

Abstract
Key points
α2δ‐1 is upregulated, promoting the interaction with NMDA receptors (NMDARs), in the hypothalamus in a rat model of hypertension.
The prevalence of α2δ‐1–bound NMDARs at synaptic sites in the hypothalamus is increased in hypertensive animals.
α2δ‐1 is essential for the increased presynaptic and postsynaptic NMDAR activity of hypothalamic neurons in hypertension.
α2δ‐1–bound NMDARs in the hypothalamus are critically involved in augmented sympathetic outflow in hypertensive animals.
Abstract
Increased glutamate NMDA receptor (NMDAR) activity in the paraventricular nucleus (PVN) of the hypothalamus leads to augmented sympathetic outflow in hypertension. However, the molecular mechanisms underlying this effect remain unclear. α2δ‐1, previously considered to be a voltage‐activated calcium channel subunit, is a newly discovered powerful regulator of NMDARs. In the present study, we determined the role of α2δ‐1 in regulating synaptic NMDAR activity of rostral ventrolateral medulla (RVLM)‐projecting PVN neurons in spontaneously hypertensive rats (SHRs). We show that the protein levels of α2δ‐1 and NMDARs in synaptosomes and the α2δ‐1–NMDAR complexes in the hypothalamus were substantially higher in SHRs than in normotensive control rats. The basal amplitude of evoked NMDAR currents and NMDAR‐mediated synaptic glutamate release in RVLM‐projecting PVN neurons were significantly increased in SHRs. Strikingly, inhibiting α2δ‐1 activity with gabapentin or disrupting the α2δ‐1–NMDAR association with an α2δ‐1 C‐terminus peptide completely normalized the amplitude of evoked NMDAR currents and NMDAR‐mediated synaptic glutamate release in RVLM‐projecting PVN neurons in SHRs. In addition, microinjection of the α2δ‐1 C‐terminus peptide into the PVN substantially reduced arterial blood pressure and renal sympathetic nerve discharges in SHRs. Our findings indicate that α2δ‐1–bound NMDARs in the PVN are required for the potentiated presynaptic and postsynaptic NMDAR activity of PVN presympathetic neurons and for the elevated sympathetic outflow in hypertension. α2δ‐1–bound NMDARs may be an opportune target for treating neurogenic hypertension.
Keywords: autonomic nervous system, sympathetic nervous system, synaptic transmission, gabapentinoids, pregabalin, synaptic plasticity
Key points
α2δ‐1 is upregulated, promoting the interaction with NMDA receptors (NMDARs), in the hypothalamus in a rat model of hypertension.
The prevalence of α2δ‐1–bound NMDARs at synaptic sites in the hypothalamus is increased in hypertensive animals.
α2δ‐1 is essential for the increased presynaptic and postsynaptic NMDAR activity of hypothalamic neurons in hypertension.
α2δ‐1–bound NMDARs in the hypothalamus are critically involved in augmented sympathetic outflow in hypertensive animals.
Introduction
Almost half of adults in the USA are considered as having high blood pressure in accordance with new guidelines from the American Heart Association and the American College of Cardiology (Reboussin et al. 2017). Hypertension is a well‐recognized risk factor for coronary artery disease, kidney failure and stroke. Essential hypertension, or hypertension of unknown cause, which accounts for more than 90% of cases of hypertension, results from a complex interaction of genetic and environmental factors. Major pathophysiological mechanisms of hypertension include activation of the sympathetic nervous system and the renin–angiotensin–aldosterone system, endothelial dysfunction, increased vascular reactivity, and vascular remodelling (Oparil et al. 2003; DiBona, 2013). The hypothalamic paraventricular nucleus (PVN) neurons that project to the rostral ventrolateral medulla (RVLM) and intermediolateral cell column in the spinal cord play a pivotal role in the augmented sympathetic outflow in spontaneously hypertensive rats (SHRs) (Allen, 2002; Li & Pan, 2007). Although increased glutamate NMDA receptor (NMDAR) activity in the PVN leads to elevated sympathetic vasomotor activity in hypertension (Li & Pan, 2007; Li et al. 2008; Qiao et al. 2017), the molecular mechanisms underlying synaptic NMDAR hyperactivity of PVN presympathetic neurons remain unclear.
α2δ‐1 was initially known as a subunit of voltage‐activated Ca2+ channels (VACCs). α2δ‐1 is also the binding site of gabapentinoids, including gabapentin and pregabalin (Gee et al. 1996; Marais et al. 2001; Fuller‐Bicer et al. 2009), which are often used for treating neuropathic pain. However, gabapentinoids have little effect on VACC activity (Rock et al. 1993; Schumacher et al. 1998; Chen et al. 2018). We recently discovered that α2δ‐1 interacts with NMDARs in the spinal cord and promotes NMDAR synaptic trafficking in neuropathic pain (Chen et al. 2018). Interestingly, α2δ‐1 is highly expressed in the hypothalamus, particularly in the PVN (Cole et al. 2005; Taylor & Garrido, 2008). Nevertheless, nothing is known about the involvement of α2δ‐1 in regulating NMDAR activity in the PVN and sympathetic outflow in hypertension.
In the present study, we demonstrate that α2δ‐1 forms a protein complex with NMDARs and plays an essential role in the increased presynaptic and postsynaptic NMDAR activity in PVN presympathetic neurons and augmented sympathetic outflow in SHRs. The results of the present study provide new insight into the molecular mechanisms underlying the elevation of sympathetic vasomotor tone in hypertension.
Methods
Ethical approval and animal models
The experimental procedures and protocols were approved by the Institutional Animal Care and Use Committee of The University of Texas MD Anderson Cancer Center (approval ref. no. 919‐RN02) and conformed with the National Institutes of Health guidelines on the ethical use of animals. Thirteen‐week‐old male normotensive Wistar‐Kyoto (WKY) rats and spontaneously hypertensive rats (SHRs) (Harlan, Indianapolis, IN, USA) were used for most of the experiments. Blood pressure was measured before the electrophysiological experiments via a non‐invasive tail‐cuff system (IITC Life Science, Inc., Woodland Hills, CA, USA) to confirm hypertension in a randomly selected group of adult SHRs and WKY rats (Table 1).
Table 1.
Arterial blood pressure of 13‐week‐old WKY rats and SHRs
| WKY | SHR | |
|---|---|---|
| SAP | 108.7 ± 2.88 | 178.57 ± 2.38*** |
| DAP | 75.57 ± 2.11 | 126.09 ± 2.57*** |
| MAP | 86.61 ± 2.10 | 143.58 ± 2.16*** |
Blood pressure was measured using a tail‐cuff system. SAP, systolic blood pressure; DAP, diastolic blood pressure; MAP, mean arterial blood pressure. WKY rats, n = 30 rats; SHRs, n = 35 rats. *** P < 0.001 compared to the WKY group.
Immunoblotting
Rats were anaesthetized with 2–3% isoflurane and then decapitated. The brain was quickly removed and placed in ice‐cold artificial CSF (aCSF) saturated with 95% O2 and 5% CO2. Hypothalamic slices were sectioned 1.08–2.12 mm caudal to the bregma, and hypothalamic tissues containing the PVN were micropunched bilaterally with a slice punch (0.5 mm diameter) following stereotactic co‐ordinates: 0.5 mm lateral to the midline and 6.5–9.0 mm ventral to the surface of the cortex (Ye et al. 2012b; Qiao et al. 2017). The tissues were frozen in liquid nitrogen and then transferred to a freezer at −80°C.
Total proteins were extracted with a RIPA lysis/extraction buffer (Thermo Fisher Scientific, Waltham, MA, USA) in the presence of a mixture containing protease inhibitor cocktail (Sigma‐Aldrich, St Louis, MO, USA). The PVN tissues were homogenized and then subjected to centrifugation at 16,000 g for 10 min to obtain the supernatant. For synaptosome isolation, the hypothalamic tissues (pooled from two rats per sample) were homogenized using 10 volumes of ice‐cold Hepes‐buffered sucrose solution (0.32 mol L−1 sucrose, 1 mmol L−1 EGTA and 4 mmol L−1 Hepes at pH, 7.4) containing the protease inhibitor cocktail. The homogenates were centrifuged at 2000 g for 10 min at 4°C to remove the nuclei and large debris. The supernatant was centrifuged at 20,000 g for 30 min to obtain the crude synaptosomes. The synaptosomal pellets were subjected to lysis via hypoosmotic shock in nine volumes of ice‐cold Hepes buffer with the protease inhibitor cocktail for 30 min. The lysates were centrifuged at 25,000 g for 45 min at 4°C to obtain the synaptosomal fraction (Chen et al. 2018), which was then dissolved in sodium dodecyl sulphate sample buffer at a final concentration of 0.25 μg μL−1 for immunoblotting. The protein concentration was determined by bicinchoninic acid assay.
The samples were subjected to 4–15% Tris‐HCl SDS‐PAGE (#456‐1086; Bio‐Rad, Hercules, CA, USA) and then transferred to a polyvinylidene difluoride membrane (EMD Millipore, Burlington, MA, USA). The membranes were treated with 5% non‐fat dry milk in Tris‐buffered saline solution at 25°C for 1 h, and then incubated in Tris‐buffered saline solution supplemented with 0.1% Triton X‐100, 1% BSA and rabbit anti‐GluN1 (#G8913; dilution 1:2000; Sigma‐Aldrich), rabbit anti‐α2δ‐1 (#C0515; dilution 1:500; Sigma‐Aldrich), rabbit anti‐GAPDH antibody (#ab37168; dilution 1:1000; Abcam, Cambridge, MA, USA) or mouse anti‐PSD‐95 (#75‐348; dilution 1:1000; NeuroMab, Davis, CA, USA) antibody overnight at 4°C. The membrane was washed three times and then incubated with horseradish peroxidase–conjugated anti‐rabbit or anti‐mouse IgG antibody (dilution 1:7500; Jackson ImmunoResearch, West Grove, PA, USA) for 1 h at room temperature. An ECL kit (Thermo Fisher Scientific) was used to detect the protein band, which was visualized and quantified with the Odyssey Fc Imager (LI‐COR Biosciences, Lincoln, NE, USA) and normalized to the GAPDH or PSD‐95 (a synaptic protein) band on the same blot.
Co‐immunoprecipitation
Rat hypothalamic slices containing the PVN were sectioned 1.08–2.12 mm caudal to the bregma. The tissues were dissected and homogenized in ice‐cold IP lysis buffer (#87788; Thermo Fisher Scientific; 25 mmol L−1 Tris‐HCl, pH 7.4, 150 mmol L−1 NaCl, 1% NP‐40, 1 mmol L−1 EDTA, 5% glycerol and the protease inhibitor cocktail) and incubated on ice for 30 min for total protein preparation. The lysates were centrifuged for 15 min at 16,000 g to obtain the supernatants. The protein sample were quantified and incubated at 4°C overnight with Protein G beads (#16‐266; EMD Millipore) prebound to rabbit anti‐GluN1 antibody (#G8913; dilution 1:100; Sigma‐Aldrich). Protein G beads prebound to rabbit IgG were used as controls. Samples were washed three times with immunoprecipitation buffer and then immunoblotted with mouse anti‐α2δ‐1 antibody (#sc‐271697; dilution 1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA). The protein bands were detected using immunoblotting analysis.
Retrograde labelling of RVLM‐projecting PVN presympathetic neurons
We performed retrograde labelling to identify RVLM‐projecting PVN neurons as described previously (Li & Pan, 2006; Qiao et al. 2017). The rats were anaesthetized with 2–3% isoflurane and placed in a stereotactic frame. A small piece of skull was removed and a microinjection pipette (tip diameter, 20–30 μm) filled with fluorescent microsphere suspension (FluoSpheres, 0.04 μm; Molecular Probes, Invitrogen, Eugene, OR, USA) was advanced into the RVLM in accordance with the stereotactic co‐ordinates: 12.5–13.0 mm caudal to the bregma, 1.9–2.0 mm lateral to the midline and 7.5–8.0 mm ventral to the dura. The FluoSpheres were pressure‐injected (Nanoject II Microinjectors; Drummond Scientific Company, Broomall, PA, USA) into the RVLM bilaterally in two separate 50 nL injections using a micromanipulator. The tracer injection was monitored through a surgical microscope. The rats were returned to their home cages for 3–5 days to allow the FluoSpheres to be transported into the PVN. The animals were inspected daily for motor activity, signs of infection, and food and water intake.
Brain slice preparation
The rats were rapidly decapitated under 2–3% isoflurane anaesthesia. The brain was quickly removed and sliced in an ice‐cold aCSF solution containing (in mmol L−1) 126 NaCl, 3 KCl, 1.5 MgCl2, 2.4 CaCl2, 1.2 NaH2PO4, 10 glucose and 26 NaHCO3 saturated with 95% O2 and 5% CO2. Coronal hypothalamic slices (300 μm thick) containing the PVN were obtained from FluoSphere‐injected rats using a vibrating microtome (VT1000 S; Leica Biosystems Inc., Buffalo Grove, IL, USA). The slices were incubated in the aCSF at 34°C for at least 1 h before recording. To confirm the injection sites of the FluoSpheres, the RVLM was sectioned at the injection level and viewed under a microscope immediately after the rat was killed. Data were excluded from the analysis if either of the injection sites were not located within the RVLM.
Electrophysiological recordings
The labelled PVN neurons were identified under an upright microscope (BX51WI; Olympus, Tokyo, Japan) with epifluorescence and infrared differential interference contrast optics. Whole‐cell, patch clamp recordings were performed in FluoSphere‐labelled PVN neurons. The hypothalamic slices were continuously perfused with aCSF (saturated by 95% O2 and 5% CO2; speed, 3.0 mL min−1) at 34°C maintained by an inline solution heater.
The miniature EPSCs (mEPSCs) were recorded in the presence of 1 μmol L−1 tetrodotoxin and 20 μmol L−1 bicuculline at a holding potential of –60 mV (Li et al. 2003; Ye et al. 2011). The recording glass pipette (4–8 MΩ) was filled with internal solution containing (in mmol L−1) 135.0 potassium gluconate, 5.0 tetraethylammonium, 2.0 MgCl2, 0.5 CaCl2, 5.0 Hepes, 5.0 EGTA, 5.0 Mg‐ATP and 0.5 Na‐GTP (adjusted to pH 7.2–7.4 with 1 mol L−1 KOH; 290–300 mosmol L−1). A sodium channel blocker, lidocaine N‐ethyl bromide (10.0 mmol L−1), was included in the pipette solution to block the firing activity of the recorded neuron.
The evoked EPSCs were elicited by electrical stimulation (0.2 ms, 0.8–1.0 mA at 0.2 Hz) through a bipolar tungsten electrode connected to a stimulator. The tip of the stimulation electrode was placed on the ventral side 150 μm from the recorded neuron (Li et al. 2003; Li et al. 2005). The internal solution contained (in mmol L−1): 110.0 Cs2SO4, 2.0 MgCl2, 0.1 CaCl2, 10.0 Hepes, 1.1 EGTA, 0.3 Na2‐GTP and 2.0 Mg‐ATP, adjusted to pH 7.25 with 1.0 mol L−1 CsOH (270–290 mosm). Evoked α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor (AMPAR)‐EPSCs were recorded at a holding potential of −60 mV in the presence of 10 μmol L−1 bicuculline, and evoked NMDAR‐EPSCs were recorded at a holding potential of +40 mV in the presence of 10 μmol L−1 bicuculline and 10 μmol L−1 6,7‐dinitroquinoxaline‐2,3‐dione. We have shown that the evoked NMDAR‐EPSCs at +40 mV were abolished by the NMDAR specific antagonist, AP5, whereas the evoked AMPAR‐EPSCs at –60 mV were completed blocked by the AMPAR antagonist 6,7‐dinitroquinoxaline‐2,3‐dione (Li et al. 2003; Li et al. 2008; Ye et al. 2011). The amplitude of AMPAR‐EPSCs recorded in labelled PVN neurons at a holding potential of –60 mV is similar in WKY rats and SHRs (Li et al. 2017). Therefore, AMPAR‐EPSCs were used to normalize the NMDAR‐EPSCs elicited under the same stimulation intensity (0.8 mA) in the present study.
To record postsynaptic NMDAR currents, we puffed NMDA (100 μmol L−1) directly onto the recorded neuron at a holding potential of –60 mV using a positive pressure system (4 psi, 50 ms; Toohey Company, Fairfield, NJ, USA). The puff pipette (10 μm tip diameter) was placed 150 μm away from the recorded neuron. Because NMDARs are voltage‐dependently blocked by Mg2+ at a negative holding potential and co‐activated by glycine, puff NMDA‐induced currents were recorded in Mg2+‐free aCSF in the presence of 10 μmol L−1 glycine and 1 μmol L−1 tetrodotoxin. Although puff application of NMDA can also result in presynaptic release of glutamate, the currents elicited by presynaptic glutamate release are very small and negligible compared to currents produced by a large amount of puff glutamate receptor agonists. Therefore, the current elicited by puff application of NMDA is generally considered to be a measure of postsynaptic NMDAR activity (Li et al. 2008; Ye et al. 2011).
PVN microinjection and recording of renal sympathetic nerve activity
Rats were anaesthetized via i.p. administration of a mixture of α‐chloralose (60 mg kg−1) and urethane (800 mg kg−1) and mechanically ventilated with trachea cannulation. The arterial blood pressure (ABP) was monitored via a catheter placed in the left femoral artery, and heart rate (HR) was extracted from the pulsatile pressure wave. A retroperitoneal incision was made, and a branch of the left renal postganglionic sympathetic nerve was isolated under a surgical microscope. The renal sympathetic nerve was cut distally to ensure that afferent activity was not recorded. The ABP, HR and renal sympathetic nerve activity (RSNA) were recorded using a 1401‐PLUS analogue‐to‐digital converter and Spike2 system (Cambridge Electronic Design, Cambridge, UK). After the brain was exposed, a microinjection pipette (tip diameter 20–30 μm) was advanced into the PVN in accordance with the stereotactic co‐ordinates: 1.6–2.0 mm caudal to the bregma, 0.5 mm lateral to the midline and 7.0–7.5 mm ventral to the dura. The microinjection was performed using a Nanoject II (Drummond Scientific Company) and monitored using a surgical microscope as described previously (Li & Pan, 2006, 2007; Qiao et al. 2017). AP5 (1.0 nmol, 50 nL) was microinjected into the PVN to observe the change in ABP and RSNA in the presence of α2δ‐1Tat peptide (50 pmol, 50 nL; Bio Basic, Markham, Ontario, Canada) or Tat‐fused control peptide (50 pmol, 50 nL). At the end of each experiment, the electrical background noise was measured after the proximal end of renal nerve was crushed at the end of each experiment. To determine the location of the injection site and diffusion of the drugs in the PVN, we included 5% rhodamine‐labelled fluorescent microspheres (0.04 μm; Molecular Probes) in the injection solution (Li & Pan, 2006, 2007; Qiao et al. 2017). Rats were excluded from the data analysis if the microinjections were misplaced outside the PVN.
Celiac ganglionectomy and ABP measurement using telemetry
Celiac ganglionectomy (CGx) or sham surgery was performed aseptically in SHRs anaesthetized with 2–3% isoflurane as described previously (Ye et al. 2011; Qiao et al. 2017). The celiac ganglion area was exposed through a midline laparotomy and identified within the area near the superior mesenteric artery and celiac artery (Hamer & Santer, 1981). For rats undergoing CGx, the celiac plexus and all visible nerves were dissected and removed as completely as possible. In sham control rats, the celiac ganglion plexus was exposed but not disrupted. A Millar catheter (Millar, Houston, TX, USA) attached to the transmitter was inserted into the abdominal aorta for continuous monitoring of ABP, and the transmitter body was implanted in an abdominal cavity. The rats were housed individually, and the ABP was measured in freely moving rats by using a telemetry system (Telemetry Research Ltd., Houston, TX, USA) (Ye et al. 2011; Ye et al. 2012a). The ABP data were recorded continuously after surgery and analysed with a data acquisition system (LabChart; AD Instruments, Sydney, Australia). Two weeks after CGx or sham surgery, the rats were anaesthetized with 2–3% isoflurane and then decapitated. The brain tissues of rats were harvested for immunoblotting analysis.
Statistical analysis
For electrophysiological recording experiments, only one neuron in each brain slice was recorded, and at least four animals were used for each recording protocol. Data are presented as the mean ± SEM. The amplitude and frequency of mEPSCs were analysed offline with peak detection software (MiniAnalysis; Synaptosoft Inc., Decatur, GA, USA). The cumulative probabilities of the amplitude and inter‐event interval were compared using the Kolmogorov–Smirnov test. Clampfit, version 10.2 (Molecular Devices, Sunnyvale, CA, USA) was used to determine the peak amplitude of evoked EPSCs and puff NMDA currents. The mean ABP, RSNA and HR were analysed using Spike2 software. The mean ABP was derived from the pulsatile ABP and calculated as the diastolic pressure plus one‐third of the pulse pressure. RSNA was rectified and integrated offline after subtracting the background noise, which was recorded after the proximal end of renal nerve was crushed at the end of each experiment. The integrated RSNA value was calculated and derived from the raw RSNA with an integrating time of 1.0 s using Spike2 software. Control values were obtained by averaging the signal over a 60 s period immediately before PVN microinjection. Response values after each intervention were averaged over 30 s when the maximal responses occurred. A two‐tailed Student's t test was used to compare two groups and one‐way ANOVA followed by the Dunnett's and Tukey's post hoc tests was used to compare more than two groups. Statistical analyses were performed using Prism, version 7 (GraphPad Software Inc., La Jolla, CA). P < 0.05 was considered statistically significant.
Results
α2δ‐1 is upregulated in the PVN in SHRs
We used immunoblotting to determine the α2δ‐1 protein levels in the PVN in WKY rats and SHRs. The α2δ‐1 protein level in the PVN was significantly greater in SHRs than in WKY rats (P = 0.0074, t 12 = 3.217, n = 7 rats in each group) (Fig. 1 A).
Figure 1. α2δ‐1 interacts with NMDARs in the hypothalamus and increases the synaptic expression of α2δ‐1–bound NMDARs in SHRs.
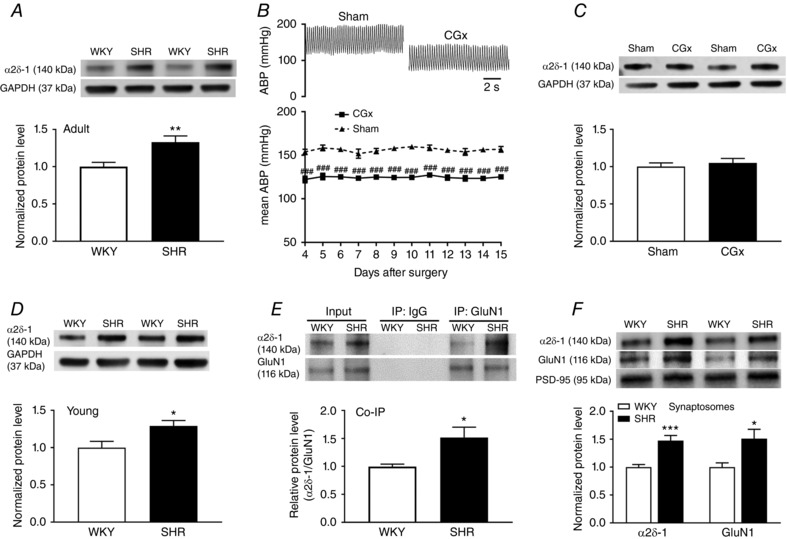
A, representative blot images and mean changes show the α2δ‐1 protein level in the PVN in adult WKY rats and SHRs (n = 7 rats per group). B, original ABP traces and summary data show the effect of CGx or sham surgery on the mean ABP in SHRs (n = 6 rats in each group). C, original blot images and summary data show the α2δ‐1 levels (normalized to GAPDH) in the PVN in SHRs subjected to CGx or sham surgery (n = 6 rats in each group). D, representative blot images and mean changes show the α2δ‐1 protein level in the PVN in young WKY rats and SHRs (n = 6 rats per group). E, representative blot images and co‐immunoprecipitation analysis show the interaction between α2δ‐1 and GluN1 in total protein extracts of hypothalamic tissues from WKY rats and SHRs (n = 6 independent experiments per group, each tissue sample was pooled from two rats). Proteins were immunoprecipitated first with an anti‐GluN1 antibody or IgG and then immunoblotted using an anti‐α2δ‐1 antibody. F, original blot images and quantification data show the protein levels of α2δ‐1 and GluN1 (normalized to PSD‐95, a synaptic protein) in hypothalamic synaptosomes in WKY rats and SHRs (n = 6 independent experiments per group, each tissue sample was pooled from two rats). * P < 0.05, ** P < 0.01, *** P < 0.001 compared to the sham group. ### P < 0.001 compared to the sham group.
To determine whether the higher α2δ‐1 protein level in the PVN of SHRs was secondary to elevated ABP in SHRs, we performed CGx (Ye et al. 2011; Qiao et al. 2017) to lower ABP in SHRs and then measured α2δ‐1 protein levels in their PVNs. CGx substantially lowered ABP in SHRs, as measured by radio telemetry, compared to the sham group (n = 6 rats in each group) (Fig. 1 B). Immunoblotting showed that lowering ABP did not significantly alter the α2δ‐1 protein level in the PVN in SHRs (Fig. 1 C). In addition, the α2δ‐1 protein level in the PVN was significantly higher in 4‐week‐old SHRs than in age‐matched WKY rats (P = 0.027, t 10 = 2.584, n = 6 rats in each group) (Fig. 1 D). These data suggest that α2δ‐1 upregulation in the PVN in SHRs is independent of ABP changes.
α2δ‐1 physically interacts with NMDARs in the hypothalamus in SHRs
We recently showed that α2δ‐1 physically interacts with NMDARs in the spinal cord (Chen et al. 2018). To determine whether α2δ‐1 and NMDARs interact in the hypothalamus, we conducted co‐immunoprecipitation analyses using total proteins extracted from the PVN of SHRs and WKY rats. The anti‐GluN1 antibody co‐precipitated with α2δ‐1 in the hypothalamic tissues obtained from these rats, whereas the irrelevant IgG did not co‐precipitate with α2δ‐1 (Fig. 1 E). Furthermore, the α2δ‐1–GluN1 complex level in the hypothalamus was much higher in SHRs than in WKY rats (P = 0.0204, t 10 = 2.752, n = 6 rats in each group) (Fig. 1 E). These results indicate that α2δ‐1 forms a protein complex in the hypothalamus and that the α2δ‐1–NMDAR interaction is enhanced in SHRs.
Synaptic α2δ‐1–bound NMDARs are increased in the hypothalamus in SHRs
α2δ‐1 primarily increases NMDAR activity by promoting synaptic targeting of NMDARs in the spinal cord after nerve injury (Chen et al. 2018). We next determined whether α2δ‐1–NMDAR complexes are increased at synaptic sites in the hypothalamus of SHRs. Immunoblotting of isolated synaptosomes showed that the protein levels of both α2δ‐1 (P = 0.0009, t 10 = 4.655) and GluN1 (P = 0.0189, t 10 = 2.797), an obligatory subunit of NMDARs, were much higher in SHRs than in WKY rats (n = 6 independent experiments, each tissue sample was pooled from two rats) (Fig. 1 F). These data suggest that the prevalence of α2δ‐1–bound NMDARs at the synaptic site in the hypothalamus is increased in SHRs.
α2δ‐1 is indispensable for enhanced postsynaptic NMDAR activity of PVN presympathetic neurons in SHRs
We then determined whether α2δ‐1 plays a role in regulating synaptic NMDAR activity of RVLM‐projecting PVN neurons in SHRs. We treated the hypothalamic slices from WKY rats and SHRs with either vehicle (aCSF) or gabapentin (100 μmol L−1), an α2δ‐1–inhibitory ligand (Gee et al. 1996; Fuller‐Bicer et al. 2009), for 60 min before electrophysiological recording. The amplitude of evoked NMDAR‐EPSCs of labelled RVLM‐projecting PVN neurons was significantly larger in SHRs (n = 8 neurons) than in WKY rats (n = 9 neurons) (P < 0.0001, F 3,30 = 25.69) (Fig. 2 A–C). Gabapentin treatment completely normalized the amplitude of evoked NMDAR‐EPSCs of labelled PVN neurons in SHRs but had no effect in WKY rats (Fig. 2 B and C). By contrast, gabapentin did not significantly alter the amplitude of evoked AMPAR‐EPSCs of labelled PVN neurons in WKY rats or SHRs. The ratio of NMDAR‐EPSCs to AMPAR‐EPSCs in vehicle‐treated slices was significantly higher in SHRs than in WKY rats (WKY, n = 9 neurons; SHR, n = 8 neurons; P < 0.0001, F 3,30 = 26.52) (Fig. 2 B–D). Gabapentin treatment also restored the ratio of NMDAR‐EPSCs to AMPAR‐EPSCs in SHRs to the level in WKY rats, although it had no such effect in WKY rats (Fig. 2 B–D).
Figure 2. α2δ‐1 is required for increased postsynaptic NMDAR activity of RVLM–projecting PVN neurons in SHRs.
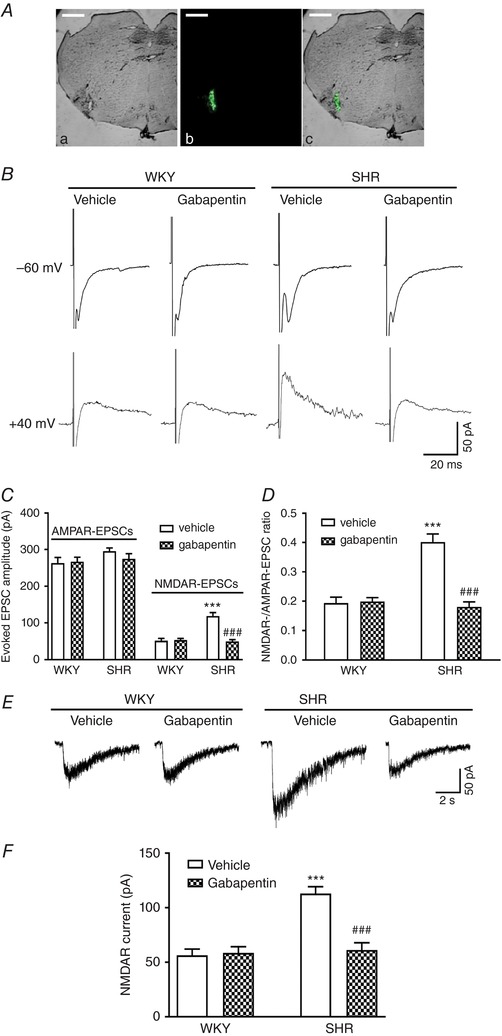
A, representative images show the microinjection site in the RVLM in bright‐field (Aa), fluorescence (Ab) and overlay images (Ac) from the same tissue section. Scale bar = 1 mm. B, representative current traces of evoked AMPAR‐EPSCs (at –60 mV) and NMDAR‐EPSCs (at +40 mV) recorded in a labelled PVN neuron treated with vehicle or 100 μmol L−1 gabapentin in slices obtained from a WKY rat and an SHR. C and D, summary of evoked AMPAR‐EPSC and NMDAR‐EPSC data (C) and the ratio of NMDAR‐EPSCs to AMPAR‐EPSCs (D) in labelled PVN neurons pretreated with vehicle or gabapentin in slices from WKY rats (n = 9 neurons in each group) or SHRs (n = 8 neurons in each group). E and F, original current traces (E) and summary data (F) show the effect of 100 μmol L−1 gabapentin on puff NMDA (100 μmol L−1) elicited currents of labelled PVN neurons recorded from WKY rats (n = 11 neurons in each group) and SHRs (vehicle, n = 12 neurons; gabapentin, n = 11 neurons). *** P < 0.001 compared to the WKY + vehicle group. ### P < 0.001 compared to the SHR + vehicle group. [Color figure can be viewed at http://wileyonlinelibrary.com]
To directly determine the role of α2δ‐1 in increased postsynaptic NMDAR activity of PVN presympathetic neurons in SHRs, we examined the effect of gabapentin on NMDAR currents elicited by puff application of NMDA (100 μmol L−1) directly to labelled PVN neurons. In vehicle‐treated slices, puff NMDA induced a significantly larger current of labelled neurons in SHRs than in WKY rats (WKY, n = 11 neurons; SHR, n = 12 neurons; P < 0.0001, F 3,41 = 20.68) (Fig. 2 E and F). Treatment with gabapentin completely restored the puff NMDA currents of labelled PVN neurons in SHRs to the levels in WKY rats (Fig. 2 E and F). These results suggest that α2δ‐1 is critically involved in increased postsynaptic NMDAR activity of PVN presympathetic neurons in SHRs.
α2δ‐1–bound NMDARs are essential for increased postsynaptic NMDAR activity of PVN presympathetic neurons in SHRs
The C‐terminus of α2δ‐1 is essential for its interaction with NMDARs, and a 30‐amino‐acid peptide (VSGLNPSLWSIFGLQFILLWLVSGSRHYLW) mimicking the transmembrane C‐terminus of α2δ‐1 fused with Tat protein (YGRKKRRQRRR; α2δ‐1Tat peptide) effectively interrupts the α2δ‐1–NMDAR interaction (Chen et al. 2018). Pretreatment with a Tat‐fused scrambled control peptide (FGLGWQPWSLSFYLVWSGLILSVLHLIRSN, 1 μmol L−1, 60 min) did not affect the evoked AMPAR‐EPSCs, evoked NMDAR‐EPSCs or the ratio of NMDAR‐EPSCs to AMPAR‐EPSCs of labelled PVN neurons from WKY rats (n = 10 neurons) or SHRs (n = 9 neurons) (Fig. 3 A–C). By contrast, α2δ‐1Tat peptide (1 μmol L−1, 60 min) fully normalized the amplitude of evoked NMDAR‐EPSCs of labelled PVN neurons in SHRs (n = 9 neurons in each group) (Fig. 3 A and B). However, α2δ‐1Tat peptide had no effect on the amplitude of evoked NMDAR‐EPSCs of labelled PVN neurons in WKY rats (Fig. 3 A and B). Also, α2δ‐1Tat peptide had no effect on the amplitude of evoked AMPAR‐EPSCs of labelled PVN neurons in WKY rats or SHRs (Fig. 3 A and B). α2δ‐1Tat peptide normalized the ratio of NMDAR‐EPSCs to AMPAR‐EPSCs in SHRs to the level in WKY rats (Fig. 3 C), although it had no effect on the ratio of NMDAR‐EPSCs to AMPAR‐EPSCs in WKY rats.
Figure 3. α2δ‐1–bound NMDARs are essential for enhanced postsynaptic NMDAR activity of PVN presympathetic neurons in SHRs.
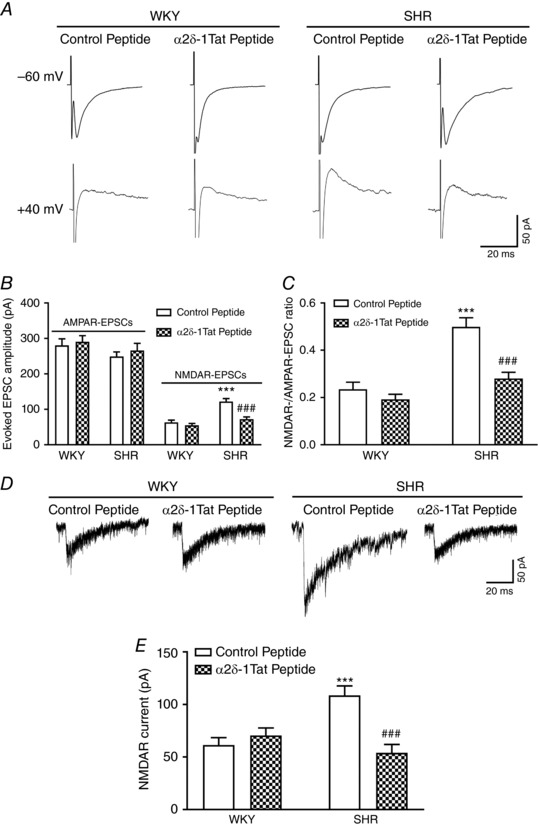
A, representative current traces of evoked AMPAR‐EPSCs (at –60 mV) and NMDAR‐EPSCs (at +40 mV) recorded in labelled PVN neurons treated with control peptide or α2δ‐1Tat peptide (1 μmol L−1) in slices from a WKY rat and an SHR. B and C, summary of evoked AMPAR‐EPSC and NMDAR‐EPSC data (B) and the ratio of NMDAR‐EPSCs to AMPAR‐EPSCs (C) in labelled PVN neurons pretreated with control peptide or α2δ‐1Tat peptide in slices from WKY rats (n = 10 neurons in each group) or SHRs (n = 9 neurons in each group). D and E, original current traces (D) and mean data (E) show the effect of 1 μmol L−1 control peptide or 1 μmol L−1 α2δ‐1Tat peptide on puff NMDA (100 μmol L−1) elicited currents of labelled PVN neurons recorded from WKY rats (n = 8 neurons in each group) and SHRs (n = 11 neurons in each group). *** P < 0.001 compared to the WKY + control peptide group. ### P < 0.001 compared to the SHR + control peptide group.
Furthermore, treatment with α2δ‐1Tat peptide had no effect on the amplitude of puff NMDA currents in WKY rats. However, α2δ‐1Tat peptide restored the amplitude of puff NMDA currents of labelled PVN neurons in SHRs (n = 11 neurons) to the level in WKY rats (n = 8 neurons) (Fig. 3 D and E). By contrast, treatment with the Tat‐fused control peptide had no effect on the amplitude of puff NMDA currents in WKY (n = 8 neurons) or SHRs (n = 11 neurons) (Fig. 3 D and E). These data indicate that α2δ‐1–bound NMDARs are crucial for increased postsynaptic NMDAR activity of PVN presympathetic neurons in SHRs.
α2δ‐1 is required for tonic activation of presynaptic NMDARs of PVN presympathetic neurons in SHRs
Presynaptic NMDARs in the PVN are normally latent, although they are tonically activated to augment glutamate release from presynaptic terminals in SHRs (Li et al. 2008; Li et al. 2017; Qiao et al. 2017). To determine whether α2δ‐1 contributes to increased presynaptic NMDAR activity in the PVN in SHRs, we recorded mEPSCs, which reflect quantal release of glutamate from presynaptic terminals (Sulzer & Pothos, 2000). The baseline frequency of mEPSCs of labelled PVN neurons was significantly higher in SHRs than in WKY rats (WKY, n = 10 neurons; SHR, n = 11 neurons; P = 0.0012, F 5,57 = 6.607) (Fig. 4), whereas the amplitude of mEPSCs did not differ significantly between the two groups. To confirm that the increase in the frequency of mEPSCs in SHRs is mediated by NMDARs, we bath‐applied a specific NMDAR antagonist, AP5 (50 μmol L−1). AP5 application readily normalized the frequency of mEPSCs of labelled PVN neurons in SHRs to the level in WKY rats (Fig. 4 C–E), whereas it had no effect in WKY rats (Fig. 4 A–E). Gabapentin (100 μmol L−1, 60 min) treatment fully restored the baseline frequency of mEPSCs of labelled PVN neurons in SHRs without changing their amplitude (n = 11 neurons) (Fig. 4 D–F). Subsequent bath application of AP5 no longer had any effect on the frequency of mEPSCs of labelled PVN neurons in SHR brain slices pretreated with gabapentin. By contrast, gabapentin had no effect on the frequency or amplitude of mEPSCs of labelled PVN neurons in WKY rats (Fig. 4 B, E and F). These findings suggest that α2δ‐1 is obligatory for tonic activation of presynaptic NMDARs of PVN presympathetic neurons in SHRs.
Figure 4. α2δ‐1 mediates increased presynaptic NMDAR activity of RVLM–projecting PVN neurons in SHRs.

A–D, original traces and cumulative probability plots show the effect of bath application of 50 μmol L−1 AP5 on the frequency and amplitude of mEPSCs of labelled PVN neurons pretreated with vehicle or 100 μmol L−1 gabapentin in slices from WKY rats and SHRs. E and F, summary data show the effects of gabapentin and AP5 on the frequency (E) and amplitude (F) of mEPSCs in labelled PVN neurons from WKY rats (vehicle, n = 10 neurons; gabapentin, n = 12 neurons) and SHRs (n = 11 neurons in each group). ** P < 0.01 compared to baseline in the WKY + vehicle group. # P < 0.05 compared to baseline in the SHR + vehicle group.
α2δ‐1–bound NMDARs mediate increased synaptic glutamate release to PVN presympathetic neurons in SHRs
We next used α2δ‐1Tat peptide to determine whether α2δ‐1–bound NMDARs are responsible for the increased presynaptic NMDAR activity in SHRs. Pretreatment with α2δ‐1Tat peptide (1 μmol L−1, 60 min) completely normalized the baseline frequency of mEPSCs of labelled PVN neurons in SHRs to the level in WKY rats (SHR + control peptide, n = 9 neurons; SHR + α2δ‐1Tat peptide, n = 10 neurons) (Fig. 5 A–E). However, α2δ‐1Tat peptide had no effect on the frequency or amplitude of mEPSCs in WKY rats. After treatment with α2δ‐1Tat peptide, further application of AP5 (50 μmol L−1) no longer had any effect on mEPSCs of labelled PVN neurons in brain slices from SHRs (Fig. 5 D–E). Treatment with the Tat‐fused control peptide had no effect on the frequency or amplitude of mEPSCs of labelled PVN neurons in brain slices from WKY rats (n = 12 neurons) or SHRs (n = 9 neurons) (Fig. 5 A, C, E and F). These data indicate that α2δ‐1–bound NMDARs are required for tonic activation of presynaptic NMDARs of PVN presympathetic neurons in SHRs.
Figure 5. α2δ‐1–bound NMDARs are essential for tonic activation of presynaptic NMDAR of PVN presympathetic neurons in SHRs.
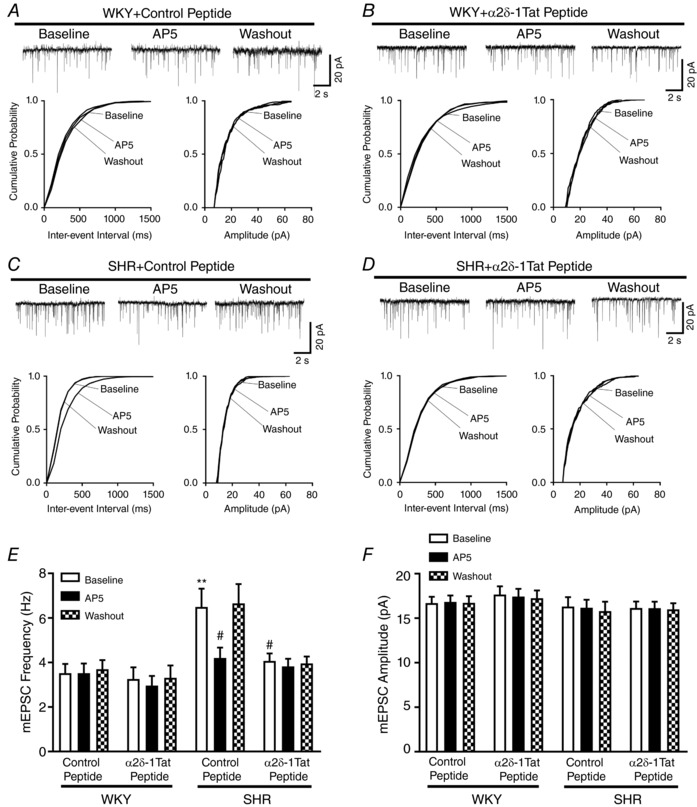
A–D, original traces and cumulative probability plots show the effect of bath application of 50 μmol L AP5 on mEPSCs of labelled PVN neurons pretreated with 1 μmol L−1 control peptide or 1 μmol L−1 α2δ‐1Tat peptide in slices from WKY rats and SHRs. E and F, group data show the effects of α2δ‐1Tat peptide and AP5 on the frequency (E) and amplitude (F) of mEPSCs in labelled PVN neurons from WKY rats (control peptide, n = 12 neurons; α2δ‐1Tat peptide, n = 11 neurons) and SHRs (control peptide, n = 9 neurons; α2δ‐1Tat peptide, n = 10 neurons). ** P < 0.01 compared to baseline in the WKY + control peptide group. # P < 0.05 compared to baseline in the SHR + control peptide group.
α2δ‐1–bound NMDARs in the PVN maintain heightened sympathetic vasomotor tone in SHRs
Because gabapentin and pregabalin bind to both α2δ‐1 and α2δ‐2 (Gee et al. 1996; Gong et al. 2001; Marais et al. 2001; Li et al. 2011), we used α2δ‐1Tat peptide to determine whether α2δ‐1–bound NMDARs in the PVN play a role in heightened sympathetic vasomotor tone in SHRs. We have shown previously that microinjection of AP5 into the PVN has no effect on the basal sympathetic vasomotor activity in normotensive WKY rats (Li & Pan, 2007). Microinjection of α2δ‐1Tat peptide (50 pmol, 50 nL) bilaterally into the PVN significantly decreased the ABP (P < 0.0001, F 2,24 = 15.67) and RSNA (P < 0.0001, F 2,24 = 20.20) in SHRs (n = 9 rats) (Fig. 6 A–C). The effect of α2δ‐1Tat peptide on the mean ABP and RSNA lasted for more than 60 min. In SHRs microinjected with α2δ‐1Tat peptide, subsequent microinjection of AP5 (1.0 nmol, 50 nL) (Li & Pan, 2007; Li et al. 2017; Qiao et al. 2017) into the PVN failed to further decrease ABP and RSNA (Fig. 6 A–C). By contrast, AP5 microinjection into the PVN significantly decreased ABP and RSNA in the SHRs subjected to a prior microinjection of Tat‐fused control peptide (50 pmol, 50 nL; n = 9 rats) (Fig. 6). These results suggest that α2δ‐1–bound NMDARs in the PVN contribute to maintaining elevated sympathetic vasomotor tone in SHRs.
Figure 6. α2δ‐1‐bound NMDARs in the PVN maintain heightened sympathetic outflow in SHRs.
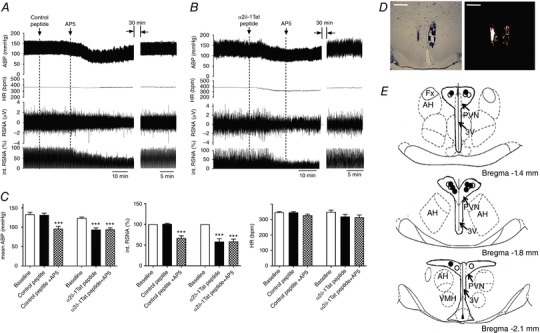
A and B, original recording traces show the effects of bilateral microinjection of control peptide (A) or α2δ‐1Tat peptide (B) followed by AP5 microinjection into the PVN on ABP, RSNA and HR in SHRs. C, summary data show changes in mean ABP, integrated RSNA (int. RSNA) and HR in response to microinjection of AP5 after microinjection of control peptide or α2δ‐1Tat peptide into the PVN in SHRs (n = 9 rats in each group). D and E, representative tissue section images (D) and schematic drawing (E) show the microinjection sites for control peptide plus AP5 (solid circle) and α2δ‐1Tat peptide plus AP5 (open circle) in the PVN in SHRs. *** P < 0.001 compared to the baseline control. 3V, third ventricle; AH, anterior hypothalamus; Fx, fornix; VMH, ventromedial hypothalamus. Scale bars = 1 mm. [Color figure can be viewed at http://wileyonlinelibrary.com]
Discussion
The present study reveals that the direct interaction between α2δ‐1 and NMDARs in the brain is of high pathophysiological relevance in dysregulation of the autonomic nervous system in hypertension. We showed that the α2δ‐1 protein level in the PVN was upregulated in SHRs and that this increase in α2δ‐1 was not altered by lowering ABP. These findings suggest that α2δ‐1 upregulation in the PVN is not a secondary response to increased ABP in SHRs but instead may contribute to hypertension development. Consistent with this notion, we found that the α2δ‐1 protein level in the PVN was significantly higher in 4‐week‐old prehypertensive SHRs than in age‐matched WKY rats. In the spinal cord, nerve injury‐induced α2δ‐1 upregulation promotes its interaction with NMDARs and the synaptic expression of α2δ‐1–bound NMDARs (Chen et al. 2018). Consistent with this concept is our finding in the present study that α2δ‐1 interacted with NMDARs in the hypothalamus and that the physical association between α2δ‐1 and NMDARs was enhanced in SHRs. In addition, we show that the protein level of α2δ‐1 and NMDARs in the hypothalamic synaptosomes was substantially increased in SHRs. Taken together, these biochemical data suggest that the prevalence of α2δ‐1–bound NMDARs in the hypothalamus is increased in SHRs, which potentiates the synaptic trafficking of α2δ‐1–NMDAR complexes in this hypertension model.
A major finding of the present study is that α2δ‐1 is required for the increased presynaptic and postsynaptic NMDAR activity in PVN presympathetic neurons in SHRs. In the present study, we found unexpectedly that inhibiting α2δ‐1 activity with gabapentin or disrupting the α2δ‐1–NMDAR association with α2δ‐1Tat peptide completely blocked the increased postsynaptic NMDAR activity and NMDAR‐mediated synaptic glutamate release to RVLM‐projecting PVN neurons in SHRs. By contrast, gabapentin and α2δ‐1Tat peptide had no effect on presynaptic or postsynaptic NMDAR activity of PVN presympathetic neurons in normotensive rats. The preferential effect of gabapentin and α2δ‐1Tat peptide on synaptic NMDAR activity in SHRs is probably a result of the low α2δ‐1 expression level in the PVN in normotensive rats. In support of this view, we have shown, in a heterologous expression system, that gabapentin inhibits NMDAR activity only when α2δ‐1 is coexpressed (Chen et al. 2018). Our present findings indicate that α2δ‐1–bound NMDARs account for most, if not all, of the increased synaptic NMDAR activity of PVN presympathetic neurons in SHRs. In addition to promoting synaptic trafficking of NMDARs, α2δ‐1 can potentiate the NMDAR activity by reducing the voltage‐dependent Mg2+ block of GluN2A‐containing NMDARs (Chen et al. 2018).
The present study provides further evidence that α2δ‐1 primarily functions as a critically important regulator of NMDARs in the central nervous system. Because we recorded mEPSCs and puff NMDA currents at a hold potential of –60 mV and in the presence of tetrodotoxin, VACCs are in a closed state in this recording condition. Therefore, α2δ‐1 probably enhances the synaptic NMDAR activity of PVN presympathetic neurons independent of VACCs. α2δ‐1 may have a minor role in regulating VACC activity because α2δ‐1 has a weak interaction with the pore‐forming VACC α1 subunit in the brain (Muller et al. 2010). Furthermore, removing α2δ‐1 has little effect on the overall VACC activity in cell lines and in hypothalamic neurons (Wu et al. 2009; Felsted et al. 2017). Previous studies have shown that protein kinases, such as casein kinase 2, calcium/calmodulin‐dependent protein kinase II and Src kinases, contribute to the NMDAR hyperactivity of PVN presympathetic neurons in SHRs (Ye et al. 2012b; Li et al. 2017; Qiao et al. 2017). However, the link between α2δ‐1–bound NMDARs and protein kinases in regulating NMDAR activity in hypertension is currently unknown. Because increased phosphorylation can strengthen protein–protein binding complexes (Nishi et al. 2011), it is possible that certain protein kinases potentiate phosphorylation of α2δ‐1 and/or NMDAR proteins to promote their physical interactions by changing their physicochemical properties, stability and dynamics.
The increased NMDAR activity in the PVN plays a dominant role in maintaining elevated sympathetic outflow in SHRs (Li & Pan, 2007; Ye et al. 2011; Qiao et al. 2017). Our findings showing that α2δ‐1–bound NMDARs in the PVN critically contributed to the increased synaptic NMDAR activity of PVN presympathetic neurons in SHRs prompted us to determine the role of α2δ‐1–bound NMDARs in the control of sympathetic outflow in hypertension. Consistent with our slice recording data is our finding that disrupting the α2δ‐1–NMDAR interaction in the PVN substantially decreased ABP and RSNA in SHRs. Importantly, subsequent microinjection of the NMDAR antagonist did not further decrease ABP and RSNA after pretreatment with α2δ‐1Tat peptide in SHRs. These results provide in vivo evidence that α2δ‐1–bound NMDARs in the PVN are critically involved in sustaining elevated sympathetic vasomotor activity in the hypertensive condition.
In summary, we present substantial new evidence indicating that α2δ‐1 plays a pivotal role in increased presynaptic and postsynaptic NMDAR activity of PVN presympathetic neurons and that α2δ‐1–bound NMDARs in the PVN are required for maintaining sympathetic vasomotor tone in SHRs (Fig. 7). Our study provides new insight into the molecular mechanism underlying elevated sympathetic outflow in an animal model of essential hypertension. Further studies are needed to determine whether α2δ‐1–bound NMDARs play a role in autonomic dysregulation in other animal models of hypertension. Because neither gabapentin, nor α2δ‐1Tat peptide affects the basal α2δ‐1–free NMDARs, targeting α2δ‐1–bound NMDARs could minimize the adverse effects associated with blocking physiological NMDARs by general NMDAR antagonists. Some clinical studies suggest that gabapentinoids can reduce the pressor response elicited by tracheal intubation and the paroxysmal sympathetic hyperactivity caused by brain injury (Fassoulaki et al. 2006; Choi et al. 2013; Doleman et al. 2016). On the basis of our findings, α2δ‐1–bound NMDARs could be targeted as a treatment for neurogenic hypertension.
Figure 7. Schematic representation shows the proposed role of α2δ‐1 in the regulation of synaptic NMDARs of PVN presympathetic neurons and sympathetic outflow in SHRs.
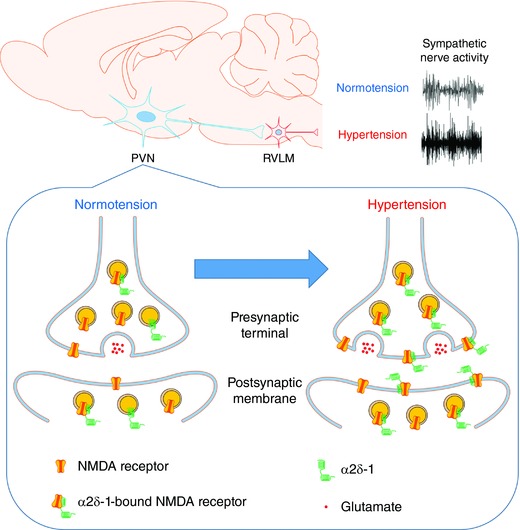
In normotensive condition, most NMDARs are not associated with α2δ‐1 in the PVN. In hypertensive condition, α2δ‐1 is upregulated and physically interacts with NMDARs to promote synaptic trafficking of α2δ‐1–bound NMDARs, which leads to increased pre‐ and postsynaptic NMDAR activity of RVLM‐projecting PVN neurons. The increased synaptic NMDAR activity in the PVN contributes to elevated sympathetic vasomotor activity in SHRs. [Color figure can be viewed at http://wileyonlinelibrary.com]
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
HM, S‐RC, HC, J‐JZ and D‐PL performed the experiments and analysed data. HM drafted and revised the manuscript. H‐LP conceived and designed the study and edited the manuscript. All authors approved the final version of the manuscript submitted for publication.
Funding
This work was supported by the National Institutes of Health (Grants HL131161 and HL142133) and the N.G. and Helen T. Hawkins Endowment (to H‐LP).
Biography
Huijie Ma received her PhD in Physiology from Hebei Medical University in China in 2010 and then continued to study the cardiac protective effect of chronic intermittent hypobaric hypoxia as a faculty member at the same university. She currently works with Dr Hui‐Lin Pan as a visiting scientist at The University of Texas MD Anderson Cancer Center in Houston, USA. She is studying the molecular and signalling mechanism governing glutamatergic synaptic plasticity of hypothalamic presympathetic neurons in hypertension. Her general research interest is to define precisely how the central nervous system regulates the sympathetic vasomotor output in cardiovascular disease conditions.

Edited by: Jaideep Bains & Katalin Toth
References
- Allen AM (2002). Inhibition of the hypothalamic paraventricular nucleus in spontaneously hypertensive rats dramatically reduces sympathetic vasomotor tone. Hypertension 39, 275–280. [DOI] [PubMed] [Google Scholar]
- Chen J, Li L, Chen SR, Chen H, Xie JD, Sirrieh RR, MacLean DM, Zhang Y, Zhou MH, Jayaraman V & Pan HL (2018). The α2δ‐1–NMDA receptor complex is critically involved in neuropathic pain development and gabapentin therapeutic actions. Cell Rep 22, 2307–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HA, Jeon SB, Samuel S, Allison T & Lee K (2013). Paroxysmal sympathetic hyperactivity after acute brain injury. Curr Neurol Neurosci Rep 13, 370. [DOI] [PubMed] [Google Scholar]
- Cole RL, Lechner SM, Williams ME, Prodanovich P, Bleicher L, Varney MA & Gu G (2005). Differential distribution of voltage‐gated calcium channel alpha‐2 delta subunit mRNA‐containing cells in the rat central nervous system and the dorsal root ganglia. J Comp Neurol 491, 246–269. [DOI] [PubMed] [Google Scholar]
- DiBona GF (2013). Sympathetic nervous system and hypertension. Hypertension 61, 556–560. [DOI] [PubMed] [Google Scholar]
- Doleman B, Sherwin M, Lund JN & Williams JP (2016). Gabapentin for the hemodynamic response to intubation: systematic review and meta‐analysis. Can J Anaesth 63, 1042–1058. [DOI] [PubMed] [Google Scholar]
- Fassoulaki A, Melemeni A, Paraskeva A & Petropoulos G (2006). Gabapentin attenuates the pressor response to direct laryngoscopy and tracheal intubation. Br J Anaesth 96, 769–773. [DOI] [PubMed] [Google Scholar]
- Felsted JA, Chien CH, Wang D, Panessiti M, Ameroso D, Greenberg A, Feng G, Kong D & Rios M (2017). Alpha2delta‐1 in SF1(+) neurons of the ventromedial hypothalamus is an essential regulator of glucose and lipid homeostasis. Cell Rep 21, 2737–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller‐Bicer GA, Varadi G, Koch SE, Ishii M, Bodi I, Kadeer N, Muth JN, Mikala G, Petrashevskaya NN, Jordan MA, Zhang SP, Qin N, Flores CM, Isaacsohn I, Varadi M, Mori Y, Jones WK & Schwartz A (2009). Targeted disruption of the voltage‐dependent calcium channel alpha2/delta‐1‐subunit. Am J Physiol Heart Circ Physiol 297, H117–H124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee NS, Brown JP, Dissanayake VU, Offord J, Thurlow R & Woodruff GN (1996). The novel anticonvulsant drug, gabapentin (Neurontin), binds to the alpha2delta subunit of a calcium channel. J Biol Chem 271, 5768–5776. [DOI] [PubMed] [Google Scholar]
- Gong HC, Hang J, Kohler W, Li L & Su TZ (2001). Tissue‐specific expression and gabapentin‐binding properties of calcium channel alpha2delta subunit subtypes. J Membr Biol 184, 35–43. [DOI] [PubMed] [Google Scholar]
- Hamer DW & Santer RM (1981). Anatomy and blood supply of the coeliac‐superior mesenteric ganglion complex of the rat. Anat Embryol (Berl) 162, 353–362. [DOI] [PubMed] [Google Scholar]
- Li DP, Atnip LM, Chen SR & Pan HL (2005). Regulation of synaptic inputs to paraventricular‐spinal output neurons by alpha2 adrenergic receptors. J Neurophysiol 93, 393–402. [DOI] [PubMed] [Google Scholar]
- Li DP, Chen SR & Pan HL (2003). Angiotensin II stimulates spinally projecting paraventricular neurons through presynaptic disinhibition. J Neurosci 23, 5041–5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP & Pan HL (2006). Plasticity of GABAergic control of hypothalamic presympathetic neurons in hypertension. Am J Physiol Heart Circ Physiol 290, H1110‐H1119. [DOI] [PubMed] [Google Scholar]
- Li DP & Pan HL (2007). Glutamatergic inputs in the hypothalamic paraventricular nucleus maintain sympathetic vasomotor tone in hypertension. Hypertension 49, 916–925. [DOI] [PubMed] [Google Scholar]
- Li DP, Yang Q, Pan HM & Pan HL (2008). Pre‐ and postsynaptic plasticity underlying augmented glutamatergic inputs to hypothalamic presympathetic neurons in spontaneously hypertensive rats. J Physiol 586, 1637–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Zhou JJ, Zhang J & Pan HL (2017). CaMKII regulates synaptic NMDA receptor activity of hypothalamic presympathetic neurons and sympathetic outflow in hypertension. J Neurosci 37, 10690–10699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Taylor CP, Weber M, Piechan J, Prior F, Bian F, Cui M, Hoffman D & Donevan S (2011). Pregabalin is a potent and selective ligand for alpha(2)delta‐1 and alpha(2)delta‐2 calcium channel subunits. Eur J Pharmacol 667, 80–90. [DOI] [PubMed] [Google Scholar]
- Marais E, Klugbauer N & Hofmann F (2001). Calcium channel alpha(2)delta subunits‐structure and gabapentin binding. Mol Pharmacol 59, 1243–1248. [DOI] [PubMed] [Google Scholar]
- Muller CS, Haupt A, Bildl W, Schindler J, Knaus HG, Meissner M, Rammner B, Striessnig J, Flockerzi V, Fakler B & Schulte U (2010). Quantitative proteomics of the Cav2 channel nano‐environments in the mammalian brain. Proc Natl Acad Sci U S A 107, 14950–14957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi H, Hashimoto K & Panchenko AR (2011). Phosphorylation in protein‐protein binding: effect on stability and function. Structure 19, 1807–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oparil S, Zaman MA & Calhoun DA (2003). Pathogenesis of hypertension. Ann Intern Med 139, 761–776. [DOI] [PubMed] [Google Scholar]
- Qiao X, Zhou JJ, Li DP & Pan HL (2017). Src kinases regulate glutamatergic input to hypothalamic presympathetic neurons and sympathetic outflow in hypertension. Hypertension 69, 154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboussin DM, Allen NB, Griswold ME, Guallar E, Hong Y, Lackland DT, Miller EPR 3rd, Polonsky T, Thompson‐Paul AM & Vupputuri S (2017). Systematic Review for the 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension, 10.1016/j.jacc.2017.1011.1004 [DOI] [Google Scholar]
- Rock DM, Kelly KM & Macdonald RL (1993). Gabapentin actions on ligand‐ and voltage‐gated responses in cultured rodent neurons. Epilepsy Res 16, 89–98. [DOI] [PubMed] [Google Scholar]
- Schumacher TB, Beck H, Steinhauser C, Schramm J & Elger CE (1998). Effects of phenytoin, carbamazepine, and gabapentin on calcium channels in hippocampal granule cells from patients with temporal lobe epilepsy. Epilepsia 39, 355–363. [DOI] [PubMed] [Google Scholar]
- Sulzer D & Pothos EN (2000). Regulation of quantal size by presynaptic mechanisms. Rev Neurosci 11, 159–212. [DOI] [PubMed] [Google Scholar]
- Taylor CP & Garrido R (2008). Immunostaining of rat brain, spinal cord, sensory neurons and skeletal muscle for calcium channel alpha2‐delta (alpha2‐delta) type 1 protein. Neuroscience 155, 510–521. [DOI] [PubMed] [Google Scholar]
- Wu ZZ, Li DP, Chen SR & Pan HL (2009). Aminopyridines potentiate synaptic and neuromuscular transmission by targeting the voltage‐activated calcium channel beta subunit. J Biol Chem 284, 36453–36461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZY, Li DP, Byun HS, Li L & Pan HL (2012a). NKCC1 upregulation disrupts chloride homeostasis in the hypothalamus and increases neuronal activity‐sympathetic drive in hypertension. J Neurosci 32, 8560–8568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZY, Li DP, Li L & Pan HL (2011). Protein kinase CK2 increases glutamatergic input in the hypothalamus and sympathetic vasomotor tone in hypertension. J Neurosci 31, 8271–8279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZY, Li L, Li DP & Pan HL (2012b). Casein kinase 2‐mediated synaptic GluN2A up‐regulation increases N‐methyl‐D‐aspartate receptor activity and excitability of hypothalamic neurons in hypertension. J Biol Chem 287, 17438–17446. [DOI] [PMC free article] [PubMed] [Google Scholar]