

Abstract
The single pass, transmembrane proteins of the cadherin family have been appreciated as important proteins that regulate intercellular adhesion. In addition to this critical function, cadherins contribute to important signalling events that control cellular homeostasis. Many examples exist of classical, desmosomal and atypical cadherins participating in the regulation of signalling events that control homeostatic functions in cells. Much of the work on cadherin mediated signalling focuses on classical cadherins or on specific disease states such as pemphigus vulgaris. Cadherin mediated signalling has been shown to play critical roles during development, in proliferation, apoptosis, disease pathobiology and beyond. It is becoming increasingly clear that cadherins operate through a range of molecular mechanisms. The diversity of pathways and cellular functions regulated by cadherins suggests that we have only scratched the surface in terms of the roles that these versatile proteins play in signalling and cellular function.
Keywords: Cadherin, Proliferation, Apoptosis, Differentiation, Adhesion, Protein Cleavage
Introduction
Cadherins serve as calcium dependent transmembrane adhesion proteins and are subdivided into classical‐, proto‐, desmosomal and atypical cadherins (Harris & Tepass, 2010). The classical and desmosomal cadherins have an ectodomain composed of five extracellular cadherin repeats, a single transmembrane region, and a cytoplasmic domain that interacts with either β‐catenin (classical cadherins) or plakoglobin (Pg) (desmosomal and classical cadherins) (Shapiro & Weis, 2009). In addition to the classical adhesive function of cadherins, they contribute to a number of other cellular processes including cellular signalling (Kovacs, 2003; Wheelock & Johnson, 2003; Heuberger & Birchmeier, 2010). Dynamic changes in cell adhesion are needed to establish and re‐establish cell–cell contacts in multiple contexts, including developmental cell movements, tissue renewal and wound repair (Harris & Tepass, 2010; Pandya et al. 2017). These processes require cells to integrate and process a variety of signals from the external as well as the internal milieu (Table 1). In many cases, the precise mechanism by which the cadherins participate in a signalling pathway to influence cellular outcome is not fully understood, even though their involvement in the pathway is clearly demonstrated. One of the most well characterized signalling functions of cadherins is their acting as scaffolds to either promote or inhibit certain signalling pathways, which most often depend on the presence of the cadherin in the junction or in its adhesion state. Through these and other mechanisms, cadherins participate in the regulation of cellular homeostatic events that encompass proliferation, differentiation and apoptosis. For the purposes of this review, we define “homeostatic pathways” as pathways that regulate proliferation, cell death or the differentiation of cells. We are also including mechanisms that may be required for cells to maintain a homeostatic state (i.e. glucose metabolism) as “non‐homeostatic” though they can play a role in homeostasis. In this review, we will discuss the emergence of distinct classes of cadherin mediated signalling mechanisms. Mechanisms that are directly dependent upon changes in the adhesion state or junctional residence of cadherins are discussed first. We will refer to these as adhesion or junctional residence dependent cadherin signalling mechanisms. Thereafter, mechanisms that are not themselves dependent upon a change in adhesion are discussed.
Table 1.
Known cadherin mediated signaling mechanisms
| Functional output | Cadherin (fragment) | Associated proteins | Effectors | Reference |
|---|---|---|---|---|
| Proliferation | E‐cadherin | β‐Catenin | TCF/LEF; fibronectin | (Solanas et al. 2008) |
| TCF/LEF; β‐catenin | (Maher et al. 2009) | |||
| β‐Catenin | Hippo; YAP | (Kim et al. 2011) | ||
| β‐Catenina | TCF/LEFb; Wnt3acWnt3a | (Howard et al. 2011) | ||
| (NTFd) | HER2/3 | ErbB | (Najy et al. 2008) | |
| (CTF2d) | p120; Kaiso | Kaisob | (Ferber et al. 2008) | |
| EGFR | EGFc; STAT5; Erk | (Perrais et al. 2007) | ||
| p120 | Rac1 | (Liu et al. 2006) | ||
| E‐cadherin/cadherin‐11 | p120; PLEKHA7 | p120; Src; p130 CAS; c‐Myc; FAK; SNAI1; | (Kourtidis et al. 2015) | |
| N‐cadherin | β‐Catenin | Akt | (Zhang et al. 2013) | |
| β‐Catenina | TCF/LEFb; Wnt3ac; TBX6 | (Zhang et al. 2013) | ||
| (CTF2d) | Cyclin D1; β‐catenin | (Shoval et al. 2007) | ||
| VE‐cadherin | TGF‐βR; ALK5; ALK1 |
PAI‐1, Id1; TGF‐βc; Smad1/5, 2, 3 |
(Rudini et al. 2008) | |
| VEGF‐RII; β‐catenin | p44/42 MAPK VEGFc; EGFc | (Grazia Lampugnani et al. 2003) | ||
| (Multiple processes affected) | VE‐cadherin/N‐cadherin | β‐Catenin |
|
(Giampietro et al. 2012) |
| Cadherin‐17 |
|
(Lin et al. 2014) | ||
| Dsg‐2 | EGFR; Dsc‐2; histone H3; Src; Erk1/2 | (Kamekura et al. 2014) | ||
| (Multiple processes affected) | MAPK; Akt; PDK‐1; GSK‐3β; PTEN; c‐Myc; NF‐κB; MEK1/2; p90RSK; Raf; STAT3; Bcl‐XL | (Brennan et al. 2007) | ||
| (ECFd) | HER2/3 | S6; mTOR; MAPK; Akt; Erk1/2 | (Kamekura et al. 2015) | |
| Dsc‐2 | EGFR; CD44; Akt; β‐cateninb | (Kolegraff et al. 2011) | ||
| Dsc‐3 | Erk1/2 | (Cui et al. 2012) | ||
| Apoptosis | E‐cadherin | DR4/5 | DISC (complex formation) | (Lu et al. 2014) |
| STAT3; Rac1; Cdc42 | (Arulanandam et al. 2009) | |||
| Notch; Bcl‐2 | (Ferreira et al. 2012) | |||
| Igf‐1r | Igf‐1c | (Bedzhov et al. 2012) | ||
| (Multiple processes affected) |
|
(Onder et al. 2008) | ||
| (Multiple processes affected) | VE‐cadherin / N‐cadherin | β‐Catenin |
|
(Giampietro et al. 2012) |
| Dsg‐1 | Caspase‐3 | (Dusek et al. 2006) | ||
| (Multiple processes affected) | MAPK; Akt; PDK‐1; GSK‐3β; PTEN; c‐Myc; NF‐κB; MEK1/2; p90RSK; Raf; STAT3; Bcl‐XL | (Brennan et al. 2007) | ||
| Differentiation | E‐cadherin | Klf4; Nanog; STAT3; Erk1/2; Akt; N‐cadherin | (Hawkins et al. 2012) | |
| STAT3; Tet1; Esrrb; Tbx3; Nanog; Klf4; Nr0B1; Nr5a2; Erk1/2 | (Segal & Ward, 2017) | |||
| Dsg‐1 | Erbin; Shoc2 | Erk1/2; K‐Ras; loricrin; keratin‐10 | (Harmon et al. 2013) | |
| EGFR; Erk1/2; lamin A/C; keratin‐10; desmocollin‐1; fillagrin; loricrin; c‐Raf; MEK1/2; EphA2; ErbB2 | (Getsios et al. 2009) | |||
| Bcre | Desmocollin‐1; MAL; SRF; keratin‐10; keratin‐1; loricrin | (Dubash et al. 2013) | ||
| Dsg‐1/Dsc‐1 |
|
(Johnson et al. 2014) | ||
| Cadherin‐11 (extracellular fragmentd – fragment is unnamed) | ADAM13; ADAM9 | ? | (McCusker et al. 2009) | |
| “Non‐homeostatic signalling”/disease | E‐cadherin | Neuregulin1e; HER2; ErbB2 | Akt; Erk; P0 | (Basak et al. 2015) |
| ROCKf; Rac1f; FAKf | ? | (Parnaud et al. 2015) | ||
| (Multiple processes affected) | β‐Catenin | NF‐κB; Wnt; c‐Myc; IκBα; GLUT1; GLUT3; GLUT4 | (Park et al. 2017) | |
| Lkb1; AMPKa | MLC; vinculin; CrkL; Abl; RhoA | (Bays et al. 2017) | ||
|
(Bae et al. 2013) | |||
| (Multiple processes affected) |
|
(Onder et al. 2008) | ||
| Twist1e |
|
(Shamir et al. 2014) | ||
| E‐cadherin/N‐cadherin | Afadin; Nectin‐2; Nectin‐3 | (Labernadie et al. 2017) | ||
| N‐cadherin (extracellular fragmentd– fragment is unnamed) | ADAM10; MMP7 | NF‐κB; Iba‐1; TNF‐α; MMP9; MCP‐1 | (Conant et al. 2017) | |
| VE‐cadherin | Notch1; LAR; Rac‐1; Trio | ? | (Polacheck et al. 2017) | |
| Dsg‐1 | Periostin | (Sherrill et al. 2014) | ||
| Dsg‐3 | E‐cadherin; actin; Rac1; RhoA; Cad42 | Src; p120; β‐catenin; MLC | (Tsang et al. 2012b) | |
|
(Tsang et al. 2012a) | |||
| Cadherin‐11 | β‐Catenin | GEF‐Trio; Rho; Rac; Cdc42 | (Kashef et al. 2009) |
aInteraction is inferred but not directly demonstrated; binferred through effects on promoter based reporter activity assay; cthe cadherin influences the outcome of this mediator's signalling, but is not activating or repressing this mediator directly; dcadherin cleavage fragment shown to mediate this signalling; ealthough no interaction is shown or inferred, the cadherin directly modulates the signalling of this protein; finhibition of this protein enhances effects of cadherin mediated signalling, although no direct binding was detected or inferred. NTF, N terminal fragment; ECF, extracellular fragment.
Cadherin mediated regulation of proliferation
Cadherins have been implicated in the regulation of cell proliferation through interaction with a wide range of signalling mediators. One example, and arguably the most classic paradigm of cadherin mediated signalling, is by modulating β‐catenin signalling (Harris & Tepass, 2010; Clevers & Nusse, 2012). The classical cadherins E‐cadherin, N‐cadherin and VE‐cadherin can prevent nuclear localization of β‐catenin by direct interaction, thereby inhibiting T‐cell factor/Lymphoid enhancer‐binding factor (TCF/LEF) mediated stimulation of proliferation (Solanas et al. 2008; Kourtidis et al. 2017). In addition to sequestration through direct binding, E‐cadherin can suppress β‐catenin signalling through other mechanisms. For example, re‐expression of E‐cadherin in SW480 colon carcinoma cells leads to increased β‐catenin phosphorylation and co‐localization at the plasma membrane with adenomatous polyposis coli (APC), thus promoting β‐catenin degradation (Maher et al. 2009). Furthermore, the authors show that neither phosphorylated β‐catenin nor members of the destruction complex co‐fractionate with E‐cadherin suggesting that regulation of β‐catenin stability by E‐cadherin can be independent of direct binding (Maher et al. 2009). However, another study found that re‐expression of E‐cadherin in this same cell line results in repression of fibronectin and Lef1 gene expression through prevention of β‐catenin nuclear localization via direct binding (Solanas et al. 2008). These data suggest that E‐cadherin can regulate the function of β‐catenin through multiple functionally distinct mechanisms within the same cell type. Additionally, depending on the circumstances and the mechanism used, E‐cadherin can either promote or repress β‐catenin signalling and therefore proliferation. Such differences highlight the intricacies and complexities of cadherin mediated signalling. Binding of E‐cadherin to β‐catenin also affects other signalling pathways in addition to TCF/LEF mediated transcriptional regulation. E‐cadherin engagement directly regulates the Hippo signalling pathway in a β‐catenin dependent manner through prevention of YES associated protein (YAP) nuclear localization, thereby inhibiting cell proliferation (Kim et al. 2011). Howard et al. noted that when comparing two common model epithelial cell lines, Madin‐Darby canine kidney (MDCK) and human embryonic kidney (HEK293), MDCK cells are more classically “epithelial” and have well developed tight and adherens junctions, whereas the HEK293 cells lack such properties. Exposure of MDCK cells to hepatocyte growth factor (HGF) results in transition from the epithelial phenotype to one resembling HEK293 cells. While treatment of standard MDCK cells with Wnt3a does not noticeably affect β‐catenin signalling, pretreatment with HGF (or treatment of HEK293 cells) increases β‐catenin signalling compared to standard MDCK cells. E‐cadherin knock‐down in the HGF‐pretreated MDCK cells or in the HEK293 cells abolishes these signalling effects (Howard et al. 2011). These findings suggest that when epithelial cells transition to a more mesenchymal phenotype, the ability of Wnt ligands to activate β‐catenin signalling is enhanced and this effect is dependent on E‐cadherin. In light of the results discussed above, the outcome of E‐cadherin on β‐catenin signalling is dependent on whether or not the cells exhibit an epithelial or a mesenchymal phenotype. During development several populations of cells undergo an epithelial to mesenchymal transition (EMT), including epiblast cells in the primitive streak (Yoshikawa et al. 1997; Chapman & Papaioannou, 1998). As part of this process, these cells switch from predominantly expressing E‐cadherin to expressing N‐cadherin with increased Wnt/β‐catenin signalling that has been linked to differentiation of these migrated cells into the mesoderm. To test the functional relevance of their in vitro findings, the authors analysed the levels of the Wnt/β‐catenin target gene Tbx6 (known to be important for the differentiation of migrated epiblasts into mesoderm) in N‐cadherin knock‐out mouse embryos (Yoshikawa et al. 1997; Chapman & Papaioannou, 1998). Reduced Tbx6 expression was observed in these knock‐out mice compared to controls, suggesting that cadherin mediated increase of Wnt/β‐catenin signalling may be required for critical steps in embryonic development (Howard et al. 2011). Together, these data suggest that through modulation of β‐catenin activity, E‐cadherin can participate in a variety of signal transduction pathways that influence cellular outcomes.
In addition to regulation of β‐catenin, E‐cadherin influences proliferation through other pathways as well. For example, engagement of E‐cadherin either through direct cell–cell contact or through application of E‐cadherin Fc‐coated beads in the absence of cell spreading enhances the number of cells in the proliferative S‐phase compared to controls. This increase in proliferation was found to be dependent upon the activation of Rac1 and was lost upon E‐cadherin knock‐down or treatment with the E‐cadherin adhesion inhibitory antibody HECD1 (Liu et al. 2006). Such observations suggest that the E‐cadherin mediated regulation of proliferation is controlled by signalling events that are directly dependent on its adhesive role in adherens junctions and not due to the steric blockage of cell spreading. Knock‐down of the adherens junction associated protein pleckstrin homology domain containing A7 (PLEKHA7) in Caco2 model intestinal epithelial cells results in increased cadherin‐11, phospho‐p130CAS, cyclin D1, SNAI1 and c‐Myc protein levels. Additional knock‐down of E‐cadherin or preventing the increase in cadherin‐11 via shRNA reverses these changes (Kourtidis et al. 2015). Although the authors focus more on the mechanism through which PLEKHA7 itself participates in this signalling pathway, it is clear that the effects of PLEKHA7 loss are dependent on the presence of cadherins.
Besides E‐cadherin, other cadherins regulate cell proliferation through a variety of mechanisms. VE‐cadherin inhibits cell growth by binding to a variety of growth factor receptors such as TGF‐βR (Rudini et al. 2008) and vascular endothelial growth factor receptor II (VEGF‐RII) (Grazia Lampugnani et al. 2003). In neural precursor cells located within the ventricular zone, N‐cadherin knock‐down results in reduced β‐catenin activity and Akt phosphorylation, as opposed to the increased β‐catenin activity seen after knock‐down of E‐cadherin and VE‐cadherin. The authors found that Akt prevented premature maturation of these neural progenitors (Zhang et al. 2013). Some unconventional or atypical cadherins can also modulate proliferative signalling. For example, cadherin‐17 knock‐down has been shown to result in decreased proliferation through reduced Erk phosphorylation (Lin et al. 2014). Even though these studies reporting detailed mechanisms by which cadherin family members other than E‐cadherin regulate proliferation are limited, it is evident that these proteins participate in these biological responses.
The role of desmosomal cadherins in proliferative signalling is complex and depends on the cell‐type and/or cadherin involved. In intestinal epithelial cells, desmocollin‐2 (Dsc‐2) knock‐down results in increased proliferation through epidermal growth factor receptor (EGFR)/β‐catenin signalling (Kolegraff et al. 2011), while desmoglein‐2 (Dsg‐2) knock‐down reduces EGFR phosphorylation and inhibits proliferation (Kamekura et al. 2014). In line with these results, expression of Dsg‐2 in the suprabasal layer of the epidermis in mice leads to epidermal hyperplasia. In western blots comparing transgenic and wild‐type mouse skin, changes in the phosphorylation of several signalling mediators known to regulate proliferation were observed. These include mitogen‐activated protein kinase (MAPK), Akt, phosphoinositide‐dependent kinase‐1 (PDK‐1), phosphatase and tensin homolog (PTEN) (inhibitory modification) and glycogen synthase kinase‐3β (GSK‐3β) as well as increased PTEN, c‐Myc and nuclear factor‐κB (NF‐κB) protein levels (Brennan et al. 2007). Exposure of intestinal epithelial cells to either interleukin‐1β (IL‐1β) or tumor necrosis factor‐α (TNF‐α) results in extracellular cleavage of Dsg‐2 by the matrix metalloproteases, matrix metallopeptidase 9 (MMP9) and A Disintegrin and metalloproteinase domain‐containing protein 10 (ADAM10). The resulting Dsg‐2 cleaved fragment reduces intercellular adhesion and promotes proliferation through activation of human epidermal growth factor receptor 2/3 (HER2/3) signalling (Fig. 1 A) (Kamekura et al. 2015). Analogous to Dsg‐2 ectodomain cleavage, ADAM15 promotes extracellular cleavage of E‐cadherin and the resulting fragment promotes HER2/3 heterodimerization and activation in breast cancer cell lines (Najy et al. 2008). The authors suggest that proliferation mediated by activation of HER2/3 signalling could be associated with the oncogenic properties of soluble E‐cadherin. Since these extracellular fragments affect both proliferation and adhesion, we have included this signalling mechanism as being adhesion dependent, despite the requirement for cadherin cleavage. Re‐expression of Dsc‐3 in Dsc‐3‐deficient lung cancer cell lines results in decreased basal levels of phosphorylated Erk1/2 and reduced proliferation (Cui et al. 2012). Thus the loss of Dsc‐3 in lung cancer cells appears to activate pro‐proliferative oncogenic Erk1/2 activity. These findings highlight a role of desmosomal cadherins that, analogous to classical cadherins, may control proliferation and invasion of tumour cells.
Figure 1. Examples of cleavage dependent cadherin mediated signalling.
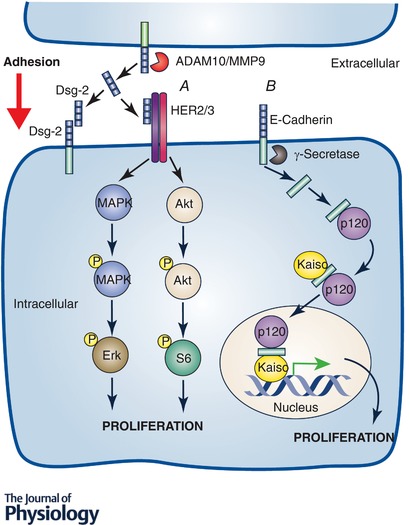
A growing number of reports have shown that cadherin protein fragments generated through regulated cleavage play important signalling roles. Many of these mechanisms are dependent upon cadherin extracellular fragment generation (A). These usually influence signalling of transmembrane receptors and/or cadherin mediated adhesion in a paracrine and/or autocrine manner. The extracellular fragment of Dsg‐2 has been shown to participate in both of these activities interfering with Dsg‐2 extracellular adhesive binding and increasing proliferation through stimulation of the HER2 and HER3 receptors (Kamekura et al. 2015). Cadherin intracellular fragments also mediate important signalling events (B). An example of this is the enhancement of E‐cadherin C‐terminal fragment 2 (CTF2) nuclear localization through binding to p120 and increased Kaiso mediated nuclear transcription (Ferber et al. 2008).
Cadherin mediated regulation of apoptosis
In addition to controlling proliferation, cadherins act as important mediators of apoptotic signalling. Much of the work into E‐cadherin mediated regulation of apoptosis has been done in the context of its role in cancer development and progression. E‐cadherin homophilic engagement in HC11 mouse breast cancer cells enhances signal transducer and activator of transcription 3 (STAT3) phosphorylation and disrupting this binding reduces pSTAT3 levels through activation of Ras‐related C3 botulinum toxin substrate 1 (Rac1) leading to increased apoptosis (Fig. 2 A) (Arulanandam et al. 2009). This increased apoptosis and reduced STAT3 activation highlights disrupting E‐cadherin engagement in breast cancer as a potentially attractive therapeutic strategy. Taken together with the findings of Liu et al. (2006), E‐cadherin adhesion‐dependent activation of Rac1 appears to both enhance proliferation and reduce apoptosis, suggesting that this is one of the E‐cadherin‐dependent mechanisms that are known to be suppressed in cancer (Liu et al. 2006). Expression of E‐cadherin in lung, colorectal and pancreatic cancer lines enhances apoptosis and caspase‐8 activation through direct interaction with the TNF‐α‐related apoptosis inducing ligand (TRAIL) receptors, death receptors 4 and 5 (DR4/5) (Lu et al. 2014). Apoptosis sensitization was enhanced by E‐cadherin adhesion and promoted formation of the death inducing signalling complex (DISC) in response to TRAIL treatment (Fig. 2 B) (Lu et al. 2014). This is an example of cadherins acting as scaffolds for transmembrane signalling proteins in addition to functioning as scaffolds for cytoplasmic/intracellular signalling proteins. E‐cadherin adhesion deficient mutant proteins were used to show that E‐cadherin mediated adhesion participates in the stimulation of Notch signalling, resulting in increased expression of the anti‐apoptotic protein Bcl‐2 (Ferreira et al. 2012). These data provide examples showing that the adhesion state of E‐cadherin, in addition to ensuring cell–cell cohesion, acts as a signal that modulates a variety of downstream signalling events to influence cellular homeostasis. Using mouse models expressing different chimeric cadherins containing portions of N‐cadherin and E‐cadherin it was shown that the extracellular domain of E‐cadherin interacts with the insulin‐like growth factor 1 (Igf‐1) receptor, which promotes the activity of this receptor and survival of trophectoderm cells during blastocyst formation (Bedzhov et al. 2012). Through its robust regulation of both cell proliferation and cell death, E‐cadherin functions to control key cellular outcomes that influence cellular homeostasis.
Figure 2. Examples of cadherin mediated signalling dependent on junctional residence and adhesive state.
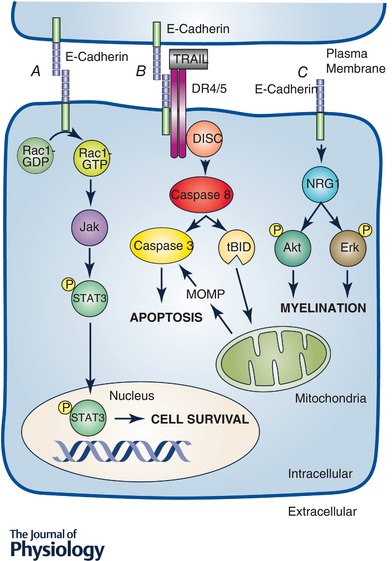
Cadherin cytoplasmic tails can act as scaffolds for intracellular signalling molecules thereby promoting distinct signalling pathways under various conditions. These scaffolding functions are often dependent upon the following: the adhesive state of the cadherin (A and B) or the presence of the cadherin within a stable junction (C). Examples of these types of mechanisms include activation of Rac1 and STAT3 signalling through E‐cadherin adhesive binding leading to repression of apoptosis (Arulanandam et al. 2009) or reduced peripheral nervous system myelination in the absence of E‐cadherin (Basak et al. 2015). There have even been a few reports of cadherin adhesive binding leading to the cadherin acting as a scaffold for and enhancing the activity of transmembrane signalling receptors such as the receptors for TNF‐α related apoptosis inducing ligand (TRAIL), death receptors 4 and 5 (DR4 and 5) leading to enhanced death inducing signalling complex (DISC) formation and apoptosis sensitivity (Lu et al. 2014). MOMP, Mitochondrial Outer Membrane Permebialization; NRG 1, Neuregulin1; tBID, trancated BH3 interacting‐domain death agonist.
The role of desmosomal cadherin mediated adhesion or junctional residence in apoptotic signalling is less well understood, but a few such mechanisms have been described. For example, expression of Dsg‐2 in suprabasal cells of the murine epidermis increases NF‐κB and Bcl‐XL protein levels. Keratinocytes derived from these mice display greater survival compared to cells derived from wild‐type mice. These effects are enhanced by epidermal growth factor (EGF) treatment and are abolished by inhibition of NF‐κB (Brennan et al. 2007). As with proliferation, other classical cadherins in addition to E‐cadherin participate in the regulation of apoptosis. In the absence of VE‐cadherin, endothelial cells express N‐cadherin through enhanced β‐catenin signalling as well as display increased apoptosis, proliferation and migration, and re‐expression of VE‐cadherin in these cells reverses these effects (Giampietro et al. 2012). The authors found that altering the balance of normal VE‐cadherin to N‐cadherin expression levels leads to vascular elongation and increased angiogenesis (Giampietro et al. 2012). Following a series of co‐expression experiments the authors suggest that VE‐cadherin and N‐cadherin play similar but divergent roles in the regulation of proliferation, apoptosis and migration of endothelial cells.
Cadherin mediated control of cellular differentiation
In addition to being important regulators of proliferation and apoptosis, cadherins also regulate cell differentiation in various contexts. Loss of E‐cadherin in in vitro cultured murine embryonic stem cells decreases Kruppel‐like factor 4 (Klf4) and Nanog expression as well as pSTAT3 levels and interaction of STAT3 with the Nanog promoter (Hawkins et al. 2012). In another study using embryonic stem cells, treatment with E‐cadherin adhesion blocking antibodies reduced the expression of several transcriptional regulators as well as ten‐eleven translocation methylcytosine dioxygenase 1 (Tet1) and estrogen‐related receptor beta (Esrrb) (Segal & Ward, 2017). These reports highlight the importance of E‐cadherin in the maintenance of the embryonic stem cell transcriptional programme and in the ability of embryonic stem cells to maintain their pluripotency and stem cell properties. Overexpression of cadherin‐11 in Xenopus embryos inhibits cranial neural crest migration and is phenocopied by ADAM13 inhibition. The authors found that ADAM13 cleaves cadherin‐11 generating an extracellular fragment and treatment with this cadherin‐11 extracellular fragment alleviates the effects of cadherin‐11 overexpression and ADAM13 inhibition on cranial neural crest migration through binding of cadherin‐11 (McCusker et al. 2009). This is another example of the effects of cadherin extracellular fragments in the modulation of cadherin mediated adhesion via competitive binding. Such observations highlight the importance of cadherin extracellular fragments functioning as signalling molecules that influence cadherin mediated adhesion. Generation of these fragments can be exquisitely regulated in order to facilitate specific functional outcomes like increased proliferation or migration of cells.
Much of the research describing roles of cadherins in the regulation of differentiation are centred on the intricately regulated expression patterns of desmosomal cadherins, which play an essential role in ensuring appropriate epidermal differentiation. This topic has been extensively discussed in previous reviews and will therefore not be detailed here (Green & Simpson, 2007; Nekrasova & Green, 2013). Exogenously expressed Dsg‐1 co‐immunoprecipitates Erbb2 interacting protein (Erbin) in normal human epidermal keratinocytes (NHEKs) (Harmon et al. 2013). Induction of differentiation in NHEKs leads to increased expression of many differentiation‐specific proteins, including loricrin and involucrin. However, after induction of differentiation in Erbin knock‐down cells, these changes are all diminished, resulting in a lack of appropriate cellular differentiation. In differentiated wild‐type NHEK cells pErk is predominantly confined to the basal layer, whereas after downregulation of Erbin, pErk was detected within the suprabasal cells of the epidermis. Dsg‐1 expression facilitates co‐immunoprecipitation of Ras–Raf scaffold protein Shoc2 with Erbin and diminishes Erbin's ability to co‐immunoprecipitate K‐Ras (Harmon et al. 2013). It is important to note that expression of wild‐type Dsg‐1 reduces pErk1/2 levels by two‐thirds, whereas expression of Dsg‐1 lacking the Erbin binding site decreases pErk1/2 levels by half. These data suggest that while the ability of Dsg‐1 to bind Erbin plays an important role in its regulation of Erk1/2 phosphorylation, this signalling function is not exclusively mediated by Erbin binding (Harmon et al. 2013). These data suggest that desmogleins can modulate binding properties of other important signalling scaffold proteins in addition to acting as a scaffold themselves. Abnormal differentiation of the suprabasal layers was observed in differentiated epidermal raft cultures derived from Dsg‐1 knock‐down NHEKs. Furthermore, inducing differentiation in Dsg‐1 knock‐down keratinocytes increases pEGFR and pErk1/2 compared to control cells. Moreover, Erk1/2 inhibition was found to reverse the effects of Dsg‐1 knock‐down on suprabasal epidermal differentiation (Getsios et al. 2009). Using NHEKs cultured in organotypic raft cultures, this same group showed that knock‐down of the guanine nucleotide exchange factor (GEF) Bcr or blockade of myelin and lymphocyte protein/serum response factor (MAL/SRF)‐induced transcriptional regulation leads to reduced expression of multiple epidermal differentiation associated proteins. Re‐expression of Dsg‐1 in these cells rescues the loss of these differentiation markers (Dubash et al. 2013). These findings indicate that Dsg‐1 dependent regulation of Erk1/2 phosphorylation, GEF Bcr and MAL/SRF‐induced transcription are critical events in epidermal differentiation and their loss results in impairment of this vital process. Exposure of NHEKs to short wave ultraviolet B (UVB) prior to the induction of differentiation caused a dose dependent decrease in Dsg‐1, Dsc‐1 and multiple keratins as well as an increased pErk which is associated with delayed/impaired epidermal differentiation. Through overexpression and knock‐down studies the authors show an influence of Dsg‐1 in protection from the adverse effects of UVB exposure on epidermal differentiation (Johnson et al. 2014). These example provide evidence for the importance of desmosomal cadherins in controlling epidermal differentiation which is vital for the defence properties of the skin.
Other cadherin signalling events
The role of cadherins in signalling extends beyond the basic homeostatic mechanisms of life, death and differentiation. In fact, recent research into cadherin mediated signalling has begun to focus more on the roles that cadherins play in other signalling pathways. Since research into these aspects of cadherin signalling is in its infancy, this section will primarily focus on some isolated examples from various systems ranging from regulation of Schwann cell mediated myelination (Basak et al. 2015) to insulin production by pancreatic β‐cells (Parnaud et al. 2015). The rate and extent of myelination in the peripheral nervous system of E‐cadherin knock‐out mice is significantly reduced compared to wild‐type animals, which would result in impaired peripheral nervous system function (Basak et al. 2015). E‐cadherin together with neuregulin 1 increases phosphorylation of HER2, Akt and Erk to facilitate myelination (Fig. 2 C) (Basak et al. 2015). Since very little is known about mechanisms that govern Schwann cell myelination, the finding that E‐cadherin and neuregulin 1 work together to enhance this process is of great interest. Pernaud et al. observed that primary human pancreatic β‐cells subjected to high glucose display a noticeable increase in insulin production when plated on E‐cadherin Fc‐coated glass compared to controls. This effect is blocked by treatment with the E‐cadherin adhesion blocking antibody DECMA‐1 (Parnaud et al. 2015). These findings suggest that engagement of E‐cadherin plays a role in insulin production in β‐cells thereby controlling overall glucose metabolism. Furthermore, a recent study by Park et al. has also supported a role of E‐cadherin in controlling glucose metabolism. Re‐expression of E‐cadherin in gastric cancer epithelial cells enhances oxidative phosphorylation and expression of glucose transporters. Glucose metabolism in cancer cells shifts from predominantly oxidative phosphorylation to anaerobic glycolysis during epithelial to mesenchymal transition (EMT), suggesting that loss of E‐cadherin may play a role in this metabolic shift. These effects are reduced following NF‐κB knock‐down, suggesting that NF‐κB participates in promoting oxidative phosphorylation upon E‐cadherin re‐expression in cancer cells (Park et al. 2017). Using MCF10A human mammary epithelial cells, Bays et al. showed that application of extracellular force to endogenous E‐cadherin through the use of E‐cadherin Fc‐coated magnetic beads stimulates AMP‐activated protein kinase (AMPK) activation as well as increased phosphorylation of myosin light chain (MLC), vinculin and CrkL. This was associated with reinforcement of the actin cytoskeleton and enhanced barrier function. Additionally, application of force to E‐cadherin increases glucose uptake and cellular ATP levels. These effects are inhibited by knock‐down of E‐cadherin, AMPK and Lkb1 as well as E‐cadherin adhesion blocking antibody treatment (Bays et al. 2017). Together, these results suggest that, in addition to its roles in adhesion and regulation of epithelial barrier properties, E‐cadherin contributes to nutrient signalling that has not been fully explored. Additionally, the work of Bays et al. highlights the diversity of cadherin mediated signalling mechanisms that are dependent on its mechanotransduction properties. Loss of E‐cadherin is known to play a role in EMT and the subsequent increased invasiveness and dissemination of metastatic cancer cells. Knock‐down of E‐cadherin in A549 non‐small cell lung carcinoma (NSCLC) cells increases invasiveness as well as enhances expression of multiple MMPs and transcriptional regulators. These effects of E‐cadherin knock‐down were reversed by mitogen‐activated protein kinase (MEK) inhibition (Bae et al. 2013). Such findings suggest that the contribution of E‐cadherin loss to EMT and tumour invasion is at least partly mediated through activation of MEK/Erk signalling.
Analogous to E‐cadherin, other classical cadherins contribute to diverse signalling events. For example, extracellular cleavage of N‐cadherin on microglial cells by ADAM10 and MMP9 and treatment of cultured primary microglial cells with the resulting protein fragment increases nuclear NF‐κB translocation as well as release of TNF‐α, MMP9 and monocyte chemoattractant protein‐1 (MCP‐1) indicating increased microglial activity (Conant et al. 2017). In light of the many examples of N‐cadherin participating in signalling mechanisms in epithelial, endothelial and neuronal cells, it will be of great interest to further explore and define the overall signalling repertoire of this protein and its control of cellular functions.
There have been recent examples of cadherin mediated signalling that involve their residence in multi‐protein complexes other than their traditional junctions. For example, Labernadie et al. showed that cancer associated fibroblasts (CAFs) exert forces on A431 human epidermal carcinoma cells in in vitro co‐culture. Co‐cultured CAFs form heterotypic N‐cadherin/E‐cadherin junctions with the A431 cells. These junctions can withstand mechanical forces and are involved in mechanotransduction cross‐talk between cancer cells and CAFs (Labernadie et al. 2017). Such cadherin mediated mechanotransduction influences CAF polarization and directional migration of cancer cells in three‐dimensional cultures led by CAF movement. E‐cadherin was required for the transduction of force between these two cell types. Thus, classical cadherins can participate in heterotypic junctions that can withstand force and these junctions can have an impact on cellular outcomes (Labernadie et al. 2017). In endothelial cells, it was shown that the transmembrane domain (TMD) of Notch1 forms a mechanosensory complex containing the transmembrane tyrosine phosphatase leukocyte common‐antigen related (LAR), GEF Trio and Rac1, which participate in the activation of Rac1 and functions to regulate the formation of adherens junctions and endothelial permeability. This complex is targeted to adherens junctions through binding of the Notch1 TMD with VE‐cadherin (Polacheck et al. 2017).
Tsang et al. examined the effects of Dsg‐3 on junctional and cytoskeletal components and observed that knock‐down of Dsg‐3 in HaCaT cells results in decreased expression of adherens junction, desmosome and actin cytoskeleton associated proteins that was linked to impaired development and function of intercellular junctions and cell polarization. In addition, Dsg‐3 knock‐down cells displayed decreased β‐catenin tyrosine phosphorylation as well as several other distinct effects including diminished re‐integration of junctional E‐cadherin and β‐catenin following calcium switch supporting its role in the assembly of intercellular junctions in HaCaT cells. This loss of Dsg‐3 impaired the formation of adherens junctions and E‐cadherin dependent Src activation (Tsang et al. 2012a, b ). In contrast, overexpression of Dsg‐3 in HaCaT and A431 cells increases the number of actin protrusions and the rate of actin turnover as well as activation of several Rho family small GTPases thereby influencing cellular polarization and tissue remodelling. When combined with Rac1 inhibition, Dsg‐3 overexpression increases A431 cell migration (Tsang et al. 2012a). The authors also described interaction between a non‐junctional pool of Triton X‐100 soluble Dsg‐3 and E‐cadherin in a calcium‐, p120‐ and plakoglobin‐dependent manner (Tsang et al. 2012b). Such observations suggest that Dsg‐3 (and potentially others desmogleins) play a much larger role in overall junctional and cytoskeletal regulation and the subsequent control of cellular polarization and tissue remodelling than previously appreciated.
Cadherin mediated signalling in the context of disease
Cadherin mediated signalling mechanisms have been shown to play an important role in multiple disease states and pathologies. One of the most well studied examples is in the context of cancer. A number of previous reports have addressed cadherin dependent regulation of cancer pathogenesis and progression that involves control of cellular proliferation, apoptosis, invasiveness, metabolism and metastasis in the context of this disease (Conacci‐Sorrell et al. 2002; Cavallaro & Christofori, 2004; Jeanes et al. 2008; Onder et al. 2008; Solanas et al. 2008; Maher et al. 2009; Bae et al. 2013; Lu et al. 2014; Park et al. 2017). Although E‐cadherin is a classic example of cadherin mediated signalling in cancer, N‐cadherin, VE‐cadherin, desmosomal cadherins and other cadherins have also been shown to participate in oncogenic signalling in cancer pathogenesis. Since we have mentioned examples of cadherin signalling in the context of oncogenesis above, and this topic has been detailed in previous reviews, we focus on cadherin contribution to other disease mechanisms in this section.
We will in large part address the role of cadherin signalling in the autoimmune skin diseases pemphigus vulgaris (PV) and pemphigus foliaceus (PF). Several signalling mechanisms have been implicated in the pathobiology of pemphigus. Pemphigus vulgaris and pemphigus foliaceus have been reported to be mediated by autoantibodies that target Dsg‐3 and Dsg‐1, respectively (Mao et al. 2011; Schulze et al. 2012). Studies using multiple systems have shown that the use of monoclonal antibodies targeting desmogleins can recapitulate pemphigus pathology (Schulze et al. 2012; Bektas et al. 2013). However, the contribution of desmogleins to these signalling events remains incompletely understood (Chernyavsky et al. 2007). We will highlight some key examples of signalling caused by pemphigus autoantibodies because this field of study has been previously reviewed more comprehensively (Sharma et al. 2007; Schmidt & Waschke, 2009; Spindler & Waschke, 2014). Following 48 h of treatment with PV autoantibodies, mouse keratinocytes display reduction in nuclear localization of plakoglobin (Pg). Plakoglobin is a member of the armadillo family of proteins and is the most closely related protein to the famous signalling mediator β‐catenin. Plakoglobin is a critical component of the desmosomal plaque and directly binds to the intracellular tails of desmogleins and desmocollins. After prolonged PV autoantibody treatment, keratinocytes display noticeably increased proliferation, c‐Myc expression and c‐Myc nuclear localization, which was shown to be reversed after plakoglobin knock‐out. Analogous increased proliferation is also seen in human PV patient skin samples (Williamson et al. 2006). NHEKs treated with PV autoantibodies display internalization and depletion of Dsg‐3, which are prevented by p38MAPK inhibition (Jolly et al. 2010). It was later shown that in response to patient derived PV autoantibody treatment p38MAPK phosphorylation occurs prior to the activation of EGFR resulting in blister formation. The PV autoantibody treatment causes an increase in pErk1/2, which is enhanced by inhibition of either p38MAPK or EGFR phosphorylation (Bektas et al. 2013). These observations suggest that some of the changes in signalling mediators observed upon PV autoantibody treatment can be antagonistic in nature. For example, the PV autoantibody induced increases in p38MAPK and EGFR dampening the increase in pErk1/2 caused by the same treatment. PV autoantibody or AK23 (a monoclonal antibody against mouse Dsg‐3 that causes PV phenotypes) treatment causes endocytosis of EGFR, which is prevented by p38MAPK inhibition resulting in reduced blister formation. Exposure of NHEKs to PV autoantibodies leads to Dsg‐3 internalization and overall loss of the protein. These effects are reduced by pre‐treatment with inhibitors of either p38MAPK or EGFR, or knock‐down of EGFR. The functional outcome of EGFR inhibition in these models was prevention of the loss of Dsg‐3 mediated adhesion resulting in decreased blister formation (Bektas et al. 2013).
Dysregulation of apoptosis is also known to be important in PV pathobiology. A mouse model often used to study these diseases is referred to as a passive transfer mouse, in which IgG or serum derived from PV or PF patients is injected into the blood stream of embryonic mice after which the mice develop symptoms and clinical manifestations of the parent disease. PV passive transfer mice show higher Bax and lower Bcl‐2 protein levels, as well as increased caspase‐3 and ‐9 activity. In addition, the PV transfer treated mice have enhanced phosphorylation of mammalian target of rapamycin (m‐TOR) and Src as well as increased blister formation and apoptosis, all of which are abolished or reduced by focal adhesion kinase (FAK) inhibition (Gil et al. 2012). Increased apoptosis was observed in the epidermis of PF passive transfer mice which led to the development of experimental PF. These effects were reversed using pan‐caspase or caspase‐3 selective inhibitors (Li et al. 2009). In biopsy samples derived from the skin surrounding the injection site of PF passive transfer mice, Lee et al. observed an increase in phospho‐p38MAPK in two peaks following treatment. The authors also described apoptosis execution beginning 30 h post‐treatment and increasing in severity over time, which is prevented by p38MAPK inhibition. Only inhibition of the first p38MAPK peak but not the second prevented blister formation in these mice (Lee et al. 2009). These data suggest that this first burst of p38MAPK activation mediates blister formation in PF. In the basal cells of PV passive transfer mice, Pretel et al. describe increased phosphorylated HER family growth factor receptors and downstream signalling molecules, including Src and m‐TOR, which were all inhibited by EGFR inhibitor treatment. The authors also found increased betacellulin, transforming growth factor‐α (TGF‐α) and EGF in these cells. Pre‐treatment with an EGFR inhibitor, a Src kinase family inhibitor, rapamycin or pan‐caspase inhibitors all prevent the apoptosis and other clinical manifestations normally seen in PV passive transfer mice (Pretel et al. 2009). These data suggest that the increased apoptosis seen in response to pemphigus autoantibody treatment is necessary for the development of clinical symptoms. Passive transfer mice have proved useful in exploring non‐apoptotic aspects of pemphigus related signalling as well. Using passive transfer of AK23 (monoclonal mouse anti‐Dsg‐3 antibody), Schulze et al. observed increased activation or expression of several signalling mediators that promote proliferation. They also observed decreased expression of total and phospho‐p38MAPK, Dsg‐1/2, Dsg‐3 and EGFR. These AK23 passive transfer mice developed typical blistering and other symptoms associated with clinical PV (Schulze et al. 2012). Ex vivo treatment of human skin tissue with PV or PF autoantibodies results in blister formation that is repressed by enhanced RhoA signalling. In HaCat cells, inhibition of p38MAPK or activation of Rho family members prevents the disruption of several desmosomal proteins and E‐cadherin normally seen in response to PV autoantibody treatment. As discussed above, disruption of these proteins is important to the development of blistering and other PV or PF symptoms. Treatment of HaCaT cells with PV or PF autoantibodies also results in reduced active RhoA, which is prevented by p38MAPK inhibition (Waschke et al. 2006). These data suggest that inhibition of RhoA occurs following p38MAPK activation and is an important step in pemphigus pathobiology through preventing RhoA activity from inhibiting the disruption of desmosomal proteins and E‐cadherin. Using a panel of PV monoclonal antibodies, only those that cause a loss of intercellular adhesion activate p38MAPK. The authors also found that inhibition of p38MAPK prevents Dsg‐3 internalization and depletion in response to the PV monoclonal antibodies (Mao et al. 2011). These data suggest that the disruption of desmoglein mediated adhesion through internalization and loss of the desmogleins themselves may be a critical factor in pathogenicity of these autoantibodies.
Cadherin mediated signalling has been shown to participate in other disease states besides cancer and pemphigus, but these are far fewer. For example, Dsg‐1 mRNA and protein expression is reduced in eosinophilic oesophagitis (EoE), that has been associated with reduced barrier integrity and adhesion (Sherrill et al. 2014). These effects are recapitulated by Dsg‐1 knock‐down of cultured oesophageal epithelial cells. Dsg‐1 knock‐down promotes pro‐inflammatory transcriptional responses as analysed by genome‐wide RNAseq (Sherrill et al. 2014). Both Dsg‐1 knock‐down in cultured oesophageal epithelial cells and in human EoE patient samples display noticeably increased mRNA expression of the integrin ligand protein periostin compared to controls (Sherrill et al. 2014). Periostin is known to be highly expressed in EoE (Dellon et al. 2016; Politi et al. 2017). These data suggest that increased periostin expression after loss of Dsg‐1 may contribute to the pathobiology of EoE. Samples from human patients with striate palmoplantar keratoderma (SPPK) arising from mutations in Dsg‐1 display increased Erk activity and reduced Dsg‐1, loricrin and keratin‐10 expression. These samples also display increased interaction of Shoc2 with K‐Ras and reduced interaction of this protein with Erbin (Harmon et al. 2013). Combined with the pemphigus examples above, desmogleins play a key role in the pathobiology of a number of disorders that include skin diseases and cancer.
Cadherin intracellular fragment signalling
The findings we have described thus far in this review highlight the diversity and importance of cadherin mediated signalling. However, most of these mechanisms are either dependent on or in some way influenced by changes in cadherin mediated adhesion in intercellular junctions. In the next section we will highlight an emerging class of cadherin mediated signalling mechanisms that do not appear to be influenced by changes in cadherin adhesion. Most of these mechanisms are based on intracellular cadherin cleavage and the generation of a functional protein fragment. For example, E‐cadherin can be cleaved intracellularly by γ‐secretase resulting in generation of the E‐cadherin C‐terminal fragment 2 (CTF2) (Marambaud et al. 2002). Expression of CTF2 results in its nuclear localization and increased proliferation, which are both enhanced by co‐expression with p120. E‐cadherin CTF2 promotes Kaiso mediated transcriptional regulation and is able to co‐immunoprecipitate both with Kaiso and p120 (Fig. 1 B) (Ferber et al. 2008). In line with the E‐cadherin CTF2, expression of N‐cadherin C‐terminal fragment 2 (CTF2) leads to N‐cadherin CTF2 nuclear localization and increased expression of β‐catenin and cyclin D1 as well as increased emigration of neural crest cells from the developing neural tube in quail embryos (Shoval et al. 2007). In response to UV induced apoptosis in keratinocytes, desmoglein‐1 is cleaved intracellularly by caspases‐3 and ‐7 and knock‐down of desmoglein‐1 protects cells from UV induced apoptosis (Dusek et al. 2006). We have recently shown that in response to TNF‐α and IFN‐γ as well as TRAIL treatment, Dsg‐2 undergoes intracellular cleavage that is mediated by caspase‐8. The resultant Dsg‐2 intracellular fragment (Dsg‐2 ICF) sensitizes intestinal epithelial cells to apoptosis through increased expression of the anti‐apoptotic Bcl‐2 family members Bcl‐XL and MCl1 (Yulis et al. 2018). Taken together, these findings highlight the importance of cadherin intracellular fragments serving as key signalling mediators.
Cadherin signalling in cells lacking intercellular adhesion
There are also a few examples of cadherin mediated signalling that remain intact in cells lacking intercellular adhesion. In immortalized untransformed human mammary epithelial cells E‐cadherin knock‐down results in increased N‐cadherin and vimentin expression as well as reduced expression of cytokeratin 18, indicative of an epithelial to mesenchymal transition. However, this was not seen after expression of adhesion blocking dominant negative E‐cadherin, indicating that the E‐cadherin dependent signalling mechanisms preventing cells from undergoing EMT in this system are independent of E‐cadherin's intercellular adhesion properties (Onder et al. 2008). In addition, E‐cadherin knock‐down results in increased motility, invasiveness and reduced sensitivity to apoptosis, all of which are rescued by expression of the adhesion blocking E‐cadherin dominant negative protein (Onder et al. 2008). While organoids generated from primary mammary epithelial cells derived from mice with inducible E‐cadherin knock‐out displayed a normal epithelial phenotype upon E‐cadherin knock‐out, the cells displayed a more classically mesenchymal phenotype. In spite of this phenotype, however, few cells disseminate out as single cells and therefore do exhibit classic invasive properties. The transcription factor Twist1 has been suggested to play an active role in promoting EMT through regulation of E‐cadherin and other mediators. Primary mammary epithelial cell organoids derived from mice with overexpression of Twist1 exhibit single cell dissemination, which was inhibited upon E‐cadherin knock‐down (Shamir et al. 2014). These data support a role of E‐cadherin–Twist signalling in the development of metastatic potential cells. Thus, E‐cadherin signalling modulates cancer phenotypes through signalling mechanisms that remain intact even in the absence of intercellular adhesion. Similarly, in Xenopus embryos knock‐down of cadherin‐11 inhibits cranial neural crest cell migration and this effect is rescued by expression of cadherin‐11 lacking the extracellular domain. However, expression of cadherin‐11 lacking the β‐catenin binding domain or the transmembrane domain does not rescue this loss of cranial neural crest cell migration. These effects are also reversed by supplementation with recombinant Rho family small GTPases or the Rho GEF Trio (Kashef et al. 2009). Although research into cadherin signalling in cells lacking intercellular adhesion is not completely understood, these examples open the door to a new frontier of possibilities regarding the breadth of cadherin mediated signalling.
Conclusion
The cadherin family of transmembrane proteins serve as important intercellular adhesion molecules. Over the years, appreciation of their function as critical signalling mediators has been steadily growing. Classical, desmosomal, atypical and unconventional cadherins all participate in many signalling cascades that influence proliferation, apoptosis and differentiation in a number of tissues and cell types as well as in disease. In more recent years, the critical roles that cadherins play in the regulation of non‐homeostatic cellular functions have emerged. Dysregulation of cadherin mediated signalling results in disease states encompassing epithelial barrier defects, inflammation and neoplasia. While great pioneering research has been performed, we have only just begun to understand the extent to which cadherins participate in signalling events. Due to their importance in intercellular adhesion, most of the cadherin mediated signalling described thus far is directly influenced by either the junctional residence or the adhesive state of the cadherin itself. Further study of these mechanisms will continue to expand our understanding of the repertoire of cadherin functions.
Additional information
Competing interests
None declared.
Author contributions
M.Y., D.H.M.K. and A.N. drafted and revised the manuscript. All authors approved the final version of this manuscript and agree to be accountable for all aspects of the work. All persons listed as authors qualify for authorship. All those who qualify for authorship are listed as authors.
Funding
The research leading to this publication was supported by the following funds: NIH DK059888 and DK055679 (to A.N.) and the People Program (Marie Curie Actions) of the European Union's Seventh Framework Program (FP7/2007‐2013) under REA grant agreement no. 608765 (to D.H.M.K.).
Acknowledgements
We would like to thank Robin Kunkel for her contributions to figure design.
Biographies
Mark Yulis received his BS in Biochemistry at Muhlenberg College in Pennsylvania and his PhD at Emory University. His studies examine mechanisms by which inflammatory mediators promote desmoglein 2 cadherin cleavage to influence epithelial apoptosis.
Dennis Kusters received his PhD in Biochemistry at the University of Maastricht in the Netherlands. For his postdoctoral training at the University of Michigan he is investigating the role of desmosomal cadherins in controlling intestinal epithelial homeostasis.
Asma Nusrat's research group investigates mechanisms by which intercellular junction proteins control epithelial homeostasis and barrier function, pathobiology of chronic mucosal inflammation and mechanisms of wound repair.

Edited by: Kim Barrett & Ole Petersen
This review was presented at the symposium ‘Gastrointestinal Tract XVII: Current Biology of the GI Tract, Mucosa, Microbiota, and Beyond’, which took place at FASEB 2017, Steamboat Springs, Colorado, USA, 30 July–4 August 2017.
References
- Arulanandam R, Vultur A, Cao J, Carefoot E, Elliott BE, Truesdell PF, Larue L, Feracci H & Raptis L (2009). Cadherin‐cadherin engagement promotes cell survival via Rac1/Cdc42 and signal transducer and activator of transcription‐3. Mol Cancer Res 7, 1310–1327. [DOI] [PubMed] [Google Scholar]
- Bae GY, Choi SJ, Lee JS, Jo J, Lee J, Kim J & Cha HJ (2013). Loss of E‐cadherin activates EGFR‐MEK/ERK signaling, which promotes invasion via the ZEB1/MMP2 axis in non‐small cell lung cancer. Oncotarget 4, 2512–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basak S, Desai DJ, Rho EH, Ramos R, Maurel P & Kim HA (2015). E‐cadherin enhances neuregulin signaling and promotes Schwann cell myelination. Glia 63, 1522–1536. [DOI] [PubMed] [Google Scholar]
- Bays JL, Campbell HK, Heidema C, Sebbagh M & DeMali KA (2017). Linking E‐cadherin mechanotransduction to cell metabolism through force‐mediated activation of AMPK. Nat Cell Biol 19, 724–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedzhov I, Liszewska E, Kanzler B & Stemmler MP (2012). Igf1r signaling is indispensable for preimplantation development and is activated via a novel function of E‐cadherin. PLoS Genet 8, e1002609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bektas M, Jolly PS, Berkowitz P, Amagai M & Rubenstein DS (2013). A pathophysiologic role for epidermal growth factor receptor in pemphigus acantholysis. J Biol Chem 288, 9447–9456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan D, Hu Y, Joubeh S, Choi YW, Whitaker‐Menezes D, O'Brien T, Uitto J, Rodeck U & Mahoney MG (2007). Suprabasal Dsg2 expression in transgenic mouse skin confers a hyperproliferative and apoptosis‐resistant phenotype to keratinocytes. J Cell Sci 120, 758–771. [DOI] [PubMed] [Google Scholar]
- Cavallaro U & Christofori G (2004). Cell adhesion and signalling by cadherins and Ig‐CAMs in cancer. Nat Rev Cancer 4, 118–132. [DOI] [PubMed] [Google Scholar]
- Chapman DL & Papaioannou VE (1998). Three neural tubes in mouse embryos with mutations in the T‐box gene Tbx6 . Nature 391, 695–697. [DOI] [PubMed] [Google Scholar]
- Chernyavsky AI, Arredondo J, Kitajima Y, Sato‐Nagai M & Grando SA (2007). Desmoglein versus non‐desmoglein signaling in pemphigus acantholysis: characterization of novel signaling pathways downstream of pemphigus vulgaris antigens. J Biol Chem 282, 13804–13812. [DOI] [PubMed] [Google Scholar]
- Clevers H & Nusse R (2012). Wnt/β‐catenin signaling and disease. Cell 149, 1192–1205. [DOI] [PubMed] [Google Scholar]
- Conacci‐Sorrell M, Zhurinsky J & Ben‐Ze'ev A (2002). The cadherin‐catenin adhesion system in signaling and cancer. J Clin Invest 109, 987–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant K, Daniele S, Bozzelli PL, Abdi T, Edwards A, Szklarczyk A, Olchefske I, Ottenheimer D & Maguire‐Zeiss K (2017). Matrix metalloproteinase activity stimulates N‐cadherin shedding and the soluble N‐cadherin ectodomain promotes classical microglial activation. J Neuroinflammation 14, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui T, Chen Y, Yang L, Knosel T, Huber O, Pacyna‐Gengelbach M & Petersen I (2012). The p53 target gene desmocollin 3 acts as a novel tumor suppressor through inhibiting EGFR/ERK pathway in human lung cancer. Carcinogenesis 33, 2326–2333. [DOI] [PubMed] [Google Scholar]
- Dellon ES, Higgins LL, Beitia R, Rusin S, Woosley JT, Veerappan R, Selitsky SR, Parker JS, Genta RM & Lash RH (2016). Prospective assessment of serum periostin as a biomarker for diagnosis and monitoring of eosinophilic oesophagitis. Aliment Pharmacol Ther 44, 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubash AD, Koetsier JL, Amargo EV, Najor NA, Harmon RM & Green KJ (2013). The GEF Bcr activates RhoA/MAL signaling to promote keratinocyte differentiation via desmoglein‐1. J Cell Biol 202, 653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusek RL, Getsios S, Chen F, Park JK, Amargo EV, Cryns VL & Green KJ (2006). The differentiation‐dependent desmosomal cadherin desmoglein 1 is a novel caspase‐3 target that regulates apoptosis in keratinocytes. J Biol Chem 281, 3614–3624. [DOI] [PubMed] [Google Scholar]
- Ferber EC, Kajita M, Wadlow A, Tobiansky L, Niessen C, Ariga H, Daniel J & Fujita Y (2008). A role for the cleaved cytoplasmic domain of E‐cadherin in the nucleus. J Biol Chem 283, 12691–12700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira AC, Suriano G, Mendes N, Gomes B, Wen X, Carneiro F, Seruca R & Machado JC (2012). E‐cadherin impairment increases cell survival through Notch‐dependent upregulation of Bcl‐2. Hum Mol Genet 21, 334–343. [DOI] [PubMed] [Google Scholar]
- Getsios S, Simpson CL, Kojima S, Harmon R, Sheu LJ, Dusek RL, Cornwell M & Green KJ (2009). Desmoglein 1‐dependent suppression of EGFR signaling promotes epidermal differentiation and morphogenesis. J Cell Biol 185, 1243–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giampietro C, Taddei A, Corada M, Sarra‐Ferraris GM, Alcalay M, Cavallaro U, Orsenigo F, Lampugnani MG & Dejana E (2012). Overlapping and divergent signaling pathways of N‐cadherin and VE‐cadherin in endothelial cells. Blood 119, 2159–2170. [DOI] [PubMed] [Google Scholar]
- Gil MP, Modol T, Espana A & Lopez‐Zabalza MJ (2012). Inhibition of FAK prevents blister formation in the neonatal mouse model of pemphigus vulgaris. Exp Dermatol 21, 254–259. [DOI] [PubMed] [Google Scholar]
- Grazia Lampugnani M, Zanetti A, Corada M, Takahashi T, Balconi G, Breviario F, Orsenigo F, Cattelino A, Kemler R, Daniel TO & Dejana E (2003). Contact inhibition of VEGF‐induced proliferation requires vascular endothelial cadherin, beta‐catenin, and the phosphatase DEP‐1/CD148. J Cell Biol 161, 793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KJ & Simpson CL (2007). Desmosomes: new perspectives on a classic. J Invest Dermatol 127, 2499–2515. [DOI] [PubMed] [Google Scholar]
- Harmon RM, Simpson CL, Johnson JL, Koetsier JL, Dubash AD, Najor NA, Sarig O, Sprecher E & Green KJ (2013). Desmoglein‐1/Erbin interaction suppresses ERK activation to support epidermal differentiation. J Clin Invest 123, 1556–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TJ & Tepass U (2010). Adherens junctions: from molecules to morphogenesis. Nat Rev Mol Cell Biol 11, 502–514. [DOI] [PubMed] [Google Scholar]
- Hawkins K, Mohamet L, Ritson S, Merry CL & Ward CM (2012). E‐cadherin and, in its absence, N‐cadherin promotes Nanog expression in mouse embryonic stem cells via STAT3 phosphorylation. Stem Cells 30, 1842–1851. [DOI] [PubMed] [Google Scholar]
- Heuberger J & Birchmeier W (2010). Interplay of cadherin‐mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb Perspect Biol 2, a002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard S, Deroo T, Fujita Y & Itasaki N (2011). A positive role of cadherin in Wnt/beta‐catenin signalling during epithelial‐mesenchymal transition. PLoS One 6, e23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanes A, Gottardi C & Yap A (2008). Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene 27, 6920–6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JL, Koetsier JL, Sirico A, Agidi AT, Antonini D, Missero C & Green KJ (2014). The desmosomal protein desmoglein 1 aids recovery of epidermal differentiation after acute UV light exposure. J Invest Dermatol 134, 2154–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly PS, Berkowitz P, Bektas M, Lee HE, Chua M, Diaz LA & Rubenstein DS (2010). p38MAPK signaling and desmoglein‐3 internalization are linked events in pemphigus acantholysis. J Biol Chem 285, 8936–8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamekura R, Kolegraff KN, Nava P, Hilgarth RS, Feng M, Parkos CA & Nusrat A (2014). Loss of the desmosomal cadherin desmoglein‐2 suppresses colon cancer cell proliferation through EGFR signaling. Oncogene 33, 4531–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamekura R, Nava P, Feng M, Quiros M, Nishio H, Weber DA, Parkos CA & Nusrat A (2015). Inflammation‐induced desmoglein‐2 ectodomain shedding compromises the mucosal barrier. Mol Biol Cell 26, 3165–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashef J, Kohler A, Kuriyama S, Alfandari D, Mayor R & Wedlich D (2009). Cadherin‐11 regulates protrusive activity in Xenopus cranial neural crest cells upstream of Trio and the small GTPases. Genes Dev 23, 1393–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N‐G, Koh E, Chen X & Gumbiner BM (2011). E‐cadherin mediates contact inhibition of proliferation through Hippo signaling‐pathway components. Proc Natl Acad Sci U S A 108, 11930–11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolegraff K, Nava P, Helms MN, Parkos CA & Nusrat A (2011). Loss of desmocollin‐2 confers a tumorigenic phenotype to colonic epithelial cells through activation of Akt/beta‐catenin signaling. Mol Biol Cell 22, 1121–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourtidis A, Lu R, Pence LJ & Anastasiadis PZ (2017). A central role for cadherin signaling in cancer. Exp Cell Res 358, 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourtidis A, Ngok SP, Pulimeno P, Feathers RW, Carpio LR, Baker TR, Carr JM, Yan IK, Borges S, Perez EA, Storz P, Copland JA, Patel T, Thompson EA, Citi S & Anastasiadis PZ (2015). Distinct E‐cadherin‐based complexes regulate cell behaviour through miRNA processing or Src and p120 catenin activity. Nat Cell Biol 17, 1145–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs EM (2003). Direct cadherin‐activated cell signaling: a view from the plasma membrane. J Cell Biol 160, 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labernadie A, Kato T, Brugues A, Serra‐Picamal X, Derzsi S, Arwert E, Weston A, Gonzalez‐Tarrago V, Elosegui‐Artola A, Albertazzi L, Alcaraz J, Roca‐Cusachs P, Sahai E & Trepat X (2017). A mechanically active heterotypic E‐cadherin/N‐cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat Cell Biol 19, 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HE, Berkowitz P, Jolly PS, Diaz LA, Chua MP & Rubenstein DS (2009). Biphasic activation of p38MAPK suggests that apoptosis is a downstream event in pemphigus acantholysis. J Biol Chem 284, 12524–12532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Zhao M, Wang J, Liu Z & Diaz LA (2009). Involvement of the apoptotic mechanism in pemphigus foliaceus autoimmune injury of the skin. J Immunol 182, 711–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Zhang C, Zhang M, Xu D, Fang Y, Zhou Z, Chen X, Qin N & Zhang X (2014). Targeting cadherin‐17 inactivates Ras/Raf/MEK/ERK signaling and inhibits cell proliferation in gastric cancer. PLoS One 9, e85296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu WF, Nelson CM, Pirone DM & Chen CS (2006). E‐cadherin engagement stimulates proliferation via Rac1. J Cell Biol 173, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Marsters S, Ye X, Luis E, Gonzalez L & Ashkenazi A (2014). E‐cadherin couples death receptors to the cytoskeleton to regulate apoptosis. Mol Cell 54, 987–998. [DOI] [PubMed] [Google Scholar]
- McCusker C, Cousin H, Neuner R & Alfandari D (2009). Extracellular cleavage of cadherin‐11 by ADAM metalloproteases is essential for Xenopus cranial neural crest cell migration. Mol Biol Cell 20, 78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher MT, Flozak AS, Stocker AM, Chenn A & Gottardi CJ (2009). Activity of the beta‐catenin phosphodestruction complex at cell‐cell contacts is enhanced by cadherin‐based adhesion. J Cell Biol 186, 219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Sano Y, Park JM & Payne AS (2011). p38 MAPK activation is downstream of the loss of intercellular adhesion in pemphigus vulgaris. J Biol Chem 286, 1283–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S & Shao Z (2002). A presenilin‐1/γ‐secretase cleavage releases the E‐cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J 21, 1948–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najy AJ, Day KC & Day ML (2008). The ectodomain shedding of E‐cadherin by ADAM15 supports ErbB receptor activation. J Biol Chem 283, 18393–18401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrasova O & Green KJ (2013). Desmosome assembly and dynamics. Trends Cell Biol 23, 537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onder TT, Gupta PB, Mani SA, Yang J, Lander ES & Weinberg RA (2008). Loss of E‐cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res 68, 3645–3654. [DOI] [PubMed] [Google Scholar]
- Pandya P, Orgaz JL & Sanz‐Moreno V (2017). Actomyosin contractility and collective migration: May the force be with you. Curr Opin Cell Biol 48, 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, Shin J‐H & Kee S‐H (2017). E‐cadherin expression increases cell proliferation by regulating energy metabolism through nuclear factor‐κB in AGS cells. Cancer Sci 108, 1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnaud G, Lavallard V, Bedat B, Matthey‐Doret D, Morel P, Berney T & Bosco D (2015). Cadherin engagement improves insulin secretion of single human beta‐cells. Diabetes 64, 887–896. [DOI] [PubMed] [Google Scholar]
- Perrais M, Chen X, Perez‐Moreno M & Gumbiner BM (2007). E‐cadherin homophilic ligation inhibits cell growth and epidermal growth factor receptor signaling independently of other cell interactions. Mol Biol Cell 18, 2013–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polacheck WJ, Kutys ML, Yang J, Eyckmans J, Wu Y, Vasavada H, Hirschi KK & Chen CS (2017). A non‐canonical Notch complex regulates adherens junctions and vascular barrier function. Nature 552, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politi E, Angelakopoulou A, Grapsa D, Zande M, Stefanaki K, Panagiotou I, Roma E & Syrigou E (2017). Filaggrin and periostin expression is altered in eosinophilic esophagitis and normalized with treatment. J Pediatr Gastroenterol Nutr 65, 47–52. [DOI] [PubMed] [Google Scholar]
- Pretel M, Espana A, Marquina M, Pelacho B, Lopez‐Picazo JM & Lopez‐Zabalza MJ (2009). An imbalance in Akt/mTOR is involved in the apoptotic and acantholytic processes in a mouse model of pemphigus vulgaris. Exp Dermatol 18, 771–780. [DOI] [PubMed] [Google Scholar]
- Rudini N, Felici A, Giampietro C, Lampugnani M, Corada M, Swirsding K, Garre M, Liebner S, Letarte M, ten Dijke P & Dejana E (2008). VE‐cadherin is a critical endothelial regulator of TGF‐beta signalling. EMBO J 27, 993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt E & Waschke J (2009). Apoptosis in pemphigus. Autoimmun Rev 8, 533–537. [DOI] [PubMed] [Google Scholar]
- Schulze K, Galichet A, Sayar BS, Scothern A, Howald D, Zymann H, Siffert M, Zenhausern D, Bolli R, Koch PJ, Garrod D, Suter MM & Muller EJ (2012). An adult passive transfer mouse model to study desmoglein 3 signaling in pemphigus vulgaris. J Invest Dermatol 132, 346–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal JM & Ward CM (2017). Novel peptides for deciphering structural and signalling functions of E‐cadherin in mouse embryonic stem cells. Sci Rep 7, 41827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamir ER, Pappalardo E, Jorgens DM, Coutinho K, Tsai WT, Aziz K, Auer M, Tran PT, Bader JS & Ewald AJ (2014). Twist1‐induced dissemination preserves epithelial identity and requires E‐cadherin. J Cell Biol 204, 839–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro L & Weis WI (2009). Structure and biochemistry of cadherins and catenins. Cold Spring Harb Perspect Biol 1, a003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Mao X & Payne AS (2007). Beyond steric hindrance: the role of adhesion signaling pathways in the pathogenesis of pemphigus. J Dermatol Sci 48, 1–14. [DOI] [PubMed] [Google Scholar]
- Sherrill JD, Kc K, Wu D, Djukic Z, Caldwell JM, Stucke EM, Kemme KA, Costello MS, Mingler MK, Blanchard C, Collins MH, Abonia JP, Putnam PE, Dellon ES, Orlando RC, Hogan SP & Rothenberg ME (2014). Desmoglein‐1 regulates esophageal epithelial barrier function and immune responses in eosinophilic esophagitis. Mucosal Immunol 7, 718–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoval I, Ludwig A & Kalcheim C (2007). Antagonistic roles of full‐length N‐cadherin and its soluble BMP cleavage product in neural crest delamination. Development 134, 491–501. [DOI] [PubMed] [Google Scholar]
- Solanas G, Porta‐de‐la‐Riva M, Agusti C, Casagolda D, Sanchez‐Aguilera F, Larriba MJ, Pons F, Peiro S, Escriva M, Munoz A, Dunach M, de Herreros AG & Baulida J (2008). E‐cadherin controls beta‐catenin and NF‐kappaB transcriptional activity in mesenchymal gene expression. J Cell Sci 121, 2224–2234. [DOI] [PubMed] [Google Scholar]
- Spindler V & Waschke J (2014). Desmosomal cadherins and signaling: lessons from autoimmune disease. Cell Commun Adhes 21, 77–84. [DOI] [PubMed] [Google Scholar]
- Tsang SM, Brown L, Gadmor H, Gammon L, Fortune F, Wheeler A & Wan H (2012a). Desmoglein 3 acting as an upstream regulator of Rho GTPases, Rac‐1/Cdc42 in the regulation of actin organisation and dynamics. Exp Cell Res 318, 2269–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang SM, Brown L, Lin K, Liu L, Piper K, O'Toole EA, Grose R, Hart IR, Garrod DR, Fortune F & Wan H (2012b). Non‐junctional human desmoglein 3 acts as an upstream regulator of Src in E‐cadherin adhesion, a pathway possibly involved in the pathogenesis of pemphigus vulgaris. J Pathol 227, 81–93. [DOI] [PubMed] [Google Scholar]
- Waschke J, Spindler V, Bruggeman P, Zillikens D, Schmidt G & Drenckhahn D (2006). Inhibition of Rho A activity causes pemphigus skin blistering. J Cell Biol 175, 721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheelock MJ & Johnson KR (2003). Cadherin‐mediated cellular signaling. Curr Opin Cell Biol 15, 509–514. [DOI] [PubMed] [Google Scholar]
- Williamson L, Raess NA, Caldelari R, Zakher A, de Bruin A, Posthaus H, Bolli R, Hunziker T, Suter MM & Muller EJ (2006). Pemphigus vulgaris identifies plakoglobin as key suppressor of c‐Myc in the skin. EMBO J 25, 3298–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa Y, Fujimori T, McMahon AP & Takada S (1997). Evidence that absence of Wnt‐3a signaling promotes neuralization instead of paraxial mesoderm development in the mouse. Dev Biol 183, 234–242. [DOI] [PubMed] [Google Scholar]
- Yulis M, Quiros M, Hilgarth R, Parkos CA & Nusrat A (2018). Intracellular Desmoglein‐2 cleavage sensitizes epithelial cells to apoptosis in response to pro‐inflammatory cytokines. Cell Death Dis 9, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Shemezis JR, McQuinn ER, Wang J, Sverdlov M & Chenn A (2013). AKT activation by N‐cadherin regulates beta‐catenin signaling and neuronal differentiation during cortical development. Neural Dev 8, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]