

Abstract
This study aimed to investigate whether pitavastatin protected against injury induced by advanced glycation end products products (AGEs) in neonatal rat cardiomyocytes, and to examine the underlying mechanisms. Cardiomyocytes of neonatal rats were incubated for 48 hours with AGEs (100μg/mL), receptor for advanced glycation end products (RAGE), antibody (1μg/mL) and pitavastatin (600 ng/mL). The levels of p62 and beclin1 were determined by Western blotting. Mitochondrial membrane potential (ΔΨm) and the generation of reactive oxygen species (ROS) were measured through the JC-1 and DCFH-DA. In the AGEs group, the expression of beclin1 was remarkably increased compared to the control group, while the expression of p62 was significantly decreased. AGEs also markedly decreased ΔΨm and significantly increased ROS compared with the control group. After treatment with RAGE antibody or pitavastatin, the level of beclin1 was markedly decreased compared with the AGEs group, but the level of p62 was remarkably increased. In the AGEs+ RAGE antibody group and AGEs+ pitavastatin group, ΔΨm was significantly increased and ROS was remarkably decreased compared with the AGEs group. In conclusion, AGEs-RAGE may induce autophagy of cardiomyocytes by generation of ROS and pitavastatin could protect against AGEs-induced injury against cardiomyocytes.
Keywords: advanced glycation end products (AGEs), receptor for advanced glycation end products (RAGE), pitavastatin, autophagy, mitochondrial oxidation, oxidative stress
Introduction
Advanced glycation end products (AGEs) are products of glycosylation reactions called the Maillard reaction, which continuously accumulate in many tissues or organs of humans and animals with the increase of age[1‒2]. AGEs play a role in the accelerated process of diabetic cardiovascular complications and atherosclerosis complications through interactions with the receptor for advanced glycation end products (RAGE)[3‒4]. Several studies have revealed that accumulation of AGEs can induce myocardial remodeling and cardiac diastolic dysfunction through oxidative stress[5‒6]. After binding to their receptors, RAGE, AGEs induce the generation of reactive oxidative species (ROS) that can lead to oxidative stress[7].
Autophagy is a self-digestion process that delivers intracellular components to lysosomes through autophagosomes, which plays an important role in clearing the damaged macromolecular materials and cell organelles[8]. It is reported that autophagic cell death is induced in human umbilical vein endothelial cells treated with AGEs[9]. Oxidative stress and mitochondria damage are closely related to the occurrence of autophagy. It is well known that ROS induce autophagy[10]. So we speculate that oxidative stress, mitochondrial dysfunction, and autophagy may be associated with the AGEs-induced myocardial injury.
Statins, 3-hydroxy-3-methylglutaryl(HMG)-CoA reductase inhibitors, are now considered a first-line pharmacologic intervention for patients with dyslipidemia and atherosclerosis. The benefits of statins are well documented. However, myotoxic side effects including myopathy or rhabdomyolysis can sometimes be severe. Pitavastatin, a new statin agent, is associated with a low incidence of adverse metabolic events related to drug-drug interactions. In addition to lipid-lowering effect, pitavastatin plays an important role as an anti-atherosclerotic, anti-oxidant, anti-inflammatory agent and vascular endothelium protection[11]. In our current study, we investigated the effects of pitavastatin on AGE-induced cardiac autophagy, which will provide an experimental basis for its application to attenuate myocardial injury.
Materials and methods
Animals
Postnatal 1-3 day Sprague-Dawley rats were from the Experimental Animal Center of Nanjing Medical University. Hearts were harvested for analysis. All experimental procedures adhered to approval of the study by local committee and the National Institutes of Health guidelines on the use of experimental animals.
Reagents
Pitavastatin was provided by Huawei Pharmaceutical Technology Development Co., (Fuzhou,China). AGE-modified bovine serum albumin (AGE-BSA) was purchased from Calbiochem (catolog number:121800, San Diego, USA). RAGE antibody was purchased from R&D (catolog number: AF1616, USA). Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were provided by Gibco (NY, USA). Collagenase II was purchased from Sigma (Santa Clara, USA). p62, beclin1, and GAPDH antibodies were obtained from CST (#5114, #3738, #2118, USA). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibody was purchased from Abcam (6721, USA). Mitochondrial membrane potential assay kit with JC-1 and Reactive Oxygen Species assay kit were from Beyotime (Shanghai, China).
Cell culture and treatment
Ventricular myocytes were obtained from 1 to 3 day old Sprague-Dawley rats. The hearts were removed quickly and washed three times with cold phosphate buffered saline (PBS). The left heart tissues were minced with scissors into 1 mm3 morsels and then digested with PBS containing 0.1% (w/v) collagenase II five to six times. Subsequently, the cell suspension was filtered with cell strainer and the mixture was centrifuged at 1,000 rpm for 10 minutes. The pellets were resuspended, transferred to culture plates, and incubated at 37°C. Fibroblasts were removed by adhesion onto the plates for 90 minutes. Non-adherent cells, neonatal rat ventricular cardiomyocytes, were plated in a culture plate and cultured in DMEM medium containing 10% (v/v) FBS, 100 U/mL penicillin, and 100μg/mL streptomycin under 5% CO2 atmosphere at 37°C. The medium was replaced every 48 hours. After 3 days' culture, when cells reached 80% confluence, cardiomyocytes were used for subsequent experiments.
Cardiomyocytes were divided into the following groups: (1) the control group; (2) the AGEs group that was treated with 100μg/mL AGE-BSA; (3) the AGEs+ RAGE antibody group that was pretreated with 1μg/mL RAGE antibody for 2 hours prior to a 48-hour exposure to AGE-BSA (100μg/mL); (4) the AGEs+ pitavastatin group that was pretreated with 600 ng/mL pitavastatin[12] for 2 hours and then cocultured with AGE; and (5) the Pit group that was treated with 600ng/ml pitavastatin. After drug administration, cardiomyocytes were incubated for 48 hours.
Total protein extraction
Cells were collected and lysed with RIPA Lysis Buffer (Beyotime) on ice, and then samples were centrifuged at 12,000 rpm for 20 minutes at 4°C. Supernatants were transferred to a new tube and the total protein concentrations were determined by a BCA Protein Assay Kit (Sigma, USA). After extraction, the protein samples were respectively stored at −20°C for further use.
Mitochondrial membrane potential (ΔΨm) assay
Cardiomyocytes were incubated with JC-1 solution for 20 minutes at 37°C.Then cells were washed with JC-1 buffer for twice and medium was added to every well. Images were collected using a fluorescence microscope, which detects J-aggregates (excitation/emission= 525/590 nm) and JC-1 monomers (excitation/emission= 490/530 nm).
Measurement of ROS
Intracellular ROS levels were assessed using DCFH-DA probe. Briefly, Cardiomyocytes were treated with 1ml DCFH-DA (10μmol/mL) for 20 minutes at 37°C. After incubation, the cells were washed with DMEM. The relative intensity of DCF fluorescence was determined at an ex/em wavelength of 488/525 nm as compared to control group cells.
Western blotting
Protein expression levels of p53, p16, p62, Beclin1 and GAPDH were determined by western blotting. After the protein samples were heat-denatured, equal amounts of protein were separated by 10% SDS-PAGE and transferred to PVDF membranes by electrophoresis. The membranes were then blocked with 5% skim milk for 2 hours and incubated with primary antibodies at 4°C overnight. Membranes were subsequently washed by TBST buffer and incubated with the horseradish peroxidase-labeled goat anti-rabbit secondary antibody for 1 hour at room temperature. After washing the membrane by TBST for three times, the protein levels were detected by chemiluminescence using an enhanced chemiluminescence (ECL) detection kit (Pierce, France). The level of GAPDH was used as an internal control.
Cell counting kit (CCK)-8 assays
Ventricular myocytes cells (100μl/well) were seeded into 96-well plate at a density of 10 × 104 cells/ml and incubated at 37°C, 5% CO2 for 3 days. The cells were then treated with Pit of various concentration (0, 100, 200, 400, 600, 800, 1,000 and 1,200 ng/mL) for 48 hours. Following Pit treatment, a CCK-8 assay kit (Dojindo, Kumamoto, Japan) was used to detect cell viabilities according to the manufacturer's instructions. In brief, 10μL CCK-8 solution was added to each well. After 1‒4 hours incubation, cell viability was determined by measuring the absorbance at 450 nm using an ELISA reader (BioTek, Winooski, VT, USA).
Statistical analysis
All data were obtained from at least 3 individual experiments. The results were presented as the mean±SEM and further analyzed by SPSS (ver.13.0). Differences between groups were determined using one-way ANOVA with the LSD multiple-comparison test when appropriate. Prism software (GraphPad) was adopted for statistical analysis. A final value of P≤0.05 was considered significant for all analyses.
Results
Pitavastatin attenuates AGEs-induced ROS production in cardiomyocytes
Intracellular ROS levels were assessed using DCFH-DA probe. Our data showed that ROS levels in the AGEs-treated group were significantly higher than that in the control group (P<0.01; Fig. 1). Pitavastatin or RAGE the antibody pretreatment resulted in significant attenuation of ROS formation in comparartion to AGEs group (P<0.01; Fig. 1).
Fig.1.
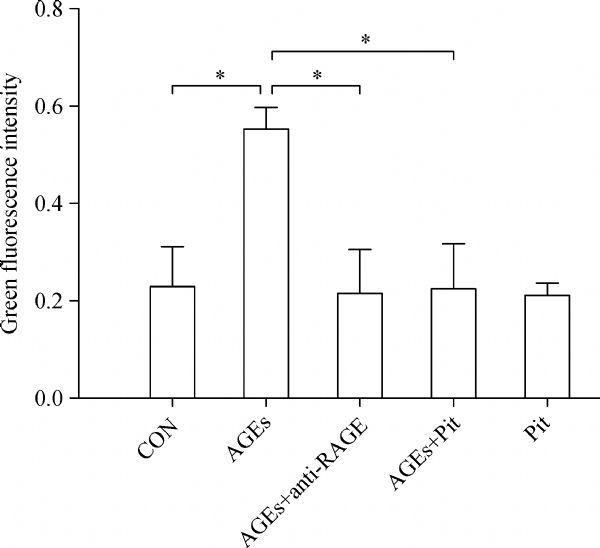
The effect of pitavastatin on AGEs-induced ROS. Intracellular ROS levels were assessed using DCFH-DA probe. Intensity alterations in ROS generation. Data represented are mean±SD of three independent experiments. *P < 0.01. AGE: advanced glycation end products; ROS: reactive oxygen species; Pit: pitavastatin.
Pitavastatin reduces AGEs-induced loss of mitochondrial membrane potential (ΔΨm)
ΔΨm was monitored by using mitochondria-specific probe JC-1. In the mitochondria of healthy cells, JC-1 forms aggregates, which emit red fluorescence. However, it remains as monomers that emit green fluorescence during the loss of ΔΨm. The intensity ratio of red to green fluorescence represents the change in ΔΨm. As shown in Fig. 2A, control cells showed mainly an intense red fluorescence coming from JC-1 aggregates. In contrast, two days of treatment with AGEs dramatically decreased ΔΨm of rat cardiomyocytes compared with control (P<0.01, Fig. 2). However, combination of AGEs and pitavastatin or RAGE antibody attenuated AGEs-induced loss of mitochondrial ΔΨm in cardiomyocytes (P<0.01, Fig. 2).
Fig.2.
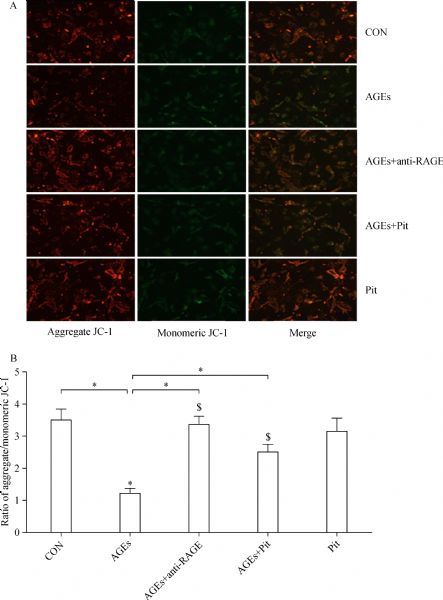
The effect of pitavastatin on AGEs-induced mitochondrial membrane potential (ΔΨm). Mitochondrial membrane potential was evaluated using JC-1 dye. Green fluorescence indicates low mitochondrial membrane potential, and red fluorescence indicates high mitochondrial membrane potential. (A) Fluorescence microscopy and (B) quantitative measurements of the fluorescence intensity uncovered alteration of intracellular mitochondrial membrane potential. Red-orange fluorescence came from JC-1 aggregates and green fluorescence came from JC-1 monomers. ΔΨm was assessed by JC-1 aggregates/JC-1 monomers ratios. Data are presented as mean±SD (n = 3 in each group). *P < 0.01; n = 3 in each group.
Pitavastatin suppresses AGEs-induced autophagy in cardiomyocytes
To determine the effects of pitavastatin on mitophagy in cardiomyocytes, we examined the expression of beclin1 and p62 by Western blotting. As shown in Fig. 3, the expression of beclin1 was significantly increased, whereas p62 was decreased in AGEs-treated cells (P<0.05). The expression of p62 significantly increased after pretreatment with pitavastatin or RAGE antibody. However, the expression of beclin1 was decreased when the cells were adminstered with pitavastatin or RAGE antibody (P<0.05).
Fig.3.
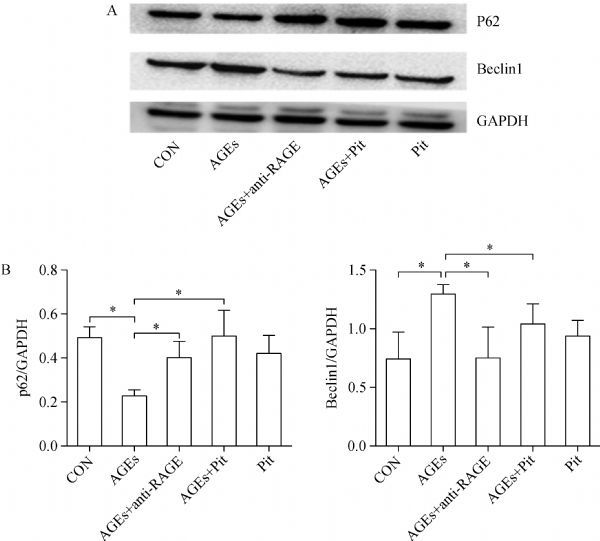
The effect of pitavastatin on AGEs-induced autophagy. A: Western blot analysis of Beclin1 and p62 protein levels. B: Protein quantification by densitometry. Data are presented as mean±SD (n = 3 in each group). *P < 0.05.
Myocardial toxicity with increasing concentrations of pitavastatin
Myocardial cells were inoculated on 96-well plates with 1 × 104 cells per well. After 72 hours, cells were treated with pitavastatin at different concentrations (0, 100, 200, 400, 600, 800, 1,000 and 1,200 ng/mL). The cell's activity was measured after culturing for 48 hours and the results are shown in Fig. 4. CCK8 assay showed that low concentration of pitavastatin (100-800 ng/mL) had no significant influence on cell viability. However, when the Pit concentration increased to 1,000 and 1,200 ng/mL, cell toxicity was evident. Therefore, the concentration of 600 ng/mL in our experiment is appropriate.
Fig.4.
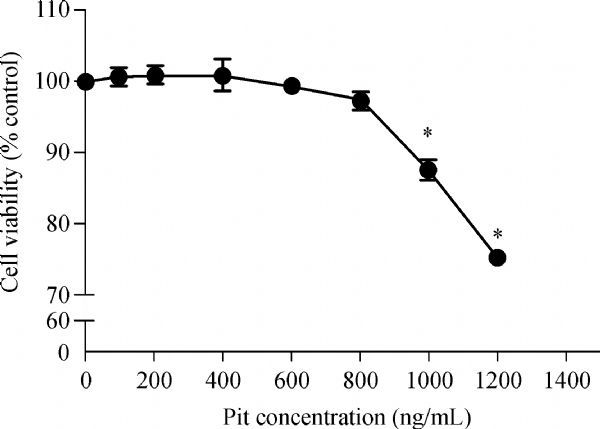
Effect of pitavastatin on rat myocardial cell activity. Data are presented as mean±SD (n = 3 in each group). *P < 0.05, versus the control group.
Discussion
Advanced glycation end products (AGEs) play an important role in the development of diabetic cardiomyopathy[13]. However, the underlying mechanisms are not completely understood. Oxidative stress plays a critical role in the development of structural and biochemical alterations of the heart[14‒16]. The detrimental effects of higher oxidative stress on cardiomyocytes include endothelial dysfunction, lipid peroxid-ation, apoptosis, activation of several inflammatory pathways etc[17‒19]. Accordingly, the inhibition of oxidative stress, an balance between formation and decomposition of ROS, is of great importance for cardiac injury. Currently, statins, in addition to their cholesterol lowering characteristics, are commonly prescribed for its antioxidant effects[20‒21]. In human umbilical vein endothelial cells, statins can decrease oxidized lowdensity lipoprotein (ox-LDL) and inhibit oxidative stress and cytotoxicity via decreasing ROS formation and downregulating NOX activity[22‒23]. In our study, we found evident increase of ROS in the AGEs group and remarkable downregulation after treatment with anti-RAGE antibody or pitavastatin, which indicates that pitavastatin may inhibit cardiac injury via downregulation of oxidative stress.
Mitochondrial membrane potential (ΔΨm) is necessary for production of energy and preservation of cellular homeostasis, which is a critical primary determinant of myocyte survival. In our study, we found that ΔΨm was downregulated after cultured cardiomyocytes were treated with AGEs. These demonstrated that AGEs can cause mitochondrial damage in cardiomyocytes. As ROS accumulation was a result of mitochondrial impairment, increased ROS, in turn, further can increase the loss of ΔΨ in the mitochondria of cardiomyocytes[24], and further attenuates the bioenergetic function of mitochondria. This causes a vicious cycle that progressively aggravates oxidative stress and mitochondrial dysfunction in tissues and organs. Our data further demonstrated that two hours of pretreatment with pitavastatin significantly attenuated the loss of ΔΨm in mitochondria of cardiomyocytes, which may imply that pitavastatin can at least partially protect the cardiomyocytes by decreasing ROS generation and preserving the integrity of mitochondrial membrane.
Autophagy is a crucial process for the removal of misfolded proteins, damaged organelles and intracellular pathogens[25]. Autophagic flux is often assessed using p62 and beclin1 Western blotting. In our study, we found that the level of beclin1 was upregulated and p62 was downregulated after cultured cardiomyocytes were treated with AGEs. Similarly, a recent research indicates that AGEs triggers autophagy via the PI3K/AKT/mTOR signaling pathway in cardiomyocytes[26]. These demonstrated that autophagic flux was enhanced by AGEs in cardiomyocytes. Nevertheless, studies have yielded conflicting results. Some experiments showed that AGEs induce autophagic inactivation in vascular smooth muscle cell or under other conditions[27‒28]. It is as though autophagy is a double-edged sword in cardiomyocyte demise and survival. However, some evidences also suggest that autophagy, in addition to its role in cell survival, can also lead to cell death[29‒31]. Patschan et al. previously reported that autophagy induced by GC, an important component of AGEs, contributes to cell death in HUVECs[32‒33]. It is implied that autophagy may promote cell death through excessive self-digestion and degradation of essential cellular constituents. Intreastingly, pitavastatin stimulated the expression of p62 and inhibited the level of Beclin1 in our study. This result indicates that the mechanisms by which pitavastatin reversed the effect of AGEs appear to involve the autophagy pathway. In previous studies, statin therapy influences autophagic events. Statins like fluvastatin, potently affect macrophage viability in vitro through the induction of apoptotic process[34]. It is plausible that the early autophagic flux induced by statin treatment may be a potential mechanism in myocardial injury process, thus explaining in part the achieved beneficial effects.
In summary, our work proved that pitavastatin effectively inhibited AGEs-induced autophagic changes in cardiomyocytes of rats, and may alleviate myocardial injury induced by AGEs via inhibiting oxidative stress, protecting mitochondria and downregulating autophagy. Taken together, these results provide the rationale for possible therapeutic strageties for the treatment of diabetic cardiomyopathy.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (NSFC 81570328, Wang Junhong) and the "Sixth-Peak Talent" of Jiangsu Province (2011WSN-029 to Prof. Guo Yan and 2013WSN-036 to Dr. Wang Junhong). Prof. Guo Yan was also support by the Health Department of Jiangsu Province (z201301)
References
- 1. Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications[J]. N Engl J Med, 1988, 318(20): 1315‒1321 10.1056/NEJM198805193182007. [DOI] [PubMed] [Google Scholar]
- 2. Yamagishi S, Takeuchi M, Inagaki Y, et al. Role of advanced glycation end products (AGEs) and their receptor (RAGE) in the pathogenesis of diabetic microangiopathy[J]. Int J Clin Pharmacol Res, 2003, 23(4): 129‒134. [PubMed] [Google Scholar]
- 3. Bucala R, Cerami A. Advanced glycosylation: chemistry, biology, and implications for diabetes and aging[J]. Adv Pharmacol, 1992, 23: 1‒34 10.1016/S1054-3589(08)60961-8. [DOI] [PubMed] [Google Scholar]
- 4. Bodiga VL, Eda SR, Bodiga S. Advanced glycation end products: role in pathology of diabetic cardiomyopathy[J]. Heart Fail Rev, 2014, 19(1): 49‒63 10.1007/s10741-013-9374-y. [DOI] [PubMed] [Google Scholar]
- 5. Guo Y, Lu M, Qian J, et al. Alagebrium chloride protects the heart against oxidative stress in aging rats[J]. J Gerontol A Biol Sci Med Sci, 2009, 64A(6): 629‒635 10.1093/gerona/glp023. [DOI] [PubMed] [Google Scholar]
- 6. Bodiga VL, Eda SR, Bodiga S. Advanced glycation end products: role in pathology of diabetic cardiomyopathy[J]. Heart Fail Rev, 2014, 19(1): 49‒63 10.1007/s10741-013-9374-y. [DOI] [PubMed] [Google Scholar]
- 7. Choi KM, Yoo HJ, Kim HY, et al. Association between endogenous secretory RAGE, inflammatory markers and arterial stiffness[J]. Int J Cardiol, 2009, 132(1): 96‒101 10.1016/j.ijcard.2007.10.047. [DOI] [PubMed] [Google Scholar]
- 8. Klettner A, Kauppinen A, Blasiak J, et al. Cellular and molecular mechanisms of age-related macular degeneration: from impaired autophagy to neovascularization[J]. Int J Biochem Cell Biol, 2013, 45(7): 1457‒1467 10.1016/j.biocel.2013.04.013. [DOI] [PubMed] [Google Scholar]
- 9. Patschan S, Chen J, Polotskaia A, et al. Lipid mediators of autophagy in stress-induced premature senescence of endothelial cells[J]. Am J Physiol Heart Circ Physiol, 2007, 294(3): H1119‒H1129 10.1152/ajpheart.00713.2007. [DOI] [PubMed] [Google Scholar]
- 10. Chen Y, Gibson SB. Is mitochondrial generation of reactive oxygen species a trigger for autophagy[J]. Autophagy, 2008, 4(2): 246‒248 10.4161/auto.5432. [DOI] [PubMed] [Google Scholar]
- 11. Ota H, Eto M, Kano MR, et al. Induction of endothelial nitric oxide synthase, SIRT1, and catalase by statins inhibits endothelial senescence through the Akt pathway[J]. Arterioscler Thromb Vasc Biol, 2010, 30(11): 2205‒2211 10.1161/ATVBAHA.110.210500. [DOI] [PubMed] [Google Scholar]
- 12. Kikuchi R, Takeshita K, Uchida Y, et al. Pitavastatin-induced angiogenesis and arteriogenesis is mediated by Notch1 in a murine hindlimb ischemia model without induction of VEGF[J]. Lab Invest, 2011, 91(5): 691‒703 10.1038/labinvest.2011.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bodiga VL, Eda SR, Bodiga S. Advanced glycation end products: role in pathology of diabetic cardiomyopathy[J]. Heart Fail Rev, 2014, 19(1): 49‒63 10.1007/s10741-013-9374-y. [DOI] [PubMed] [Google Scholar]
- 14. Wojciechowska C, Romuk E, Tomasik A, Skrzep-Poloczek B, Nowalany-Kozielska E, Birkner E, et al. Oxidative stress markers and C-reactive protein are related to severity of heart failure in patients with dilated cardiomyopathy[J]. Mediators Inflamm 2014; 2014: 147040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wojciechowska C, Romuk E, Tomasik A, Skrzep-Poloczek B, Nowalany-Kozielska E, Birkner E, et al. Oxidative stress markers and C-reactive protein are related to severity of heart failure in patients with dilated cardiomyopathy[J]. Mediators Inflamm 2014; 2014: 147040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rueckschloss U, Villmow M, Klockner U. NADPH oxidase-derived superoxide impairs calcium transients and contraction in aged murine ventricular myocytes[J]. Exp Gerontol, 2010, 45(10): 788‒796 10.1016/j.exger.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 17. Qin F, Siwik DA, Pimentel DR, et al. Cytosolic H2O2 mediates hypertrophy, apoptosis, and decreased SERCA activity in mice with chronic hemodynamic overload[J]. Am J Physiol Heart Circ Physiol, 2014, 306(10): H1453‒H1463 10.1152/ajpheart.00084.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Piarulli F, Sartore G, Ceriello A, et al. Relationship between glyco-oxidation, antioxidant status and microalbuminuria in type 2 diabetic patients[J]. Diabetologia, 2009, 52(7): 1419‒1425 10.1007/s00125-009-1367-y. [DOI] [PubMed] [Google Scholar]
- 19. Stocker R, Keaney JJ. Role of oxidative modifications in atherosclerosis[J]. Physiol Rev, 2004, 84(4): 1381‒1478 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- 20. Cangemi R, Loffredo L, Carnevale R, et al. Early decrease of oxidative stress by atorvastatin in hypercholesterolaemic patients: effect on circulating vitamin E[J]. Eur Heart J, 2007, 29(1): 54‒62 10.1093/eurheartj/ehm565. [DOI] [PubMed] [Google Scholar]
- 21. Miyamoto-Sasaki M, Yasuda T, Monguchi T, et al. Pitavastatin increases HDL particles functionally preserved with cholesterol efflux capacity and antioxidative actions in dyslipidemic patients[J]. J Atheroscler Thromb, 2013, 20(9): 708‒716 10.5551/jat.17210. [DOI] [PubMed] [Google Scholar]
- 22. Kojanian H, Szafran-Swietlik A, Onstead-Haas LM, et al. Statins prevent dextrose-induced endoplasmic reticulum stress and oxidative stress in endothelial and HepG2 cells[J]. Am J Ther, 2016, 23(6): e1456‒e1463. [DOI] [PubMed] [Google Scholar]
- 23. Zhang L, Guo R, Cui L. Simvastatin attenuates oxidized low density lipoprotein induced oxidative stress in human umbilical vein endothelial cells via downregulating NADPH oxidase activity[J]. Zhonghua Xin Xue Guan Bing Za Zhi, 2014, 42(1): 43‒47. [PubMed] [Google Scholar]
- 24. Akar FG, Aon MA, Tomaselli GF, et al. The mitochondrial origin of postischemic arrhythmias[J]. J Clin Invest, 2005, 115(12): 3527‒3535 10.1172/JCI25371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang H, Guo M, Chen JH, et al. Osteopontin knockdown inhibits alphav,beta3 integrin-induced cell migration and invasion and promotes apoptosis of breast cancer cells by inducing autophagy and inactivating the PI3K/Akt/mTOR pathway[J]. Cell Physiol Biochem, 2014, 33(4): 991‒1002 10.1159/000358670. [DOI] [PubMed] [Google Scholar]
- 26. Hou X, Hu Z, Xu H, et al. Advanced glycation endproducts trigger autophagy in cadiomyocyte via RAGE/PI3K/AKT/mTOR pathway[J]. Cardiovasc Diabetol, 2014, 13(1): 78 10.1186/1475-2840-13-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ma M, Guo X, Chang Y, et al. Advanced glycation end products promote proliferation and suppress autophagy via reduction of Cathepsin D in rat vascular smooth muscle cells[J]. Mol Cell Biochem, 2015, 403(1‒2): 73‒83 10.1007/s11010-015-2338-x. [DOI] [PubMed] [Google Scholar]
- 28. Liu WJ, Shen TT, Chen RH, et al. Autophagy-lysosome pathway in renal tubular epithelial cells is disrupted by advanced glycation end products in diabetic nephropathy[J]. J Biol Chem, 2015, 290(33): 20499‒20510 10.1074/jbc.M115.666354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword[J]. Science, 2004, 306(5698): 990‒995 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death[J]. Cell Death Differ, 2005, 12(Suppl 2): 1509‒1518 10.1038/sj.cdd.4401751. [DOI] [PubMed] [Google Scholar]
- 31. Baehrecke EH. Autophagy: dual roles in life and death[J]. Nat Rev Mol Cell Biol, 2005, 6(6): 505‒510 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- 32. Patschan S, Chen J, Polotskaia A, et al. Lipid mediators of autophagy in stress-induced premature senescence of endothelial cells[J]. Am J Physiol Heart Circ Physiol, 2007, 294(3): H1119‒H1129 10.1152/ajpheart.00713.2007. [DOI] [PubMed] [Google Scholar]
- 33. Patschan S, Goligorsky MS. Autophagy: The missing link between non-enzymatically glycated proteins inducing apoptosis and premature senescence of endothelial cells[J]. Autophagy, 2008, 4(4): 521‒523 10.4161/auto.5904. [DOI] [PubMed] [Google Scholar]
- 34. Croons V, De Meyer I, Houten SM, et al. Effect of statins on the viability of macrophages and smooth muscle cells[J]. J Cardiovasc Pharmacol, 2010, 55(3): 269‒275 10.1097/FJC.0b013e3181d0bec3. [DOI] [PubMed] [Google Scholar]