

Abstract
The enzyme sphingomyelinase (SMase) is an important biomarker for several diseases such as Niemann Pick’s, atherosclerosis, multiple sclerosis, and HIV. We present a two-component colorimetric SMase activity assay that is more sensitive and much faster than currently available commercial assays. Herein, SMase-triggered release of cysteine from a sphingomyelin (SM)-based liposome formulation with 60 mol % cholesterol causes gold nanoparticle (AuNP) aggregation, enabling colorimetric detection of SMase activities as low as 0.02 mU/mL, corresponding to 1.4 pM concentration. While the lipid composition offers a stable, nonleaky liposome platform with minimal background signal, high specificity toward SMase avoids cross-reactivity of other similar phospholipases. Notably, use of an SM-based liposome formulation accurately mimics the natural in vivo substrate: the cell membrane. We studied the physical rearrangement process of the lipid membrane during SMase-mediated hydrolysis of SM to ceramide using small- and wide-angle X-ray scattering. A change in lipid phase from a liquid to gel state bilayer with increasing concentration of ceramide accounts for the observed increase in membrane permeability and consequent release of encapsulated cysteine. We further demonstrated the effectiveness of the sensor in colorimetric screening of small-molecule drug candidates, paving the way for the identification of novel SMase inhibitors in minutes. Taken together, the simplicity, speed, sensitivity, and naked-eye readout of this assay offer huge potential in point-of-care diagnostics and high-throughput drug screening.
Keywords: liposome, gold nanoparticle, sphingomyelinase, colorimetric detection, sphingomyelin, cholesterol
Sphingomyelins (SMs) are an important class of phospholipids, constituting 2–15% of the total phospholipid content of mammalian tissues.1 SMs and other sphingolipids are widely reported to induce structural rigidity in cell membranes and are key features of lipid rafts. Recently, it has become evident that their role is much more than a structural one.2 Specifically, the hydrolysis of SM by the enzyme SMase leads to the production of ceramide, which is a downstream cell-signaling molecule responsible for a host of pathologies.3−5 Therefore, changes in SMase activity are associated with several diseases such as Niemann Pick’s,6,7 atherosclerosis,8 depression,9,10 multiple sclerosis, and HIV.10,11
Upregulated SMase activity provides a reliable biomarker target for improved diagnostic and prognostic tools, which could significantly improve patient outcome. Likewise, in the pharmaceutical industry, high-throughput assays to identify novel SMase inhibitors are crucial for developing effective treatments. A quick and convenient method to determine SMase activity and inhibition is highly desirable in the development of point-of-care sensors for disease detection and treatment. However, current SMase activity and inhibition assays rely on complicated multistep enzymatic reactions and expensive equipment to perform read-outs11−13 and the use of radioactive markers14 or non-natural substrates,15 and often measure SMase concentration rather than activity.16 Similarly, a simple assay to screen for SMase inhibitors rapidly would allow the as-yet untapped potential for SMase inhibitors as therapeutics to be realized.
Liposome aggregation and phosphate quantification-based assays to study the relative activity of SMase on membrane bilayers of different compositions are well known.17,18 These methods are often used to study the relative activity of SMase to, for example, different lipid compositions. However, these assays typically use enzyme activities of >1 U/mL and are orders of magnitude less sensitive than required for applications in quantifying biologically relevant SMase activities. Liposomes formulated from both natural19−21 and synthetic lipids22 have been shown to release their cargo in the presence of a target protein, causing a colorimetric response. Release of encapsulated contents from liposomes can also be triggered by osmotic shock, as used in detecting pathogens with attomolar sensitivity.23 The activity of SMase on membrane bilayers is largely influenced by their lipid composition. Mixtures of SM with cholesterol (Chol)18 and/or phospholipids such as phosphatidylcholines (PC) and phosphatidylethanolamines (PE) have been shown to be substantially more active toward SMase than bilayers comprising pure SM.17 The origin of this increase in activity is a change in physical state from a gel (pure SM) to a fluid membrane bilayer.24 Ternary mixtures with coexisting liquid and gel phases have been shown to further increase membrane bilayer activity. The gel phase sequesters the ceramide product, depleting its concentration in the liquid-ordered phase and reducing the inhibitory effect it has on SMase,25 whereas the presence of cholesterol “dissolves” SM into an enzyme-accessible liquid ordered phase. However, PC- and PE-containing mixtures are not ideal for SMase-selective assays since such phospholipids are also active toward other phospholipases. Additionally, the reaction product of SM hydrolysis, ceramide, has a large influence on SMase activity due to its impact on lipid phase behavior. A review article by Goñi etal. provides a nice discussion on the subject.26
Aggregation of gold nanoparticles (AuNPs) and the resulting color changes provide a useful transduction mechanism for biodetection, and numerous optical sensing platforms have been developed based on the aggregation principle.27−29 The color changes induced by aggregation of plasmonic NPs originate from the coupling between the localized surface plasmon resonance (LSPR) of the NPs, resulting in lower energy excitation levels and red-shifted absorbance bands.30,31 Different types of aggregation mechanisms have been reported32 including the bridging of NPs by complementary recognition complexes, hydrogen bonds, donor–acceptor interactions,33,34 and host–guest interactions.35,36 The combination of NP-based aggregation assays with liposomal platforms offers a useful system for the sensitive detection of proteins. We hypothesized that combining liposomes and AuNP aggregation would allow detection of very low SMase activity by the naked eye.
Herein, we present an efficient colorimetric assay that measures activity of SMase on liposome membrane bilayers. Using lipids formulated into liposomes instead of suspensions of free lipids used in currently available assays37 helps mimic the natural environment for SMase reactivity, i.e., the cell membrane. We prepared robust liposomes containing cysteine, which is released in the presence of active SMase. The released cysteine causes rapid aggregation of gold nanoparticles, leading to a colorimetric read-out visible by the naked eye. To gain insight into the origins of membrane reorganization leading to cysteine release, we carried out small- and wide-angle X-ray scattering (SAXS and WAXS, respectively) measurements of bulk lipid mixtures. These measurements showed a phase transition between 0 and 50% conversion of SM to ceramide from a fluid to gel phase, suggesting changes in permeability across the bilayer during this lipid rearrangement process. This straightforward assay allows us to achieve naked eye sensitivity down to 0.08 mU/mL SMase, corresponding to a concentration of 5.5 pM, which is as sensitive as the current commercially available absorbance-based assays. Quantification based on AuNP absorbance spectra achieves a sensitivity of 0.02 mU/mL SMase. Finally, we demonstrated the utility of the platform to rapidly detect SMase inhibitors at physiologically relevant concentrations.
Results and Discussion
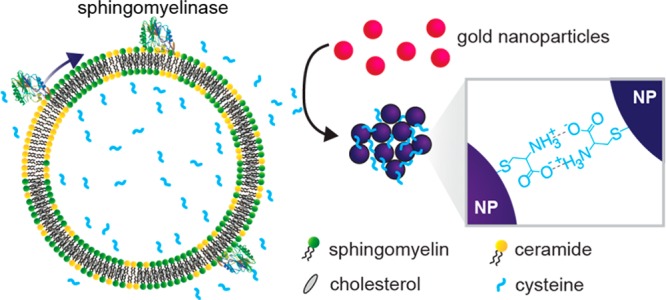
A schematic of the assay design is shown in Figure 1. In the presence of active SMase, the SM component of SM:Chol liposomes is enzymatically hydrolyzed into ceramide and phosphocholine. This process causes the release of cysteine from these liposomes as their membranes undergo a fluid to gel phase transition. The released cysteine causes aggregation of AuNPs, leading to a colorimetric change due to the LSPR effect of the AuNPs. In the absence of enzyme activity the AuNP dispersion appears red, whereas in the presence of SMase activity a blue color is observed with an enzyme concentration-dependent tonality variation, visible by the naked eye. This assay strategy combining the amplification from the liposomes and sensitive color change from AuNP LSPR shifts provides a robust and sensitive platform for SMase detection.
Figure 1.

Assay schematic. (Top) Hydrolysis of the phosphate–oxygen bond of SM by SMase to produce ceramide and phosphocholine (not shown). (Bottom) Action of SMase on BSM:Chol liposomes encapsulating cysteine leads to a composition-driven membrane phase change with the formation of ceramide-rich gel domains and partitioning of cholesterol into remaining SM domains. This causes cysteine leakage and aggregation of subsequently added gold nanoparticles by formation of hydrogen bonds between cysteine molecules on adjacent nanoparticles. Graphic of SMase enzyme structure is adapted from Ago etal.38
Cysteine-Mediated AuNP Aggregation
Cysteine initiates the aggregation of AuNPs through interparticle zwitterionic electrostatic interactions and the formation of hydrogen bonds.39,40 We used citrate-stabilized AuNPs to perform the cysteine-mediated particle aggregation, studied through transmission electron microscopy (TEM) and ultraviolet/visible spectrometry (UV/vis). As shown in Figure 2a, ∼13 nm diameter AuNPs are well dispersed in solution, but form large aggregates (Figure 2b) upon addition of cysteine. Accordingly, a color tonality change from the red (dispersed AuNPs) to a blue color (aggregated AuNPs) is observed (Figure 2c). Particle aggregation was also evident in the UV/vis spectra. Multiple modes of coupling are expected due to the complexity of the aggregates formed, with the absorbance and intensity of the aggregated system dependent on the degree of aggregation and orientation of the individual particles within the aggregate. Upon increasing cysteine concentration and particle aggregation, the 525 nm peak (dispersed AuNPs) broadens to form a shoulder at 640 nm, followed by generation of a new absorption peak at around 660 nm (Figure 2d). In these aggregated systems, the first peak near 525 nm can be attributed to the quadrupole plasmon excitation in coupled spheres, while the broader peak with longer wavelength is due to the dipole plasmon resonance of the gold nanoparticles.41,42 Determining the ratios between the areas under the curve of UV/vis spectral regions corresponding to dispersed and aggregated AuNPs provides a reliable calibration of analyte concentrations43,44 (see the Supporting Information for discussion, Figures S1 and S2). For simplicity, we used changes in the ratio between absorbance at 640 and 525 nm to accurately capture changes in absorbance even at lower cysteine concentrations (SI, Figure S3). Notably, the aggregation of the AuNPs begins at 0.4 μM cysteine concentration and reaches a saturation value at 2 μM cysteine for a fixed 15 min time interval (SI, Figure S3). Therefore, we recorded all subsequent experiments at this time point. The dynamic range of colorimetric detection of cysteine concentration is dependent on several parameters including particle concentration and buffer conditions.
Figure 2.
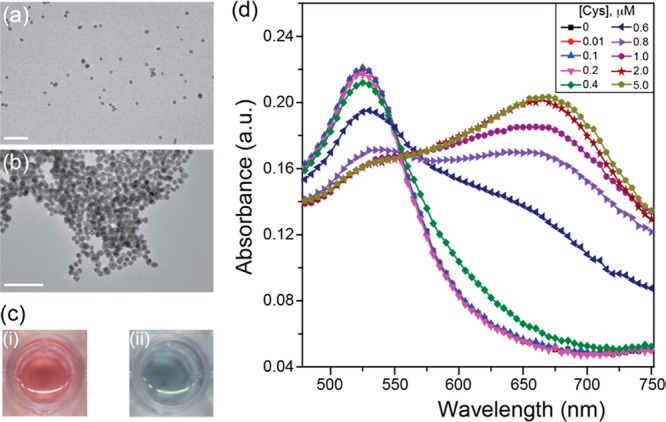
Cysteine-mediated gold nanoparticle aggregation. TEM of AuNPs incubated for 5 min in (a) 10% v/v DPBS or (b) 10% v/v DPBS spiked with 5 μM cysteine. Scale bar 100 nm. (c) Photographs of wells containing (i) AuNPs in DPBS or (ii) AuNPs in DPBS spiked with 5 μM cysteine. (d) Representative absorbance spectra of AuNP incubated for 15 min in DPBS or DPBS spiked with 0.01 to 5 μM cysteine. Characteristic emergence of a broad peak at around 660 nm is observed.
SMase-Sensitive Liposome Formulations
One of the most important requirements for small molecule (such as cysteine) containing liposome-based assays is liposomal formulations that are intrinsically nonpermeable and stable over a period of time. Initially, we focused on producing nonleaky membrane compositions, varying the mol percentages of SM extracted from bovine brain SM (BSM) and Chol within the liquid ordered state. We prepared cysteine-containing liposomes using conventional lipid film hydration and extrusion through polycarbonate membranes of varying pore sizes. We then removed free cysteine molecules from the liposome suspension by filtration through a Sephadex G-100 column. AuNP aggregation induced by cysteine released from the liposomes provided a straightforward assay for optimizing liposome formulations.
Initial screening of different BSM:Chol ratios showed that liposomes formulated from lipid films with a BSM:Chol molar ratio of 30:70 and 60:40 were intrinsically leaky. On mixing these liposomes with AuNPs, we observed an immediate color change of the solution from red to blue. On the other hand, lipid films with BSM:Chol molar ratios of 40:60 and 50:50 both formed stable liposomes that retained their encapsulated cysteine over a number of days. However, the 50:50 mol % mixtures had a slightly higher background leakage, resulting in an absorbance ratio of 640 nm/525 nm greater than 0.4 upon mixing liposomes with AuNPs without SMase (Figure 3a). The higher background gave rise to a slightly purple tonality and made naked eye detection more difficult than in the case of liposomes formulated from BSM:Chol 40:60 mol % lipid films, which under the same conditions showed absorbance ratios below 0.4 and appeared red in color. Additionally, the dynamic range for 40:60 mol % liposomes was slightly higher than that of 50:50 mol %. Therefore, BSM:Chol 40:60 mol % appeared to be the optimal lipid composition.
Figure 3.

Effect of liposome lipid composition, cysteine concentration, and diameter. (a) Effect of liposome lipid composition: 100 nm liposomes formulated from lipid films with BSM:Chol 40:60 (red) or 50:50 (black) mol % mixtures and 100 mM encapsulated cysteine. (b) Chol mol % of liposome formulations prepared from BSM:Chol films with 40, 50, 60, and 70 mol % Chol, as quantified by 1H NMR. Error bars are the standard deviation of the two integrated proton ratios calculated for each spectrum. (c) Effect of concentration of encapsulated cysteine: 100 nm liposomes formulated from BSM:Chol 40:60 mol % lipid films with 50, 100, or 200 mM encapsulated cysteine. (d) Effect of liposome diameter: 100 or 200 nm liposomes formulated from lipid films with BSM:Chol 40:60 mol % and 100 mM encapsulated cysteine. Liposome concentration is normalized to total volume of encapsulated cysteine based on NTA size measurements. In all cases, liposomes were incubated for 1 h with SMase in the reaction buffer, followed by addition of AuNPs. Absorbance spectra were recorded at 15 min after AuNP addition. The responses in (a), (c), and (d) are the average of three independent measurements, and the error bars represent the standard deviation.
While previous studies have reported the membrane phase behavior of EggSM:Chol lipid mixtures up to 50 mol % Chol, to our knowledge there are no previous reports of BSM:Chol 40:60 mol % vesicles. In order to confirm that liposomes with such a lipid composition can indeed be formed from BSM:Chol 40:60 mol % films, we characterized the liposomes formulated from BSM:Chol 60:40, 50:50, 40:60, and 30:70 mol % films using 1H NMR. We prepared liposomes by hydrating lipid films with water, vortexing, and extruding, as described earlier. The liposome samples were lyophilized and redissolved in CDCl3 before measuring their 1H NMR spectra. We then calculated two ratios for each spectrum by comparing areas under peaks for Chol at 0.68 (3H)45 and 1.84 ppm (3H)46 and BSM at 5.70 ppm (1H).47 For full NMR spectra and integrals, see Figures S7–S13. It can be observed that the calculated Chol mol % in the liposome samples agreed quite well with that of the lipid films containing 40, 50, and 60 mol % Chol (Figure 3b). Specifically, we observed Chol content increasing steadily at respectively 43.3 ± 3.5, 55.8 ± 0.1, and 64.7 ± 1.1 mol %. The slight discrepancy between calculated and measured Chol content is not surprising given the pretreatment of the sample and known measurement uncertainty of quantitative NMR, even under extensively optimized conditions.48 Notably, liposomes formulated from 70 mol % Chol lipid films had significantly less Chol than predicted at only 61.7 ± 0.5 mol % Chol. Therefore, the saturation limit of Chol in the BSM membrane bilayer appears to be between 60 and 70 mol %. Above the membrane saturation limit, crystals of excess Chol are removed during the extrusion process. This observation is supported by SAXS/WAXS studies (Figures 7 and S6), which show a single liquid ordered (Lo) phase for BSM:Chol 40:60 mol % lipid films, with no observed formation of Chol crystals, which would be expected above the saturation limit of Chol in BSM.
Figure 7.

SAXS/WAXS characterization of BSM:BC:Chol mixtures. Bulk SAXS and WAXS (inset) measurements and graphical illustration of lipid packing of (a) BSM:Chol 40:60 mol % and (b) BSM:BC:Chol 20:20:60 mol %. Blue lipids represent BSM, yellow lipids represent BC, and gray represents Chol.
To achieve high sensitivity, the optimal liposome formulation is expected to depend on the concentration of encapsulated cysteine. We prepared 100 nm liposomes formulated from BSM:Chol 40:60 mol % lipid films with 50, 100, or 200 mM encapsulated cysteine. While no difference was observed between the 50 and 100 mM cysteine liposomes, the 200 mM cysteine liposomes were sensitive down to lower SMase activities (Figure 3c). The background 640/525 nm ratio of >0.4 gave rise to a purple-colored solution, making the distinction between background and low SMase activities difficult by eye. Therefore, we considered cysteine concentrations of 100 mM to be most suitable for this colorimetric sensor.
We also tested the effect of liposome size on the SMase assay sensitivity. We formulated liposomes by hydrating BSM:Chol 40:60 mol % lipid films with 100 mM buffered cysteine solution, followed by vortexing and then extrusion through polycarbonate membranes with 100 or 200 nm pore sizes. We normalized liposome concentrations measured by nanoparticle tracking analysis (NTA) to contain the same total volume of encapsulated cysteine. Liposome hydrodynamic radii, determined using dynamic light scattering (DLS), gave intensity-based measurements reporting average diameters of 122.0 nm (PDI 0.10) and 192.8 nm (PDI 0.15) for liposomes extruded through 100 and 200 nm pore sizes, respectively. Therefore, the final concentration of 100 nm liposomes was 8.5 × 1010 particles/mL (140 pM), and that of 200 nm liposomes was 1.8 × 1010 particles/mL (30 pM). We observed that the 100 nm liposomes have a limit of detection (at three times higher than the standard deviation of the control) of 0.02 mU/mL compared with 0.5 mU/mL in the case of 200 nm liposomes (Figure 3d). The 200 nm liposomes contain 4 times as many lipids per liposome compared to the 100 nm ones. Therefore, to achieve the same change in the overall membrane composition, 4 times as many SM molecules must be hydrolyzed. In these assay conditions the SMase and liposome concentrations are within the same order of magnitude as each other. Hence, over the same incubation time, fewer 200 nm liposomes reach the required conversion of SM to ceramide to induce a phase change with consequent cysteine release.
Following the above optimizations, we concluded that the BSM:Chol 40:60 mol % liposomes with 100 nm diameter and containing 100 mM encapsulated cysteine (hereafter, referred to as BSM:Chol 40:60 liposomes) were the most suitable liposomes for the final SMase assays. Further characterizations of the BSM:Chol 40:60 liposomes showed predominantly unilamellar spherical vesicles as observed in CryoTEM (Figure 4a) and a monodisperse population with mean hydrodynamic diameter of 120.1 ± 2.4 nm (Figure 4b, blue trace), measured by NTA. Liposome size and concentration were stable over 1 h in the reaction buffer comprising Dulbecco’s phosphate-buffered saline (DPBS), 1 mM MgSO4, and 1 mM CaCl2 (Figure 4b, black trace). We found these concentrations of MgSO4 and CaCl2 to be optimal for naked-eye detection of SMase using the liposome–AuNP hybrid platform (SI, Figure S4). At liposome concentrations up to around 8.5 × 1010 particles/mL, there was no observable cysteine leakage upon 1 h incubation with DPBS including 1 mM MgSO4 and 1 mM CaCl2. However, above this threshold, evolution of a purple color was observed (Figure S5). Notably, the liposomes were stable in DPBS at 4 °C for at least one month (Figure 4c) with no observed cysteine leakage, indicating the possibility of long-term storage for commercial applications.
Figure 4.
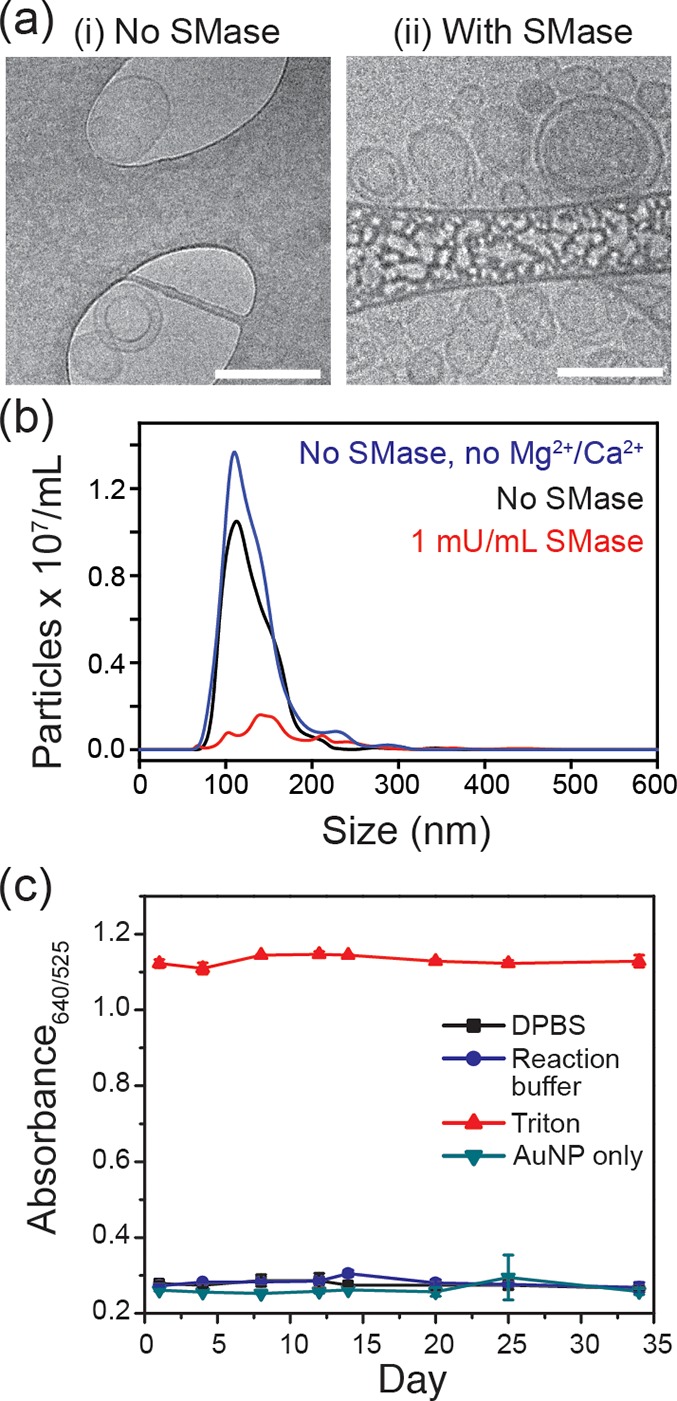
BSM:Chol liposomes formulated from a 40:60 mol % lipid film and their sensitivity to SMase. (a) CryoTEM images of liposomes after 1 h incubation in (i) reaction buffer (Dulbecco’s phosphate-buffered saline, DPBS, with Mg2+/Ca2+); or (ii), SMase (1 mU/mL SMase in reaction buffer). Scale bar is 200 nm. (b) NTA traces of liposomes incubated for 1 h in DPBS (blue), reaction buffer (black), or reaction buffer and 1 mU/mL SMase (red). (c) Stability of liposomes over time.
We studied enzymatic hydrolysis-induced changes to size and morphology of the BSM:Chol 40:60 liposomes in the reaction buffer containing 1 mU/mL SMase, with a final liposome concentration of 8.5 × 1010 particles/mL. As shown in Figure 3b (red trace), we observed a distinct drop in the liposome concentration measured by NTA after 1 h of incubation with SMase. CryoTEM of this sample confirmed a huge variation in liposome size and morphology, with liposomes from 20 to 200 nm diameter observed and the evolution of elongated rod-like structures (Figure 4a). The TEM grid was also significantly less populated with liposomes than the sample without SMase, despite both samples having the same initial liposome concentration. These observations confirm the disruption of liposomes upon addition of SMase on this BSM:Chol 40:60 liposome formulation.
Specificity and Selectivity of the Liposome-Based Assay
We observed cysteine release as a function of SMase activity via changes in the AuNP LSPR, with an increase in the 640 nm peak intensity and a decrease at 525 nm. To confirm that the cysteine release from the BSM:Chol 40:60 liposomes is specific to SMase-mediated cleavage of SM, we performed control experiments with denatured SMase. Liposomes that had been incubated with either active SMase or the surfactant Triton X100 (positive control) led to a blue color, with a 640/525 nm absorbance ratio greater than 1 (Figure 5a). However, incubation of the liposomes with SMase that had been denatured by heating to 85 °C for 30 min (dSMase) led to an absorbance ratio only slightly above the baseline of the intrinsic AuNP absorption ratio. While the slight increase in absorbance ratio compared to the baseline was possibly due to a combination of incomplete enzyme denaturation and increased scattering from the dSMase/AuNP/liposome agglomerates, it is much less than the increase observed with the same concentration of active SMase. In control experiments, AuNPs incubated with SMase or dSMase in the absence of liposomes showed absorbance ratios of <0.4 and appeared red (Figure 5a). Likewise, AuNPs and liposomes co-incubated without SMase appeared red. Therefore, it can be inferred that the release of cysteine is driven by the hydrolysis of BSM within the liposome membranes by active SMase.
Figure 5.

Specificity and selectivity of SMase assay with BSM:Chol 40:60 liposomes. (a) Absorbance ratio 15 min after AuNP addition to samples of (A–G) reaction buffer, 1 mU/mL SMase, 1 mU/mL denatured SMase, liposomes only, liposomes and 1 mU/mL SMase, liposomes and 1 mU/mL denatured SMase, and liposomes and 0.02 v/v % Triton X100. Reaction buffer was DPBS with 1 mM MgSO4 and 1 mM CaCl2. BSM:Chol 40:60 liposomes were used. (b) Specificity of the assay toward SMase. Absorbance ratios of mixtures of BSM:Chol 40:60 liposomes incubated for 1 h at rt with (L–R) SMase, phospholipase A2 from snake, phospholipase A2 from bee, phospholipase C, phospholipase D, lysozyme, BSA, and control (reaction buffer). Incubation times were 1 h. The responses are the average of three independent measurements, and the error bars represent the standard deviation.
We assessed selectivity of the liposome formulation toward SMase by screening activity toward a range of other commonly occurring phospholipases. As shown in Figure 5b, we observed no activity even at up to 10 mU/mL enzyme activities of snake and bee phospholipase A2 (PLA2) and phospholipase D (PLD). While phospholipase C (PLC) showed almost no activity at 1 mU/mL, activity at 10 mU/mL was similar to that of SMase. Specific phospholipase C inhibitors such as 1,10-phenanthroline could overcome any eventual interference of phospholipase C activity in human patient samples. As expected, there was no change in absorption of the control (no protein) baseline on incubation of liposomes with lysozyme (Lys) and bovine serum albumin (BSA), confirming that cysteine release is not induced by protein adsorption onto the liposome surface, but rather by the SMase-induced hydrolysis of SM.
Colorimetric Detection of SMase
Following the characterization of the liposomal system, we tested the sensitivity of the plasmonic assay toward SMase using the optimum BSM:Chol 40:60 liposome formulation. For these liposomes, total release of encapsulated contents leads to a final cysteine concentration of 10 μM in the reaction buffer. This concentration ensures that even a small percentage of released cysteine results in complete aggregation of AuNPs (cf. Figure S3). We incubated BSM:Chol liposomes in the reaction buffer containing 0.005 to 20 mU/mL SMase for 1 h at room temperature followed by the addition of AuNPs. After 15 min of AuNP incubation, we determined the limit of detection to be 0.02 mU/mL (1.4 pM enzyme concentration) using the ratio between absorption at 640 and 525 nm on a plate reader (Figure 6a). Here, the limit of detection is the lowest concentration of SMase that yields a signal 3 times higher than the standard deviation of the control. It should be noted that the SMase activity/concentration we report is based on the stock concentration provided by the commercial supplier. Our assay achieves better sensitivity than the commercially available assay kits, in a shorter time frame, at room temperature, and without multiple enzymatic amplification steps. In addition to sensitive absorbance-based measurements, we were able to distinguish clinically relevant SMase activities as low as 0.08 mU/mL by the naked eye. The high sensitivity of this assay is attributed to the specificity of SMase toward the liposomes, signal amplification from the release on the order of 106 cysteine molecules per liposome, efficient aggregation of AuNPs by the released cysteine, and the favorable plasmonic properties of AuNPs.
Figure 6.

Detection of SMase activity using AuNP aggregation. (a) SMase-assay sensitivity. Absorbance ratios of mixtures of BSM:Chol 40:60 liposomes and varying activities of SMase incubated at rt for 1 h. (b) Assay time-dependence. Absorbance ratios of liposomes incubated at rt with SMase (0–1 mU/mL) added at different time points. The lines are drawn to guide the eye. The absorbance ratios were read 15 min after addition of AuNPs. The responses are the average of at least three independent measurements, and the error bars represent the standard deviation.
Our group and others have previously shown a lag-burst effect on interaction of phospholipase A2 with liposomes.19,49 Such lag phases have been reported previously for SM-containing liposomes in the presence of SMase, where the length of the lag phase is inversely proportional to the final enzyme activity50,51 and a marked decrease in lag time is observed from the gel to fluid state.17 Here, we also observe this phenomenon on incubating SMase at 0, 0.1, 0.5, and 1 mU/mL activities with BSM:Chol 40:60 liposomes, whereby we added AuNPs at varying time points between 0 and 1 h. The time dependence of the extent of aggregation was inversely related to the SMase activity (Figure 6b). Specifically, we observed a lag time of 45 min in the 0.1 mU/mL sample before aggregation. We did not observe any time lag on direct addition of even very low concentrations of cysteine to AuNPs (Figure S3), where any aggregation is immediate.
Characterization of BSM:Chol 40:60 Membrane Phase Behavior
Changes in lipid membrane stability and permeability have been attributed to changes in the lipid ordering. In particular, membranes are known to become significantly more permeable during fluid–gel structural transitions. Therefore, to characterize the mechanistic origins of the observed liposome destabilization and cysteine leakage, we used SAXS and WAXS to study the phase behavior of bulk lipid mixtures containing 60 mol % cholesterol and varying molar ratios of BSM:BC from BSM:Chol 40:60 mol % to BSM:BC:Chol 20:20:60 mol % at 25 °C, where BC is bovine ceramide. Start and end points are shown in Figure 7a and b with sequential data available in Figure S6. The start and end point lipid mixtures represent the initial liposome lipid membrane composition and lipid membrane composition after 50% conversion of BSM to ceramide, respectively.
The BSM:Chol 40:60 mol % mixture (Figure 7a) shows predominantly a single lamellar phase with a lattice parameter of 66.7 ± 0.1 Å. The splitting of the second-order peak shows evidence of a minor coexisting structure, which we attribute to the fact that BSM is a natural mixture of lipids predominantly (>50%) composed of 18:0 SM, with the next largest component (>21%) being 24:1 SM. These components have their main gel–fluid transitions at 44.7 and 24.1 °C, respectively,52 with the BSM mixture having a broad transition temperature of 35–40 °C.53 The WAXS pattern of BSM:Chol 40:60 mol % shows a broad fluid peak centered at 4.7 Å. This is in good agreement with previous WAXS data on BSM:Chol 60:40 mol % mixtures in the fluid state, which showed a broad fluid peak centered at 4.5 Å at 25 °C.54 Here, the authors also showed that there is an increase in the d-spacing with increased cholesterol content, which provides an explanation for the slight difference between the two values. While pure BSM is in a gel state at 25 °C, the addition of >25 mol % cholesterol to SM has been previously shown to cause a transition from a bilayer gel state to the Lo fluid state.55,56 BSM:Chol mixtures above 50 mol % Chol exist exclusively in the Lo state, as seen in the BSM:Chol data (Figure 7a). Activity of SMase on liposomes formulated from mixtures of EggSM and Chol is also influenced by this transition. Specifically, increased SMase activity was observed in EggSM:Chol 50:50 mol % at 37 °C, thought to be in part due to the presence of the Lo phase, because SMase activity on EggSM in the gel state is significantly lower.57
Increasing the content of ceramide to mimic the effect of SMase at a constant Chol mol % led to an increase in the lattice parameter of 0.6 Å from 0 to 30% conversion of BSM to BC. This agrees well with previous reports of egg SM and egg ceramide mixtures, where increasing amounts of ceramide increased the observed lattice parameter.58 As the ceramide content increases from BSM:BC:Chol 40:0:60 to 20:20:60 mol %, a gelling of the bilayer also occurs. This is characterized by a reduction in the fluid peak in the WAXS data (Figure 7b inset). Cholesterol crystals also become visible from 10 mol % conversion of BSM to BC with the peaks growing in intensity relative to the lamellar phase as the content of ceramide increases. The cholesterol crystals are characterized by peaks centered at 34 and 17 Å (SAXS) and multiple sharp peaks in the wide angle region (WAXS Figure 7b inset). These are in good agreement with previous literature reports characterizing cholesterol crystal structure in isolation and lipid systems.52,59
The gelling of the bilayer with increasing ceramide content indicates a shift from a fluid lamellar phase toward a disrupted ripple phase coexisting with cholesterol crystals increasingly visible in both the SAXS and WAXS data as the mol % of ceramide increases (Figures 7b and S6). Pure SM extracts from milk, brain, and egg have all shown ripple phase formation below 37 °C and a single fluid phase above the Tm.60 However, the ripple phase can be difficult to resolve.60 Recently, the formation of the ripple phase has also been shown in EggSM and egg ceramide (EggCer) mixtures, where the ripple phase was noted up to EggSM:EggCer 80:20 mol % below 37 °C and at atmospheric pressure.58 The incorporation of ceramide caused an increase in the ripple period and thereby a broadening of the ripple. Other research has suggested that there may be some phase coexistence in the gel phase, although distinguishing between a poorly resolved ripple and a gel phase coexistence can be complex.61 In our case, the lack of a strong gel peak in the WAXS implies that the dominant phase is the disrupted ripple rather than coexisting gel phases. However, it is possible that at this lipid composition there is some weak coexistence of Lo with the ripple gel phase, which is not possible to resolve because of the broadening of the ripple gel pattern. The ripple phase we report in the BSM:BC:Chol 20:20:60 mol % mixture is poorly resolved and disrupted, as indicated by the lack of a sharp peak in the WAXS data at 4.2 Å; therefore accurate lattice parameter determination is not possible. We can estimate the lattice parameter by assuming that the angle between the a and b lattice parameters is 90° so that a is given by the d spacing of the 10 peak and is approximately 67.2 ± 0.4 Å.60 The 01 peak is broad, suggesting that the ripple is almost flat, and hence we are unable to estimate the ripple period. The poor resolution of the ripple phase has been reported previously for other SM extracts, where highly resolved diffraction patterns of BSM have only been achieved at 62 °C and 100 MPa.60 The WAXS data are a further corroboration of a single ripple phase, since a fluid–gel coexistence in the BSM:BC:Chol mixture would exhibit two SAXS lattice parameters—unless they were identical—and the gel would be characterized by a sharp peak in the WAXS region, corresponding to the ceramide in a gel state.58,62
These observed changes in the phase behavior of the binary and ternary mixtures agree well with previous reports on the effects of cholesterol and ceramides on SM phase behavior. While Chol lowers the gel–fluid transition temperature in SM mixtures, SMase causes an increase in the ceramide content of the bilayer, leading to an eventual increase in the gel–fluid transition temperature and transition of the bilayer back to a gel state. This is supported by previous DSC data showing that the gel–fluid melting temperatures of mixtures of 18:0 and 24:1 SM with 10 mol % of their respective ceramides showed a series of complex endotherms with increased gel–fluid transitions when compared to SM alone.53,58
Bulk and vesicular measurements are generally accepted to be in good agreement; hence we can infer that the change in phase behavior due to the increase in BC content within the bilayer would alter the behavior and stability of extruded liposomes, leading to potential changes in leakage across the bilayer. NTA measurements of particle concentration quantification and CryoTEM images support this hypothesis. At significant SMase-mediated conversion of BSM to BC, global liposome structural rearrangement and a range of liposome sizes and morphologies are observed by cryoTEM, and NTA shows a significant drop in particle concentration (see Figure 4a and b). Here, a large release of encapsulated cysteine is inevitable.
Detection of SMase Inhibitors
The ability to rapidly and sensitively measure inhibitor efficacy has huge implications in identifying new SMase inhibitors and monitoring in vivo drug concentrations in patients undergoing SMase inhibitor based treatments. In particular, patients being treated for depression are at elevated risk of overdose, and it is crucial to quickly identify such cases so they can be treated in a timely manner. We therefore investigated the usefulness of our optimized assay for detecting small-molecule SMase inhibitors. Three known small-molecule SMase inhibitors, desipramine, cambinol, and altenusin, were incubated at different concentrations with a fixed activity of SMase (1 mU/mL) for 15 min prior to 1 h of incubation with BSM:Chol 40:60 liposomes. We selected this SMase activity since it is the minimum activity required for complete AuNP aggregation under the assay conditions. To quantify the extent of inhibition, we recorded absorbance spectra 15 min after addition of AuNPs and calculated the 640/525 nm absorbance ratio. We observed that all three inhibitors completely inhibited SMase activity at concentrations above 5 μM, whereas we observed no inhibition below around 10 nM concentrations (Figure 8). The relative half-maximal inhibitory concentration (IC50) values that we observed are in line with relative reported inhibitor activities. We obtained the IC50 values by fitting the data to the monophasic dose–response curve:
where x0 is the concentration corresponding to IC50. Notably, the range in which desipramine concentration gives a linear response to SMase activity covers its in vivo concentrations in both therapeutic and toxic concentrations. Unlike the traditional methods, our assay screened the drug candidates much quicker (in 90 min, compared to 3–4 h for conventional assays).
Figure 8.
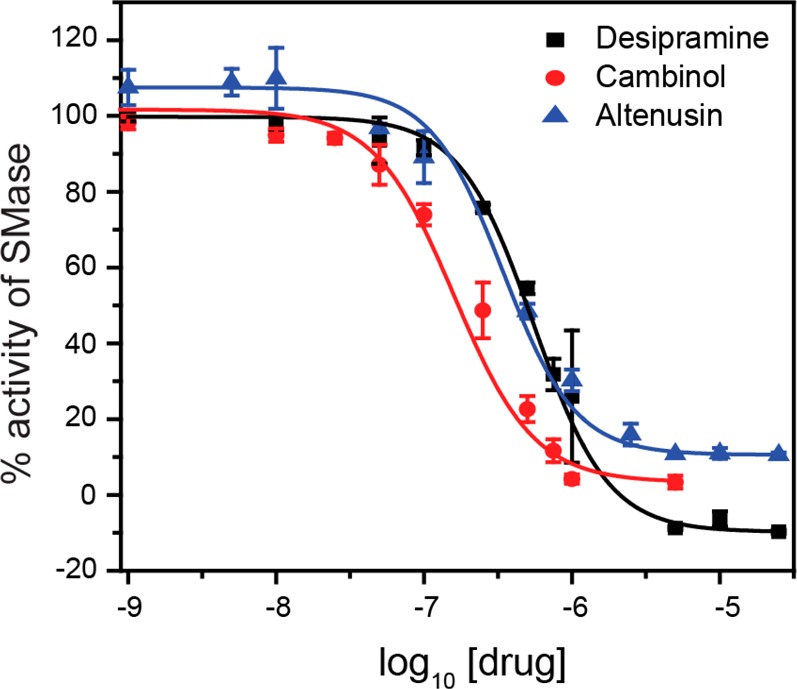
Screening of SMase inhibitors. The inhibitors desipramine, cambinol, and altenusin were screened using the optimized assay conditions using BSM:Chol 40:60 liposomes. The responses are the average of three independent measurements, and the error bars represent the standard deviation.
Conclusions
In summary, we demonstrated here a highly sensitive colorimetric enzyme assay using AuNPs and cysteine-encapsulating liposomes. The use of liposomes allows the investigation of enzymes acting on a biologically relevant substrate, while the use of cysteine to induce aggregation of AuNPs offers a specific and robust transduction mechanism. Using liposomes formulated exclusively from naturally occurring lipids to detect SMase offers a cell-membrane-mimicking environment for the enzymatic activity. Using this assay, we were able to detect SMase enzyme concentrations as low as 1.4 pM at room temperature, which outperforms the commercially available colorimetric assays. Even higher sensitivity could be achieved with an extended incubation time and lower particle concentrations. Notably, the assay provides a rapid change in color tonality as a function of SMase concentration, providing an efficient means of detecting the enzyme in point-of-care settings. We have investigated the underlying mechanism of cargo release using SAXS and WAXS to characterize changes in liposome lipid phase behavior and observed a fluid–gel structural transition below 50% conversion of BSM to ceramide. At this transition, it appears that the liposome membrane becomes significantly more permeable. In addition to determining the SMase activity, we have also shown the capability of this assay to rapidly screen SMase inhibitors using colorimetric responses. These results highlight the advantage of this hybrid approach utilizing two very different nanomaterials to develop colorimetric sensors for membrane-active enzymes. The current assay is amenable to different enzymes requiring appropriate substrates on the liposomal platform. We expect that this system will have applications in both point-of-care diagnostic testing and high-throughput pharmaceutical screening of potential drug candidates.
Materials and Methods
AuNPs
The AuNPs with an average diameter of 13 nm were prepared using the citrate reduction method.63 Briefly, a solution of sodium citrate (10 mL; 38 mM) was added to a rapidly stirred boiling aqueous solution of HAuCl4 (100 mL; 1 mM). After 30 min of reflux, the red mixture was allowed to cool to room temperature. Then, 50 mL of AuNPs was mixed with 2 mL of 1 wt % surfactant Tween-20 to yield well-dispersed AuNPs that were collected by filtering through a 0.45 μm membrane and stored in a refrigerator at 4 °C. The concentration of the prepared AuNP dispersion was determined with UV–vis spectrometry reported previously and found to be 12 nM. It should be noted that the Tween-20 additive stabilizes the individual as well as the aggregated AuNPs. The AuNP stock solution was diluted in DPBS buffer before adding to the assay solutions.
Lipid Films
SM from bovine brain (Avanti Polar Lipids, Alabaster, AL, USA) and cholesterol (Sigma) were dissolved in methanol and chloroform, respectively, at 10 mg/mL concentrations. Solutions of BSM:Chol were mixed in ratios of 70:30, 60:40, 50:50 and 40:60 mol % and aliquoted into 1.75 mL glass vials, with a final concentration of 2 mg (2.7 μmol) BSM per vial. Solvents were evaporated using a stream of nitrogen gas to give a homogeneous lipid film and dried further for 12 h in vacuo, then stored sealed in a nitrogen atmosphere at −20 °C until liposome formulation.
Liposome Formulation
DPBS (1/3×) was prepared by diluting 1 mL of DPBS without magnesium and calcium (Gibco) in 2 mL of pure water (Gibco). Cysteine solutions of 50, 100 or 200 mM were prepared by diluting cysteine hydrochloride (Sigma) in 1/3× DPBS to 8.8, 17.6 or 35.1 mg/mL, respectively. The lipid film was hydrated with 1 mL of this cysteine solution for 30 min and agitated on a vortex shaker for 6 × 10 s, after which the lipid film had fully detached from the vial walls to form a homogeneous lipid suspension. In the case of liposomes formulated from BSM:Chol 40:60 mol % lipid films (BSM:Chol 40:60 liposomes), the 30 min incubation followed by 6 × 10 s vortex shaker agitation was repeated twice before the lipid film was fully detached from the vial sides. The suspension was extruded 31 times through a 200 or 100 nm pore size polycarbonate membrane (Whatman Nuclepore) using a microextruder (Avanti Polar Lipids), affording monodisperse, unilamellar liposomes. The cysteine/DPBS external media was exchanged for DPBS on a Sephadex G50 (medium) column, affording the pure liposome formulation at approximately 0.5 mg/mL SM concentration. Liposome particle concentrations were characterized by NTA measurements. Particle concentration varied very little from batch to batch and was around 2 × 1012 particles/mL.
Assay Protocol
SMase from Bacillus cereus was diluted in DPBS with 1 mM magnesium sulfate and 1 mM calcium chloride to 2× the final assay concentration from a 1:1 DPBS:glycerol 12.5 U/mL stock solution. Liposomes were diluted to 1.7 × 1011 (100 nm diameter) or 3.6 × 1010 (200 nm diameter) particles/mL in DPBS with 1 mM magnesium sulfate and 1 mM calcium chloride. In a 96-well plate, 70 μL of liposome suspension was added to 70 μL of enzyme solutions at varying concentrations and incubated at rt for 1 h. AuNPs (60 μL) were added, and absorption at 525 and 640 nm was measured every minute until 20 min using a SpectraMax M5, at which point absorbance spectra were measured (525–700 nm, Δ 5 nm). SMase from B. cereus was purchased from Sigma and is structurally and mechanistically similar to mammalian neutral SMase.64,65
NMR Measurements
Lipid films were prepared in glass vials with BSM:Chol ratios of 60:40, 50:50, 40:60, and 30:70 mol % and 2 mg total BSM, as described above. These films were hydrated with 1 mL of pure water (Gibco) for 30 min and agitated on a vortex shaker for 6 × 10 s. The incubation/vortex step was repeated three times before complete detachment of the lipid from the vial wall. Lipid suspensions were extruded 31 times through a 100 nm polycarbonate membrane, as described above, lyophilized, and dissolved in 0.5 mL of deuterated chloroform (Sigma). 1H NMR spectra were recorded on a Bruker AV-400 (400 MHz) spectrometer and analyzed using MestReNova v12.0. After automatic phase correction and ablative baseline correction (15 points, 20 passes), the peaks used for analysis were integrated over fixed regions around the peak maxima. Chemical shifts are quoted in ppm on the δ scale, using residual solvent as the internal standard (1H NMR: CDCl3 = 7.26).
SAXS/WAXS Measurements
Brain SM, brain ceramide, and cholesterol were obtained from Avanti Polar Lipids. These are supplied in lyophilized powder form with a purity of >99%, so no further purification was necessary. Lipids were stored at −20 °C and defrosted before use. Mixtures of BSM:Chol at 40:60 mol % and BSM:BC:Chol at 36:4:60, 32:8:60, 28:12:60, and 20:20:60 mol % were codissolved in chloroform, which was then evaporated under a stream of nitrogen. Samples were then lyophilized overnight for a minimum of 12 h to remove any residual solvent. Mixtures were hydrated in pure water (Gibco) w/w to 60 wt %, sealed, and heat cycled between −196 and 60 °C a minimum of 10 times. SAXS/WAXS data were obtained at 25 °C at Diamond Light Source, UK, using beamline I22, with samples mounted in glass capillaries (Capillary Tube Supplies Ltd., SGCT 1.5 mm) at an X-ray wavelength of 0.6902 Å and a sample to detector distance of 3.7 m. Images were analyzed using the AXcess software package. Briefly, the two-dimensional SAXS and WAXS images were radially integrated to give one-dimensional diffraction patterns. The Bragg peaks were then fitted using Gaussian functions and indexed by comparison to characteristic peak spacings from known lipid structures.
Inhibitor Screening
Desipramine, cambinol, and altenusin were dissolved in DMSO, and stock solutions were prepared, followed by serial dilutions. In a 96-well clear microplate, 50 μL of 2.8 mU/mL SMase was incubated with 20 μL of inhibitors at different concentrations at room temperature for 15 min. Then, 70 μL of 1.7 × 1011 particles/mL liposomes was added to the wells and incubated for 1 h at room temperature. Next, 60 μL of AuNPs was added, and the absorbance after 15 min of incubation was recorded. Data fitting was performed in Origin 10 software.
Acknowledgments
M.N.H. acknowledges support from the FP7 Marie Curie Intra-European Fellowship “SMase LIPOSOME” [626766] and the Swiss National Science Foundation [P300PA_171540 and 406240_147493], S.R. from the FP7 Marie Curie International Incoming Fellowship “FunStar Capsule” [629218], H.B. from the H2020 through the Individual Marie Skłodowska-Curie Fellowship “SmartCubes” [703666], and U.K. from the Deutsche Forschungsgemeinschaft [KA 4370/1-1]. M.M.S. and S.R. acknowledge support from the i-sense EPSRC IRC in Early Warning Sensing Systems for Infectious Diseases [EP/K031953/1] and the EPSRC grant “Biofunctionalized nanomaterials for ultrasensitive biosensing” [EP/K020641/1]. M.M.S. acknowledges the ERC Seventh Framework Programme Consolidator grant “Naturale CG” [616417]. H.B. and N.B. acknowledge the “Capitals” EPSRC Programme Grant [EP/J017566/1]. We acknowledge Diamond Light Source for provision of synchrotron beamtime [SM14217 and SM14792], and we would like to thank Dr. Andy Smith for assistance using beamline I22. M.N.H. also thanks Dr. Derrick Roberts and Dr. Christopher Wood for their helpful discussions concerning the NMR data analysis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.8b03308.
Spectroscopic data analysis, cysteine-mediated aggregation, effect of liposome concentration on background stability, stability of liposomes over time, effect of Ca2+/Mg2+ on assay sensitivity, SAXS/WAXS diffraction patterns of all studied compositions, further materials and methods, annotated NMR spectra (PDF). Raw data is available at DOI: 10.5281/zenodo.1293380.
Author Contributions
¶ M. N. Holme and S. Rana contributed equally to the work.
The authors declare no competing financial interest.
Supplementary Material
References
- Slotte J. P.; Ramstedt B. The Functional Role of Sphingomyelin in Cell Membranes. Eur. J. Lipid Sci. Technol. 2007, 109, 977–981. 10.1002/ejlt.200700024. [DOI] [Google Scholar]
- Futerman A. H.; Hannun Y. A. The Complex Life of Simple Sphingolipids. EMBO Rep. 2004, 5, 777–782. 10.1038/sj.embor.7400208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan C. M.; Rawat S. S.; Puri A.; Wang J. M.; Ruscetti F. W.; Blumenthal R. Ceramide, a Target for Antiretroviral Therapy. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 15452–15457. 10.1073/pnas.0402874101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. G.; Junk P.; Huwiler A.; Burkhardt C.; Wallerath T.; Pfeilschifter J.; Forstermann U. Dual Effect of Ceramide on Human Endothelial Cells - Induction of Oxidative Stress and Transcriptional Upregulation of Endothelial Nitric Oxide Synthase. Circulation 2002, 106, 2250–2256. 10.1161/01.CIR.0000035650.05921.50. [DOI] [PubMed] [Google Scholar]
- Pan W.; Yu J.; Shi R.; Yan L.; Yang T.; Li Y.; Zhang Z.; Yu G.; Bai Y.; Schuchman E. H.; He X.; Zhang G. Elevation of Ceramide and Activation of Secretory Acid Sphingomyelinase in Patients with Acute Coronary Syndromes. Coron. Artery Dis. 2014, 25, 230–235. 10.1097/MCA.0000000000000079. [DOI] [PubMed] [Google Scholar]
- Jones I.; He X.; Katouzian F.; Darroch P. I.; Schuchman E. H. Characterization of Common Smpd1Mutations Causing Types a and B Niemann-Pick Disease and Generation of Mutation-Specific Mouse Models. Mol. Genet. Metab. 2008, 95, 152–162. 10.1016/j.ymgme.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth B.; Erdos M.; Szekely A.; Ritli L.; Bagossi P.; Sumegi J.; Marodi L. Molecular Genetic Characterization of Novel Sphingomyelin Phosphodiesterase 1 Mutations Causing Niemann-Pick Disease. JIMD Rep. 2011, 3, 125–129. 10.1007/8904_2011_80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornhuber J.; Rhein C.; Muller C. P.; Muhle C. Secretory Sphingomyelinase in Health and Disease. Biol. Chem. 2015, 396, 707–736. 10.1515/hsz-2015-0109. [DOI] [PubMed] [Google Scholar]
- Muehle C.; Huttner H. B.; Walter S.; Reichel M.; Canneva F.; Lewczuk P.; Gulbins E.; Kornhuber J. Characterization of Acid Sphingomyelinase Activity in Human Cerebrospinal Fluid. PLoS One 2013, 8, e62912. 10.1371/journal.pone.0062912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulbins E.; Palmada M.; Reichel M.; Luth A.; Bohmer C.; Amato D.; Muller C. P.; Tischbirek C. H.; Groemer T. W.; Tabatabai G.; Becker K. A.; Tripal P.; Staedtler S.; Ackermann T. F.; van Brederode J.; Alzheimer C.; Weller M.; Lang U. E.; Kleuser B.; Grassme H.; et al. Acid Sphingomyelinase-Ceramide System Mediates Effects of Antidepressant Drugs. Nat. Med. 2013, 19, 934–938. 10.1038/nm.3214. [DOI] [PubMed] [Google Scholar]
- Figuera-Losada M.; Stathis M.; Dorskind J. M.; Thomas A. G.; Bandaru V. V. R.; Yoo S.-W.; Westwood N. J.; Rogers G. W.; McArthur J. C.; Haughey N. J.; Slusher B. S.; Rojas C. Cambinol, a Novel Inhibitor of Neutral Sphingomyelinase 2 Shows Neuroprotective Properties. PLoS One 2015, 10, e0124481. 10.1371/journal.pone.0124481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M.; Liu K.; Southall N.; Marugan J. J.; Remaley A. T.; Zheng W. A High-Throughput Sphingomyelinase Assay Using Natural Substrate. Anal. Bioanal. Chem. 2012, 404, 407–414. 10.1007/s00216-012-6174-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghomashchi F.; Barcenas M.; Turecek F.; Scott C. R.; Gelb M. H. Reliable Assay of Acid Sphingomyelinase Deficiency with the Mutation Q292k by Tandem Mass Spectrometry. Clin. Chem. 2015, 61, 771–772. 10.1373/clinchem.2014.236448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B.; Hannun Y. A. Sphingomyelinase Assay Using Radiolabeled Substrate. Methods Enzymol. 2000, 311, 164–167. 10.1016/S0076-6879(00)11077-8. [DOI] [PubMed] [Google Scholar]
- He X. X.; Chen F.; Dagan A.; Gatt S.; Schuchman E. H. A Fluorescence-Based, High-Performance Liquid Chromatographic Assay to Determine Acid Sphingomyelinase Activity and Diagnose Types a and B Niemann-Pick Disease. Anal. Biochem. 2003, 314, 116–120. 10.1016/S0003-2697(02)00629-2. [DOI] [PubMed] [Google Scholar]
- Legnini E.; Orsini J. J.; Muehl A.; Johnson B.; Dajnoki A.; Bodamer O. A. Analysis of Acid Sphingomyelinase Activity in Dried Blood Spots Using Tandem Mass Spectrometry. Ann. Lab. Med. 2012, 32, 319–323. 10.3343/alm.2012.32.5.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Arguello M. B.; Veiga M. P.; Arrondo J. L.; Goñi F. M.; Alonso A. Sphingomyelinase Cleavage of Sphingomyelin in Pure and Mixed Lipid Membranes. Influence of the Physical State of the Sphingolipid. Chem. Phys. Lipids 2002, 114, 11–20. 10.1016/S0009-3084(01)00195-5. [DOI] [PubMed] [Google Scholar]
- Contreras F. X.; Sot J.; Ruiz-Arguello M. B.; Alonso A.; Goni F. M. Cholesterol Modulation of Sphingomyelinase Activity at Physiological Temperatures. Chem. Phys. Lipids 2004, 130, 127–134. 10.1016/j.chemphyslip.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Aili D.; Mager M.; Roche D.; Stevens M. M. Hybrid Nanoparticle-Liposome Detection of Phospholipase Activity. Nano Lett. 2011, 11, 1401–1405. 10.1021/nl1024062. [DOI] [PubMed] [Google Scholar]
- Liu N. J.; Chapman R.; Lin Y.; Mmesi J.; Bentham A.; Tyreman M.; Abraham S.; Stevens M. M. Point of Care Testing of Phospholipase A2 Group Iia for Serological Diagnosis of Rheumatoid Arthritis. Nanoscale 2016, 8, 4482–4485. 10.1039/C5NR08423G. [DOI] [PubMed] [Google Scholar]
- Liu N. J.; Chapman R.; Lin Y. Y.; Bentham A.; Tyreman M.; Philips N.; Khan S. A.; Stevens M. M. Phospholipase A2 as a Point of Care Alternative to Serum Amylase and Pancreatic Lipase. Nanoscale 2016, 8, 11834–11839. 10.1039/C6NR03376H. [DOI] [PubMed] [Google Scholar]
- Ewing H.; Fernández-Vega V.; Spicer T. P.; Chase P.; Brown S.; Scampavia L.; Roush W. R.; Riley S.; Rosen H.; Hodder P.; Lambeau G.; Gelb M. H. Fluorometric High-Throughput Screening Assay for Secreted Phospholipases A2 Using Phospholipid Vesicles. J. Biomol. Screening 2016, 21, 713–721. 10.1177/1087057116646742. [DOI] [PubMed] [Google Scholar]
- Bui M.-P. N.; Ahmed S.; Abbas A. Single-Digit Pathogen and Attomolar Detection with the Naked Eye Using Liposome-Amplified Plasmonic Immunoassay. Nano Lett. 2015, 15, 6239–6246. 10.1021/acs.nanolett.5b02837. [DOI] [PubMed] [Google Scholar]
- Silva L. C.; Futerman A. H.; Prieto M. Lipid Raft Composition Modulates Sphingomyelinase Activity and Ceramide-Induced Membrane Physical Alterations. Biophys. J. 2009, 96, 3210–3222. 10.1016/j.bpj.2008.12.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ale E. C.; Maggio B.; Fanani M. L. Ordered-Disordered Domain Coexistence in Ternary Lipid Monolayers Activates Sphingomyelinase by Clearing Ceramide from the Active Phase. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 2767–2776. 10.1016/j.bbamem.2012.06.017. [DOI] [PubMed] [Google Scholar]
- Goñi F. M.; Montes L. R.; Alonso A. Phospholipases C and Sphingomyelinases: Lipids as Substrates and Modulators of Enzyme Activity. Prog. Lipid Res. 2012, 51, 238–266. 10.1016/j.plipres.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Howes P. D.; Chandrawati R.; Stevens M. M. Bionanotechnology. Colloidal Nanoparticles as Advanced Biological Sensors. Science 2014, 346, 1247390. 10.1126/science.1247390. [DOI] [PubMed] [Google Scholar]
- Saha K.; Agasti S. S.; Kim C.; Li X.; Rotello V. M. Gold Nanoparticles in Chemical and Biological Sensing. Chem. Rev. 2012, 112, 2739–2779. 10.1021/cr2001178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aili D.; Gryko P.; Sepulveda B.; Dick J. A. G.; Kirby N.; Heenan R.; Baltzer L.; Liedberg B.; Ryan M. P.; Stevens M. M. Polypeptide Folding-Mediated Tuning of the Optical and Structural Properties of Gold Nanoparticle Assemblies. Nano Lett. 2011, 11, 5564–5573. 10.1021/nl203559s. [DOI] [PubMed] [Google Scholar]
- Ghosh S. K.; Pal T. Interparticle Coupling Effect on the Surface Plasmon Resonance of Gold Nanoparticles: From Theory to Applications. Chem. Rev. 2007, 107, 4797–4862. 10.1021/cr0680282. [DOI] [PubMed] [Google Scholar]
- Su K. H.; Wei Q. H.; Zhang X.; Mock J. J.; Smith D. R.; Schultz S. Interparticle Coupling Effects on Plasmon Resonances of Nanogold Particles. Nano Lett. 2003, 3, 1087–1090. 10.1021/nl034197f. [DOI] [Google Scholar]
- Howes P. D.; Rana S.; Stevens M. M. Plasmonic Nanomaterials for Biodiagnostics. Chem. Soc. Rev. 2014, 43, 3835–3853. 10.1039/C3CS60346F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balogh D.; Zhang Z.; Cecconello A.; Vavra J.; Severa L.; Teply F.; Willner I. Helquat-Induced Chiroselective Aggregation of Au Nps. Nano Lett. 2012, 12, 5835–5839. 10.1021/nl303179s. [DOI] [PubMed] [Google Scholar]
- Jiang Y.; Zhao H.; Zhu N.; Lin Y.; Yu P.; Mao L. A Simple Assay for Direct Colorimetric Visualization of Trinitrotoluene at Picomolar Levels Using Gold Nanoparticles. Angew. Chem., Int. Ed. 2008, 47, 8601–8604. 10.1002/anie.200804066. [DOI] [PubMed] [Google Scholar]
- Liu J.; Mendoza S.; Román E.; Lynn M. J.; Xu R.; Kaifer A. E. Cyclodextrin-Modified Gold Nanospheres. Host–Guest Interactions at Work to Control Colloidal Properties. J. Am. Chem. Soc. 1999, 121, 4304–4305. 10.1021/ja990330n. [DOI] [Google Scholar]
- Shenton W.; Davis S. A.; Mann S. Directed Self-Assembly of Nanoparticles into Macroscopic Materials Using Antibody–Antigen Recognition. Adv. Mater. 1999, 11, 449–452. . [DOI] [Google Scholar]
- He X. X.; Chen F.; McGovern M. M.; Schuchman E. H. A Fluorescence-Based, High-Throughput Sphingomyelin Assay for the Analysis of Niemann-Pick Disease and Other Disorders of Sphingomyelin Metabolism. Anal. Biochem. 2002, 306, 115–123. 10.1006/abio.2002.5686. [DOI] [PubMed] [Google Scholar]
- Ago H.; Oda M.; Takahashi M.; Tsuge H.; Ochi S.; Katunuma M.; Miyano M.; Sakurai J. Structural Basis of the Sphingomyelin Phosphodiesterase Activity in Neutral Sphingomyelinase from Bacillus Cereus. J. Biol. Chem. 2006, 281, 16157–16167. 10.1074/jbc.M601089200. [DOI] [PubMed] [Google Scholar]
- Lu C.-H.; Wang Y.-W.; Ye S.-L.; Chen G.-N.; Yang H.-H. Ultrasensitive Detection of Cu2+ with the Naked Eye and Application in Immunoassays. NPG Asia Mater. 2012, 4, e10. 10.1038/am.2012.18. [DOI] [Google Scholar]
- Xiao Q.; Shang F.; Xu X.; Li Q.; Lu C.; Lin J.-M. Specific Detection of Cysteine and Homocysteine in Biological Fluids by Tuning the Ph Values of Fluorosurfactant-Stabilized Gold Colloidal Solution. Biosens. Bioelectron. 2011, 30, 211–215. 10.1016/j.bios.2011.09.013. [DOI] [PubMed] [Google Scholar]
- Jensen T.; Kelly L.; Lazarides A.; Schatz G. C. Electrodynamics of Noble Metal Nanoparticles and Nanoparticle Clusters. J. Cluster Sci. 1999, 10, 295–317. 10.1023/A:1021977613319. [DOI] [Google Scholar]
- Ghosh S. K.; Pal T. Interparticle Coupling Effect on the Surface Plasmon Resonance of Gold Nanoparticles: From Theory to Applications. Chem. Rev. 2007, 107, 4797–4862. 10.1021/cr0680282. [DOI] [PubMed] [Google Scholar]
- de laRica R.; Fratila R. M.; Szarpak A.; Huskens J.; Velders A. H. Multivalent Nanoparticle Networks as Ultrasensitive Enzyme Sensors. Angew. Chem., Int. Ed. 2011, 50, 5704–5707. 10.1002/anie.201008189. [DOI] [PubMed] [Google Scholar]
- Laromaine A.; Koh L.; Murugesan M.; Ulijn R. V.; Stevens M. M. Protease-Triggered Dispersion of Nanoparticle Assemblies. J. Am. Chem. Soc. 2007, 129, 4156–4157. 10.1021/ja0706504. [DOI] [PubMed] [Google Scholar]
- Iida T.; Tamura T.; Matsumoto T. Proton Nuclear Magnetic Resonance Identification and Discrimination of Side Chain Isomers of Phytosterols Using a Lanthanide Shift Reagent. J. Lipid Res. 1980, 21, 326–338. [PubMed] [Google Scholar]
- Sawan S. P.; James T. L.; Gruenke L. D.; Craig J. C. Proton Nmr Assignments for Cholesterol. Use of Deuterium Nmr as an Assignment Aid. J. Magn. Reson. 1979, 35, 409–413. 10.1016/0022-2364(79)90063-5. [DOI] [Google Scholar]
- Adosraku R. K.; Choi G. T. Y.; Constantinou-Kokotos V.; Anderson M. M.; Gibbons W. A. Nmr Lipid Profiles of Cells, Tissues, and Body Fluids: Proton Nmr Analysis of Human Erythrocyte Lipids. J. Lipid Res. 1994, 35, 1925–1931. [PubMed] [Google Scholar]
- Malz F.; Jancke H. Validation of Quantitative Nmr. J. Pharm. Biomed. Anal. 2005, 38, 813–823. 10.1016/j.jpba.2005.01.043. [DOI] [PubMed] [Google Scholar]
- Tabaei S. R.; Rabe M.; Zetterberg H.; Zhdanov V. P.; Hook F. Single Lipid Vesicle Assay for Characterizing Single-Enzyme Kinetics of Phospholipid Hydrolysis in a Complex Biological Fluid. J. Am. Chem. Soc. 2013, 135, 14151–14158. 10.1021/ja4046313. [DOI] [PubMed] [Google Scholar]
- Urbina P.; Flores-Diaz M.; Alape-Giron A.; Alonso A.; Goni F. M. Effects of Bilayer Composition and Physical Properties on the Phospholipase C and Sphingomyelinase Activities of Clostridium Perfringens Alpha-Toxin. Biochim. Biophys. Acta, Biomembr. 2011, 1808, 279–286. 10.1016/j.bbamem.2010.08.011. [DOI] [PubMed] [Google Scholar]
- Urbina P.; Flores-Diaz M.; Alape-Giron A.; Alonso A.; Goñi F. M. Phospholipase C and Sphingomyelinase Activities of the Clostridium Perfringens Alpha-Toxin. Chem. Phys. Lipids 2009, 159, 51–57. 10.1016/j.chemphyslip.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Gater D. L.; Seddon J. M.; Law R. V. Formation of the Liquid-Ordered Phase in Fully Hydrated Mixtures of Cholesterol and Lysopalmitoylphosphatidylcholine. Soft Matter 2008, 4, 263–267. 10.1039/B710726A. [DOI] [PubMed] [Google Scholar]
- Jimenez-Rojo N.; Garcia-Arribas A. B.; Sot J.; Alonso A.; Goñi F. M. Lipid Bilayers Containing Sphingomyelins and Ceramides of Varying N-Acyl Lengths: A Glimpse into Sphingolipid Complexity. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 456–464. 10.1016/j.bbamem.2013.10.010. [DOI] [PubMed] [Google Scholar]
- Quinn P. J.; Wolf C. Thermotropic and Structural Evaluation of the Interaction of Natural Sphingomyelins with Cholesterol. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 1877–1889. 10.1016/j.bbamem.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Oldfield E.; Chapman D. Effects of Cholesterol and Cholesterol Derivatives on Hydrocarbon Chain Mobility in Lipids. Biochem. Biophys. Res. Commun. 1971, 43, 610–616. 10.1016/0006-291X(71)90658-9. [DOI] [PubMed] [Google Scholar]
- Maulik P. R.; Shipley G. G. N-Palmitoyl Sphingomyelin Bilayers: Structure and Interactions with Cholesterol and Dipalmitoylphosphatidylcholine. Biochemistry 1996, 35, 8025–8034. 10.1021/bi9528356. [DOI] [PubMed] [Google Scholar]
- Contreras F. X.; Sot J.; Ruiz-Argüello M. B.; Alonso A.; Goñi F. M. Cholesterol Modulation of Sphingomyelinase Activity at Physiological Temperatures. Chem. Phys. Lipids 2004, 130, 127–134. 10.1016/j.chemphyslip.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Barriga H. M. G.; Parsons E. S.; McCarthy N. L. C.; Ces O.; Seddon J. M.; Law R. V.; Brooks N. J. Pressure–Temperature Phase Behavior of Mixtures of Natural Sphingomyelin and Ceramide Extracts. Langmuir 2015, 31, 3678–3686. 10.1021/la504935c. [DOI] [PubMed] [Google Scholar]
- Craven B. M. Crystal Structure of Cholesterol Monohydrate. Nature 1976, 260, 727–729. 10.1038/260727a0. [DOI] [PubMed] [Google Scholar]
- Shaw K. P.; Brooks N. J.; Clarke J. A.; Ces O.; Seddon J. M.; Law R. V. Pressure-Temperature Phase Behaviour of Natural Sphingomyelin Extracts. Soft Matter 2012, 8, 1070–1078. 10.1039/C1SM06703F. [DOI] [Google Scholar]
- Quinn P. J.; Wolf C. Hydrocarbon Chains Dominate Coupling and Phase Coexistence in Bilayers of Natural Phosphatidylcholines and Sphingomyelins. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 1126–1137. 10.1016/j.bbamem.2008.12.011. [DOI] [PubMed] [Google Scholar]
- Shah J.; Atienza J. M.; Duclos R. I. Jr; Rawlings A. V.; Dong Z.; Shipley G. G. Structural and Thermotropic Properties of Synthetic C16:0 (Palmitoyl) Ceramide: Effect of Hydration. J. Lipid Res. 1995, 36, 1936–1944. [PubMed] [Google Scholar]
- Wang F.; Liu X.; Lu C.-H.; Willner I. Cysteine-Mediated Aggregation of Au Nanoparticles: The Development of a H2o2 Sensor and Oxidase-Based Biosensors. ACS Nano 2013, 7, 7278–7286. 10.1021/nn402810x. [DOI] [PubMed] [Google Scholar]
- Goñi F. M.; Alonso A. Sphingomyelinases: Enzymology and Membrane Activity. FEBS Lett. 2002, 531, 38–46. 10.1016/S0014-5793(02)03482-8. [DOI] [PubMed] [Google Scholar]
- Hofmann K.; Tomiuk S.; Wolff G.; Stoffel W. Cloning and Characterization of the Mammalian Brain-Specific, Mg(2+)-Dependent Neutral Sphingomyelinase. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 5895–5900. 10.1073/pnas.97.11.5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.