

Abstract
Key points
Endothelial dysfunction is an early hallmark of multiple disease states that also display sex differences with respect to age of onset, frequency and severity.
Results of in vivo studies of basal and stimulated microvascular barrier function revealed sex differences that are difficult to ascribe to specific cells or environmental factors.
The present study evaluated endothelial cells (EC) isolated from macro‐ and/or microvessels of reproductively mature rats under the controlled conditions of low‐passage culture aiming to test the assumption that EC phenotype would be sex independent.
The primary finding was that EC, regardless of where they are derived, retain a sex‐bias in low‐passage culture, independent of varying levels of reproductive hormones. The implications of the present study include the fallacy of expecting a universal set of mechanisms derived from study of EC from one sex and/or one vascular origin to apply uniformly to all EC under unstimulated conditions, and no less in disease.
Abstract
Vascular endothelial cells (EC) are heterogeneous with respect to phenotype, reflecting at least the organ of origin, location within the vascular network and physical forces. As an independent influence on EC functions in health or aetiology, susceptibility, and progression of dysfunction in numerous disease states, sex has been largely ignored. The present study focussed on EC isolated from aorta (macrovascular) and skeletal muscle vessels (microvascular) of age‐matched male and female rats under identical conditions of short‐term (passage 4) culture. We tested the hypothesis that genomic sex would not influence endothelial growth, wound healing, morphology, lactate production, or messenger RNA and protein expression of key proteins (sex hormone receptors for androgen and oestrogens α and β; platelet endothelial cell adhesion molecule‐1 and vascular endothelial cadherin mediating barrier function; αvβ3 and N‐cadherin influencing matrix interactions; intracellular adhesion molecule‐1 and vascular cell adhesion molecule‐1 mediating EC/white cell adhesion). The hypothesis was rejected because the EC origin (macro‐ vs. microvessel) and sex influenced multiple phenotypic characteristics. Statistical model analysis of EC growth demonstrated an hierarchy of variable importance, recapitulated for other phenotypic characteristics, with predictions assuming EC homogeneity < sex < vessel origin < sex and vessel origin. Furthermore, patterns of EC mRNA expression by vessel origin and by sex did not predict protein expression. Overall, the present study demonstrated that accurate assessment of sex‐linked EC dysfunction first requires an understanding of EC function by position in the vascular tree and by sex. The results from a single EC tissue source/species/sex cannot provide universal insight into the mechanisms regulating in vivo endothelial function in health, and no less in disease.
Keywords: Endothelial cell, Microvascular function, macrovascular function, wound healing, Sexual dimorphism, Metabolism, Reproductive Hormones
Key points
Endothelial dysfunction is an early hallmark of multiple disease states that also display sex differences with respect to age of onset, frequency and severity.
Results of in vivo studies of basal and stimulated microvascular barrier function revealed sex differences that are difficult to ascribe to specific cells or environmental factors.
The present study evaluated endothelial cells (EC) isolated from macro‐ and/or microvessels of reproductively mature rats under the controlled conditions of low‐passage culture aiming to test the assumption that EC phenotype would be sex independent.
The primary finding was that EC, regardless of where they are derived, retain a sex‐bias in low‐passage culture, independent of varying levels of reproductive hormones. The implications of the present study include the fallacy of expecting a universal set of mechanisms derived from study of EC from one sex and/or one vascular origin to apply uniformly to all EC under unstimulated conditions, and no less in disease.
Introduction
Endothelial dysfunction often precedes manifestation of multiple chronic conditions, ranging from cardiovascular diseases, such as peripheral vascular disease, stroke and myocardial infarction, to metabolic diseases including type 2 diabetes, to autoimmune rheumatic disease such as scleroderma. Consequently, many studies focus on endothelial cell (EC) biology by isolating EC from the multicellular environment of intact vessels and then maintaining and studying them under the controlled conditions of cell culture. Valuable insight has been gained with respect to EC signalling and molecular mechanisms regulating the function of these cells in health and disease. Mismatches, however, exist between the results of these studies and the behaviour of EC in situ. Furthermore, there are tacit assumptions that, until tested, call into question the appropriateness of drawing universal conclusions from isolated cell, single vessel or organ studies.
One untested assumption has been that the sex of the animal or human model is without influence on phenotype. Over the last 15 years, evidence of sex differences has emerged regarding fundamental properties ascribed primarily to the endothelium, most notably barrier function, for which permeability responses of in situ and isolated microvessels to a variety of vasoactive mediators differ in males and females of the same species (Huxley et al. 2004, 2007; Huxley, 2007; Huxley & Wang, 2010; Wang et al. 2010; Panazzolo et al. 2013). These studies have been limited in the ability to drill down to the scale of cellular signalling mechanisms responsible for sexual dimorphism, reflecting the difficulty of visualizing intracellular function in the small number of endothelial cells that make up a microvessel segment in vivo. Of note, many of the diseases displaying early endothelial dysfunction mentioned above also display sexual dimorphism with respect to occurrence and manifestation. For example, the incidence and progression of human cardiovascular disease, from the macrovasculature to the microvasculature, differs by sex (Zimmerman & Sullivan, 2013). At the macrovascular level, atherosclerosis occurs more often with greater ill effects in males, whereas pulmonary arterial hypertension occurs with higher frequency in females (de Jesus Perez, 2011). Vascular disorders, such as Hypothernar hammer syndrome (Hui‐Chou & McClinton, 2015), occur more frequently in males, whereas up to 80% Raynaud's disease patients are female (Fraenkel, 2002). The existence of sex differences has resulted in much of the work focussing on the role reproductive sex hormones and few studies have considered the role of EC being XX or XY. To rectify the lack of fundamental knowledge of basal genomic sex in EC physiology and to gain insight into the mechanisms responsible for our observed sexual dimorphism in in situ and in isolated microvessel permeability and its control, the present study evaluated the hypothesis that basal endothelial cell phenotype is the same for age‐matched, sexually mature males and females.
A second confounding factor often overlooked in the interpretation of results from studies of cultured EC has been the organ or vessel location from which the cells were isolated. For decades, it has been known that EC phenotype can vary by organ (e.g. brain, heart, kidney or lung), vessel size (conduit, feed or microvessel) and position within the microvascular network (arteriole, capillary or venule) (Garlanda & Dejana, 1997; Craig et al. 1998; Stevens et al. 2001; Tse & Stan, 2010; Aird, 2015) because it is well understood that the endothelium, by virtue of its existence at the interface between flowing blood and metabolizing tissue, is subjected to differing environmental conditions (Aird, 2001, 2007a, 2015). Despite these data, it is not uncommon for the results from studies using human umbilical vein endothelium (HUVEC), for example, to be interpreted as universally applicable to endothelial cell physiology. Until recently, it was assumed that the aforementioned differences were diminished under the conditions of cell culture (Aird, 2005).
The notion that genomic sex and also whether the EC are derived from XX or XY animals could be important and distinct from the contributions of the reproductive hormones originates from studies on sexually immature animals. We observed sex‐specific responses under basal and activated conditions in juvenile rats when reproductive hormones were low and unchanging (Wang, 2005; Sasaki et al. 2006; Wang & Huxley, 2006; Sasaki, 2007). These data, consistent with a genomic contribution to sex differences, infer that the assumption of the basal properties of EC from males and females being equivalent, even in the absence of reproductive hormones, may be inappropriate.
Cell culture, despite its limitations (Durr et al. 2004; Aird, 2005), was chosen to evaluate whether selected phenotypes of macrovascular and microvascular endothelium from males and females were equivalent in the absence of changing sex hormones. In this model, the EC, once isolated, could be maintained under identical conditions with respect to environment, passage number, plating conditions, cell density, substrate, and nutrient content to limit and match multiple variables. EC were isolated from aorta (conduit, macrovessel) and skeletal muscle (microvessel) of age‐matched, sexually mature, Sprague–Dawley rats. The physiological functions assessed were EC morphology, growth, metabolism and wound healing. In addition, mRNA and protein expression of selected sex hormone receptors and barrier, adhesion, and integrin proteins were measured and compared.
Multiple differences in several EC functions between aorta and skeletal muscle microvessels (SKMs) were found, with some expected and some not. EC heterogeneity of function between the two vascular sources differed significantly by sex, demonstrating outright rejection of the hypothesis that endothelial phenotype, even from the same source maintained under identical conditions, does not differ by sex. Finally, one interpretation of the statistical model analysis of the data for EC growth was consistent with an hierarchy of importance of variables such that model has predictions assuming EC homogeneity < grouping by sex < grouping by vessel origin < grouping by sex and vessel origin. This hierarchy was recapitulated for several of the phenotypic characteristics tested. The implication of this work is that future studies of EC function require understanding of basal characteristics not only by position in the vascular tree, but also by sex. Only once these data are known will it be possible to assess accurately EC dysfunction and its impact on males and females. Scaling from the results in culture to the intact vasulature will require an assessment of function under more physiological conditions of changing solute concentrations, shear stress, transmural fluid flux and flow, in addition to ageing, circadian rhythms and exposure to reproductive hormones. Furthermore, the results from a given EC tissue source/species/sex cannot be expected to provide universally applicable answers regarding the mechanisms regulating endothelial function in health, and no less in disease.
Methods
To define clearly the mechanisms underlying endothelial function in health and disease, it is imperative to know first whether the phenotype of EC from males and females exposed to identical conditions differs in the absence of changing but measured levels of reproductive hormones. We chose to work with EC isolated from aorta and skeletal muscle of age‐matched, sexually mature Sprague–Dawley rats under conditions of cell culture at the same early passage, exposed to identical media and environment. The phenotypic characteristics covered in the present study included aspects of morphology, growth, wound healing, metabolism, and gene and protein expression of selected markers. Conditions of cell culture, limitations not withstanding, facilitated sufficient cell numbers to carry out the present study using a minimum number of animals.
Ethical approval
The protocols for the handling, care and subsequent endothelial cell harvest from Sprague–Dawley rats of both sexes were approved by the University of Missouri‐Columbia Institutional Animal Care and Use Committee in accordance with the National Institute of Health's Guide for the Care and Use of Laboratory Animals and conform with the guidelines outlined in Grundy (2015).
Animals
Young sexually mature (>63 days, generally 67—83 days old) male (275–375 g) and female (200–300 g) Sprague–Dawley rats (Envigo, Huntingdon, Cambridgeshire, UK) were used for these studies. The protocols for the rat handling and endothelial cell harvest were approved by the University of Missouri‐Columbia Institutional Animal Care and Use Committee in acordance with the National Institute of Health's Guide for the Care and Use of Laboratory Animals. The anaesthetized rats were killed following tissue harvest with an overdose of the anaesthetic Inactin hydrate C‐IIIN (Thiobarbital, Sigma‐Aldrich, St Louis, MO, USA) followed by exsanguination.
General tissue preparation
To lessen the likelihood of mixing the sexes, increase the total tissue harvest and randomize the populations of cells, two rats of the same sex were weighed and then anaesthetized by i.m. injection of Inactin (130 mg kg–1). Once sedated fully, fur and skin were removed from the anterior abdominal wall. Abdominal skeletal muscle was isolated using the methods described by Wang et al. (2010). Briefly, the abdominal free wall was removed, rinsed in Dulbecco's phosphate buffered saline (D‐PBS, calcium and magnesium‐free, with gentamicin and amphotericin B) and then placed into Hank's balanced salt solution supplemented with L‐glutamine, Na pyruvate, 20 mm Hepes, gentamicin and amphotericin B. Next, microvascular networks were isolated from the external and internal free wall layers of the muscle by dissecting away fascia, fat, nerves and myocytes. The endothelial cells were then prepared in a multistep process combining enzymatic digestion (Wang et al. 2010), centrifugation, re‐suspension and selection with platelet endothelial cell adhesion molecule‐1 (PECAM‐1, CD‐31)‐coated magnetic beads (MagnaBind Goat Anti‐Mouse; Thermo Scientific, Waltham, MA, USA) before initial plating into tissue culture flasks coated with 0.2% w/v gelatin (bovine Type B; Sigma‐Aldrich). Aortas from the same rats were treated in the same manner as the SKMs, except that the aortic endothelial layer was removed by direct application of the enzymatic cocktail to the excised vessel that had been opened and pinned flat on a Sylgard 184 (Dow Corning, Midland, MI, USA) coated dish. The next day, for each preparation, the media was exchanged with fresh D‐PBS to remove debris, dead cells and excess magnetic beads. Once multiple cell colonies were established, they were removed from the flask by trypsinization and subjected to a second round of CD‐31 selection to ensure purity of the EC type. Cells at this stage were used for the measures of mRNA. The culture medium consisted of phenol red‐free M199 (Gibco®; Thermo Scientific) containing 20 mm sodium pyruvate, GlutaMAX (1 × final), 1 U of heparin (sodium salt, Grade I‐A, Porcine Intestinal Mucosa; Sigma‐Aldrich), 5.24 × 10−2 mm gentamicin (Fresenius Kabi USA, Lake Zurich, IL, USA), fetal bovine serum (FBS) (20% v/v F6178; Sigma‐Aldrich) and 50 mg of endothelial cell growth supplement (Corning, Bedford, MA, USA). Phenol red was omitted from the media to eliminate contributions of a weak phyto‐oestrogen. Independent laboratory analysis (LabCorp, Burlington, NC, USA) of the 20% v/v FBS used in all of these studies revealed no detectable insulin, <0.1 ng mL–1 progesterone, 7.3 ng dL–1 testosterone, 24 pg mL–1 total oestrogens, <0.1 ng mL–1 growth hormone and <80 μg L–1 insulin‐like growth factor (IGF) binding protein‐3.
Except for the study of EC growth and wound healing, once the cells were ∼80% confluent, they were again washed, trypsinized and replated for a total of four passages to obtain sufficient cell numbers. An aliquot was removed for cell counting (Cellometer Auto T4; Nexcelom Bioscience, St Lawrence, MA, USA) in accordance with the manufacturer's instructions to prepare the cells freezing at –70°C with transfer to liquid nitrogen after at least 24 h at a concentration of 2 × 105 cell mL–1. Unless specified otherwise, all studies were performed on EC at passage 4.3–4.4 (four passages to freeze down, three or four passages following removal from liquid nitrogen). The results represent a total of eight preparations (16 pairs of rats) over 4 years.
Endothelial cell culture and identification
EC verification of primary, pre‐freezing and early passage (4.4) cells was achieved by staining for PECAM‐1, vascular endothelial cadherin (VE‐CAD), von Willebrand's factor, uptake of acetylated‐low density lipoprotein, binding of isolectin IB‐4 from Griffonia simplifolica and capillary‐like tube formation on a Matrigel (Corning) coated surface as described by Wang et al. (2010) (Fig. 1 A–E). Identification of cell sex was confirmed by the presence (male) or absence (female) of the testis‐determining SRY gene by PCR (Fig. 1 F). As a laboratory standard, EC verification and SRY determinations were performed after each EC isolation procedure prior to freezing.
Figure 1. Verification of endothelial cell phenotype and sex.
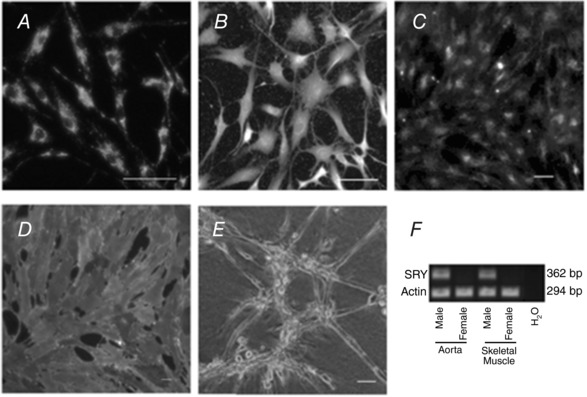
Immunofluorescence staining of the endothelial cells was positive for PECAM‐1 in (A) and von Willebrand's factor in (B). The EC took up acetylated‐low density lipoprotein (Ac‐LDL) (C), stained positively with the lectin Griffonia simplicifolia IB4 (GS‐IB4) (D) and formed a tube‐like structure when plated on Matrigel (E). Scale bar = 50 μm. Male sex was confirmed using quantitative RT‐PCR for the presence of the sex‐determining region Y (SRY) protein (lanes 1 and 3) vs. its absence in endothelial cells from females (lanes 2 and 4). Actin was used as the loading control, whereas sterile water was used as a negative control (F).
The aim was to provide a consistent environment for the isolated EC for the conduct of all subsequent experiments to determine whether sex was an independent determinant of endothelial cell phenotype.
Cell morphology: EC size and spread
The size of freely suspended endothelial cells was determined from counts of freshly dispersed cells using a Cellometer Auto T4 imaging cytometer and associated Cellometer Auto software (Nexcelom Bioscience).
The extent of EC spread (planar area measurements) and nuclear area were determined using fluorescence images of EC grown to confluence, permeabilized and stained with labelled antibodies used for in‐cell westerns. The primary antibodies were visualized via green immunofluorescence staining (Alexa 488, excitation 499 nm; emission 520 nm) and 4',6‐diamidino‐2‐phenylindole (DAPI) nuclear staining at blue emission wavelengths (excitation 354 nm; emission 461 nm), respectively.
Fluorescence imaging procedure
The final fixed, permeabilized and stained cells were imaged with a confocal laser‐scanning microscope (TCP SP8 MP; Leica Microsystems, Wetzlar, Germany) in the Molecular Cytology Core of the University of Missouri‐Columbia using a constant set of settings [laser intensity, shutter speed, gain, threshold, region of interest (ROI), etc., optimized for viewing with a 20× and a 63× oil immersion lens, respectively]. Images were taken at an excitation of 605–700 nm (to target tubulin protein fluorescence) and again using the automated excitation wavelength setting (350 ± 10 nm) for the nuclear stain DAPI, and stored for offline analysis of cell number, cell area and nuclear area. The stored images were analysed using ImageJ (NIH, Bethesda, MD, USA) (Schneider et al. 2012) to obtain the corrected total cell fluorescence of eight‐bit grey scale images. From these images, two areas were determined: the area of EC cytoplasm and the area of nuclei in a defined ROI that was used for all analyses. The area of cytoplasm, or area of nuclear staining, was determined using a threshold mask within the ROI. Cell number within the ROI was obtained from a count of areas staining with DAPI. Background intensity was subtracted for each image after a measure of light intensity over a blank portion of the same slide under the same magnification and settings.
Growth curves
Six 96‐well plates (leaving blank the perimeter cells) were coated with gelatin (see procedures with respect to general tissue preparation) and then seeded with 105 cells well–1 in culture medium containing 10% v/v FBS (triplicates of male aorta, female aorta, male SKM and female SKM) and incubated at 37°C with 18.6% O2, 5% CO2 and balance nitrogen. At 24 h intervals, one plate was removed from the incubator; once the media was drained, it was wrapped in foil and frozen at –70°C. Once six plates were collected, they were thawed cell lysis buffer and diluted with CyQUANT stock added to the wells in accordance with the instructions provided with the CyQuant Cell Proliferation kit (Invitrogen, Carlsbad, CA, USA). Fluorescence intensity was measured for each well along with a set of control samples because the assay is based on the binding of a cellular nucleic acid‐avid dye to form a complex for which the fluorescence intensity increases and correlates linearly with cell number (Jones et al. 2001).
Lactate metabolism
A colourimetric approach was used to detect lactate, the end product of glycolysis. The assay kit (glycolysis cell‐based assay kit #600450; Cayman Chemical Company, Ann Arbor, MI, USA), containing lactate dehydrogenase to catalyse lactate oxidation to form pyruvate, also contained a tetrazolium substrate that formed a coloured (formazan) product on binding the NADH produced by the coupled reaction.
Cells for the glycolysis assay were grown for 48 h on 96‐well plates following seeding at 105 cells well–1 (triplicates wells for each tissue and sex) in Cellgro media (Corning) plus 10% v/v FBS, washed with PBS and then provided with Cellgro media plus 4% w/v BSA (A4362; Sigma‐Aldrich) to incubate for 4 h at 37°C. At this point, the kit instructions were followed to determine lactate content from absorbance readings converted to lactate concentration from a standard curve. Lactate production was expressed on a per cell basis as a result of measured sex and tissue differences in EC volume and spread.
Wound healing
EC for each of the four groups were grown after harvest in 25 mm2 gelatin‐coated dishes; on attainment of confluence, they were scratched with a pipette tip in two parallel lines. They were then imaged on a black and white charge‐coupled device camera in four separate marked locations at 0, 2, 4, 6, 22 and 24 h following wounding. Imaging for wound area was analysed using ImageJ calibrated against the image of a stage micrometer. The data from the four areas were averaged for a total of six replicates for each sex and tissue.
mRNA and protein expression
RNA was extracted (RNeasy Mini Kit; Qiagen, Valencia, CA, USA) from rat primary EC cultures (single passage following isolation, removal of debris and an additional selection with PECAM coated beads) for first‐strand DNA synthesis (High‐Capacity cDNA Reverse Transcription Kit; Applied Biosystems, Foster City, CA, USA). Quantitative RT‐PCR was performed using the CFX96 Real‐Time PCR Detection System (Bio‐Rad, Hercules, CA, USA) with SYBR green intercalating dye (Takara SYBR Premix Ex Taq; Takara‐Clontech, Mountain View, CA, USA). Primer sequences obtained from Roche's Universal ProbeLibrary (https://lifescience.roche.com/en_gb/brands/universal‐probe‐library.html) [the NCBI RefSeq numbers used were NM_017008 GAPDH, NM_012502.1 (AR), NM_012689.1 (ERa), NM_012754.1 (ERb), NM_031591.1 (PECAM1), NM_012967.1 (ICAM1), NM_012889.1 (VCAM1), NM_031333.1 (N‐Cad) and NM_001107407.1 (VE‐Cad)] were synthesized by Integrated DNA Technologies (Coralville, IA, USA). Several of the genes used commonly for a ‘housekeeping function’ displayed differences by sex (data not shown). Because we found that glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH; Integrated DNA Technologies) did not display sexual dimorphism for the cells and sites chosen, this protein was used to facilitate calculation of the relative mRNA expression levels by the 2−ΔΔC T method (Livak & Schmittgen, 2001).
Flow cytometry was used to measure protein content quantitatively. Accordingly, confluent cells were detached by trypsin digestion, counted and re‐suspended in 2% v/v FBS in PBS to a count of ∼1–5 × 106 cells mL–1 for preparation of control and experimental samples. Three controls were used for each vessel network origin/sex: a negative control with no antibody, a negative control with secondary antibody to mouse and a negative control with secondary antibody to rabbit. The cells were then fixed in 0.02% v/v paraformaldehyde in PBS prior to permeabilization with 0.1% v/v Triton X‐100 in PBS. Cells were first exposed to primary antibody, incubated and washed. The process was repeated in the dark for the addition of the secondary antibody. All primary antibodies except for αvβ3, which was labelled with a fluorescence probe, used a secondary fluorescence‐labelled antibody that was either anti‐mouse or anti‐rabbit. All flow cytometry, performed within 2 h of the final wash and with samples placed on ice, used a LSR Fortessa X‐20 counter (Becton‐Dickinson Biosciences, Franklin Lakes, NJ, USA) in the Cell Immunology Core at the University of Missouri.
The flow cytometry data were analysed using FlowJo software (FlowJo LLC, Ashland, OR, USA). Minimal gating was performed to eliminate signal from potential debris; gating was applied uniformly to the negative controls and target proteins. Figure 2 provides an example of the frequency histogram of the gated data for αvβ3‐labelled Alexa Fluor‐647 fluorescence providing the relative intensity per cell (each point), with all experiments being performed in duplicate on a second batch of cells at passage 4.4. Protein expression by EC from the males (blue) and females (red) were then compared with their respective negative controls (green). Accordingly, median fluorescence intensity of the negative control was subtracted from the median fluorescence intensity of experimental group. Medians were used as these graphs are logarithmic (i.e. the populations were not normally distributed).
Figure 2. Flow cytometry was used to analyse protein expression.
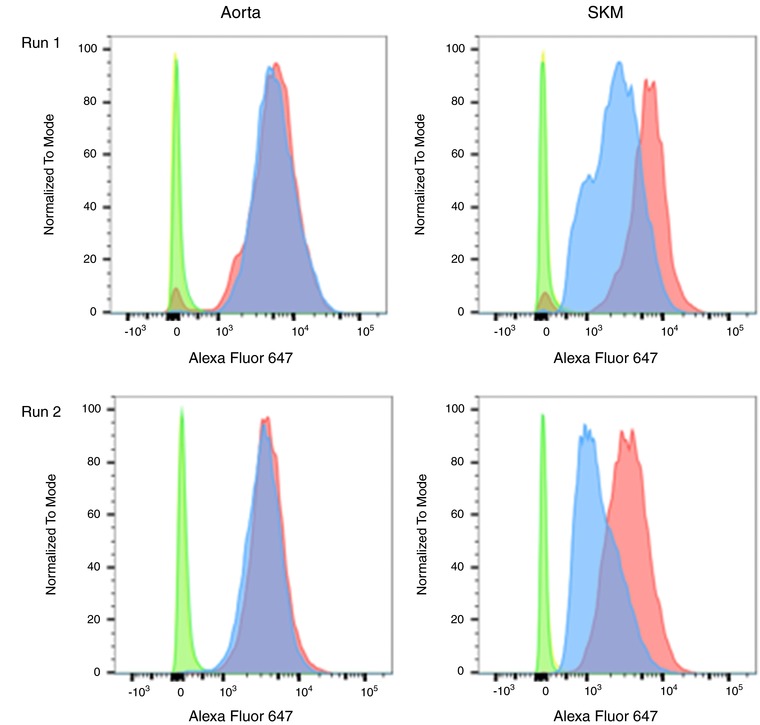
Measures of αvβ3 are provided as an example of the approach used to assess protein levels in the EC for the male and female endothelial cells from the microvasculature (aorta) and microvasculature (skeletal muscle). In Run #1, all eight measures were performed under the same conditions on the same day: four of the relative fluorescences of the secondary antibody labelled with Alexa Fluor‐647 to αvβ3 measurements for male (blue) and female (pink) aorta; male (blue) and female (FS, pink) skeletal muscle (SKM) and their four associated negative controls (green and yellow peaks). Run #2 was performed in the same manner on a separate population of cells at a later date in the same manner.
Statistical analysis
Given the differences in cell volume (Fig. 3 A) and area of spread (Fig. 3 B), it became evident that the majority of the data and analyses needed to be on a per cell basis. Statistical analysis was conducted using Prism, version 5 and 6 (GraphPad Software Inc., San Diego, CA, USA) and Enterprise Guide, version 6.1 (SAS Institute, Cary, NC, USA). The D'Agastino‐Pearson omnibus normality test was first performed to assess normality. The non‐parametric Wilcoxon rank sum test was used to analyse distributions that significantly departed from normality (Daniel, 1990). The data are reported as the median and interquartile range. P < 0.05 was considered statistically significant. In a secondary analysis, individual growth models were evaluated using mixed models with random linear and quadratic effects, as well as grouping variables.
Figure 3. Sex influences EC size and spread.
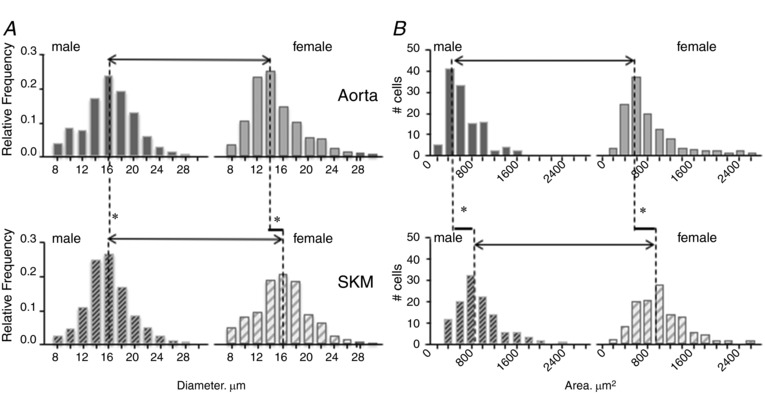
Sex and position in the vasculature influence endothelial cell size when free in solution (A) and following attainment of confluence on gelatin (B). A, data are frequency histograms of EC diameter determined by cytometry repeated for six preparations, illustrating the smaller diameter of the aortic EC from females relative to the other three groups. B, frequency diagram of the area of 119 confluent EC per sex and tissue origin from measures of surface area following staining with fluorescence‐labelled tubulin. A sex difference in the macrovascular EC was found, with the cells from males being ∼15% smaller than those from females. Although no sex differences were noted with spreading in the EC of microvascular origin, male and female microvascular EC spread ∼50% and ∼46% further, respectively than those of macrovascular origin.
Results
Multiple differences were observed reflecting position in the vasculature (macrovascular aorta vs. SKM) and sex (male vs. female) despite maintaining constant and identical conditions for all of the EC phenotypes reported.
In all cases, cells from males could be distinguished from those of females by the PCR presence of the SRY gene (Fig. 1 F). This technique was used to confirm the genomic‐sex of the cells following isolation and at multiple times during the conduct of these studies as an internal control to ensure that the cultures had not been mislabelled with respect to sex. Regardless of sex, all of the cells were confirmed to be of endothelial origin by the presence of CD31 (PECAM‐1), VE‐cadherin, von Willebrand's factor, binding of isolectin IB‐4 from Griffonia simplifolica, uptake of acetylated‐low density lipoprotein, and formation of capillary‐like tubes on surfaces coated with Matrigel (Fig. 1 A–E).
Following vessel isolation and EC preparation, free, unattached aortic EC diameter was found to differ by sex [male > female; median (Q1–Q3) 16.1 (13.9–18.6) μm, n = 1669 cells, P < 0.001 vs. 14.0 (12.2–16.9) μm, n = 1829 cells, P < 0.001] (Fig. 3 A). By contrast, a sexual dimorphism in cell size was not observed for skeletal muscle microvascular EC [male = female, 15.6 (13.8–17.7) μm, n = 2300 cells, vs. 16.0 (13.4–18.3) μm, n = 1758 cells, P = 0.32, respectively, from six preparations] (Fig. 3 A). In addition, cell size differed by vessel location with the macrovascular EC isolated from aorta being significantly smaller than those from SKM (P < 0.001).
The morphology of cells plated on gelatin at a density of 105 cells mL–1, regardless of area that they were to cover, differed by sex and by vessel of origin (Fig. 3 B). The surface areas of confluent EC from six gelatin‐coated coverslips for each of the four groups were determined using NIH ImageJ examining tubulin staining of the cytoplasm and DAPI staining of the nuclei. The average planar area of the male aortic endothelial cells was 576 (426–802) μm2 (n = 119), which is significantly smaller than aortic EC from females [657 (506–994) μm2, n = 119, P = 0.02]. Similar to free cell size, SKM EC area of spread did not display a sexual dimorphism (P = 0.13), although SKM EC from both sexes spread over a greater area than aortic EC of either sex [P < 0.01, male SKM EC were 858 (669–1115) μm2 and female SKM EC were 951 (715–1277) μm2, respectively]. Independently (Fig. 4), nuclear area was found to differ by sex (P < 0.001) for the macrovascular endothelial cells, being 172 (159–209) μm2 and 136 (133–139) μm2 for male and female aortic EC (Fig. 4 A), respectively. Although no sex difference was observed for SKM nuclear area by sex [138 (120–141) μm2 and 170 (157–204) μm2 for male and female SKM (Fig. 4 B), respectively, P = 0.07], the nuclear area of the male SKM EC [138 (120–141) μm2] was significantly smaller than that for male aorta EC [172 (159–209) μm2] (P < 0.001).
Figure 4. Differences in EC nuclear size by sex and by origin.
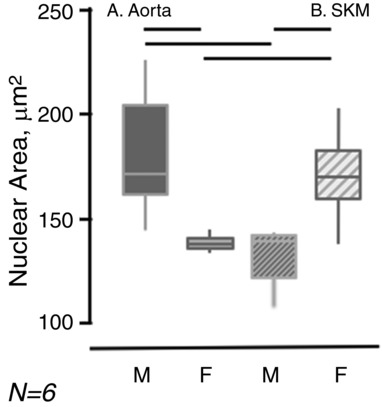
The areas of confluent EC nuclei, identified by DAPI staining, were determined in six sets of cell preparations. The data are represented by bar and whisker plots with the solid horizontal line being the median, the bar extending from the 25th to 50th percentile and the vertical lines extending from the 5th to 95th percentiles. The solid horizontal bars identify significant differences (P < 0.05).
Growth
Although EC were isolated from a pair of animals of one sex from two sites at the same time and were treated using identical selection, washing, growth media, incubation times, etc., their responses once plated differed significantly by sex and depending on whether they were of macro‐ or microvascular origin. With respect to sex, EC from aorta, whether male or female, grew at the same rate having increased by 10‐fold by day 6 (Fig. 5 A). By contrast, by day 6 (Fig. 5 B), microvascular SKM EC from males increased by 20‐fold, whereas SKM EC from females increased by 7‐fold (P < 0.01, n = 6).
Figure 5. Cell growth of macrovascular EC is sex independent, whereas male microvascular EC grow faster than female microvascular EC.
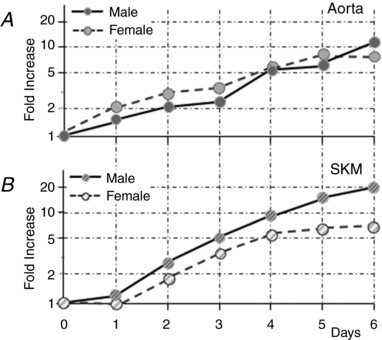
The average fold‐increase of EC plated on gelatin was tracked for six preparations of EC over 6 days. Aortic EC, regardless of whether from males or females, had increased in number by 10‐fold. By contrast, the microvascular EC did not begin to increase in number for 1 day following seeding and, although the EC of the females had increased by 7‐fold some 5 days later, the EC of the males had increased in number by 20‐fold. No error bars are plotted because the magnitude was smaller than the size of the symbols.
Quadratic growth models with the grouping variables of sex or vessel origin provided the best fit, indicating that EC from females grow at a different rate than those from males and that macrovascular EC from the aorta grow at a different rate than microvascular SKM EC. Overall, the rate of growth ranked (highest to lowest) male SKM > male aorta = female aorta > female SKM. The full model analysis also revealed a hierarchy of predictive ability such that assuming a general homogenous population of endothelial cells was less predictive than grouping by sex, which was less predictive than grouping by vessel of origin. The best model fit was obtained when both sex and vessel of origin were combined (Kemp, 2017).
Metabolism
The amount of lactate produced by confluent EC was measured and found to not differ by sex or by vessel type [male aorta 0.19 × 10−9 m cell–1 (0.12–0.30); female aorta 0.25 × 10−9 m cell–1 (0.15–0.38), P = 0.13; male SKM 0.23 × 10−9 m cell–1 (0.13–0.35); female SKM 0.17 × 10−9 m cell–1 (0.13–0.30), P = 0.50, n = 21, respectively] (Fig. 6 A and B).
Figure 6. Lactate production on a per cell basis does not differ by sex or cell origin.
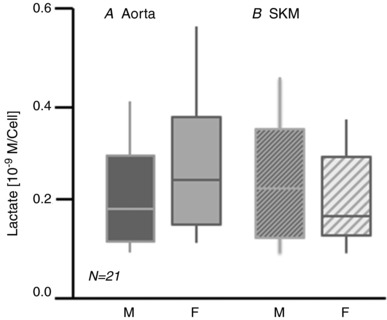
The data are median lactate levels (solid horizontal line) for 21 preparations of confluent EC over the course of these studies. The solid bars extend from the 25th to 50th percentile; the vertical lines extend from the 5th to 95th percentile.
Wound healing
Although newly plated aortic EC of both sexes grew at the same rate (Fig. 5 A), the same behaviour was not recapitulated when the rates of wound healing were determined (Fig. 7 A). Although SKM microvascular EC growth rates differed by sex (Fig. 5 B), wound healing was sex independent (Fig. 7 B). Although male and female macrovessel EC grew at the same rate, wounds of the confluent aortic male EC took significantly longer to close (45 h) relative to aortic EC from females (38 h). The aortic macrovascular EC from males also filled the wound more slowly than the microvascular SKM EC from males (40 h) or females (39 h), respectively. In all cases, there was a significant latency of 4 h before the wounds began to close (n = 6) (Fig. 7).
Figure 7. Healing of scratch‐wounded confluent primary EC is slower for macrovascular EC of males than macrovascular EC of females or microvascular EC of either sex.
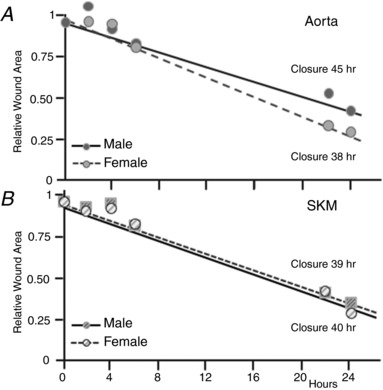
The area of four scratch wounds per dish of confluent EC of macro‐ and microvessel origin of both sexes was monitored at 0, 2, 4, 6, 22 and 24 h following injury in six sets of preparations. The average relative area for each time point is plotted and the slopes compared. Unlike the cell growth curves, the macrovascular EC demonstrate a sex difference, with the wounds of male aortic EC closing more slowly. The microvascular EC of both sexes were indistinguishable from themselves or the aortic EC from females.
mRNA
mRNA for 10 proteins was determined using quantitative real‐time RT‐PCR; to facilitate presentation and comparison, the results are grouped.
Sex steroid receptors
First, because the present study focused on sex differences, in addition to using the presence and absence of mRNA for the SRY testis‐determining gene to confirm the inherent genomic sex of the endothelial cells (Fig. 1 F), we probed for mRNA for the three primary steroid receptors: the X chromosome‐linked androgen receptor (AR) and the two oestrogen receptors, (ER)α and ERβ, respectively. Although mRNA was detected for all three receptors the relative expression varied by receptor, vessel origin, and sex (Fig. 8 A). Overall, mRNA levels for AR and ERα were highest in the macrovascular EC from male aorta, whereas mRNA for ERβ was highest in the microvascular EC of male SKM. Although present in the macro‐ and microvessel EC of females, the mRNA levels for all three sex steroid receptors were significantly lower or did not differ from that of males.
Figure 8. Sex and tissue origin influences independently EC sex hormone receptor mRNA and protein expression.
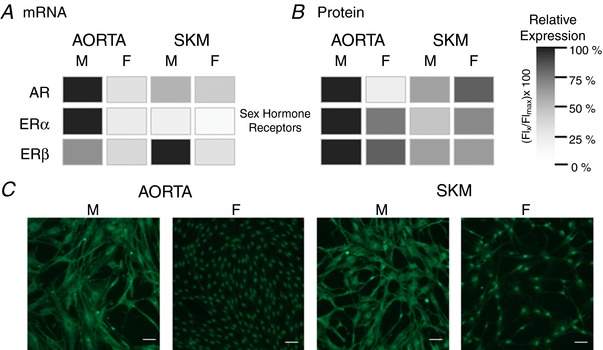
Patterns of messenger RNA (A) and protein expression (B) are not recapitulated for the primary sex hormone receptors. A, relative expression of AR, ERα and ERβ mRNA determined by quantitative RT‐PCR are given for the EC from aorta and skeletal muscle of males and females. The sample with the highest expression level was set at 100% with the other samples compared to that level. B, the same groups are represented except, in this case, it is protein expression determined by flow cytometry. Immunocytochemistry of ERβ demonstrates that sex and tissue differences in protein expression can reflect not only total protein, but also the distribution within the endothelial cells. In these examples (C), ERβ is distributed mainly in the cytosolic compartment of the EC of males and restricted to the nuclear compartment of the EC females, regardless of whether being of macrovascular (aorta) or microvascular (skeletal muscle) origin. Scale bar = 10 μm.
EC junctional proteins
Given that regulation of exchange is one of the primary functions of the endothelium and also given the data demonstrating sex differences in basal arteriole and venular permeability to protein (Huxley et al. 2007) and permeability responses to a variety of stimuli (Huxley et al. 2004; Wang et al. 2005) in intact microvessels, we measured mRNA for the junctional proteins, PECAM‐1 (CD31) and VE‐CAD (CD144) (Fig. 9 Aa). Although mRNA for PECAM‐1 was highest in microvascular SKM EC from males, it was low and indistinguishable in the remaining three groups. As a result, no sex difference was detected for EC of aorta, and the difference was profoundly higher for microvascular SKM EC of males relative to females. mRNA levels of VE‐CAD, also associated with the control of vascular permeability (Gavard, 2014), were highest in EC from female aorta and relatively low in the EC from either site in males.
Figure 9. Key EC protein expression differs by sex and tissue of origin.
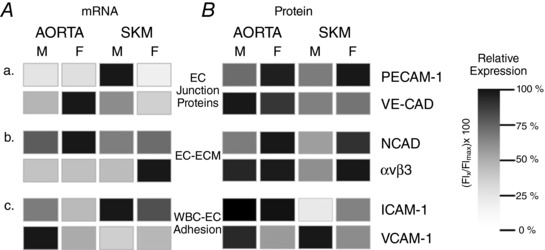
A, influence of both sex and tissue of origin on EC mRNA levels relative to the relative protein expression (B) of selected EC proteins for selected targets involved with endothelial cell functions. Aa and Ba, two proteins associated with barrier function, PECAM and VE‐CAD. Ab and Bb, NCAD and αvβ3, two proteins associated with EC/matrix interactions. Ac and Bc, two proteins associated with white blood cell/endothelial cell interactions, ICAM‐1 and VCAM‐1. A, the sample with the highest mRNA expression was set at 100% with the other samples compared to that level. B, the highest protein expression was set at 100%, with the other samples compared to that level. Notably, the patterns of protein expression also differ from the patterns of mRNA expression for the same targets.
Endothelial cell‐extracellular matrix (ECM) interactions (EC‐ECM)
The ability of the EC to migrate, spread, and respond to mechanical stimuli is related to the presence of integrins such as αvβ3. mRNA levels for αvβ3 were highest and most pronounced in EC from SKM microvessels of females; they were lower and not different in EC from the macrovasculature of either sex, and no macro‐/microvessel difference was discernable for EC from the males (Fig. 9 Ab). We also targeted neural cadherin (N‐CAD, CD325), a protein found in EC whose function has been more difficult to discern but considered to share signalling pathways with VE‐CAD and to be involved with microvessel tube formation and pericyte recruitment (Navarro et al. 1998; Tillet et al. 2005; Giampietro et al. 2012; Zheng et al. 2016) mRNA levels for N‐CAD were highest in the EC from female aorta. The relative expression hierarchy (strongest to weakest) was female aorta > male aorta > female SKM > male SKM (Fig. 9 Ab).
Endothelial cell‐white blood cell adhesion molecules (EC‐WBC)
Given that acute and chronic inflammation have are associated with loss of barrier function and edema formation; that chronic inflammation is associated with disease progression; and that immune mediated diseases are more prevalent in females than males (Casimir & Duchateau, 2011; Claesson‐Welsh, 2015), EC‐WBC adhesion proteins might provide insight with respect to the basis for sex differences in inflammatory states. The two proteins probed were the cell adhesion molecules, intracellular adhesion molecule‐1 (ICAM‐1, CD54) and vascular cell adhesion molecule‐1 (VCAM‐1, CD106), respectively. Levels of ICAM‐1 message were highest in microvessel EC from male SKM and slightly lower in female SKM EC; message levels were lower in the macrovascular EC for both sexes; the mRNA levels of aortic EC were higher for males than females (Fig. 9 Ac). VCAM‐1 mRNA had a completely different pattern, with message levels being highest in macrovascular EC from male aorta and low levels that did not differ among the remaining three groups.
Protein expression
Initially, immunocytochemistry was performed to image and localize the three steroid receptors and the six EC‐associated proteins. As shown in Fig. 8 B for ERβ, differences exist in protein labelling for macro‐ and microvessel EC of males and females, respectively. The difficulty was that analysis of the immunocytochemistry images was problematic given a number of factors, including EC morphology, cell density within the ROI and protein distribution within the cell, to name but a few. In particular, the staining pattern of ERβ is notable (Fig. 8 C) and distinguished from the other proteins (data not shown). In both macro‐ and microvessel EC from females, the majority of ERβ staining is in the region of the cell nucleus. By contrast, for male EC of both tissue sources, there is a significant cytosolic ERβ staining with little staining in the region of the nucleus.
Because of the heterogeneity of distribution, cell morphology and ability to analyse sufficiently large numbers of cells, we moved to flow cytometry to assess protein expression in EC at passage 4.4 in the four groups. As in the case of the immunocytochemistry, the EC were fixed and permeabilized. Consequently, it was total protein that was measured regardless of distribution within the cell; furthermore, because the method measures fluorescence for each cell and cell sample sizes were in the tens of thousands, inherent heterogeneity amongst EC was taken into account in the final sample distribution. Again, the results are presented by groups of proteins to facilitate comparison with the mRNA data.
Sex steroid receptors
Figure 8 B illustrates the relative expression of the sex steroid receptor protein expression as measured by flow cytometry. Figure 8 shows that, except for AR of the male and female macrovascular EC, mRNA levels (Fig. 8 A) bore little resemblance to the presence and relative levels of the protein expression (Fig. 8 B). Overall, AR, ERα and ERβ protein expression was highest in EC of male aorta, whereas, in EC from female aorta, the levels of all three steroid receptors were lower, with the levels of AR being negligible. Staining for AR and ERα in EC from female microvessel, although lower than in EC from male aorta, tended to be higher than in EC from male microvessels. ERβ levels were comparable in the EC from males and females. The main distinction for ERα and ERβ was that their protein levels were higher in macro‐ than microvessel EC.
EC junctional proteins
PECAM‐1 protein expression was significantly higher in the EC from females than those from males of both macro‐ and microvessel origin (Fig. 9 Ba). Although no sexual differences in VE‐CAD protein expression were observed, for both sexes, the expression of VE‐CAD was higher in EC from aorta than SKM.
ECM
NCAD expression (Fig. 9 Bb) displayed a sexual dimorphism similar to that observed for PECAM‐1 (Fig. 9 Ba), with levels being higher in EC from females relative to males, regardless of whether of macro‐ or microvascular origin. No significant differences were observed between tissue sources. Of the proteins studied, integrin αvβ3 levels were fairly similar in EC from aorta of both sexes and in EC from female SKM, although they were significantly lower in microvascular SKM EC from males (Fig. 9 Bb).
EC‐WBC adhesion
We found that macro‐ and microvascular EC levels of ICAM‐1 differed, with protein expression being highest in EC of macrovascular origin. By contrast, VCAM‐1 varied by sex for both macro‐ and microvascular EC, with protein expression in males exceeding that of the females (Fig. 9 Bc).
Discussion
The primary question addressed in the present study was whether it was correct to assume the absence of sex differences in the endothelial phenotype of healthy, reproductively mature males and females. Undeniably, it cannot be assumed that male and female EC of a given origin, exposed to identical conditions, are identical. The confounding aspect of the data is that it is not readily apparent which endothelial functions will or will not display sexual dimorphism. The second major finding was that, if a phenotype differed by EC origin, it did not necessarily differ by sex and vice versa. The third major finding was that measures of messenger RNA were not surrogates for protein expression by vascular origin or by sex; consequently, using changes in mRNA as being indicative of changes in EC function is probably confusing and incorrect. A major implication of the findings is that, given the sexual dimorphism in receptor and protein expression, there are probably not only organ‐ and location differences in sensitivity or responses to the sex hormones, but also sex differences in cell signalling mechanisms within and amongst EC. Finally, the observed similarities and differences were not recapitulated across the phenotypic characteristics surveyed, making it clear that ‘healthy endothelial function’ is achieved through an assemblage of mechanisms influenced by sex in addition to anatomical location.
That endothelial cell phenotype can vary by organ/tissue of origin and by vessel size (usually reflecting position in the vascular network) has been recognized for many decades. These differences probably reflect the endothelium, by virtue of its existence at the interface between flowing blood and metabolizing tissue, being subjected to differing environmental conditions (Gerritsen, 1987; Aird, 2005, 2007a, 2015). Until recently, it was assumed that these phenotypic differences would be lessened in the conditions of culture and that sexual dimorphism would not be a factor of importance. Over the last decade and a half, data documenting sex differences in fundamental properties ascribed primarily to the endothelium have emerged. Among the notable examples are data on endothelial barrier function, where permeability responses to a variety of vasoactive mediators differ between age‐matched animals of both sexes (Huxley & Wang, 2010). Furthermore, sex differences have been demonstrated in sexually immature animals under basal and activated conditions when reproductive hormones were low and unchanging (Wang et al. 2005; Sasaki, 2007). When the endothelium from the microvessels that displayed sex differences in barrier function was removed to culture, sexual dimorphism persisted at the level of cell signalling (Wang et al. 2010).
The findings in the present study are of fundamental importance in the quest for an understanding of the mechanistic basis for endothelial cell dysfunction in disease and for designing targeted therapy. The existence of sex differences in the state of health provides clues about the mechanistic bases for sex differences with regard to the susceptibility, onset and manifestation of disease. Because vascular disease incidence and severity points to EC dysfunction and multiple examples of male/female differences in both vascular disease incidence and progression exist (Hall et al. 2013; Arnetz et al. 2014), it is imperative that questions of sex differences in basal endothelial phenotype are answered before meaningful results and subsequent translation to the clinical setting can be attained.
Furthermore, given that the incidence of cardiovascular disease prior to menopause is lower in women than men and becomes equivalent, or worse, for females relative to males following this transition (Oda et al. 2006; Kautzky‐Willer et al. 2016) the prevalent focus has been on the reproductive hormones, especially oestrogen. Nevertheless, data exist indicating that cells from males and females are not equivalent even before the sex hormones are changing, implicating a role for genomic sexual dimorphism in the vascular system, a concept embraced by neurobiologists (Hughes et al. 2013; Friedrich et al. 2015; Zarros et al. 2015) and also more recently in the cardiovascular system (Regitz‐Zagrosek & Kararigas, 2017).
Much of the data on EC cell biology and physiology are derived from specialized vascular beds. The vast majority are of EC from two sources, HUVEC and bovine aorta (BAEC). HUVEC, derived from the vessel carrying oxygen‐ and nutrient‐rich blood from the placenta to the fetus, are interesting because the cells studied to date represent those pooled from both sexes. Amongst recent studies of HUVEC separated by sex, several differences have become apparent with respect to tolerance to hypoxia, mRNA expression, responses to shear stress, cell proliferation and receptor expression (Addis et al. 2014; Annibalini et al. 2014; Lorenz et al. 2015; Zhang & Lingappan, 2017). The data reported by Lorenz et al. 2015 demonstrate that HUVEC from females possess greater transcriptional levels of genes related to the immune response, and that they also have a greater capacity to form tubes and tolerate the stress of serum deprivation better than their male counterparts. These phenotypic characteristics are notable because it has also been shown that mortality is lower in premature females than males (Stark et al. 2008).
The second source of EC, bovine aorta, are often derived from slaughterhouse material and are probably of mixed sex or predominantly that of young males, depending on the geographical location of the slaughterhouse. For the present study, we chose EC from the aorta and SKMs. The aortic EC provided a point of comparison for the phenotype of a high‐pressure conduit vessel, access to the considerable literature on BAEC and the significant literature on human aortic EC (HAEC). SKMs were chosen for several reasons. First, skeletal muscle represents the largest organ of the body and regulation of its vasculature has been the focal point for decades of study on blood flow control in health and disease. With respect to barrier function, solutes and water traverse between blood and metabolizing tissue across the smallest vessels of the vasculature. Furthermore, it is in arterioles and venules from skeletal muscle (abdominal free wall) where sex‐specific differences, with respect to both basal permeability properties and permeability responses, have been observed (Wang, 2005; Sarelius et al. 2006; Wang & Huxley, 2006). The use of EC from this source facilitates the design of future in vivo and ex vivo studies. The vast majority of in situ studies of blood flow control have been conducted on arterioles of males (primarily from musculus cremaster, which is derived anatomically from the abdominal free wall) (McVay & Anson, 1940). Observations of capillary rarefaction in response to diabetes and in hypertension have been made in skeletal muscle. For example, although it is reported widely that microvessel rarefaction occurs in these disease states, studies have focused primarily on male animals. The sole comparative study of rarefaction in both sexes demonstrated that, when rarefaction occurred in females of the same species and experimental model (reduced renal mass), it was in fewer muscle beds, much later in time, and at lower systemic blood pressures than in males (Papanek et al. 1998). Skeletal muscle, a highly metabolic tissue, also represents the largest source of EC in the body by virtue of the large number of microvessels residing in the tissue. An implication of this density of vessels is that the SKM vasculature represents a reservoir or source of inflammatory and vasoactive mediators/cytokines. We have evidence for sex differences in skeletal muscle arterioles and venules that persist in microvascular EC isolated from these vessels (Wang et al. 2010). Finally, a substantive literature exists on white blood cell and platelet interactions with venular EC in male models (Sarelius & Duling, 1982; Damon & Duling, 1984; Vicaut et al. 1994; Sarelius et al. 2006). In that light, it notable that ligands identified with EC‐WBC interactions, such as the integrins, ICAM‐1 and VCAM‐1, displayed differences with respect to vessel location and sex. We found that ICAM‐1 varied by EC vessel of origin, with much higher protein expression in EC from aorta than SKM microvessels (Fig. 9). By contrast, VCAM‐1 varied by sex, where protein expression in male EC exceeded that of the females (Fig. 9) for both macro‐ and microvascular origin. The VCAM‐1 data are also interesting because a previous study using HUVEC of unknown sex demonstrated a marked increase in VCAM‐1 expression following exposure to androgen (McCrohon et al. 1999). The study by McCrohon et al. (1999) suggested that the sensitivity to androgen may render arterial EC prone to development of atherosclerosis; the present data might infer that EC from males are themselves more atherogenic. Whether or not there were sex differences in HUVEC expression of VCAM‐1, per se, is not known. It is well known that loss of barrier function occurs in inflammatory states and that inflammatory processes appear to differ by sex even prior to adolescence (Casimir et al. 2010; Casimir & Duchateau, 2011). It therefore stands to reason that the observed differences in EC‐WBC adhesion protein expression provide insight into the basis for sex differences in inflammatory states. Whether sexual dimorphism with respect to white blood cell/endothelial cell interactions will exist in vivo is a supposition that required testing.
Ideally, the phenotypic assessments carried out for the present study, especially those measuring receptor expression and endothelial morphology, would be conducted using endothelial cells in situ. In that case, mature EC sit on their complex biological matrix, possess an intact glycocalyx, and experience the force of blood flow over their surface and transmural flux of fluid and solute. Unfortunately, with presently available techniques, it is not yet possible to obtain sufficient in vivo data for comparison between the sexes given the small number of endothelial cells in a given vessel segment. It is well documented that EC phenotype can and does change with passage under cultured conditions (Durr et al. 2004); we also know that EC are difficult to characterize in vivo given the inherent heterogeneity amongst EC and the dynamic nature of the environment within which EC exist. Furthermore, the present study was conducted under conditions of low and unchanging levels of reproductive hormones and growth factors as a means of controlling multiple variables. Obviously, in intact tissue, these elements will probably modify EC phenotype in ways not addressed in the present study and the global applicability of the results to human health and disease remains to be determined more fully. With these caveats, we focused on the two components: macro‐ and microvessel origin by isolating EC from aorta and skeletal muscle, respectively, and sex of age‐matched, sexually mature Sprague–Dawley rats under conditions of culture at early passage (4.4).
Existing data on sex differences of EC phenotype are conflicting. Most studies focused on a single phenotypic feature or expression of a few proteins. In several studies, if one feature did not differ by sex, it was then assumed that no other features would differ by sex. Often, it is stated that no sex differences were found when the sample sizes were small and of insufficient statistical power to discern differences. Given these difficulties, a range of phenotypic characteristics was examined: morphology, growth, metabolism, cytoskeletal mechanics, protein expression, and signalling and inflammatory markers, under the same set of defined conditions to facilitate comparisons. Striking differences in phenotype were noted for most of the characteristics tested with the exception of metabolism, where overall lactate production did not differ by sex but by vascular tissue for male EC, and for the aortic endothelial cells growth curves.
EC morphology
It is well known that EC size can vary with position in the vascular tree (Sarelius et al. 2006) and that the morphology of aortic and venous endothelial cells differ (Wagner et al. 1988). What has not been studied is whether EC morphology differs by sex or by age. We found that female aortic endothelial cells following isolation were smaller in size than those of males (macro‐ or microvessel) and those from SKM of the same sex (Fig. 3 A). Once the cells settled and grew on gelatin, their degree of spreading, as indicated by the areas that they covered, differed by sex and by source (Fig. 3 B). What happens when EC of either sex are exposed to native matrix or whether the composition of the matrix itself differs by sex has not been ascertained. In males, both Wagner et al. (1988) and Sarelius et al. (2006) found that the area of EC in situ differed with position in the vasculature (arterial vs. venous). The implications of our data with respect to endothelial size are shown in Fig. 10. In the simplest model, if the EC are spheres of the median volume and settle into squares of their median area, a thickness and a perimeter can be calculated. Basically, the result is that aortic EC of males would be the thickest and no significant difference would exist between the other three groups. If solute permeability is considered to be the diffusion coefficient per unit depth (thickness), all else being equal, the permeability of aortic EC in males would be predicted to be approximately half that of the female aorta or SKM microvessels of either sex.
Figure 10. Sex‐ and tissue‐differences in EC volume and spreading.
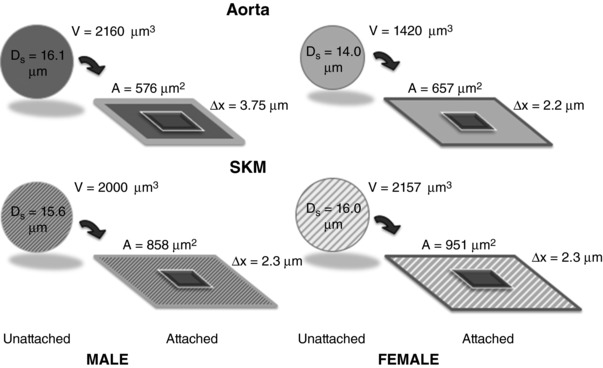
Illustration of the implications of sex‐ and tissue‐differences in EC volume and spreading on EC thickness, ∆x, a determinant of barrier permeability.
Of greater importance, the density of cells required to cover the vessel wall will differ should the behaviour observed ex vivo be recapitulated in vivo. A 100,000 μm2 (0.1 mm2) area, corresponding a 1 mm in length and 62 μm in diameter microvessel segment, for example, would contain ideally 174 male aortic EC; 152 female aortic EC, 117 male SKM EC or 105 female SKM EC, respectively. These differences in cell densities would influence the interpretation of the lactate data (Fig. 6) where no significant differences in lactate production were noted on a per cell basis. Given the potential density differences, lactate production would be predicted to be 14%, 49% and 65% higher in the male aorta than female aorta, male SKM, or female SKM, respectively. This level of interpretation should make it clear that the next step will be to conduct these studies in intact vessels under more physiological conditions.
Reproductive hormone receptors
All three sex hormone receptors were present on the macro‐ and microvascular endothelium, albeit at different levels depending on whether it was mRNA or protein expression that was being measured. mRNA for AR and ERα and protein expression for AR, ERα and ERβ were highest in EC from the aortas of male rats (Fig. 8 A and B). AR, located on the X‐chromosome, is present in both males and females to varying degrees. AR, identified on EC of multiple organs in humans, including brain, heart, umbilical vein, dermis, salivary gland and bone, as well as prostate, ovary and myometrium (Torres‐Estay et al. 2015, 2017), has been implicated as mediating angiogenesis in both sexes (Yoshida et al. 2013). In one study, although AR was identified on macrovascular EC (HAEC, sex not stated), no AR immunostaining was observed in skeletal muscle EC (Torres‐Estay et al. 2017); by contrast, in another AR, staining on capillaries of musculus vastis lateralis was observed in muscle biopsies from human males (Sinha‐Hikim et al. 2004). AR was present in replicate samples EC of both macro‐ and microvessel origin, with the sex differences being reversed. Consequently, our data suggest that differences in sex, in addition to anatomical location, could account for the apparent disparities in the literature.
Similarly, the primary ERs, ERα and ERβ, varied by both macro‐ and microvessel and by sex, making it difficult to ascribe a universal set of roles for either or both receptors. These data illustrate the complexity of the ER family. In addition to ERα and ERβ, multiple isoforms exist because of the ability to hetero‐ or homodimerize (Nilsson et al. 2001; Leung et al. 2006; Vrtačnik et al. 2014). With respect to EC, ERα and ERβ have been shown to possess redundant effects. In one study, both ERα and ERβ ligation reduced inflammatory cytokine actions mediated by EC, stabilized the extracellular matrix and inhibited endothelin production (Evans et al. 2002), all considered to be vascular protective responses. The two receptors also can have remarkable disparate effects. ERβ has been shown to be down‐regulated when EC are exposed to lipopolysaccharide, whereas ERβ mRNA increased 8‐fold after stimulation with 17β‐oestradiol, possibly showing ERβ downregulation during inflammation, and a rescue effect by oestrogen (Holm et al. 2010). 17β‐oestradiol, for example, will induce vascular re‐endothelialization via ERα exclusively (Toutain et al. 2009). In general, oestrogens appear to enhance EC nitric oxide production, promote angiogenesis and wound healing, have pro‐inflammatory effects (possibly in conflict with what was found above) and, at the same time, exert anti‐atherogenic effects (Cid et al. 2002). It is possible that the conflicting results arise as a result of interactions with the dimerized receptors or isoforms of the receptors.
Our data with respect to ERα protein expression differ from the results reported for HUVEC (sex not identified: Hervé et al. 2006; Toth et al. 2008; sex identified: Annibalini et al. 2014) where the receptor expression was not detectable. In the study by Annibalini et al. (2014), cloned HUVEC were used and low levels of mRNA for ERα were detected, although protein expression was not; these EC were grown in media containing charcoal stripped FBS. The FBS (20% v/v) used in the present study, from a single lot and not charcoal stripped, was confirmed to have low levels of the reproductive hormones (7.3 ng dL–1 testosterone, 24 pg mL–1 total oestrogens, progesterone <0.1 ng mL–1, respectively). It was also determined that the FBS had no detectable insulin, <0.1 ng mL–1 growth hormone and <16 μg L–1 insulin‐like growth factor binding protein‐3. We chose to not charcoal strip the FBS because EC fail to grow well for longer than 1 day. When heat‐inactivated FBS was used, Klinge et al. (2005) demonstrated the presence of both ERα and ERβ protein in HUVEC, human microvascular endothelial cell (HMEC) and BAEC (sex not identified) in ratios of 1.59, 1.28 and 1.42, respectively (Klinge et al. 2005). Similarly, Tschugguel et al. (2003) found both message and protein for both ER subtypes in human umbilical arterial endothelial cells (HUAEC, sex not identified).
Consistent with previous studies of HUVEC (Wagner et al. 2001; Hervé et al. 2006; Simard et al. 2011; Addis et al. 2014) and HUAEC (Tschugguel et al. 2003; Simard et al. 2011), mRNA levels for the sex hormone receptors were generally low, with the exception of AR and ERα in male aortic EC and ERβ in male SKM EC. Our data contrast with the findings of Klinge et al. (2005) where levels of ERα and ERβ did not differ between BAEC (macrovessel) and HMEC (microvessel) (sex not identified). With respect to sex hormone receptor protein expression, levels were greatest for all three receptors in EC from male aorta. Studies in primary culture fetal ovine intrapulmonary arterial EC (sex unidentified) demonstrate the presence of both ERs and their dynamic and interrelated expression in response to changes in reproductive hormones levels (Ihionkhan et al. 2002).
Immunostaining demonstrated sex hormone receptor distribution in the cytosolic and nuclear regions indicative of the receptors playing non‐genomic actions in addition to a classical role in nuclear transcription (SimonciniI et al. 2004; Kim et al. 2009). Notable in the present study and different from the other proteins probed, the distribution of ERβ within the EC differed by sex. As shown in Fig. 8 C, immunostaining of ERβ was brightest in the vicinity of the nucleus (classical ER) of the EC of both macro‐ and microvascular origin in females. By contrast, fluorescent product was distributed to both the cytoplasm and nucleus of the EC from both positions in the network of males. The functional impact of this distinct sexual dimorphism in ERβ distribution warrants further study. The flow cytometry data (summarized in Fig. 8 B) illustrate the differences in total protein and not the location of the protein stained. Finally, the important consequence of these differences in receptor composition and density by sex and by vessel location infers differences for both variables with respect to cell signalling and the physiological responses to the sex hormones in the intact vasculature.
EC heterogeneity: roles of source and of sex
Most published studies on endothelial cell heterogeneity have focused on organ or position in the circulation (Bottaro et al. 1986; Gerritsen, 1987; Kumar et al. 1987; Garlanda & Dejana, 1997; Craig et al. 1998; Murphy et al. 1998; Thorin & Shreeve, 1998; Aird 2007a, 2007b, 2012; Hewett, 2009; Matheeussen et al. 2011; Augustin & Koh, 2017) and have described multiple features reiterated in the present study, particularly as they relate to macro‐ vs. microvessel origin. None have investigated the contribution of sexual dimorphism beyond the work of Laughlin et al. (2003) and Simmons et al. (2012) focusing on porcine skeletal muscle conduit vessel function. In addition to male/female differences, we have identified multiple macro‐ and microvessel differences in EC protein expression that are summarized without regard to sex in Fig. 11 A. A strong predominance of protein expression was revealed in EC of aortic origin for all but AR, PECAM‐1 and VCAM‐1.
Figure 11. Ignoring sex (A) of tissue of origin (B) provides vastly differing data interpretations.
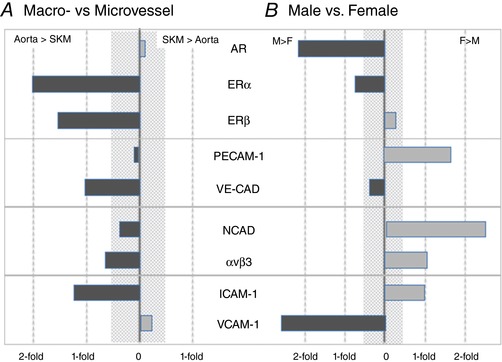
Overall assessment of the contributions of tissue origin (macro‐ vs. microvessel) in the absence of sex (A) and sex (male vs. female) in the absence of tissue origin (B) to protein expression. The shaded area represents differences of 50% or less, which is often difficult to detect reliably. For the proteins surveyed, overall expression levels appear to be higher in EC from the microvasculature and provide one image. In EC from males, androgen receptor (AR), ERα and VCAM levels are higher than females, ERβ and VE‐CAD display no sexual dimorphism, whereas PECAM‐1, NCAD, αvβ3 and ICAM‐1 levels are higher in EC from females relative to males. [Color figure can be viewed at http://wileyonlinelibrary.com]
When the data are segregated by sex without regard to macro/microvessel origin (Fig. 11 B), a different array of similarities and differences is observed. In particular, the expression of AR and VCAM‐1 is more than 2‐fold higher in EC of males relative to females, whereas N‐CAD and PECAM‐1 are more highly expressed in EC from females. ERα was expressed more prominently in male EC; by contrast, avβ3 and ICAM‐1 expression was higher in female EC and no sex differences were observed for ERβ or VE‐CAD. The findings, as shown in Fig. 11, are consistent with the earlier analysis of the EC growth curves where a quadratic growth model grouped by sex and tissue type confirmed a significant difference in growth between sex within cell type, as well as a difference between cell type within sex. Both sets of data (i.e. protein expression and EC growth) make it clear that ignoring EC sex is problematic and probably leads to an erroneous understanding of EC biology.
We posited that the area over which the EC spread would correlate with the pattern of the integrin αvβ3 expression given that the integrins mediate cell interactions with the extracellular matrix and focal adhesion formation. Unfortunately, although the ratio of relative αvβ3 fluorescence intensity to median cell area of microvascular EC of males and females matched, the same could not be said for male and female macrovascular EC (1.50, 0.64, 0.98 and 1.00 for male aorta, male SKM, female aorta and female SKM, respectively). Figure 12 provides summary of the differences in cell dimension and the relative distributions of the target proteins, illustrating the variety of parameters that differ by sex and by position in the vascular tree. Indeed, none of the phenotypic characteristics of EC growth, wound healing, lactate production, cell size or area follow the patterns of mRNA or protein expression uniformly. In many cases, it was either EC of macro‐ or microvascular origin without regard to sex or EC from a given sex independent of position in the vasculature that would match these parameters, but not both. This outcome indicates that the underlying cell signalling and molecular mechanisms regulating endothelial phenotype are complex and should not be considered in isolation and that they also require additional study.
Figure 12. Cartoon of EC protein expression, size and thickness illustrating the influence of tissue origin and sex on EC phenotype.
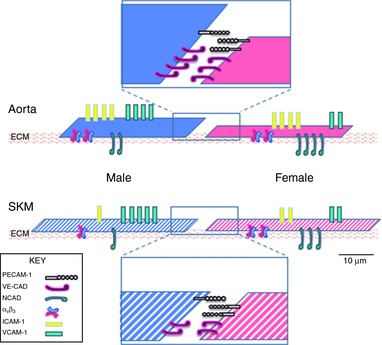
Solid colours indicate the macrovascular EC and striped colours indicate the microvascular EC. Solid blue indicates male aortic EC; solid magenta indicates female aortic EC; striped blue indicates male SKM EC; and striped magenta indicates female SKM EC. Cells are drawn to illustrate the calculated length of one side and cell depth in a simplistic model from Fig. 10. Using the relative expression heat maps from Figs 8 and 9, rough approximations of whole number protein expression were made. Cell junctions were enlarged to view junctional proteins. Scale bar = 10 μm.
Implications of EC heterogeneity by source and by sex
The results of the present study demonstrated differences between aorta and SKMs with regard to several endothelial functions; some expected and some not. Furthermore, the heterogeneity in EC function between the two tissue sources differed significantly by sex, demonstrating outright rejection of the hypothesis that endothelial phenotype, even from the same source maintained under identical conditions, is independent of sex. The implication for future studies of EC function requires an understanding of basal EC characteristics not only by vascular location, but also by sex. Furthermore, the results from a given EC source/sex cannot be expected to provide a universal answer to the mechanisms regulating endothelial function in health, and no less in disease.
The present study was undertaken as a first step with respect to determining the mechanisms accounting for the sex differences observed in vivo (Huxley et al. 2004, 2007; Wang, 2005; Wang et al. 2005; Huxley & Wang, 2010). The untested presumption has been that normal, healthy, basal endothelial functions sex‐insensitive. Although it is well known that endothelial phenotype varies by tissue and by position in the vasculature, it is not unusual that the overarching conclusions pertaining to endothelial biology are derived from a single source of cultured EC, often without information regarding time in culture, as well as passage number, and often in the presence of different media and exposed to differing stressors (e.g. serum starving, serum‐free media, charcoal stripped FBS). Only rarely and very recently has the sex of the cells been considered. Consequently, the presumption has been that the behaviours of cells from males reflect those from females in health and disease. The data and analyses of the present study show that failure to consider either EC source or sex is problematic and that the next step in the study of the influence of sex on endothelial function needs to focus on cell signalling.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
VHH, JW and SK were responsible for the study conception and design. CS, SB, SK, YY, SS and KS were responsible for data acquisition. VHH, SK, SB, CS, YY, SS and KS were responsible for data analysis. VHH, SK and JW were responsible for data interpretation. IZ, SK and VHH were responsible for statistical analysis. VHH, JW, SK and SB were responsible for drafting and revision. All authors approved the final version of the manuscript submitted for publication, and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship and all those who qualify for authorship are listed.
Funding
Supported by National Institutes of Health R01 DK095501‐01A1, VHH PI.
Biography
Virginia H. Huxley obtained her PhD in Biophysics from University of Virginia in 1980 amd then went on to complete postdoctoral Fellowships at Memorial Sloan‐Kettering and the University of California‐Davis with Professor F. E. Curry, where she began her studies on the control of microvascular permeability. Since 1984, she has been at the University of Missouri‐Columbia pursuing this subject in amphibian, rodent and porcine models. It was during this time that she and her laboratory began to focus on the fundamental mechanisms by which males and females, regardless of species, regulate volume and solute homeostasis differently.

Edited by: Laura Bennet & Fernando Santana
This is an Editor's Choice article from the 1 September 2018 issue.
References
- Addis R, Campesi I, Fois M, Capobianco G, Dessole S, Fenu G, Montella A, Cattaneo MG, Vicentini LM & Franconi F (2014). Human umbilical endothelial cells (HUVECs) have a sex: characterisation of the phenotype of male and female cells. Biol Sex Differ 5, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aird WC (2001). Vascular bed‐specific hemostasis: role of endothelium in sepsis pathogenesis. Crit Care Med 29, S28–S35. [DOI] [PubMed] [Google Scholar]
- Aird WC (2005). Spatial and temporal dynamics of the endothelium. J Thromb Haemost 3, 1392–1406. [DOI] [PubMed] [Google Scholar]
- Aird WC (2007a). Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res 100, 158–173. [DOI] [PubMed] [Google Scholar]
- Aird WC (2007b). Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res 100, 174–190. [DOI] [PubMed] [Google Scholar]
- Aird WC (2012). Endothelial cell heterogeneity. Cold Spring Harb Perspect Med 2, a006429–a006429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aird WC (2015). Endothelium and haemostasis. Hamostaseologie 35, 11–16. [DOI] [PubMed] [Google Scholar]
- Annibalini G, Agostini D, Calcabrini C, Martinelli C, Colombo E, Guescini M, Tibollo P, Stocchi V & Sestili P (2014). Effects of sex hormones on inflammatory response in male and female vascular endothelial cells. J Endocrinol Invest 37, 861–869. [DOI] [PubMed] [Google Scholar]
- Arnetz L, Ekberg N & Alvarsson M (2014). Sex differences in type 2 diabetes: focus on disease course and outcomes. Diabetes, Metab Syndr Obes Targets Ther 7, 409–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin HG & Koh GY (2017). Organotypic vasculature: from descriptive heterogeneity to functional pathophysiology. Science 357, 2379. [DOI] [PubMed] [Google Scholar]
- Bottaro D, Shepro D & Hechtman HB (1986). Heterogeneity of intimal and microvessel endothelial cell barriers in vitro. Microvasc Res 32, 389–398. [DOI] [PubMed] [Google Scholar]
- Casimir GJA & Duchateau J (2011). Gender differences in inflammatory processes could explain poorer prognosis for males. J Clin Microbiol 49, 478–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casimir GJA, Heldenbergh F, Hanssens L, Mulier S, Heinrichs C, Lefevre N, Désir J, Corazza F & Duchateau J (2010). Gender differences and inflammation: an in vitro model of blood cells stimulation in prepubescent children. J Inflamm (Lond) 7, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid MC, Schnaper HW & Kleinman HK (2002). Estrogens and the vascular endothelium. Ann NY Acad Sci 966, 143–157. [DOI] [PubMed] [Google Scholar]
- Claesson‐Welsh L (2015). Vascular permeability – the essentials. Ups J Med Sci 120, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig LE, Spelman JP, Strandberg JD & Zink MC (1998). Endothelial cells from diverse tissues exhibit differences in growth and morphology. Microvasc Res 55, 65–76. [DOI] [PubMed] [Google Scholar]
- Damon DH & Duling BR (1984). Distribution of capillary blood flow in the microcirculation of the hamster: an in vivo study using epifluorescent microscopy. Microvasc Res 27, 81–95. [DOI] [PubMed] [Google Scholar]
- Daniel WW (1990). Applied Nonparametric Statistics, 2nd edn PWS‐Kent, Boston, MA. [Google Scholar]
- Durr E, Yu J, Krasinska KM, Carver LA, Yates JR, Testa JE, Oh P & Schnitzer JE (2004). Direct proteomic mapping of the lung microvascular endothelial cell surface in vivo and in cell culture. Nat Biotechnol 22, 985–992. [DOI] [PubMed] [Google Scholar]
- Evans MJ, Harris HA, Miller CP, Karathanasis SK & Adelman SJ (2002). Estrogen receptors alpha and beta have similar activities in multiple endothelial cell pathways. Endocrinology 143, 3785–3795. [DOI] [PubMed] [Google Scholar]
- Fraenkel L (2002). Raynaud's phenomenon: epidemiology and risk factors. Curr Rheumatol Rep 4, 123–128. [DOI] [PubMed] [Google Scholar]
- Friedrich V, Bi W & Sehba FA (2015). Sexual dimorphism in gene expression after aneurysmal subarachnoid hemorrhage. Neurol Res 37, 1054–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlanda C & Dejana E (1997). Heterogeneity of endothelial cells: specific markers. Arterioscler Thromb Vasc Biol 17, 1193–1202. [DOI] [PubMed] [Google Scholar]
- Gavard J (2014). Endothelial permeability and‐VE cadherin: a wacky comradeship. Cell Adhes Migr 8, 158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerritsen ME (1987). Functional heterogeneity of vascular endothelial cells. Biochem Pharmacol 36, 2701–2711. [DOI] [PubMed] [Google Scholar]
- Giampietro C, Taddei A, Corada M, Sarra‐Ferraris GM, Alcalay M, Cavallaro U, Orsenigo F, Lampugnani MG & Dejana E (2012). Overlapping and divergent signaling pathways of N‐cadherin and VE‐cadherin in endothelial cells. Blood 119, 2159–2170. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JR, Wiechmann AR, Johnson LA, Edwards M, Barber RC, Winter AS, Singh M & O'Bryant SE (2013). Biomarkers of vascular risk, systemic inflammation, and microvascular pathology and neuropsychiatric symptoms in Alzheimer's disease. J Alzheimer's Dis 35, 363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervé M a J, Meduri G, Petit FG, Domet TS, Lazennec G, Mourah S & Perrot‐Applanat M (2006). Regulation of the vascular endothelial growth factor (VEGF) receptor Flk‐1/KDR by estradiol through VEGF in uterus. J Endocrinol 188, 91–99. [DOI] [PubMed] [Google Scholar]
- Hewett PW (2009). Vascular endothelial cells from human micro‐ and macro‐vessels: isolation, characterisation and culture. In Angiogenesis Protocols. Methods in Molecular Biology(Methods and Protocols), vol 467, ed. Murray C. & Martin S., pp. 95–111. Humana Press, Clifton, NJ. [DOI] [PubMed] [Google Scholar]
- Holm A, Andersson KE, Nordström I, Hellstrand P & Nilsson B‐O (2010). Down‐regulation of endothelial cell estrogen receptor expression by the inflammation promoter LPS. Mol Cell Endocrinol 319, 8–13. [DOI] [PubMed] [Google Scholar]
- Hughes EL, Cover PO, Buckingham JC & Gavins FNE (2013). Role and interactions of annexin A1 and oestrogens in the manifestation of sexual dimorphisms in cerebral and systemic inflammation. Br J Pharmacol; 169, 539–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui‐Chou HG & McClinton MA (2015). Current options for treatment of Hypothenar hammer syndrome. Hand Clin 31, 53–62. [DOI] [PubMed] [Google Scholar]
- Huxley VH (2007). Sex and the cardiovascular system: the intriguing tale of how women and men regulate cardiovascular function differently. Adv Physiol Educ 31, 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxley VH & Wang J (2010). Cardiovascular sex differences influencing microvascular exchange. Cardiovasc Res 87, 230–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxley VH, Wang J & Whitt SP (2004). Sexual dimorphism in the permeability response of coronary microvessels to adenosine. Am J Physiol Heart Circ Physiol 288, H2006–H2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxley VH, Wang JJ & Sarelius IH (2007). Adaptation of coronary microvascular exchange in arterioles and venules to exercise training and a role for sex in determining permeability responses. Am J Physiol Heart Circ Physiol 293, H1196–H1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihionkhan CE, Chambliss KL, Gibson LL, Hahner LD, Mendelsohn ME & Shaul PW (2002). Estrogen causes dynamic alterations in endothelial estrogen receptor expression. Circ Res 91, 814–820. [DOI] [PubMed] [Google Scholar]
- de Jesus Perez VA (2011). Making sense of the estrogen paradox in pulmonary arterial hypertension. Am J Respir Crit Care Med 184, 629–630. [DOI] [PubMed] [Google Scholar]
- Jones LJ, Gray M, Yue ST, Haugland RP & Singer VL (2001). Sensitive determination of cell number using the CyQUANT cell proliferation assay. J Immunol Methods 254, 85–98. [DOI] [PubMed] [Google Scholar]
- Kautzky‐Willer A, Harreiter JJ & Pacini G (2016). Sex and gender differences in risk, pathophysiology and complications of type 2 diabetes mellitus. Endocr Rev 37, 278–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp SS (2017). Endothelial phenotype differs by both sex and vessel function (conduit vs. exchange). MSc. Thesis University of Missouri‐Columbia.
- Kim KH, Moriarty K & Bender JR (2009). Vascular cell signaling by membrane estrogen receptors. Steroids 73, 864–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinge CM, Blankenship KA, Risinger KE, Bhatnagar S, Noisin EL, Sumanasekera WK, Zhao L, Brey DM & Keynton RS (2005). Resveratrol and estradiol rapidly activate MAPK signaling through estrogen receptors alpha and beta in endothelial cells. J Biol Chem 280, 7460–7468. [DOI] [PubMed] [Google Scholar]
- Kumar S, West DC & Ager A (1987). Heterogeneity in endothelial cells from large vessels and microvessels. Differentiation 36, 57–70. [DOI] [PubMed] [Google Scholar]
- Laughlin MH, Welshons W V., Sturek M, Rush JWE, Turk JR, Taylor JA, Judy BM, Henderson KK & Ganjam VK (2003). Gender, exercise training, and eNOS expression in porcine skeletal muscle arteries. J Appl Physiol 95, 250–264. [DOI] [PubMed] [Google Scholar]
- Leung Y‐K, Mak P, Hassan S & Ho S‐M (2006). Estrogen receptor (ER)‐β isoforms: a key to understanding ER‐β signaling. Proc Natl Acad Sci U S A 103, 13162–13167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K & Schmittgen T (2001). Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐delta delta C(T)) method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Lorenz M, Koschate J, Kaufmann K, Kreye C, Mertens M, Kuebler WM, Baumann G, Gossing G, Marki A, Zakrzewicz A, Miéville C, Benn A, Horbelt D, Wratil PR, Stangl K & Stangl V (2015). Does cellular sex matter? Dimorphic transcriptional differences between female and male endothelial cells. Atherosclerosis 240, 61–72. [DOI] [PubMed] [Google Scholar]
- Matheeussen V, Baerts L, De Meyer G, De Keulenaer G, Van der Veken P, Augustyns K, Dubois V, Scharpé S & De Meester I (2011). Expression and spatial heterogeneity of dipeptidyl peptidases in endothelial cells of conduct vessels and capillaries. Biol Chem 392, 189–198. [DOI] [PubMed] [Google Scholar]
- McCrohon JA, Jessup W, Handelsman DJ & Celermajer DS (1999). Androgen exposure increases human monocyte adhesion to vascular endothelium and endothelial cell expression of vascular cell adhesion molecule‐1. Circulation 99, 2317–2322. [DOI] [PubMed] [Google Scholar]
- McVay CB & Anson BJ (1940). Aponeurotic and fascial continuities in the abdomen, pelvis and thigh. Anat Rec 76, 213–231. [Google Scholar]
- Murphy HS, Bakopoulos N, Dame MK, Varani J & Ward PA (1998). Heterogeneity of vascular endothelial cells: differences in susceptibility to neutrophil‐mediated injury. Microvasc Res 56, 203–211. [DOI] [PubMed] [Google Scholar]
- Navarro P, Ruco L & Dejana E (1998). Differential localization of VE‐ and N‐cadherins in human endothelial cells: VE‐cadherin competes with N‐cadherin for junctional localization. J Cell Biol 140, 1475–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson S, Mäkelä S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M & Gustafsson J‐Å (2001). Mechanisms of estrogen action. Physiol Rev 81, 1535–1565. [DOI] [PubMed] [Google Scholar]
- Oda E, Abe M, Kato K, Watanabe K, Veeraveedu PT & Aizawa Y (2006). Gender differences in correlations among cardiovascular risk factors. Gend Med 3, 196–205. [DOI] [PubMed] [Google Scholar]
- Panazzolo DG, Silva LH, Cyrino FZ, Sicuro FL, Kraemer‐Aguiar LG & Bouskela E (2013). Gender differences in microcirculation: observation using the hamster cheek pouch. Clinics (Sao Paulo) 68, 1537–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanek PE, Rieder MJ, Lombard JH & Greene AS (1998). Gender‐specific protection from microvessel rarefaction in female hypertensive rats. Am J Hypertens 11, 998–1005. [DOI] [PubMed] [Google Scholar]
- Regitz‐Zagrosek V & Kararigas G (2017). Mechanistic pathways of sex differences in cardiovascular disease. Physiol Rev 97, 1–37. [DOI] [PubMed] [Google Scholar]
- Sarelius IH & Duling BR (1982). Direct measurement of microvessel hematocrit, red cell flux, velocity, and transit time. Am J Physiol Heart Circ Physiol 243, H1018–H1026. [DOI] [PubMed] [Google Scholar]
- Sarelius IH, Kuebel JM, Wang J & Huxley VH (2006). Macromolecule permeability of in situ and excised rodent skeletal muscle arterioles and venules. Am J Physiol Heart Circ Physiol 290, H474–H480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki R (2007). Roles of sex and insulin on microvascular exchange function. PhD Thesis University of Missouri‐Columbia.
- Sasaki R, Whitt SP & Huxley VH (2006). Permeability response of the rat mesenteric microvasculature to insulin. Clin Hemorheol Microcirc 34,259–63. [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS & Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard M, Drolet R, Blomquist CH & Tremblay Y (2011). Human type 2 17beta‐hydroxysteroid dehydrogenase in umbilical vein and artery endothelial cells: differential inactivation of sex steroids according to the vessel type. Endocrine 40, 203–211. [DOI] [PubMed] [Google Scholar]
- Simmons GH, Padilla J & Laughlin MH (2012). Heterogeneity of endothelial cell phenotype within and amongst conduit vessels of the swine vasculature. Exp Physiol 9, 1074–1082. [DOI] [PubMed] [Google Scholar]
- Simoncini IT, Mannella P, Fornari L, Caruso A, Varone G & Genazzani AR (2004). Genomic and non‐genomic effects of estrogens on endothelial cells. Steroids 69, 537–542. [DOI] [PubMed] [Google Scholar]
- Sinha‐Hikim I, Taylor WE, Gonzalez‐Cadavid NF, Zheng W & Bhasin S (2004). Androgen receptor in human skeletal muscle and cultured muscle satellite cells: up‐regulation by androgen treatment. J Clin Endocrinol Metab 89, 5245–5255. [DOI] [PubMed] [Google Scholar]
- Stark MJ, Clifton VL & Wright IMR (2008). Sex‐specific differences in peripheral microvascular blood flow in preterm infants. Pediatr Res 63, 415–419. [DOI] [PubMed] [Google Scholar]
- Stevens T, Rosenberg R, Aird WC, Quertermous T, Johnson FL, Garcia JGN, Hebbel RP, Tuder RM & Garfinkel S (2001). NHLBI workshop report: endothelial cell phenotypes in heart, lung, and blood diseases. Am J Physiol Cell Physiol 281, C1422–C1433. [DOI] [PubMed] [Google Scholar]
- Thorin E & Shreeve SM (1998). Heterogeneity of vascular endothelial cells in normal and disease states. Pharmacol Ther 78, 155–166. [DOI] [PubMed] [Google Scholar]
- Tillet E, Vittet D, Féraud O, Moore R, Kemler R & Huber P (2005). N‐cadherin deficiency impairs pericyte recruitment, and not endothelial differentiation or sprouting, in embryonic stem cell‐derived angiogenesis. Exp Cell Res 310, 392–400. [DOI] [PubMed] [Google Scholar]
- Torres‐Estay V, Carreno D V., Fuenzalida P, Watts A, San Francisco IF, Montecinos VP, Sotomayor PC, Ebos J, Smith GJ & Godoy AS (2017). Androgens modulate male‐derived endothelial cell homeostasis using androgen receptor‐dependent and receptor‐independent mechanisms. Angiogenesis 20, 25–38. [DOI] [PubMed] [Google Scholar]
- Torres‐Estay V, Carreno D V., San Francisco IF, Sotomayor P, Godoy AS & Smith GJ (2015). Androgen receptor in human endothelial cells. J Endocrinol 224, R131–R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth B, Saadat G, Geller A, Scholz C, Schulze S & Friese K (2008). Human umbilical vascular endothelial cells express estrogen receptor beta (ER) and progesterone receptor A (PR‐A), but not ER and PR‐B. Histochem Cell Biol 130, 399–405. [DOI] [PubMed] [Google Scholar]
- Toutain CE, Filipe C, Billon A, Fontaine C, Brouchet L, Guéry JC, Gourdy P, Arnal JF, Lenfant F, Guery JC, Gourdy P, Arnal JF & Lenfant F (2009). Estrogen receptor α expression in both endothelium and hematopoietic cells is required for the accelerative effect of estradiol on reendothelialization. Arterioscler Thromb Vasc Biol 29, 1543–1550. [DOI] [PubMed] [Google Scholar]
- Tschugguel W, Dietrich W, Zhegu Z, Stonek F, Kolbus A & Huber JC (2003). Differential regulation of proteasome‐dependent estrogen receptor α and β turnover in cultured human uterine artery endothelial cells. J Clin Endocrinol Metab 88, 2281–2287. [DOI] [PubMed] [Google Scholar]
- Tse D & Stan R V. (2010). Morphological heterogeneity of endothelium. Semin Thromb Hemost 36, 236–245. [DOI] [PubMed] [Google Scholar]
- Vicaut E, Hou X, Decuypère L, Taccoen A & Duvelleroy M (1994). Red blood cell aggregation and microcirculation in rat cremaster muscle. Int J Microcirc Exp 14, 14–21. [DOI] [PubMed] [Google Scholar]
- Vrtačnik P, Ostanek B, Mencej‐Bedrač S & Marc J (2014). The many faces of estrogen signaling. Biochem Med 24, 329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AH, Schroeter MR & Hecker M (2001). 17beta‐estradiol inhibition of NADPH oxidase expression in human endothelial cells. FASEB J 15, 2121–2130. [DOI] [PubMed] [Google Scholar]
- Wagner WH, Henderson RM, Hicks HE, Banes AJ & Johnson G (1988). Differences in morphology, growth rate, and protein synthesis between cultured arterial and venous endothelial cells. J Vasc Surg 8, 509–519. [PubMed] [Google Scholar]
- Wang J (2005). Modulation of coronary and skeletal muscle exchange by adensoine: role of adenosine receptors. PhD Thesis University of Missouri‐Columbia.
- Wang J, Bingaman S & Huxley VH (2010). Intrinsic sex‐specific differences in microvascular endothelial cell phosphodiesterases. Am J Physiol Heart Circ Physiol 298, H1146–H1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J & Huxley VH (2006). Adenosine A 2A receptor modulation of juvenile female rat skeletal muscle microvessel permeability. Am J Physiol Heart Circ Physiol 291, H3094–H3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Whitt SP, Rubin LJ & Huxley VH (2005). Differential coronary microvascular exchange responses to adenosine: roles of receptor and microvessel subtypes. Microcirculation 12, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S, Aihara K ‐i., Ikeda Y, Sumitomo‐Ueda Y, Uemoto R, Ishikawa K, Ise T, Yagi S, Iwase T, Mouri Y, Sakari M, Matsumoto T, Takeyama K ‐i., Akaike M, Matsumoto M, Sata M, Walsh K, Kato S & Matsumoto T (2013). Androgen receptor promotes sex‐independent angiogenesis in response to ischemia and is required for activation of vascular endothelial growth factor receptor signaling. Circulation 128, 60–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarros A, Iraci N, Liu J, Rubin JB, Warrington NM & Sun T (2015). Targeting brain tumor cAMP: the case for sex‐specific therapeutics. Front Pharmacol 6, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y & Lingappan K (2017). Differential sex‐specific effects of oxygen toxicity in human umbilical vein endothelial cells. Biochem Biophys Res Commun 486, 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Zhang Y, Barutello G, Chiu K, Arigoni M, Giampietro C, Cavallo F & Holmgren L (2016). Angiomotin like‐1 is a novel component of the N‐cadherin complex affecting endothelial/pericyte interaction in normal and tumor angiogenesis. Sci Rep 6, 30622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman MA & Sullivan JC (2013). Hypertension: what's sex got to do with it? Physiology 28, 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]