Scholar]
- Schmidt PH, Lee JR, Joshi V, Playford RJ, Poulsom R, Wright NA & Goldenring JR (1999). Identification of a metaplastic cell lineage associated with human gastric adenocarcinoma. Lab Invest 79, 639–646. [PubMed] [Google Scholar]
- Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, Gorman DM, Bazan JF & Kastelein RA (2005). IL‐33, an interleukin‐1‐like cytokine that signals via the IL‐1 receptor‐related protein ST2 and induces T helper type 2‐associated cytokines. Immunity 23, 479–490. [DOI] [PubMed] [Google Scholar]
- Schwartz C, O'Grady K, Lavelle EC & Fallon PG (2016). Interleukin 33: an innate alarm for adaptive responses beyond Th2 immunity‐emerging roles in obesity, intestinal inflammation, and cancer. Eur J Immunol 46, 1091–1100. [DOI] [PubMed] [Google Scholar]
- Seyfizadeh N, Seyfizadeh N, Gharibi T & Babaloo Z (2015). Interleukin‐13 as an important cytokine: a review on its roles in some human diseases. Acta Microbiol Immunol Hung 62, 341–378. [DOI] [PubMed] [Google Scholar]
- Shi G, DiRenzo D, Qu C, Barney D, Miley D & Konieczny SF (2013). Maintenance of acinar cell organization is critical to preventing Kras‐induced acinar‐ductal metaplasia. Oncogene 32, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyken SJ & Locksley RM (2013). Interleukin‐4‐ and interleukin‐13‐mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol 31, 317–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RH ( 2015). From reflux esophagitis to Barrett's esophagus and esophageal adenocarcinoma. World J Gastroenterol 21, 5210–5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis VG, Sousa JF, LaFleur BJ, Nam KT, Weis JA, Finke PE, Ameen NA, Fox JG & Goldenring JR (2013). Heterogeneity in mouse spasmolytic polypeptide‐expressing metaplasia lineages identifies markers of metaplastic progression. Gut 62, 1270–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills‐Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL & Donaldson DD (1998). Interleukin‐13: central mediator of allergic asthma. Science 282, 2258–2261. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Goldenring JR, Kaminishi M & Lee JR (2002). Identification of spasmolytic polypeptide expressing metaplasia (SPEM) in remnant gastric cancer and surveillance postgastrectomy biopsies. Dig Dis Sci 47, 573–578. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen JN, Dong M, Zhang ZG, Zhang YW, Wu JY, Du H, Li HG, Huang Y & Shao CK (2017). Clinical significance of spasmolytic polypeptide‐expressing metaplasia and intestinal metaplasia in Epstein‐Barr virus‐associated and Epstein‐Barr virus‐negative gastric cancer. Hum Pathol 63, 128–138. [DOI] [PubMed] [Google Scholar]