

Abstract
Nonmuscle myosin-IIA (NMHC-IIA) heavy chain phosphorylation has gained recognition as an important feature of myosin-II regulation. In previous work, we showed that phosphorylation on S1943 promotes myosin-IIA filament disassembly in vitro and enhances EGF-stimulated lamellipod extension of breast tumor cells. However, the contribution of NMHC-IIA S1943 phosphorylation to the modulation of invasive cellular behavior and metastasis has not been examined. Stable expression of phosphomimetic (S1943E) or non-phosphorylatable (S1943A) NMHC-IIA in breast cancer cells revealed that S1943 phosphorylation enhances invadopodia function, and is critical for matrix degradation in vitro and experimental metastasis in vivo. These studies demonstrate a novel link between NMHC-IIA S1943 phosphorylation, the regulation of extracellular matrix degradation and tumor cell invasion and metastasis.
Keywords: Myosin-II, Phosphorylation, Invasion, Matrix degradation, Invadopodia
1. Introduction
Heightened cell motility in tumor cells is a key phenotypic change that contributes to invasion and metastasis. Within three-dimensional environments, tumor cells exhibit a variety of migratory behaviors, including both mesenchymal and amoeboid migration [1]. Notably, interconversion between these modalities is an important mediator of metastatic spread, as cancer cells must penetrate a variety of extracellular matrix and tissue barriers and traverse endothelial layers in order to disseminate to distant sites. The ability of tumor cells to overcome these migratory barriers requires precise tuning of the actomyosin cytoskeleton and myosin-dependent cellular contractility [2–6]. However, the molecular mechanisms modulating myosin-II function during these processes are not well defined.
Vertebrates express three nonmuscle myosin heavy chain genes; Myh9 (NMHC-IIA), Myh10 (NMHC-IIB) and Myh14 (NMHC-IIC) [7]. Each heavy chain consists of a globular N–terminal motor domain; a neck domain that binds an essential light chain (ELC) and a regulatory light chain (RLC); a coiled-coil rod domain; and a short C-terminal segment, termed the tailpiece. The heavy chains form a homodimer, which in a complex with two ELCs and two RLCs, is termed the myosin- II monomer. The three myosin-II isoforms exhibit different actin-activated MgATPase activities and duty ratios [8–12] and distinct patterns of tissue/cell expression [13,14], and they have non-redundant as well as overlapping functional roles in vivo [10,15].
Recent studies with nonmuscle myosin-II suggest that irrespective of RLC phosphorylation, folded myosin-II monomers assemble into antiparallel folded dimers and tetramers that unfold and polymerize into filaments [16]. Notably, RLC phosphorylation is thought to weaken interactions between the RLC and the folded myosin-II tail, which facilitates unfolding of the compact 10S structure and polymerization into filaments [16]. Whereas RLC phosphorylation promotes the assembly of myosin-II into filaments, phosphorylation of the myosin-II coiled-coil and C-terminal tailpiece promotes filament disassembly. Multiple kinases phosphorylate the coiled-coil and tailpiece sites in vitro, including the transient receptor potential melastatin 7 (TRPM7), members of the protein kinase C (PKC) family and casein kinase 2 (CK2) [17]. In particular, phosphorylation on S1943 of the NMHC-IIA C-terminal tailpiece has been shown to regulate myosin-IIA filament assembly and localization in vivo [18,19]. Moreover, NMHC-IIA S1943 phosphorylation is upregulated during TGF-p-mediated epithelial-mesenchymal transition in mammary epithelial cells [20], and substitution of S1943 with alanine attenuates the invasion of breast tumor cells into a collagen gel, at least in part via the stabilization of cellular protrusions [21]. In addition, NMHC-IIA S1943 phosphorylation is associated with invadopodia formation on gelatin high density fibrillar collagen [22]. Together these observations suggest that phosphorylation on NMHC-IIA S1943 is critical for 3D invasion.
To further examine the role of NMHC-IIA S1943 phosphorylation in regulating the invasive properties of tumor cells, we produced breast cancer cells that stably express wild-type, phosphomimetic (S1943E) or non-phosphorylatable (S1943A) NMHC-IIA. Using these cell lines, we now demonstrate that S1943 phosphorylation is critical for invadopodia maturation, the secretion of matrix metalloproteinases, and matrix degradation, all of which are required for tumor metastasis. These data suggest that NMHC-IIA S1943 phosphorylation contributes to tumor cell invasion and metastasis via the regulation of extracellular matrix degradation.
2. Materials and methods
2.1. Myosin-IIA constructs
A pcDNA3.1 construct encoding the full-length mouse nonmuscle myosin-IIA heavy chain with an N-terminal Flag tag was a gift from Dr. Anna Savoia (University of Trieste, Trieste, Italy) [23]. A DNA fragment encoding full length mouse nonmuscle myosin-IIA heavy chain (residues 1–1960) was subcloned in frame into the Kpnl and Xbal sites of pEGFP-C3 (Clontech, Palo Alto, CA) and will be hereafter referred to as green fluorescent protein (GFP)-NMHC-IIA. Using the Quick Change XL site-directed mutagenesis kit (Stratagene, La Jolla, CA), S1943 was substituted with alanine or glutamic acid in the full-length GFP-NMHC- IIA. All constructs were confirmed by DNA sequencing. Human GFP- tagged wild-type and S1943A NMHC-IIA constructs were prepared as described previously [18].
2.2. Cell culture
MDA-MB-231, MDA-MB-157, MDA-MB-468, and MCF-7 cells were obtained from the American Type Culture Collection. MDA-MB-361 and T47D cells were a gift from Dr. Paraic Kenny (Kabara Cancer Research Laboratory, Gundersen Medical Foundation). Cells were maintained as monolayer cultures in DMEM containing 10% FBS at 37 °C with 5% C02. MCF7 lines were supplemented with 10 μg/ml insulin. HEK-293T and mouse mammary E0771 cells were grown in DMEM containing 10% FBS and RPMI containing 10% FBS and 10 mM HEPES, respectively. S100A4−/− bone marrow-derived macrophages (BMMs) were maintained as described previously [24].
2.3. Antibodies and reagents
For invadopodia assays, the FISH (Tks5) antibody was obtained from Santa Cruz, and the cortactin antibody was obtained from Millipore. For immunoblotting, the human NMHC-IIA and NMHC-IIB C- terminal antibodies were produced in-house [25], and the NMHC-IIA pS1943 antibodies were produced in collaboration with Millipore and Cell Signaling Technology. The NMHC-IIA 2B3 monoclonal antibody from Abeam was used to recognize the exogenously expressed mouse GFP-NMHC-IIA. The β-actin and vinculin antibodies were purchased from Sigma. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylte- trazolium bromide) was obtained from Invitrogen. Calf intestine phosphatase (CIP) and lambda protein phosphatase (lambda PP) were obtained from New England Biolabs. shRNAs against the human NMHC- IIA were obtained from Open Biosystems (sh5: clone TRC 0000029465, 5’CGCATCAACTTTGATGTCAAT3’ and sh7: clone TRC0000029467, 5’CCGCGAAGTCAGCTCCCTAAA3’). The control shRNA (shC) was obtained from Sigma (MISSION® pLKO.l-puro non-target shR NA control plasmid). Recombinant human proMMP9 was from R&D Systems (cat #911-MP-010) and recombinant human MMP2 was from Calbiochem (cat #PF023).
2.4. Cell Line construction
MDA-MB-231 NMHC-IIA knockdown cells were generated with a lentivirus expressing shRNA against human NMHC-IIA (Open Biosystems clone TRC0000029467), and infected cells were selected using 2 μg/ml puromycin. Loss of NMHC-IIA expression in stable knockdown lines was confirmed by immunoblotting. Stable knockdown cells were transfected with mouse wild-type, S1943A, or S1943E GFP- NMHC-IIA using Lipofectamine 2000 (Life Technologies), and selected using 800 μg/ml G418 (Fisher). G418-resistant cells were sorted by FACS to enrich for GFP-NMHC-IIA cells and clonal lines were selected on the basis of robust expression of GFP-NMHC-IIA and correct localization to stress fibers.
2.5. Myosin-IIA rod phosphorylation
Myosin-IIA rod constructs (NMHC-IIA1338−1960) were bacterially expressed and purified as described previously [26]. The myosin-IIA rods were phosphorylated with PKCα or CK2 as described previously [27]. Briefly, myosin-IIA rods were preincubated with 140 nM PKCα (Invitrogen) in a buffer containing 100 μg/ml phosphatidylserine, 20 μg/ml 1,2-dioleoyl-sn-glycerol (Avanti Polar Lipids), 20 mM Tris-HCl (pH 7.5), 200 mM NaCl, 0.5 mM DTT, 2mM ATP, and phosphatase inhibitor cocktails (Sigma) at room temperature for 20 min. Phosphorylation was initiated by the addition of MgCl2 and CaCl2 to concentrations of 5 mM and 0.3 mM, respectively, and the reactions were allowed to proceed for approximately 2 h at 30 °C. Alternatively, myosin-IIA rods were incubated with 5000 units of CK2 (New England BioLabs) in 20 mM Tris-HCl (pH 7.5), 200 mM NaCl, 0.2 mM DTT, 0.5 mM ATP, 10 mM MgCl2, and phosphatase inhibitor cocktails as described above, at 30 °C for 2 h.
2.6. Immunoblots and characterization of pS1943 NMHC-IIA antibodies
Millipore pS1943 NMHC-IIA antibody: Cells were lysed on ice in 2X modified Sample Buffer (4% SDS, 124 mM Tris, 20% glycerol, 5mM DTT, 2 mM EDTA, pH 6.8) and then boiled. Lysate concentrations were determined using the BioRad DC Assay. Thirty micrograms of protein from whole cell lysates and 0.5 μg purified and phosphorylated myosin- IIA rods [27] were separated on 8% SDS-PAGE gels. After transferring to nitrocellulose membranes, membranes were treated with calf intestinal phosphatase (CIP), followed by treatment with lambda phosphatase for 1 h at room temperature. Alternatively, membranes were treated with buffer under identical conditions. The membranes were then simultaneously immunoblotted with a β-actin monoclonal antibody and either the pS1943 myosin-IIA rabbit antibody from Millipore or an antibody that recognizes total myosin-IIA. The immunoreactive proteins were detected using a LiCor Odyssey v.3.0.
Whole cell lysates from the panel of breast cancer cell lines were prepared by lysing 1 × 106 cells in 100 μl of 2X Laemmli Sample Buffer on ice, boiling and running equal volumes on an 8% SDS-PAGE gel.
Cell Signaling Technology pS1943 NMHC-IIA antibody: Cell lysates were prepared as described above. Mouse lung tissue was homogenized in 2X modified Sample Buffer (described above) in an Eppendorf tube on ice and boiled. Protein concentration was determined using the BioRad DC Assay. Thirty micrograms of protein from whole cell lysates from parental HEK-293T cells, HEK-293T cells transfected with human GFP-tagged wild-type or S1943A NMHC-IIA constructs, E0771 cells, S100A4−/− BMMs, and mouse lung tissue were separated on parallel 6% SDS-PAGE gels and transferred to nitrocellulose membranes. The membranes were incubated with antibodies against vinculin, NMHC-IIA pS1943 (Cell Signaling Technology) and total NMHC-IIA (2B3 monoclonal from Abeam). The immunoreactive proteins were detected using a LiCor Odyssey v.3.0.
Unstained slides of sections from paraffin embedded lungs and tumors from MDA-MB-231–3475 orthotopic xenografts in SCID mice were a gift from Dr. Rachel Hazan (Albert Einstein College of Medicine). The slides were immunostained with an antibody against either NMHC-IIA pS1943 (1:100 Cell Signaling antibody) or total NMHC-IIA (10 μg/ml, in-house antibody) using 3,3’-diaminobenzidine (DAB) detection and counterstained with hematoxylin. The slides were scanned using a Perkin Elmer P250 High Capacity Slide Scanner.
2.7. Proliferation assays
2D cell proliferation was evaluated using the Vibrant MTT Cell Proliferation Assay Kit (Thermo Fisher) in accordance with the manufacturer’s protocol. Briefly, 2 × 103 cells per well were plated in a 96- well plate. Fresh DMEM, 10% FBS was added and 1.2 mM MTT was add to each well. After three hours, the cells were lysed in 0.01 M HC1, 0.1% SDS, incubated for an additional 4 h and the absorbance at 570 nm was measured. Three independent experiments were performed in duplicate and cell proliferation was evaluated at 24, 48, 72, and 96 h after plating. The rate of proliferation was derived from the slopes of plots of absorbance versus time.
For 3D cell proliferation assays, cells expressing wild-type or mutant NMHC-MIIA were trypsinized, counted, centrifuged for 5 min at 600 x g, and resuspended in ice cold Matrige:fetal bovine serum (3:1 vol:vol) at 15,000 cells/50 μl. 50 μl of the Matrigel-cell mixture for each cell line was plated in triplicate in poly-HEMA-coated white opaque 96-well dishes on ice. The plates were incubated at 37 °C for 30 min, and 100 μl of complete media per well was added. The cells were incubated for 0–3 days, with a media change on Day 2. At each time point, 50 μl of media was removed, and cells were assayed by the addition of 100 μl Cell-Titer Glo 3D reagent (Promega). The samples were mixed on a rotary shaker at 200 rpm for 5 min, and luminescence was read after 30 min on a SpectraMax M3 plate reader (500 m s acquisition time).
2.8. Gelatin degradation and invadopodia maturation assays
MDA-MB-231 lines were plated on glass coverslips coated with Oregon Green 488-conjugated gelatin, as previously described [28]. Briefly, coverslips were washed with 0.2 N HC1 and sterilized with 70% ethanol prior to treating with 50 μg/ml poly-L-lysine (Gibco) for 10 min. The coated coverslips were treated with 0.5% glutaraldehyde for 10 min, washed with PBS and then coated with 200 μg/ml Oregon Green 488-conjugated gelatin (Life Technologies) for 15 min. Finally, the coverslips were treated with 0.1 M glycine for 10 min. 4 × 104 tumor cells were plated on the coverslips in DMEM containing 10% FBS and incubated for 7 or 17 h. Cells were fixed in 3.7% formaldehyde in PBS and stained with either rhodamine phalloidin (Molecular Probes) or co-immunostained with FISH (Tks5) and cortactin antibodies. Cells were imaged with a PlanApo 60X, 1.4 NA objective (Olympus), and at least 50 cells per condition per experiment were examined. Cells were analyzed using ImageJ software to determine areas of degradation and co-localization of Tks5 and cortactin. Mature invadopodia were defined by the co-localization of Tks5 and cortactin with gelatin degradation. Precursor invadopodia were defined by the co-localization of Tks5 and cortactin in regions without gelatin degradation.
2.9. Gelatin zymography
To prepare conditioned media from MDA-MB-231 lines, 60 mm tissue culture dishes were plated with 1.1 × 106 tumor cells, which were allowed to attach overnight in DMEM containing 10% FBS. The cells were washed with PBS, 2 ml of serum-free DMEM was added and the cells were incubated for 17 h. Conditioned medium was collected, filtered with a 0.22 μm Millex-GV filter unit and concentrated 15-fold with an Amicon Ultra Centrifugal Filter (3000 Da molecular weight cut off). Samples were diluted in non-reducing sample buffer and run on a 7% Tris-Tricine gel containing 0.1% gelatin for 2h (15 min at 100 V, 105 min at 130 V). Gels were then washed twice for 30 min each in 2.5% Triton-XlOO, and incubated overnight at 37°C in developing buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 5mM CaCl2, 0.02% NaN3, and 5 μM ZnCl2.) Gels were Coomassie stained for one hour, and then destained until areas of enzyme activity appeared.
2.10. MMP9 activity assay
MMP9 activity in conditioned media collected from MDA-MB-231 cells (as described above) was determined using the MMP9 Biotrak Activity Assay (GE Healthcare Life Sciences) following the manufacturers protocol. To measure total MMP9 activity (pro and active MMP9), 1 mM p-aminophenylmercuric acetate (APMA) was added to the samples. A MMP9 standard curve (0.0625–20 ng/ml) was used to determine the concentration of active MMP9 in the conditioned media.
2.11. Experimental metastasis assay
All experiments were performed according to protocols approved by the Animal Welfare Committee at the Albert Einstein College of Medicine. MDA-MB-231 cells (5 × 105 cells/100 μl per mouse) were injected into the lateral tail vein of 8-week old female SCID mice. After six or eight weeks, the mice were euthanized. The lungs were excised, fixed in 10% buffered formalin, paraffin embedded, sectioned, and stained with hematoxylin and eosin or immunostained with a human- specific pan-cytokeratin antibody (4.5 μg/ml, Leica cat #AE1/AE-L-CE) using DAB detection. The slides were scanned using a Perkin Elmer P250 High Capacity Slide Scanner. The metastatic lesions in lungs 6 weeks after cell injection were analyzed by tracing the lungs and me- tastases using 3D Histech CaseViewer 1.4 software. The metastatic lesions in lungs 8 weeks after cell injection were analyzed using image analysis software developed for DAB immunohistochemistry [29].
3. Results
3.1. Detection of NMHC-IIA S1943 phosphorylation in breast cancer cells
Using [32P]-orthophosphate labeling approaches, our previous studies identified S1943 as a site of NMHC-IIA phosphorylation in MDA- MB-231 cells [18]. To determine if S1943 phosphorylation is observed in other breast carcinoma cells, we first confirmed the specificity and selectivity of available pS1943 antibodies. We used shRNAs directed against NMHC-IIA, which do not affect the expression of NMHC-IIB (data not shown); we previously showed that MDA-MB-231 cells do not express NMHC-IIC [24]. We observed that loss of the pS1943 NMHC-IIA signal in whole cell lysates paralleled the loss in total NMHC-IIA expression. This is consistent with the finding that approximately 45–60% of total NMHC-IIA is phosphorylated on S1943 in MDA-MB-231 cells [18]. Treatment with calf intestinal and lambda phosphatases completely eliminated the pS1943 signal, but did not affect the detection of total NMHC-IIA (Fig. 1A). To further confirm specificity of the antibody for pS1943, we stoichiometrically phosphorylated purified myosin-IIA rod constructs with either PKC or CK2, which phosphorylate S1916 and S1943, respectively [27]. The pS1943 antibody reacted only with CK2- phosphorylated myosin-IIA rods, but not with unphosphorylated or PKC-phosphorylated rods, and phosphatase treatment resulted in a complete loss of pS1943 antibody reactivity (Fig. 1B). A second pS1943 NMHC-IIA antibody recognized both endogenous NMHC-IIA from HEK- 293T cells and exogenously expressed GFP-tagged human wild-type NMHC-IIA, but not GFP-tagged S1943A NMHC-IIA (Supplemental Fig. 1). Notably, the second pS1943 NMHC-IIA antibody did not react with mouse NMHC-IIA. Altogether, these data demonstrate that pS1943 antibodies specifically detect pS1943 NMHC-IIA.
Fig. 1.
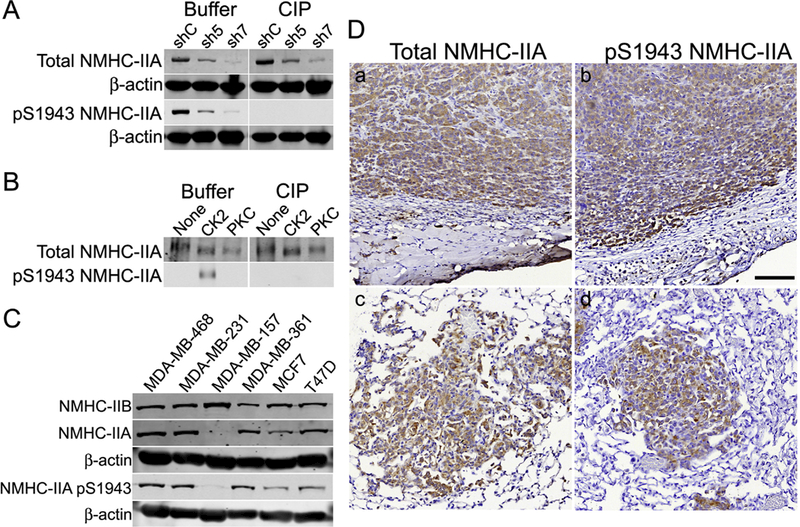
Characterization of pS1943 NMHC-IIA antibodies. (A) MDA-MB-231 cells were transduced with lentivirus expressing control (shC) or shRNAs (sh5 and sh7) against human NMHC-IIA. Buffer- or calf intestinal and lambda phosphatase (ClP)-treated immunoblots were blotted for total NMHC-IIA, pS1943 NMHC-IIA (Millipore antibody) or β-actin. (B) Buffer- or CIP-treated immunoblots (total and pS1943 NMHC-IIA - Millipore antibody) of unphosphorylated, S1943-phos- phorylated (CK2) and S1916-phosphorylated (PKC) myosin-IIA rods. (C) Representative immunoblots showing expression of total NMHC-IIA, NMHC-IIB, pS1943 NMHC-IIA (Millipore antibody) or β-actin in a panel of breast cancer cell lines. (D) Immunohistochemistry of MDA-MB-231–3475 orthotopic tumors (a, b) and spontaneous lung metastatic nodules (c, d), showing expression of total NMHC-IIA or pS1943 NMHC-IIA (Cell Signaling Technology antibody), which appears as a brown stain. Scale bar = 100 μm.
We examined pS1943 NMHC-IIA expression in hormone receptor positive and triple negative (estrogen receptor, progesterone receptor and Her2 negative) breast cancer cell lines. In all cell lines expressing myosin-IIA, we observed S1943 phosphorylation (Fig. 1C). To evaluate NMHC-IIA phosphorylation in vivo, we compared total and pS1943 NMHC-IIA staining in tumors and spontaneous lung metastases derived from orthotopic xenografts of MDA-MB-231 3475, a lung tropic MDA- MB-231 sublime [30]. Immunohistochemistry of formalin-fixed, paraffin-embedded material revealed robust pS1943 staining throughout the primary tumor and in the metastatic lesions (Fig. 1D).
3.2. NMHC-IIA phosphorylation regulates invadopodia-mediated gelatin degradation
To test the contribution of S1943 phosphorylation to the invasive properties of breast tumor cells, we first generated MDA-MB-231 cells in which endogenous NMHC-IIA was partially knocked down by transduction with lentivirus expressing an shRNA targeting human NMHC-IIA. Since proteomic studies have shown that NMHC-IIA constitutes 82% of the total myosin-II heavy chain in MDA-MB-231 cells [31], and complete loss of NMHC-IIA could lead to defects in cytokinesis, we expressly chose an shRNA that produced a partial knockdown of NMHC-IIA expression. Stable NHMC-IIA knockdown cells were then transfected with cDNA constructs encoding GFP-tagged murine wild- type, S1943A or S1943E NMHC-IIA. The combined expression of the exogenous murine GFP-NMHC-IIA constructs and endogenous NMHC- IIA yielded total NMHC-IIA levels that were comparable to parental MDA-MB-231cells (Fig. 2). The levels of S1943 phosphorylation in the residual endogenous NMHC-IIA were similar in wild-type, S1943A and S1943E NMHC-IIA cell lines (Supplemental Fig. 2). Thus, rescue of the knockdown by expression of mutant NMHC-IIA resulted in a net decrease in S1943 phosphorylation for the S1943A mutant, and an effective increase in S1943 phosphorylation for the phosphomimetic S1943E mutant.
Fig. 2.
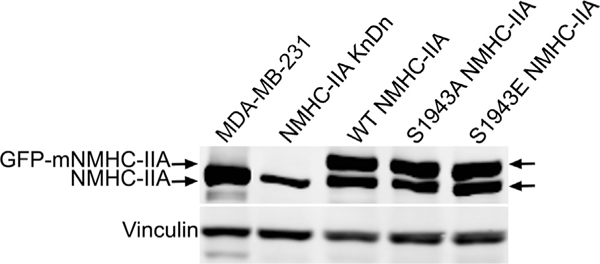
Development of NMHC-IIA knockdown-replace cell lines. Representative immunoblots showing expression of endogenous NMHC-IIA, murine GFP-tagged NMHC-IIA and vinculin in parental MDA-MB-231 cells and stable NMHC-IIA knockdown cells.
Previous studies have shown that the matrix-degrading activity of invadopodia is regulated via a myosin-II-FAK-Cas pathway [3], and a recent phosphoproteomic study identified NMHC-IIA S1943 phosphorylation in MDA-MB-231 cells under conditions of invadopodia induction [22]. To investigate the regulatory role of S1943 phosphorylation in invadopodia function, we assayed the ability of cells expressing wild- type or mutant NMHC-IIA to degrade matrix. Areas of degradation were defined by the colocalization of dark spots in the fluorescent gelatin with punctate F-actin structures (Fig. 3A). Seventeen hours after plating on a gelatin substrate, approximately 90% of cells expressing wild-type or S1943E NMHC-IIA had degraded the matrix, as compared to 39% of S1943A NMHC-IIA expressing cells (Fig. 3B). Moreover, the total de- gradative area under cells expressing S1943A NMHC-IIA was reduced by 14-fold and 19-fold relative to cells expressing wild-type or S1943E NMHC-IIA, respectively (Fig. 3C). In these assays we also detected a trend toward increased degradation area in cells expressing S1943E NMHC-IIA as opposed to wild-type NMHC-IIA. While this difference was not statistically significant, degradation under a cell can plateau after long plating times. To determine if cells expressing S1943E NMHC-IIA had increased degradative capacity, we measured matrix degradation seven hours after plating on a gelatin substrate. While there was no statistically significant difference in the percent of cells exhibiting matrix degradation (Fig. 4A), we observed a 5.8-fold increase in the total degradative area under S1943E NMHC-IIA expressing cells as compared to cells expressing wild-type NMHC-IIA. These data support a role for NMHC-IIA S1943 phosphorylation as a positive regulator of matrix degradation.
Fig. 3.
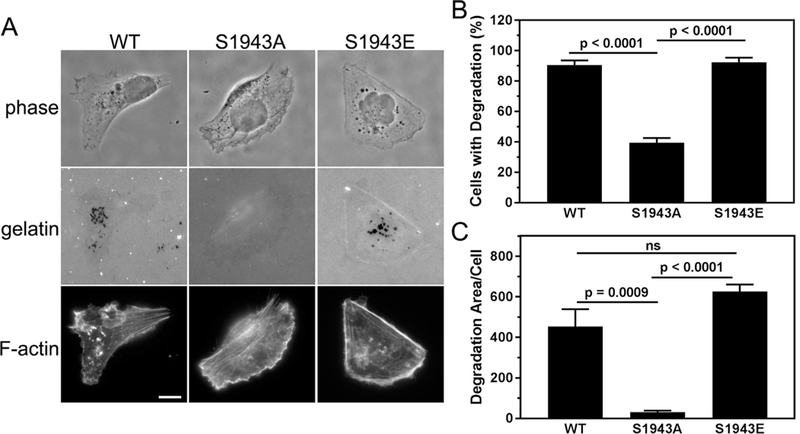
Cells deficient for S1943 NMHC-IIA phosphorylation exhibit defects in matrix degradation. (A) Representative images of MDA-MB-231 cells expressing wild- type, S1943A or S1943E NMHC-IIA that were plated on Oregon Green 488-gelatin for 17 h. Areas of degradation appear as dark spots that colocalize with punctate F- actin structures. Bar = 10 μm. (B) Percentage of cells that degrade the gelatin and (C) the average degradation area per cell (pixels/cell). Data represent the mean ± SEM from 4 independent experiments, > 50 cells per condition per experiment. Statistical analyses were performed using ANOVA.
Fig. 4.
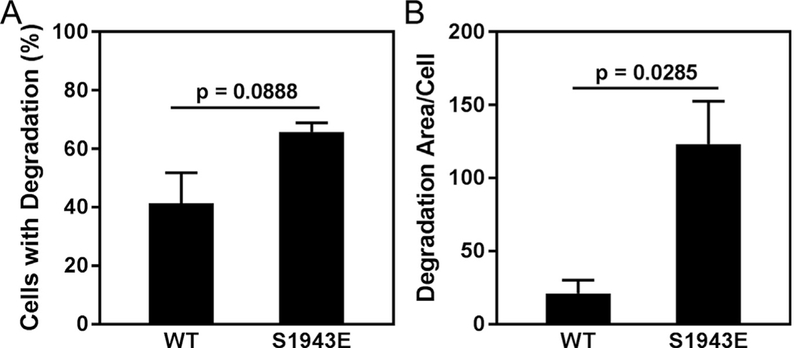
S1943 NMHC-IIA phosphorylation enhances matrix degradation. MDA-MB-231 cells expressing wild-type or S1943E NMHC-IIA were plated on Oregon Green 488- gelatin for 7 h and then evaluated for matrix degradation. (A) Percentage of cells that degrade the gelatin, and (B) the average degradation area per cell (pixels/cell). Data represent the mean ± SEM from 3 independent experiments, > 50 cells per condition per experiment. Statistical analyses were performed using a Student’s t-test.
Invadopodia form in a step-wise manner. Invadopodium precursors, which are characterized as actin/cortactin/Tks5 punctae, progress into mature invadopodia that are defined by actin/cortactin/Tks5 punctae that co-localize with areas of matrix degradation [32,33]. Studies by Weaver and colleagues have shown that despite the attenuation of matrix degradation in cells treated with blebbistatin or inhibitors of kinases that mediate RLC phosphorylation, cortactin and F-actin-posi- tive punctae persist [3]. These data suggest that inhibition of myosin-II motor function negatively affects invadopodium maturation but not precursor formation. We found that MDA-MB-231 cells expressing either wild-type or mutant NMHC-IIA had similar numbers of invadopodium precursors (colocalization of cortactin and Tks5 in punctae in the absence of matrix degradation; Fig. 5A, rectangles and Fig. 5B) [33]. However, when we measured the colocalization of cortactin and Tks5 with sites of gelatin degradation (mature, degradation-competent invadopodia; Fig. 5A, squares), we observed an approximately 8-fold decrease in the number of mature invadopodia in cells expressing S1943A NMHC-IIA (Fig. 5C).
Fig. 5.
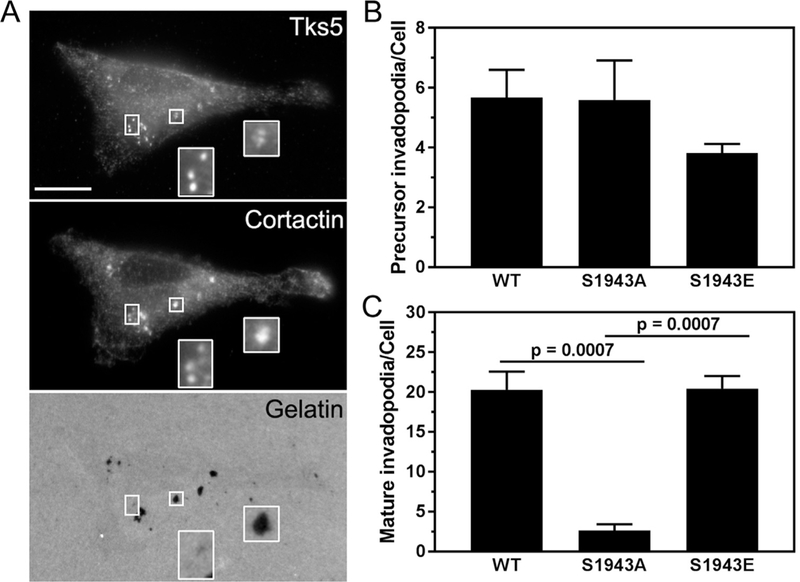
Cells deficient for S1943 NMHC-IIA phosphorylation form fewer mature invadopodia. (A) Representative images of MDA- MB-231 cells expressing wild-type, S1943A or S1943E NMHC-IIA that were plated on Oregon Green 488-gelatin for 17 h and stained for the invadopodia markers Tks5 and cortactin. Bar = 10 pm. Insets show magnified images of invadopodium precursors (rectangles) and mature invadopodia (squares). (B and C) Quantification of the number of (B) invadopodium precursors and (C) mature invadopodia per cell. Data represent the mean ± SEM from 3 independent experiments, ≥ 50 cells per condition per experiment. Statistical analyses were performed using ANOVA.
Maturation is characterized by elongation of the invadopodium followed by MMP recruitment and ECM degradation [34,35]. Given the alterations we observed in invadopodium maturation in cells expressing S1943A NMHC-IIA, we examined MMP secretion. Gelatin zymography of conditioned medium from the three cell lines revealed a MMP9 band that co-migrated with the proMMP9 standard. The zymogram showed reduced proMMP9 secretion by cells expressing S1943A NMHC-IIA and elevated proMMP9 secretion by cells expressing S1943E NMHC-IIA, as compared to cells expressing wild type NMHC-IIA (Fig. 6A); the active form of MMP9 was not detected. In contrast, we observed similar levels of proMMP2 in the three cell lines (Fig. 6A). As an alternative method to assess the levels of total and active MMP9 activity secreted by the cells, we used a MMP9 activity assay that monitors MMP9 activity through cleavage of a chromogenic peptide substrate [36]. Active MMP9 in the conditioned medium is measured directly, and total MMP9 (active and pro) is determined after treatment with p-amino- phenylmercuric acetate (APMA), which activates proMMP9. There was a 2-fold increase in total MMP9 in the medium from S1943E NMHC-IIA cells as compared to wild-type NMHC-IIA cells, whereas there was 4- fold less MMP9 in the medium from S1943A NMHC-IIA cells (Fig. 6B). Consistent with the zymography experiments, the level of active MMP9 in conditioned medium was approximately 10-fold less than total MMP9. Nonetheless, we observed a trend toward increased levels of active MMP9 in the conditioned medium from cells expressing S1943E NMHC-IIA as compared to cells expressing wild-type or S1943A NMHC- IIA (Fig. 6C). These data indicate that the effects of NMHC-IIA S1943 phosphorylation on invadopodium maturation correlate with altered MMP9 secretion.
Fig. 6.
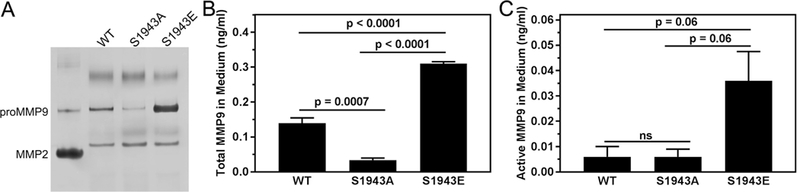
S1943 NMHC-IIA phosphorylation regulates MMP9 secretion. (A) Gelatin zymography of conditioned medium from MDA-MB-231 cells expressing wild-type, S1943A or S1943E NMHC-IIA. Standards are human proMMP9 and active MMP2. (B and C) Biotrak MMP9 activity assay of (B) total MMP9 and (C) active MMP9 in conditioned medium from MDA-MB-231 cells expressing wild-type, S1943A or S1943E NMHC-IIA. Data represent the mean ± SEM from 3 independent experiments. Statistical analyses were performed using ANOVA.
3.3. NMHC-IIA phosphorylation is required for experimental lung metastasis
Given that NMHC-IIA phosphorylation regulates the formation of mature, degradation-competent invadopodia in vitro, we wanted to evaluate the effects of NMHC-IIA phosphorylation on extravasation in an experimental metastasis assay. To visualize metastatic lesions, IHC on lung sections was performed with a human-specific pan-cytokeratin antibody that showed little reactivity with naive SCID lung (Supplemental Fig. 3). The lungs from mice injected with cells expressing wild-type or S1943A NMHC-IIA had metastatic nodules throughout the lung parenchyma (Fig. 7A, a and b - arrowheads, Supplemental Fig. 4). However, the lungs from mice injected with S1943E NMHC-IIA cells showed metastatic foci primarily localized along the pleura with occasional small nests of cells in the parenchyma (Fig. 7A, c - arrowheads, Supplemental Fig. 4). Six weeks after injection, there was a 2.5–4.7-fold increase in the number of lung metastases in mice injected with S1943E NMHC-IIA expressing cells as compared to cells expressing wild-type or S1943A NMHC-IIA (Fig. 7B). In addition, the total metastatic area per lung was 3.8–18-fold greater for cells expressing S1943E NMHC-IIA relative to wild-type and S1943A NMHC-IIA cells, respectively (Fig. 7B). However, there was no statistically significant difference in the number or size of metastases produced by S1943A NMHC- IIA expressing cells as compared to wild-type NMHC-IIA expressing cells.
Fig. 7.
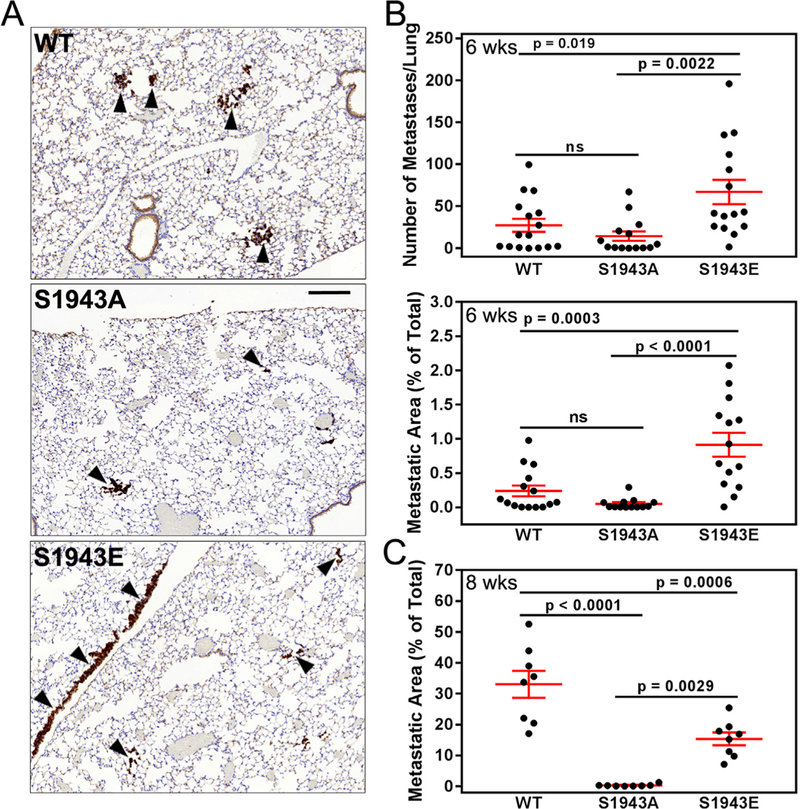
S1943 NMHC-IIA phosphorylation regulates extravasation in vivo. (A) Pan-cytoker- atin stained lung sections from SCID mice injected with MDA-MB-231 cells expressing wild- type, S1943A or S1943E NMHC-IIA six weeks after injection of tumor cells. Arrowheads indicate metastatic foci. Bar = 200 μm. (B) The number and percent area of metastases per lung section six weeks after injection of tumor cells. Data represent the mean ± SEM from two independent experiments with a total of 15, 13, and 14 mice for wild-type, SI943A, or S1943E NMHC-IIA cells, respectively. Statistical analyses were performed using ANOVA. (C) The percent area of metastases per lung section eight weeks after injection of tumor cells. Data represent the mean ± SEM from 8 mice each for wild-type, S1943A or S1943E NMHC-IIA cells, respectively. Statistical analyses were performed using ANOVA.
To amplify potential differences between the metastatic capabilities of wild-type and S1943A NMHC-IIA cells, we examined the lungs of animals eight weeks after tumor cell injection. These studies revealed a 100-fold reduction in the metastasis area produced by S1943A NMHC- IIA expressing cells as compared to wild type NMHC-IIA cells (Fig. 7C). The reduction in metastatic foci for S1943A NMHC-IIA expressing cells was not due to alterations in cell proliferation, as in vitro 3D proliferation assays indicated that S1943A and S1943E NMHC-IIA expressing cells have similar proliferative capacities (Supplemental Figs. 5A and B). Moreover, given the very significant defects in invadopodia maturation observed for S1943A NMHC-IIA expressing cells in vitro, reduced extravasation and tumor cell seeding in vivo likely account for the reduced number of metastatic nodules. Surprisingly, at 8 weeks the metastatic area produced by cells expressing S1943E NMHC-IIA was less than that produced by wild-type NMHC-IIA cells, although still 46-fold more than S1943A NMHC-IIA cells (Fig. 7C). This may reflect the propensity of the S1943E NMHC-IIA tumors to grow along the pleural surfaces (Supplemental Fig. 6), which may constrain tumor invasion into the lung parenchyma.
4. Discussion
Phosphoproteomic and biochemical studies have documented phosphorylation on SI943 of the NMHC-IIA in a number of cancer cell types (e.g., breast, cervical and glioblastoma) [18,20,37–39]. Cellular studies have shown that SI943 phosphorylation 1) increases the turnover of myosin-IIA filaments [40], 2) mediates the recruitment of myosin-IIA to the lamella during cell spreading and 3) enhances EGF-stimulated lamellipod extension [18,19,21]. More recent studies indicate that myosin-IIA SI943 phosphorylation changes during durotaxis of mesenchymal stem cells [40]. In soft collagen gels, myosin-IIA shows high levels of S1943 phosphorylation and is diffusely localized, and a NMHC-IIA phosphomimetic (S1943D) exhibits increased mobility (as measured by fluorescence recovery after photobleaching) and increased solubility [40]. In stiff 3D collagen gels, myosin-IIA is organized into stress fibers and shows reduced levels of SI943 phosphorylation. Moreover, invasion into stiff gels is inhibited by a phosphomimetic S1943D NMHC-IIA. In contrast, work by Egelhoff and colleagues has shown that in breast tumor cells, NMHC-IIA phosphorylation is critical for stabilizing protrusions as well as for cellular invasion into stiff, but not soft collagen gels [21]. While both of these studies support a role for NMHC-IIA phosphorylation in sensing ECM stiffness, they also suggest that the regulation of cellular behavior by NMHC-IIA S1943 phosphorylation depends on the cell type as well as the unique environments they encounter. Importantly, the latter studies suggest that S1943 phosphorylation could be an important regulatory feature that allows tumor cells to efficiently navigate the dense ECM within the tumor microenvironment. Consistent with this idea, invadopodia activity has been shown to depend on matrix rigidity [3,41], and our data links S1943 phosphorylation to invadopodium maturation.
To address how myosin-IIA S1943 phosphorylation regulates the invasive capacity of breast tumor cells, we established MDA-MB-231 cell lines in which we knocked down endogenous NMHC-IIA and replaced it with exogenously expressed GFP-tagged wild-type or mutant NMHC-IIA. Our previous in vitro biochemical studies showed that substitution of S1943 with either an aspartic or a glutamic acid inhibits salt-dependent myosin-IIA assembly in a comparable manner to S1943 phosphorylation [18,27]. However, while phosphorylation on S1943 inhibits the binding of S100A4 [27], a calcium-dependent regulator of myosin-IIA assembly [42], substitution of S1943 with aspartic/glutamic acid (or alanine) does not affect S100A4 binding [18]. This means that charge replacement of S1943 does not mimic all aspects of heavy chain phosphorylation at this site. As a consequence, studies with cells expressing S1943E NMHC-IIA do not reflect changes in the interaction of S100A4 with myosin-IIA that would occur upon S1943 phosphorylation.
Our previous studies demonstrated that 45–60% of endogenous NMHC-IIA is phosphorylated on S1943 [18]. Given that our MDA-MB- 231 cells express S1943E NMHC-IIA at 50–60% of endogenous NMHC- IIA levels (Fig. 2), the expression level of phosphomimetic NMHC-IIA is comparable to endogenous levels of pS1943-NMHC-IIA in wild-type cells. However, the residual endogenous NMHC-IIA in the S1943E NMHC-IIA cells can still be phosphorylated, so the net effective level of NMHC-IIA phosphorylation (S1943E plus pS1943-NMHC-IIA) will be increased as compared to cells expressing wild-type NMHC-IIA. Furthermore, as discussed above, the S1943E mutant can still bind S100A4, which might further promote filament disassembly. As a consequence, the behavior of cells expressing the S1943E mutant is likely due to a combination of this effective increase in NMHC-IIA phosphorylation, plus the binding of S100A4 to both S1943E and residual unpho- sphorylated endogenous NMHC-IIA, which all lead to increases in myosin-IIA filament disassembly.
Mutation of S1943 profoundly affected the degradative capacity of breast tumor cells, as S1943A NMHC-IIA expression severely limited the formation of mature, degradation competent invadopodia. While S1943E NMHC-IIA expression did not appear to impact invadopodium maturation, it nonetheless increased the degradative capacity of breast tumor cells. Myosin-II does not localize to invadopodia, but rather has been reported to be enriched in ring-like structures that surround invadopodia in breast carcinoma cells [3]. A similar localization of myosin-II has been observed in podosomes [43–45], matrix degrading structures that are predominantly found in cells of monocytic lineage. Although myosin-IIA is not a component of invadopodia or podosomes, actomyosin contractility regulates the activity of both structures [3,44]. In particular, myosin-IIA-mediated contractility controls the oscillatory behavior of the podosome core and thus podosome protrusive activity [44,46]. While the regulation of myosin-II activity in podosomes and invadopodia has been attributed to the modulation of RLC phosphorylation [3,46], our studies clearly demonstrate that myosin-IIA heavy chain phosphorylation is an important modulator of myosin-IIA function in invadopodia.
Our studies also reveal changes in MMP9 secretion and activity in cells expressing myosin-IIA S1943 mutants, but not by MMP2. Overexpression of MMP9 is sufficient to promote invasion in ovarian tumor cells [47], and MMP9 overexpression is correlated with poor prognoses in breast cancer patients [48]. In addition, shRNA-mediated knockdown of MMP9 in MDA-MB-231 cells attenuates in vitro invasion and blocks spontaneous metastasis to the lung in vivo [49]. Studies in other cell types suggest that MMP2 and MMP9 reside in distinct vesicle populations [50,51]. Furthermore, a siRNA screen of Rab family members, which are important regulators of membrane transport, primarily identified candidate proteins that effected secretion of MMP2 or MMP9, but not both [35]. Together, these observations suggest that the molecular machinery responsible for mediating the subcellular transport of MMPs could be different for MMP2 and MMP9 containing vesicles.
Cells expressing S1943E NMHC-IIA exhibited elevated MMP9 secretion, whereas those expressing S1943A NMHC-IIA showed reduced MMP9 secretion. While a reduction in MMP9 secretion in cells expressing the S1943A mutant could be due to defects in invadopodium assembly, invadopodium maturation was not enhanced by the S1943E mutants. Thus, the increased MMP9 secretion observed in S1943E NMHC-IIA cells suggests a more direct role for S1943 phosphorylation in the transport and/or delivery of MMP vesicles to invadopodia. Myosin-IIA has been identified as a Rab6 effector [52,53], and has been reported to mediate vesicle transport as well as vesicle shedding, including MMP-containing vesicles [54–56]. While myosin-IIA filaments are generally considered the functional cellular form of myosin-IIA, the identification of activated myosin-IIA monomers in cells (i.e. RLC- phosphorylated with an extended conformation) suggests that this monomeric species could have unique cellular functions, such as vesicular trafficking [57]. Additionally, in macrophages, myosin-IIA has been reported to directly bind the microtubule motor KIF1C [58], a mediator of vesicle transport. We have been unable to detect a KIF1C/ myosin-IIA complex in MDA-MB-231 cells (unpublished observations). Nonetheless, the association of myosin-IIA with other transport complexes, and the regulation of this interaction by S1943 phosphorylation, could provide a mechanism for the modulation of vesicle transport and/ or release.
With respect to filamentous myosin-IIA, studies on purified soluble, single-headed myosin-IIA demonstrated that the motor has a low duty ratio, and is therefore not expected to move processively [8]. However, recent work suggests that when co-polymerized with myosin-IIB, or under high viscosity conditions that mimic the cytosol, myosin-IIA filaments move processively along F-actin [59]. Moreover, studies showing that ADP release from the myosin-IIA motor is only modestly affected by resistive loads suggest that this motor is more suited for rapid, short-lived contractile events such as moving cargo [60,61]. Increased myosin-IIA filament dynamics due to S1943 phosphorylation could therefore enhance vesicle trafficking. Conversely, a reduction in net pS1943-NMHC-IIA in the S1943A NMHC-IIA cells, and the resultant increase in myosin-IIA assembly [18,19], could limit filament dynamics required for trafficking of MMP containing vesicles.
Loss of S1943 phosphorylation in S1943A NMHC-IIA cells led to decreases in experimental metastasis, whereas the phosphomimetic S1943E NMHC-IIA mutant led to increased experimental metastasis at 6 weeks, but not at 8 weeks. While the reason for this difference is not apparent, it may be due to the selective growth of the S1943E NMHC- IIA tumors along the pleural surfaces of the lung lobes. It is possible that by 8 weeks, a requirement for these pleural surfaces becomes limiting for tumor spread. Thus, the S1943E NMHC-IIA tumors might establish themselves more rapidly that wild-type NMHC-IIA tumors, leading to increased tumor volume at 6 weeks, but show a more limited ability to invade the lung parenchyma that is manifested at 8 weeks.
Recently, actomyosin contractility has been recognized as an important regulator of multiple steps in the metastatic cascade [5], and we now show that NMHC-IIA S1943 phosphorylation is a critical enhancer of matrix degradation and tumor metastasis. This suggests that inhibition of the kinase that phosphorylates this site could be an effective strategy for limiting myosin-dependent contractility and interfering with myosin-dependent events in metastasis. While the kinase responsible for mediating S1943 phosphorylation in vivo has yet to be established [17], this pathway and its contribution to multiple steps in the metastatic cascade bears further investigation.
5. Conclusions
This study shows that phosphorylation on S1943 on the NMHC-IIA enhances invadopodia function, and is critical for matrix degradation in vitro and experimental metastasis in vivo. This work has revealed a novel link between NMHC-IIA S1943 phosphorylation, the regulation of extracellular matrix degradation, and tumor cell invasion and metastasis.
Supplementary Material
Acknowledgements
We thank members of the Bresnick and Backer labs for helpful discussions, and Dr. Rachel Hazan for providing sections from paraffin embedded lungs and tumors from MDA-MB-231–3475 orthotopic xenografts.
Funding
This work was supported by NIH grants P01 CA100324 (A.R. Bresnick, J.M. Backer, J.S. Condeelis, L.E. Norwood Toro and Y. Wang) and R01 GM119524 (A.R. Bresnick, J.M. Backer), the Animal Barrier and Histotechnology and Comparative Pathology Core of the Einstein Cancer Center (P30 CA013330) and shared instrumentation NIH grant 1S100D019961–01.
Abbreviations:
- NMHC-IIA
nonmuscle myosin-IIA heavy chain
- ELC
essential light chain
- RLC
regulatory light chain
- TRPM7
transient receptor potential melastatin 7
- PKC
protein kinase C
- CK2
casein kinase 2
- CIP
calf intestinal phosphatase
- MMP9
matrix metalloproteinase 9
- IHC
immunohistochemistry
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at http://dx.doi.Org/10.1016/j.yexcr.2018.06.028.
References
- [1].Te Boekhorst V, Preziosi L, Friedl P, Plasticity of cell migration in vivo and in silico, Annu. Rev. Cell Dev. Biol. 32 (2016) 491–526. [DOI] [PubMed] [Google Scholar]
- [2].Wyckoff JB, Pinner SE, Gschmeissner S, Condeelis JS, Sahai E, ROCK- and myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo, Curr. Biol. 16 (2006) 1515–1523. [DOI] [PubMed] [Google Scholar]
- [3].Alexander NR, Branch KM, Parekh A, Clark ES, Iwueke IC, Guelcher SA, Weaver AM, Extracellular matrix rigidity promotes invadopodia activity, Curr. Biol. 18 (2008) 1295–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Doyle AD, Wang FW, Matsumoto K, Yamada KM, One-dimensional topography underlies three-dimensional fibrillar cell migration, J. Cell Biol. 184 (2009) 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rodriguez-Hernandez I, Cantelli G, Bruce F, Sanz-Moreno V, Rho ROCK and actomyosin contractility in metastasis as drug targets, F1000Research 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Doyle AD, Carvajal N, Jin A, Matsumoto K, Yamada KM, Local 3D matrix microenvironment regulates cell migration through spatiotemporal dynamics of contractility-dependent adhesions, Nat. Commun. 6 (2015) 8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Conti MA, Adelstein RS, Nonmuscle myosin II moves in new directions, J. Cell Sci. 121 (2008) 11–18. [DOI] [PubMed] [Google Scholar]
- [8].Kovacs M, Wang F, Hu A, Zhang Y, Sellers JR, Functional divergence of human cytoplasmic myosin II: kinetic characterization of the non-muscle IIA isoform, J. Biol. Chem. 278 (2003) 38132–38140. [DOI] [PubMed] [Google Scholar]
- [9].Rosenfeld SS, Xing J, Chen LQ, Sweeney HL, Myosin IIB is unconventionally conventional, J. Biol. Chem. 278 (2003) 27449–27455. [DOI] [PubMed] [Google Scholar]
- [10].Heissler SM, Manstein DJ, Nonmuscle myosin-2: mix and match, Cell Mol. Life Sci. (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Heissler SM, Manstein DJ, Comparative kinetic and functional characterization of the motor domains of human nonmuscle myosin-2C isoforms, J. Biol. Chem. 286 (2011) 21191–21202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chinthalapudi K, Heissler SM, Preller M, Sellers JR, Manstein DJ, Mechanistic insights into the active site and allosteric communication pathways in human nonmuscle myosin-2C, Elife 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kawamoto S, Adelstein RS, Chicken nonmuscle myosin heavy chains: differential expression of two mRNAs and evidence for two different polypeptides, J. Cell Biol. 112 (1991) 915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Golomb E, Ma X, Jana SS, Preston YA, Kawamoto S, Shoham NG, Goldin E, Conti MA, Sellers JR, Adelstein RS, Identification and characterization of nonmuscle myosin II-C, a new member of the myosin II family, J. Biol. Chem. 279 (2004) 2800–2808. [DOI] [PubMed] [Google Scholar]
- [15].Ma X, Adelstein RS, The role of vertebrate nonmuscle Myosin II in development and human disease, Bioarchitecture 4 (2014) 88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Billington N, Wang A, Mao J, Adelstein RS, Sellers JR, Characterization of three full-length human nonmuscle myosin II paralogs, J. Biol. Chem. 288 (2013) 33398–33410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dulyaninova NG, Bresnick AR, The heavy chain has its day: regulation of myosin- II assembly, Bioarchitecture 3 (2013) 77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dulyaninova NG, House RP, Betapudi V, Bresnick AR, Myosin-IIA heavy-chain phosphorylation regulates the motility of MDA-MB-231 carcinoma cells, Mol. Biol. Cell 18 (2007) 3144–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Breckenridge MT, Dulyaninova NG, Egelhoff TT, Multiple regulatory steps control mammalian nonmuscle myosin II assembly in live cells, Mol. Biol. Cell 20 (2009) 338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Beach JR, Hussey GS, Miller TE, Chaudhury A, Patel P, Monslow J, Zheng Q, Keri RA, Reizes O, Bresnick AR, Howe PH, Egelhoff TT, Myosin II isoform switching mediates invasiveness after TGF-beta-induced epithelial-mesenchymal transition, Proc. Natl. Acad. Sci. USA 108 (2011) 17991–17996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rai V, Thomas DG, Beach JR, Egelhoff TT, Myosin IIA Heavy Chain, Phosphorylation mediates adhesion maturation and protrusion in three dimensions, J. Biol. Chem. 292 (2017) 3099–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Artym VV, Swatkoski S, Matsumoto K, Campbell CB, Petrie RJ, Dimitriadis EK, Li X, Mueller SC, Bugge TH, Gucek M, Yamada KM, Dense fibrillar collagen is a potent inducer of invadopodia via a specific signaling network, J. Cell Biol. 208 (2015) 331–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].D’Apolito M, Guarnieri V, Boncristiano M, Zelante L, Savoia A, Cloning of the murine non-muscle myosin heavy chain IIA gene ortholog of human MYH9 responsible for May-Hegglin, Sebastian, Fechtner, and Epstein syndromes, Gene 286 (2002) 215–222. [DOI] [PubMed] [Google Scholar]
- [24].Li ZH, Dulyaninova NG, House RP, Almo SC, Bresnick AR, S100A4 regulates macrophage chemotaxis, Mol. Biol. Cell 21 (2010) 2598–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li ZH, Bresnick AR, The S100A4 metastasis factor regulates cellular motility via a direct interaction with myosin-IIA, Cancer Res. 66 (2006) 5173–5180. [DOI] [PubMed] [Google Scholar]
- [26].Dulyaninova NG, Hite KM, Zencheck WD, Scudiero DA, Almo SC, Shoemaker RH, Bresnick AR, Cysteine 81 is critical for the interaction of S100A4 and myosin-IIA, Biochemistry 50 (2011) 7218–7227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dulyaninova NG, Malashkevich VN, Almo SC, Bresnick AR, Regulation of myosin-IIA assembly and Mtsl binding by heavy chain phosphorylation, Biochemistry 44 (2005) 6867–6876. [DOI] [PubMed] [Google Scholar]
- [28].Starnes TW, Cortesio CL, Huttenlocher A, Imaging podosome dynamics and matrix degradation, Methods Mol. Biol. 769 (2011) 111–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Law AMK, Yin JXM, Castillo L, Young AIJ, Piggin C, Rogers S, Caldon CE, Burgess A, Millar EKA, O’Toole SA, Gallego-Ortega D, Ormandy CJ, Oakes SR, Andy’s algorithms: new automated digital image analysis pipelines for FIJI, Sci. Rep. 7 (2017) 15717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J, Genes that mediate breast cancer metastasis to lung, Nature 436 (2005) 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Betapudi V, Gokulrangan G, Chance MR, Egelhoff TT, A proteomic study of myosin II motor proteins during tumor cell migration, J. Mol. Biol. 407 (2011) 673–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Beaty BT, Sharma VP, Bravo-Cordero JJ, Simpson MA, Eddy RJ, Koleske AJ, Condeelis J, Betal integrin regulates Arg to promote invadopodial maturation and matrix degradation, Mol. Biol. Cell 24 (2013) 1661–1675 (S1661–1611). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Oser M, Yamaguchi H, Mader CC, Bravo-Cordero JJ, Arias M, Chen X, Desmarais V, van Rheenen J, Koleske AJ, Condeelis J, Cortactin regulates cofilin and N-WASp activities to control the stages of invadopodium assembly and maturation, J. Cell Biol. 186 (2009) 571–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Clark ES, Whigham AS, Yarbrough WG, Weaver AM, Cortactin is an essential regulator of matrix metalloproteinase secretion and extracellular matrix degradation in invadopodia, Cancer Res. 67 (2007) 4227–4235. [DOI] [PubMed] [Google Scholar]
- [35].Jacob A, Jing J, Lee J, Schedin P, Gilbert SM, Peden AA, Junutula JR, Prekeris R, Rab40b regulates trafficking of MMP2 and MMP9 during invadopodia formation and invasion of breast cancer cells, J. Cell Sci. 126 (2013) 4647–4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Verheijen JH, Nieuwenbroek NM, Beekman B, Hanemaaijer R, Verspaget HW, Ronday K, Bakker AH, Modified proenzymes as artificial substrates for proteolytic enzymes: colorimetric assay of bacterial collagenase and matrix metalloproteinase activity using modified pro-urokinase, Biochem J. 323 (Pt 3) (1997) 603–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M, Global, in vivo, and site-specific phosphorylation dynamics in signaling networks, Cell 127 (2006) 635–648. [DOI] [PubMed] [Google Scholar]
- [38].Kozuka-Hata H, Nasu-Nishimura Y, Koyama-Nasu R, Ao-Kondo H, Tsumoto K, Akiyama T, Oyama M, Phosphoproteome of human glioblastoma initiating cells reveals novel signaling regulators encoded by the transcriptome, PLoS One 7 (2012) e43398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ivkovic S, Beadle C, Noticewala S, Massey SC, Swanson KR, Toro LN, Bresnick AR, Canoll P, Rosenfeld SS, Direct inhibition of myosin II effectively blocks glioma invasion in the presence of multiple motogens, Mol. Biol. Cell 23 (2012) 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Raab M, Swift J, Dingal PC, Shah P, Shin JW, Discher DE, Crawling from soft to stiff matrix polarizes the cytoskeleton and phosphoregulates myosin-II heavy chain, J. Cell Biol. 199 (2012) 669–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Parekh A, Ruppender NS, Branch KM, Sewell-Loftin MK, Lin J, Boyer PD, Candiello JE, Merryman WD, Guelcher SA, Weaver AM, Sensing and modulation of invadopodia across a wide range of rigidities, Biophys. J. 100 (2011) 573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Li ZH, Spektor A, Varlamova O, Bresnick AR, Mtsl regulates the assembly of nonmuscle myosin-IIA, Biochemistry 42 (2003) 14258–14266. [DOI] [PubMed] [Google Scholar]
- [43].Rhuwania R, Cornfine S, Fang Z, Kruger M, Luna EJ, Linder S, Supervillin couples myosin-dependent contractility to podosomes and enables their turnover, J. Cell Sci. 125 (2012) 2300–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].van den Dries K, Meddens MR, de Keijzer S, Shekhar S, Subramaniam V, Figdor CG, Cambi A, Interplay between myosin IIA-mediated contractility and actin network integrity orchestrates podosome composition and oscillations, Nat. Commun. 4 (2013) 1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gawden-Rone C, Zhou Z, King E, Prescott A, Watts C, Lucocq J, Dendritic cell podosomes are protrusive and invade the extracellular matrix using metalloproteinase MMP-14, J. Cell Sci. 123 (2010) 1427–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Labernadie A, Rouissou A, Delobelle P, Ralor S, Voituriez R, Proag A, Fourquaux I, Thibault C, Vieu C, Poincloux R, Charriere GM, Maridonneau- Parini I, Protrusion force microscopy reveals oscillatory force generation and me- chanosensing activity of human macrophage podosomes, Nat. Commun. 5 (2014) 5343. [DOI] [PubMed] [Google Scholar]
- [47].Cowden Dahl KD, Symowicz J, Ning Y, Gutierrez E, Fishman DA, Adley RP, Stack MS, Hudson LG, Matrix metalloproteinase 9 is a mediator of epidermal growth factor-dependent e-cadherin loss in ovarian carcinoma cells, Cancer Res. 68 (2008) 4606–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ren F, Tang R, Zhang X, Madushi WM, Luo D, Dang Y, Li Z, Wei K, Chen G, Overexpression of MMP family members functions as prognostic biomarker for breast cancer patients: a systematic review and meta-analysis, PLoS One 10 (2015) e0135544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mehner C, Hockla A, Miller E, Ran S, Radisky DC, Radisky ES, Tumor cell- produced matrix metalloproteinase 9 (MMP-9) drives malignant progression and metastasis of basal-like triple negative breast cancer, Oncotarget 5 (2014) 2736–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Schnaeker EM, Ossig R, Ludwig T, Dreier R, Oberleithner H, Wilhelmi M, Schneider SW, Microtubule-dependent matrix metalloproteinase-2/matrix me- talloproteinase-9 exocytosis: prerequisite inhuman melanoma cell invasion, Cancer Res. 64 (2004) 8924–8931. [DOI] [PubMed] [Google Scholar]
- [51].Sbai O, Ould-Yahoui A, Ferhat L, Gueye Y, Bernard A, Charrat E, Mehanna A, Risso HJ, Chauvin JP, Fenouillet E, Rivera S, Khrestchatisky M, Differential vesicular distribution and trafficking of MMP-2, MMP-9, and their inhibitors in astrocytes, Glia 58 (2010) 344–366. [DOI] [PubMed] [Google Scholar]
- [52].Miserey-Lenkei S, Chalancon G, Rardin S, Formstecher E, Goud R, Echard A, Rab and actomyosin-dependent fission of transport vesicles at the Golgi complex, Nat. Cell Biol. 12 (2010) 645–654. [DOI] [PubMed] [Google Scholar]
- [53].Majeed W, Liu S, Storrie B, Distinct sets of Rab6 effectors contribute to ZWIO-and COG-dependent Golgi homeostasis, Traffic 15 (2014) 630–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wakana Y, van Galen J, Meissner F, Scarpa M, Polishchuk RS, Mann M, Malhotra V, A new class of carriers that transport selective cargo from the trans Golgi network to the cell surface, EMBO J. 31 (2012) 3976–3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lee CP, Chiang SL, Ko AM, Liu YF, Ma C, Lu CY, Huang CM, Chang JG, Kuo TM, Chen CL, Tsai EM, Ko YC, ALPK1 phosphorylates myosin IIA modulating TNF-alpha trafficking in gout flares, Sci. Rep. 6 (2016) 25740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Schlienger S, Campbell S, Claing A, ARF1 regulates the Rho/MLC pathway to control EGF-dependent breast cancer cell invasion, Mol. Biol. Cell 25 (2014) 17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Shutova MS, Spessott WA, Giraudo CG, Svitkina T, Endogenous species of mammalian nonmuscle myosin IIA and HR include activated monomers and heteropolymers, Curr. Biol. 24 (2014) 1958–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kopp P, Lammers R, Aepfelbacher M, Woehlke G, Rudel T, Machuy N, Steffen W, Linder S, The kinesin KIF1C and microtubule plus ends regulate podosome dynamics in macrophages, Mol. Biol. Cell 17 (2006) 2811–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Pathan-Chhatbar S, Taft MH, Reindl T, Hundt N, Latham SL, Manstein DJ, Three mammalian tropomyosin isoforms have different regulatory effects on nonmuscle myosin-28 and filamentous beta-actin in vitro, J. Biol. Chem. 293 (2018) 863–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kovacs M, Thirumurugan K, Knight PJ, Sellers JR, Load-dependent mechanism of nonmuscle myosin 2, Proc. Natl. Acad. Sci. USA 104 (2007) 9994–9999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Melli L, Billington N, Sun SA, Bird JE, Nagy A, Friedman TB, Takagi Y, Sellers JR, Bipolar filaments of human nonmuscle myosin 2-A and 2-B have distinct motile and mechanical properties, Elife 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.