

Abstract
Current strategies for predicting carcinogenic mode of action for non-genotoxic chemicals are based on identification of early key events in toxicity pathways. The goal of this study was to evaluate short-term key event indicators resulting from exposure to androstenedione (A4), an androgen receptor agonist and known liver carcinogen in mice. Liver cancer is more prevalent in men compared to women, but androgen-related pathways underlying this sex difference have not been clearly identified. Short-term hepatic effects of A4 were compared to reference agonists of the estrogen receptor (ethinyl estradiol, EE) and glucocorticoid receptor (prednisone, PRED). Male B6C3F1 mice were exposed for 7 or 28 days to A4, EE, or PRED. EE increased and PRED suppressed hepatocyte proliferation, while A4 had no detectable effects. In a microarray analysis, EE and PRED altered >3000 and >670 genes, respectively, in a dose-dependent manner, whereas A4 did not significantly alter any genes. Gene expression was subsequently examined in archival liver samples from male and female B6C3F1 mice exposed to A4 for 90 days. A4 altered more genes in females than males and did not alter expression of genes linked to activation of the mitogenic xenobiotic receptors AhR, CAR, and PPARα in either sex. A gene expression biomarker was used to show that in female mice, the high dose of A4 activated the growth hormone-regulated transcription factor STAT5b, which controls sexually dimorphic gene expression in the liver. These findings suggest that A4 induces subtle age-related effects on STAT5b signaling that may contribute to the higher risk of liver cancer in males compared to females.
Keywords: mode of action, key events, liver carcinogenesis, androstenedione, ethinyl estradiol, prednisone, nuclear receptor, sexual dimorphism, STAT5b, aryl hydrocarbon receptor, constitutive activated receptor, peroxisome proliferator-activated receptor α
INTRODUCTION
Pathway information organized by mode of action (MOA) has played a central role in the assessment of chemical carcinogens for over a decade (Boobis et al., 2006; EPA 2005). In practice, however, use of the MOA framework has been largely retrospective, following identification of a cancer signal in long-term carcinogenicity or epidemiologic studies. An alternative prospective approach to the classical two-year rodent bioassay was proposed by Cohen (2004), in which potential carcinogenicity of a chemical would be based in part on short-term exposures and measurement of cancer risk indicators such as DNA-reactivity and increases in cell proliferation. These evaluations may also include in vitro assays, computerized models, and short-term (up to 13 weeks) bioassays as needed. A number of more recent reviews have expanded on opportunities for integrating alternative methods into screening for cancer hazard (Luijten et al., 2016; Simon et al., 2014). Moving forward, a central challenge of toxicological science is to identify shorter-term biomarkers that can be used to predict potential health hazards and inform risk assessment in the absence of traditional toxicity data. This type of approach relies strongly on the mapping of early chemical effects to key events in known biological constructs, called adverse outcome pathways (AOPs) (Villenueve et al., 2014a; Villenueve et al., 2014b), which are intended to integrate newer types of mechanistic information into chemical safety assessments and facilitate the transition toward more prospective molecular endpoints.
In the current case study, we examined early markers of steroid effects in the liver mediated by androgen, estrogen, and glucocorticoid receptors. Our primary focus was growth factor pathways mediated by androgen activity. Androgenic agents include natural steroid hormones, hormonal therapies, off-market athletic supplements, and intermediates in the production of various chemical and pharmaceutical products (Dodge and Hoagland, 2011). The primary health effects of androgens are mediated through the androgen receptor (AR), a classical nuclear receptor that regulates transcriptional responses in the liver following ligand binding and transactivation (Davey and Grossman, 2016). An important but poorly understood health concern for androgenic agents is increased risk of liver cancer (Giannitrapani et al., 2006; IARC 2007). Liver cancer is among the leading causes of cancer mortality worldwide, and although a variety of factors contribute to its development, including hepatitis B or hepatitis C virus infections, alcohol consumption, smoking and obesity, the sex differences in disease rate cannot be ignored (Giannitrapani et al., 2006; Zhang et al,, 2015). Epidemiologic studies indicate that the incidence of hepatocellular carcinoma is more common in men compared to women (>2:1) and correlates positively with serum androgens (De Maria et al., 2002; NTP 2010). There is also evidence, albeit limited, for increased risk of liver cancer with use of androgenic steroids, for example, among bodybuilders and patients with anemia (IARC 2007; NTP 2010). While gender differences in liver cancer in humans may be impacted by social or behavioral factors (e.g. differences in alcohol consumption or life style choices that lead to higher incidence of hepatitis infection), these factors are not sufficient to explain the sex differences observed in animal models (Shepard et al., 2005; Wilsnack et al., 2010). In experimental studies, there is a higher background incidence of liver cancer in many strains of male mice compared to female mice; decreased incidence of liver cancer in male mice following castration, knockout of the AR gene, or treatment with an AR antagonist; and increased incidence of liver cancer in ovariectomized female mice treated with AR agonists (e.g., Ma et al., 2014). In addition, the androgenic agents androstenedione (A4) and oxymetholone both resulted in increased incidence of liver cancer in long-term rodent carcinogenicity studies of the National Toxicology Program (NTP) (Blystone et al., 2011; NTP 1999; NTP 2010). A4 increased hepatocellular adenomas or carcinomas in both male and female B6C3F1 mice and preneoplastic hepatocellular foci in male F344/N rats (Blystone et al., 2011; NTP 2010). Based on the 2-year gavage studies, the NTP concluded that there was clear evidence of carcinogenicity for A4 in male and female mice. The carcinogenic effects in mice were observed in the absence of overt cytotoxicity or mutagenicity, suggesting a mitogenic role for AR signaling. Currently, however, there is limited understanding of growth factor pathways induced by AR activity in the liver
Other steroid receptors may also influence proliferative responses in the liver. Estrogen receptor (ER) activity in particular may interact with AR signaling and contribute to sexually dimorphic effects (Giannitrapani et al., 2006; Li et al. 2012). The role for ER in hepatocellular carcinogenesis is complex, however. Epidemiologic studies of estrogen-containing hormone therapies in menopausal women have reported lower risk of liver cancer (e.g., Fernandez et al., 2003), while other reports indicate higher liver cancer risk for oral contraceptive use in premenopausal women (Giannitrapani et al., 2006). Experimental evidence on estrogens and liver cancer in rodents is also mixed. Early studies reported promotional effects of estrogens on liver cell growth (dependent on the type of estrogen and study model) (e.g., Reznik-Schuller 1979), while more recent work has pointed to protective effects of estrogen mediated by anti-inflammatory mechanisms (reviewed in Shi et al., 2014). In this study, we evaluated early effects of ethinyl estradiol (EE), which is widely prescribed as part of many oral contraceptive regimens. EE was negative for liver tumors in previous mouse carcinogenicity studies (IARC 2007; Schuppler and Gunzel, 1979) and reduced the incidence (Lee et al., 1989; Standeven et al., 1994) and cell proliferation (Moser et al., 1996) of preneoplastic foci in mice initiated with diethylnitrosamine (DEN), indicating that it is not a liver carcinogen in mice and suggesting that it may have anti-proliferative effects. For this work we predicted that EE would interact with AR-mediated pathways and potentially oppose androgenic effects in the liver.
A third major steroid hormone pathway in the liver is mediated by the glucocorticoid receptor (GR), which has broad roles in energy metabolism, immunity, and cell fate determination (Cain and Cidlowski, 2015). In the liver, there is limited evidence that GR may have a protective role in cancer risk. First, Dillberger et al. (1992) found that prednisone (PRED), a GR agonist, significantly decreased the incidence of hepatocellular tumors in male and female mice. Second, combined deletion of mouse hepatic GR and the transcription factor STAT5, which controls growth hormone-regulated gene expression, increased incidence of hepatocellular carcinomas (Mueller et al., 2011). Third, a recent study by Matthews et al. (2015) found that an intact GR ligand-binding domain was necessary for mitotic progression, that GR heterozygous mice compared to wild-type mice had an increase in liver tumors, and that GR gene expression was reduced in a panel of human tumors, including those from the liver. Collectively, these findings suggest that expression and activation of GR may have protective effects on liver cancer incidence.
In the current study, we evaluated short-term effects of steroidal agents in the liver and investigated pathways by which A4 may induce liver cancer. We compared A4 effects to those induced by the reference agonists for ER (EE) and GR (PRED). Endpoints included liver histopathology, cell proliferation, markers of xenobiotic receptor activation, and transcriptomic profiles. Prior studies of A4 have shown a lack of mutagenicity in genotoxicity assays, and inconsistent evidence of liver cytotoxicity at carcinogenic doses (NTP 2010). Based on this evidence, we hypothesized that A4 would induce a direct mitogenic effect and a male-specific transcriptomic profile.
MATERIALS AND METHODS
Chemicals.
Androstenedione (A4) (Steraloids, Newport, RI; 98% purity, lot number B1141) was suspended in 0.5% aqueous methylcellulose (Sigma-Aldrich, St. Louis, MO) and administered by gavage. Ethinyl estradiol (EE) (Sigma-Aldrich, >98%% purity, lot number SLBF2546V) and prednisone (PRED) (Sigma-Aldrich, >98% purity, lot number BCBK19224V) were added to AIN93G rodent diet (TestDiet, Richmond, IN). Control AIN93G diet was processed in the same way as diets with chemicals added. Phenobarbital (PB) (Sigma-Aldrich, 99.8% purity, lot number SLBF7347V) was administered in the drinking water as a non-steroidal mitogenic positive control.
A4 purity determination.
The purity of an A4 solution prepared from the Steraloids standard was confirmed to be at least 98.9% by comparison of the HPLC/UV peak area obtained on an Agilent 1200 Series HPLC (Santa Clara, CA) to the peak area from an A4-certified reference standard (Cerilliant, Round Rock, TX, 99.8% purity, lot number FE08201302) at the same concentration. HPLC method parameters are provided in Supplemental Table 1.
Study design.
The study design is analogous to that used by Lake et al. (2016). Weanling B6C3F1 male mice were acquired at ~21 days of age due to the difficulty of obtaining 60-day old age-matched mice. Mice were obtained from Charles River Laboratories (Raleigh, NC) and were maintained according to the guidelines of the National Research Council Guide for the Care and Use of Laboratory Animals (NRC, 2011) at the U.S. EPA AAALAC-accredited animal facility (Research Triangle Park, NC). Mice were group-housed to fulfill social housing recommendations from the NRC guideline (NRC, 2011), AAALAS, and our Institutional Animal Care and Use Committee. All procedures involving animals were conducted under a protocol approved by the Institutional Animal Care and Use Committee of the U.S. EPA National Health and Environmental Effects Research Laboratory (NHEERL). Upon receipt, mice were randomly placed (4 per cage) in solid-bottom cages on Alpha-dri® bedding (Sheppard Specialty Papers) with nestlets (Anacare) and nylabones for enrichment. Mice were allowed ad libitum access to feed and water and housed under standard conditions (21 ± 1°C room temperature, 50 ± 10% humidity, and 12 hr on/12 hr off light cycles). AIN93G was used as the control and base diets for incorporation of chemicals administered in the feed, because it is free of soybean meal and soy protein constituents and thus has low to no phytoestrogens (Thigpen et al., 1999; Thigpen et al., 2013).
Body weight and consumption of feed were monitored during acclimation and treatment periods. At the time of experimentation, cages were randomly assigned to treatment groups and total body weight per cage was analyzed to ensure treatment groups did not differ significantly at the start of dosing. Mice were aged to approximately 60 days before the experiment began.
Mice were exposed for 7 days to A4 administered in 0.5% aqueous methylcellulose by gavage (10 ml/kg) at the following dose levels (0, 2, 10, 20, 50 mg/kg/day) (n=16/dose group). Control mice (0 mg A4/kg/day) received the appropriate amount of gavage vehicle. Other groups received either control diet or diets containing EE (1, 2.5, 5, 10 ppm) or PRED (1.25, 2.5, 5, 25 ppm) at one of four doses (n=16/dose group). The levels in the diet correspond to nominal intake levels of 0.2, 0.5, 1, and 2 mg/kg/day for EE and 0.25, 0.5, 1, and 5 mg/kg/day for PRED, respectively (Dillberger et al., 1992; Tyl et al., 2008). Dose ranges were established based on previous rodent carcinogenicity studies, which showed a significant increase in liver tumor incidence for A4 but not EE or PRED (Dillberger et al., 1992; IARC 2007; NTP 2010). Additional dose groups of mice (n=16/group) were exposed for 7 days to PB in the drinking water (with AIN93G diet control feed) at 600 ppm as a reference non-steroid positive mitogenic control group. Separate groups of mice were treated for 28 days. These mice were (1) fed either a control diet or diets containing high doses of EE (10 ppm) or PRED (25 ppm), (2) administered A4 (50 mg/kg/day in methylcellulose) or methylcellulose, or (3) maintained on PB in the drinking water at 600 ppm (n=16–24/group).
Sample sizes varied depending on the assay measured and sample availability. Group sample sizes for body and liver weights and clinical chemistry were n=10–16/group. Subsets of liver samples were analyzed for liver cell proliferation (n=8–10/group), histopathology (n=6–14/group), global gene expression (n=4/group), targeted gene expression (n=6–8/group), and hepatic cytochrome P450 (CYP) enzyme activity (n=5–8/ group).
Tissue collection.
Necropsies were carried out on unfasted mice in 4 blocks at 7 days of exposure and 2 blocks at 28 days following euthanasia by carbon dioxide. Mice were randomly selected for termination, with necropsies starting at ~9:30 a.m. and ending no later than ~1:30 p.m. Blood was collected by cardiac puncture, and serum was harvested from the blood and frozen at −80°C. After weighing the livers, the left lateral, caudate, and right medial liver lobes were trimmed, and portions of each lobe were either snap-frozen in liquid nitrogen or fixed in fresh 4% paraformaldehyde. After 18–24 hrs, fixed samples were transferred to 70% ethanol and used for histopathology and immunohistochemistry (IHC). Frozen samples were stored at −80°C and used for gene expression and enzyme activity assays.
Hepatic cytochrome P450 enzyme activity.
Frozen liver samples were thawed on ice and homogenized in buffer. Microsomes were isolated for enzyme activity analysis according to Guengerich et al. (1982). Centrifugation steps were conducted at 4°C and total protein was quantitated with bovine serum albumin (BSA) standards. Enzyme activity was measured, without reference to treatment group identification, using O-deethylation of ethoxyresorufin (EROD) for CYP1A, pentoxyresorufin O-depentylation (PROD) for CYP2B, and benzyloxyresorufin O-debenzylation (BROD) for CYP3A. A Spectramax Gemini SX plate reader (Molecular Devices, Sunnyvale, CA) was used to measure the fluorescence activity of the enzyme reaction. The excitation and emission wavelengths were set at 544 and 590 nm, respectively. Stock solutions of substrates were prepared in methanol at 420 μM benzyloxyresorufin and ethoxyresorufin and 4200 μM for pentoxyresorufin. A stock solution of 1 mM resorufin, the enzymatic product, was also prepared in methanol. Stock solutions were stored at −20oC. The reaction mixture consisted of 0.05 M Tris buffer, pH 8, 0.05–0.1 mg microsomal protein, and either 4.5 μM ethoxyresorufin, 4.5 μM benzyloxyresorufin, or 0.9 μM pentoxyresorufin. Samples were run in triplicate in an opaque 96-well plate. The sample plate was pre-incubated in the plate reader at 37oC for 3–5 min. The reaction was initiated by the addition of 25 μl of stock NADPH (100 mM NADPH was prepared in 0.05 M Tris buffer, pH 8). The total volume of incubate was 235 μl. The production of resorufin was monitored for 5 min at 37oC. Data were collected at 36 sec intervals. Because of the large number of samples, analysis was conducted over 3 days (7-day Blocks 1 and 2; 7-day Blocks 3 and 4; 28-day Blocks 1 and 2). A standard curve of 0 – 50 pmol resorufin was developed for each analysis. The standard curve was forced through zero. The r2 for the standard curve was: 7-day Blocks 1 and 2, 0.9986; 7-day Blocks 3 and 4, 0.9994; 28-day Blocks 1 and 2, 0.9993. Results were calculated as pmol of resorufin formed per mg of protein per min. A Bio-Rad (Richmond, CA) protein assay kit with BSA as the protein standard was used to measure the concentration of microsomal protein.
Serum liver enzyme chemistry
Alanine aminotransferase (ALT) and sorbitol dehydrogenase (SDH) activity levels were measured blindly as serum indicators of liver cytotoxicity using automated procedures in the NHEERL Clinical Chemistry Research Core Laboratory on a Konelab Arena 30 Clinical Chemistry Analyzer (ThermoFisher Scientific, Waltham, MA).
Liver histopathology.
Fixed liver samples were embedded in paraffin, sectioned at 6 μm, and stained with hematoxylin and eosin (H&E) using standard histologic procedures. Liver sections were then evaluated via light microscopy by a board-certified study pathologist at Experimental Pathology Laboratories, Inc. (EPL) (Research Triangle Park, NC). Histopathological review was performed according to best practices for nonclinical safety studies, as described by Crissman et al. (2004). Histopathological diagnoses were based on standard criteria and nomenclature (Thoolen et al., 2010). Severity of non-neoplastic lesions was qualitatively scored using a 0–5 scale (0=absent, 1=minimal, 2=mild, 3=moderate, 4=moderately severe, 5=severe). Severity index was calculated as the average severity score per group for a given change. All pathology data, tabulations, and observations were recorded under GLP-compliant conditions.
Ki67 IHC and labeling indices.
Liver cell proliferation was evaluated by IHC staining for the Ki67 antigen (Wood et al., 2015). The labeling procedure included a rabbit monoclonal primary antibody for Ki67 (Clone SP6, ThermoFisher Scientific), antigen retrieval with citrate buffer, biotinylated goat anti-rabbit secondary antibody as a linking reagent (BA-6100, Vector Laboratories, Burlingame, CA), peroxidase-conjugated avidin as the label (ABC Complex Vector Kit, Vector Laboratories), and 3,3’-diaminobenzidine (DAB) as the chromogen (Biocare Medical, Concord, CA) (Lake et al., 2016). Anti-Ki67 antibodies were diluted 1:500 with diluent buffer (S0809, Dako, Carpinteria, CA), and immunostained slides were counterstained with hematoxylin (QS, Vector Laboratories). Negative and positive control slides were run with each IHC batch. Negative staining slides used the same protocol as for study slides except with Tris-buffered saline in place of the primary antibody; positive control slides used mouse intestine tissues in place of liver.
Nuclear labeling for Ki67 was quantified by a computer-assisted counting technique using an automated procedure of cell selection (Lake et al., 2016). At least three fields per liver (≥1 per lobe) were digitally imaged at 20× objective magnification using a Nikon DS-Fi2 color camera and Nikon Ti inverted microscope (Nikon Instruments, Inc.). Following image capture, nuclei were counted using NIS Elements software (v4.13), and counting was performed in a blinded manner. Binary layers generated from thresholding individual color components were used to identify liver cell nuclei based on size, shape, and intensity criteria. Customized macros were used to sort cells into four categories based on label (positive or negative) and size/shape (large round nuclei indicating mature hepatocytes or “other” smaller cells). At least 1000 cells were counted per liver, and counts were analyzed as a percentage of the total number of cells examined to generate a labeling index (LI) for each sample.
Gene expression microarrays.
Global gene expression was evaluated by microarray analysis. Total RNA was isolated from ~20 mg of frozen liver tissue homogenized in RNAzol®RT (Molecular Research Center, Cincinnati, OH), purified with the RNeasy MinElute column protocol (Qiagen GmbH, Hilden, Germany), evaluated for integrity using an Agilent RNA 6000 Nano chip on an Agilent 2100 Bioanalyzer (Agilent Technologies GmbH, Berlin, Germany), and quantitated using the NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, DE). RNA samples were isolated and purified at Expression Analysis Genomic Services, Q2 Solutions (Durham, NC). After initial randomization, samples were hybridized in a blinded manner onto Mouse Ref-8 V2.0 Expression BeadChip arrays (Illumina, San Diego, CA) in the NHEERL Genomics Research Core Laboratory using standard Illumina protocols. Arrays were scanned, and raw data (.idat files) were obtained using Illumina iScan software (v3.3.28) and analyzed using Illumina GenomeStudio® Data Analysis Software. Array data are publicly available at Gene Expression Omnibus (GEO), accession number GSE84590.
Targeted gene expression.
Relative expression of 11 gene targets indicative of sex-specific expression (Ahnak, Cyp2c29, Elovl3, Hsd3b5, Mup4, Mup5, Nat8, Prkaa2, Scara5, Stbd1, and Susd4) were measured using reverse transcriptase quantitative PCR (qPCR). Genes were derived from the STAT5b biomarker (Oshida et al., 2016a) and from the literature (Clodfelter et al., 2006). RNA was isolated as described for the array analysis with additional steps using TurboDNase treatment (ThermoFisher Scientific,) and Qiagen MinElute column purification before cDNA preparation. Total RNA was quantitated using the Qubit RNA assay (ThermoFisher Scientific). The cDNA was prepared using the BioRad iScript™ cDNA synthesis kit (BioRad, Hercules, CA). qPCR was carried out using BioRad PrimePCR SYBR assays. Three reference genes (Hprt, Ppia, and Tbp) and PCR quality controls were also assayed for each sample. Samples were loaded in technical duplicate for each gene target using the Qiagen QiAgility robotic workstation and measured on a BioRad CFX384 real-time qPCR machine using manufacturer’s recommendations for PrimePCR assays. Melt-curve analysis was used to verify single products for each amplified target. During analysis, cycle threshold (Ct) values were determined using the BioRad CFX Manager software (v3.0.12224.1015). Relative quantification of gene expression to controls, normalized to reference gene expression, was assessed using the 2-ΔΔCt method (Livak and Schmittgen, 2001). Statistical significance of expression values for 7-day control vs. treatment samples were determined by one-way ANOVA and Tukey-Kramer post hoc analysis with multiple test correction. Significantly different expression in 28-day control vs. treatment samples were determined by a two-sided, non-parametric Mann Whitney test using qBase+ software (Biogazelle, Zwijnaarde, Belgium).
TempO-Seq analysis of archival liver samples.
Changes in gene expression patterns were analyzed using targeted RNA-sequencing (TempO-Seq, BioSpyder Technologies, Inc., Carlsbad, CA) of formalin-fixed paraffin-embedded (FFPE) liver samples from study TR-560 of the NTP (http://ntp.niehs.nih.gov/results/pubs/longterm/reports/longterm/tr500580/listedreports/tr560/index.html), in which male and female B6C3F1 mice were treated by gavage with 0, 10, 20, and 50 mg/kg of A4 for 90 days. A total of 32 samples were analyzed, 16 each from males and females (n=4/dose group). The TempO-Seq platform measures a predefined set of 3099 mouse genes that represent a surrogate whole transcriptome assay called the “S1500” (https://federalregister.gov/a/2015-08529). Two sections 10 μm in thickness were collected from each tissue block, placed in a microfuge tube, and processed according to BioSpyder recommendations. Briefly, 76 μL of lysis buffer was added to each tube and overlaid with 150 μL mineral oil. Samples were heated to 75°C until all paraffin had melted (~30 min), and the paraffin separated into the mineral oil was removed. Proteinase-K was added to the remaining tissue/lysis buffer at a final concentration of 1 μg/mL, incubated for 1 hr at 37°C, and inactivated at 95°C for 15 min. Any remaining tissue was pelleted by centrifugation at 13,000 × g for 15 min. To hybridize detector oligonucleotides to mRNA targets, 2 μL of lysate was transferred to a well in a 96-well plate and combined with 2 μL of annealing mix (1× annealing buffer and 1 nM detector oligonucleotide pool). Samples were incubated at 75°C for 10 min, then ramped down to 45°C at a rate of 0.5°C per min, and held overnight at 45°C. Excess oligonucleotides were digested by adding 24 μL of nuclease mix (1× nuclease buffer and 3’exo-nuclease enzyme) to each sample and incubated at 37°C for 1.5 hr. Detector oligonucleotides were ligated to generate a pool of amplification templates that share PCR primer annealing sites by adding 24 μL of ligation mix (1x ligation buffer and ligase) to each sample and incubating at 37°C for 1 hr, followed by 80°C for 30 min to inactivate ligase. Ten μL of each sample was amplified in a pre-made qPCR plate as follows: 37°C for 10 min, 95°C for 2 min, 6 cycles of 95°C for 30 sec, 54°C for 30 sec, 72°C for 2 min, 16 cycles of 95°C for 30 sec, 66°C for 30 sec, 72°C for 2 min, and a final step of 72°C for 2 min. During product amplification, each sample well was assigned a specific barcoded primer pair, allowing for sample identification following sequencing. Sample amplicons were pooled and purified using a PCR clean-up kit (Clontech, Mountain View, CA). Libraries were sequenced at MOGene, LC (St. Louis, MO) using a NextSeq Mid-Output Sequencing System (Illumina, San Diego, CA).
Statistical analyses.
Total body weights by cage were analyzed by analysis of variance (ANOVA) using the SAS GLM procedure (SAS v9.4) prior to the start of dosing. Terminal measurements for body weight, relative liver weight, clinical chemistries (ALT and SDH), body weight change, and enzymatic activity data were analyzed as dependent variables in 7- and 28-day two-way ANOVA models using the SAS MIXED procedure. To determine the scale of analysis for each variable, model residuals for both linear and log10-scaled dependent variables were evaluated for normality (Shapiro-Wilk test in SAS UNIVARIATE) and homogeneity of variance (Levene’s test in SAS GLM). Separate two-way main-effects ANOVA with factors for block and dose group were performed for each dependent variable for each of the four chemicals for each dosing duration time. A Bonferroni multiple-comparison adjustment was used for the ANOVA F-tests for the EE, PRED, and PB treatments which shared control animals but not for the A4 study which had its own control animals. Where an F-test was declared significant, Dunnett’s pairwise tests for differences in least-squares mean between each dose group and the corresponding control group are also reported using a statistical significance threshold of α=0.05. The P450 activity values from one mouse treated for 7 days with prednisone at 1.25 ppm were dropped from the analysis because it was an outlier.
Histopathology incidence data were analyzed by comparing results for each treatment group to the respective control group at 7 or 28 days using a one-sided Fisher’s exact test. Pairwise p-values for all incidence data were adjusted using a Bonferroni multiple test correction for the number of direct comparisons within each treatment arm. An adjusted α level of 0.05 was used to determine a significant difference between groups. A Cochran-Armitage trend test with Bonferroni adjustment was used to evaluate dose response trends for incidence of histopathology outcomes. Severity scores of non-neoplastic histopathology outcomes are presented only as descriptive data.
For microarray data, raw gene expression intensities were quantile-normalized using Illumina GenomeStudio®. Principal Component Analysis (PCA) was performed to examine within and across group profiles in Partek Genomics Suite. ANOVA analysis was also performed in Partek Genomics Suite, with false discovery rate testing (Benjamini–Hochberg test) and a p ≤ 0.05 for significance. For TempO-Seq data, sequencing readouts were demultiplexed to generate FASTQ files. Genes that were differentially expressed between each A4 dose group and control, and between the male and female control groups were identified with a Wald test followed by a Benjamini-Hochberg multiple test correction (false discovery rate q-value < 0.1) in the DESeq2 R package (Love et al., 2014). Heat maps were generated using Eisen Lab Treeview software (http://rana.lbl.gov/EisenSoftware.htm). Pathway analysis was performed on differentially expressed genes in Ingenuity Pathway Analysis (Qiagen Bioinformatics, Redwood City, CA).
Gene expression correlation analysis.
Lists of statistically significant, differentially expressed genes (DEGs) were compared to each other using tools in a commercially available database provided by Illumina BaseSpace Correlation Engine, also called NextBio (www.nextbio.com). This database contains over 128,000 lists of statistically filtered genes from over 20,200 microarray studies carried out in 16 species (as of June, 2016). The normalized ranking approach used by NextBio enables comparability across data from different studies, microarray platforms, and analysis methods by removing dependence on absolute values of fold-change, and minimizing some of the effects of the normalization methods used, while accounting for the level of genomic coverage by the different platforms. The Running Fisher algorithm using a Fisher’s exact test generates a p-value and correlation direction (positive or negative) for each pairwise comparison. A description of criteria used to identify datasets in GEO suitable for analysis and details of the microarray processing steps in the NextBio pipeline are summarized in Oshida et al. (2015c) and are discussed in greater detail in Kupershmidt et al. (2010). All statistically filtered gene lists generated as part of this study were uploaded into NextBio after filtering for │fold-change│ ≥ 1.2. We have previously used the database and computational tools to accurately identify factors (e.g., chemicals, genes, diets) that activate or suppress other transcription factors important in chemical-induced liver carcinogenesis, such as aryl hydrocarbon receptor (AhR), constitutive activated receptor (CAR), peroxisome proliferator-activated receptor (PPAR) α, and STAT5b (Oshida et al., 2015a-c, 2016a,b).
RESULTS
Liver weight and serum markers.
Treatment with A4 did not significantly alter terminal body weight, the ratio of liver weight (LW) to body weight (BW), or serum activity of ALT or SDH at 7 or 28 days (Table 1, Supplemental Table 2). EE resulted in lower terminal body weight at 7 and 28 days and higher LW/BW and ALT at 28 days suggestive of an effect on the liver. PRED resulted in decreased body weight gain at the two highest doses at 7 days and decreased body weight gain, increased relative LW/BW, and increased ALT at 28 days. PB resulted in higher LW/BW at 7 and 28 days and higher ALT and SDH at 28 days. As these modest increases in serum ALT and/or SDH were observed in the absence of cytotoxicity (based on histopathological analysis), they are likely associated with microsomal enzyme induction, which can occur earlier in the disease process and be a more sensitive indicator of liver cell injury compared to histopathology (Hall et al., 2012).
Table 1.
Terminal Body Weight, Relative Liver Weights and Clinical Chemistries Following 7 and 28 Days of Exposure to Androstenedione (A4), Ethinyl Estradiol (EE), Phenobarbital (PB) and Prednisone (PRED).
| Terminal Body Weight gm |
Relative Liver Weight % |
ALTb IU/L |
SDHc IU/L |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Day | Chemical | Dosea | n |
LS-Mean (SE) |
n |
LS-Mean (SE) |
n |
LS-Mean (SE) |
n |
LS-Mean (SE) |
| 7 | A41 | 0 | 11 | 31.30 (0.79) |
11 | 4.17 (0.11) |
6 | 52.57 (28.95) |
6 | 19.54 (12.54) |
| 2 | 13 | 31.80 (0.72) |
13 | 4.24 (0.10) |
7 | 55.21 (27.08) |
8 | 36.05 (10.91) |
||
| 10 | 15 | 31.09 (0.67) |
15 | 4.22 (0.09) |
10 | 80.49 (22.36) |
10 | 39.02 (9.68) |
||
| 20 | 15 | 30.62 (0.67) |
15 | 4.38 (0.09) |
9 | 44.17 (23.84) |
9 | 15.82 (10.33) |
||
| 50 | 15 | 31.99 (0.67) |
15 | 4.26 (0.09) |
10 | 37.20 (22.36) |
10 | 17.57 (9.68) |
||
| EE | 0 | 16 | 33.25 (0.52) |
16 | 4.48 (0.36) |
9 | 56.99 (73.94) |
10 | 25.97 (24.37) |
|
| 1 | 16 | 24.38*** (0.52) |
16 | 4.56 (0.36) |
10 | 52.96 (70.22) |
10 | 20.78 (24.37) |
||
| 2.5 | 16 | 24.28*** (0.52) |
16 | 4.48 (0.36) |
10 | 87.75 (70.22) |
9 | 14.84 (25.94) |
||
| 5 | 16 | 24.09*** (0.52) |
16 | 4.66 (0.36) |
11 | 321.32 (66.87) |
10 | 68.91 (24.37) |
||
| 10 | 16 | 25.04*** (0.52) |
16 | 4.81 (0.36) |
8 | 29.82 (78.95) |
8 | 65.05 (27.13) |
||
| PB | 0 | 16 | 33.25 (0.52) |
16 | 4.48 (0.10) |
9 | 69.69 (26.15) |
10 | 30.70 (7.14) |
|
| 600 | 16 | 32.43 (0.52) |
16 | 6.03*** (0.10) |
10 | 105.73 (24.86) |
10 | 33.19 (7.14) |
||
| PRED | 0 | 16 | 33.25 (0.53) |
16 | 4.48 (0.09) |
9 | 69.99 (13.76) |
10 | 30.86 (5.17) |
|
| 1.25 | 16 | 31.94 (0.53) |
16 | 4.05** (0.09) |
9 | 68.77 (13.77) |
10 | 24.29 (5.17) |
||
| 2.5 | 16 | 32.44 (0.53) |
16 | 4.22 (0.09) |
10 | 58.92 (13.07) |
10 | 23.95 (5.17) |
||
| 5 | 16 | 30.31*** (0.53) |
16 | 4.18 (0.09) |
10 | 70.47 (13.07) |
10 | 23.33 (5.17) |
||
| 25 | 16 | 29.62*** (0.54) |
16 | 4.55 (0.09) |
10 | 92.01 (13.07) |
10 | 35.85 (5.17) |
||
| 28 | A42 | 0 | 13 | 34.73 (0.76) |
13 | 4.12 (0.14) |
9 | 37.97 (4.29) |
9 | 28.75 (16.95) |
| 50 | 20 | 34.43 (0.61) |
20 | 4.21 (0.11) |
13 | (37.21) (3.49) |
14 | 45.21 (13.38) |
||
| EE | 0 | 16 | 38.43 (0.61) |
16 | 4.02 (0.13) |
10 | 43.14 (28.34) |
10 | 29.15 (8.47) |
|
| 10 | 21 | 21.77*** (0.54) |
21 | 6.73*** (0.11) |
12 | 125.58** (25.63) |
13 | 41.99 (7.38) |
||
| PB | 0 | 16 | 38.43 (0.70) |
16 | 4.02 (0.21) |
10 | 36.54 (8.08) |
10 | 26.52 (5.02) |
|
| 600 | 183 | 35.81* (0.67) |
18 | 6.17*** (0.20) |
11 | 79.02** (7.65) |
11 | 45.51* (4.75) |
||
| PRED | 0 | 16 | 38.43 (0.69) |
16 | 4.02 (0.11) |
10 | 36.36 (5.13) |
10 | 26.65 (2.83) |
|
| 25 | 194 | 31.45*** (0.64) |
19 | 4.42* (0.10) |
12 | 72.37*** (4.64) |
12 | 33.03 (2.56) |
||
n reduced to less than 16 per dose/group due to deaths over the course of the 7 dosing period.
7 mice in the control group and 4 mice in the 50 mg/kg/day dose group died over the course of 28 day dosing period.
2 mice died over the course of the 28 day dosing period.
1 mouse died over the course of the 28 day dosing period.
Least-squares (LS) means and standard errors (SE) by dose for 7- and 28-day steroid exposed mice.
p < 0.05,
p < 0.01,
p < 0.001.
Dose is the gavage dose in mg chemical/kg body weight for A4, dietary concentration (ppm in diet) for EE and PRED, and ppm in drinking water for PB.
Alanine aminotransferase
Sorbitol dehydrogenase
Liver histopathology.
No significant histopathological effects of A4 or PRED were observed at 7 or 28 days (Supplemental Table 3). Treatment with EE for 7 days resulted in bile duct proliferation at the three lowest doses, but not the high dose, and oval cell proliferation at doses ≥ 2.5 ppm. Treatment with EE for 28 days resulted in midzonal to periportal hypertrophy and bile duct and oval cell proliferation. PB induced minimal to mild centrilobular hypertrophy at 7 and 28 days, consistent with the effects observed in a prior study by our group using similar methods (Lake et al., 2016). Representative images of select histopathological findings in the liver are provided in Supplemental Figure 1.
Liver cell proliferation.
Steroid effects on liver cell proliferation were determined by Ki67 labeling. A4 did not alter cell proliferation at 7 days at any dose (Figure 1A). For EE, higher LIs were observed at all doses at 7 days, but this effect was statistically significant only for small cells at 2.5 and 10 ppm (Figure 1B). PRED resulted in lower LIs (large cells only) at 7 days at the 1.25, 5, and 25 ppm doses (Figure 1C), while PB resulted in significantly higher LIs at 7 days (large, small, and all cells) (Figure 1A). At 28 days, there were no effects of A4 or PRED (Figure 1D), while EE resulted in significantly higher cell proliferation (large, small, and all cells) which was comparable in magnitude to that seen at 7 days, suggesting a sustained compensatory proliferative effect secondary to cytotoxicity. PB also resulted in higher Ki67 LI at 28 days, but this effect (4.8% LI, all cells) was diminished compared to 7 days (8.2% LI), consistent with an early mitogenic burst (Wood et al., 2015). Representative images of Ki67 immunolabeling are provided in Supplemental Figure 2.
Figure 1. Labeling index (LI) for the proliferation marker Ki67 in the livers of mice treated with A4, EE, or PRED.

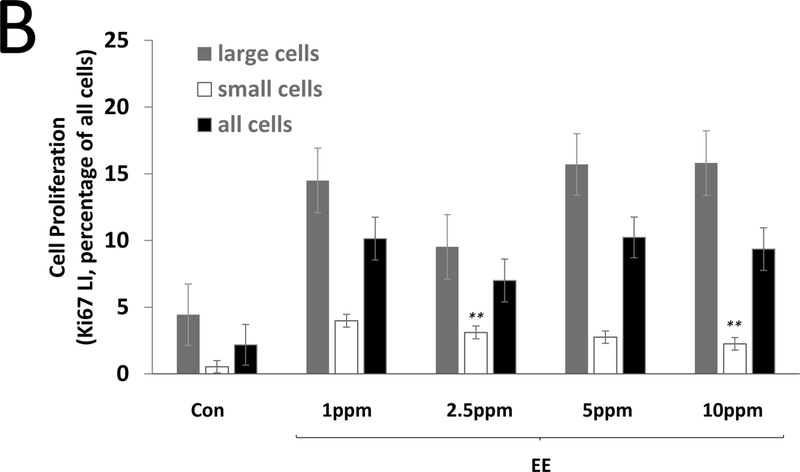
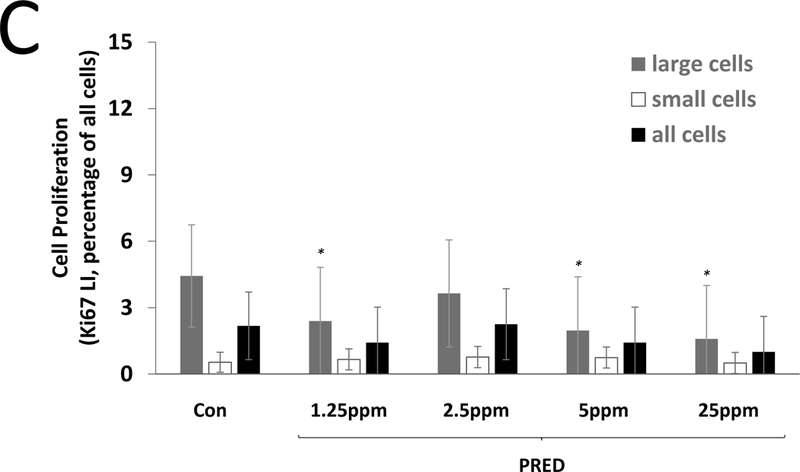
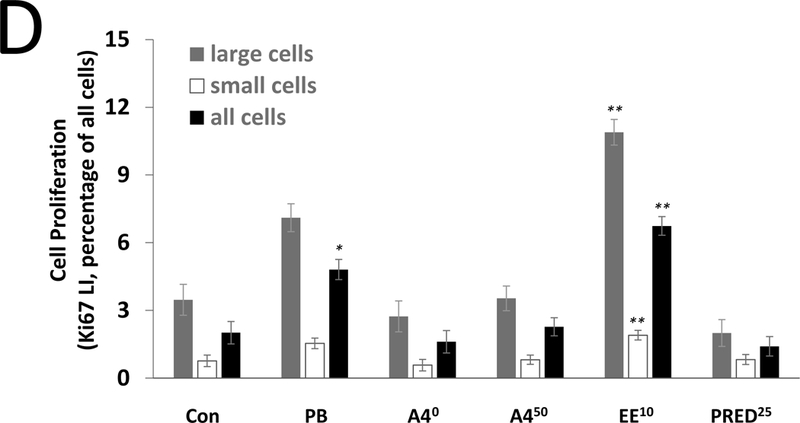
Ki67 LI is shown for large cells (grey), small cells (white), and all cells combined (black) after 7 days (A-C) and 28 days (D) of exposure. Significance is indicated by *p < 0.05 and **p < 0.01 compared to the respective control (Con) group. The 0 mg/kg A4 group indicates gavage control.
CYP450 enzyme activity.
We measured the activities of EROD, PROD, and BROD, which provide indicators of AhR, CAR, and Pregnane X Receptor (PXR) activation, respectively (Burke and Mayer, 1974; Burke et al., 1985; Hagemeyer et al., 2010; Lehmann et al., 1998; Ma and Lu, 2007; Yoshinari et al., 2001). No significant changes were observed in enzyme activities following A4 exposure at 7 or 28 days (Table 2). EE significantly decreased BROD (0.4- to 0.6-fold), EROD (0.5- to 0.7-fold), and PROD (0.3- to 0.4-fold) activities following 7 days of exposure and EROD (0.7-fold) and PROD (0.4-fold) activities following 28 days of exposure. EE has been shown to inactivate CYP2B (i.e., PROD) and CYP3A (i.e., BROD) in a mechanism-based manner (Guengerich, 1988; Kent et al., 2002). PRED at 25 ppm significantly increased BROD (9.9- and 16.4-fold, respectively) and PROD (4.2- and 13.3-fold) activities after 7 and 28 days, respectively. PRED effects corresponded to increases in genes encoding CYP2B and CYP3A family members (Supplemental Table 4). PRED has been shown to induce CYP3A (Deb et al., 2012; Pichard et al., 1992). In contrast, PRED decreased EROD (0.5- to 0.8-fold and 0.8-fold) activity following 7 and 28 days, respectively, at doses >1.25 ppm. PB exposure for 7 and 28 days resulted in significant increases in BROD (6.1- and 15.4-fold, respectively), EROD (3.0- and 4.0-fold), and PROD (5.1- and 11.1-fold) activities.
Table 2.
17Hepatic P-450 Enzyme Activity Levels and Fold Changes Following 7 and 28 Days of Exposure to Androstenedione (A4), Ethinyl Estradiol (EE), Phenobarbital (PB) and Prednisone (PRED).
| BROD pmole/mg protein/min | EROD pmole/mg protein/min | PROD pmole/mg protein/min | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Day | Chemical | Dosea | n | Least Square Mean (SE) |
Fold Change | n | Least Square Mean (SE) |
Fold Change | n | Least Square Mean (SE) |
Fold Change |
| 7 | A4 | 0 | 5 | 3.63 (0.41) |
5 | 22.75 (1.49) |
5 | 2.62 (0.31) |
|||
| 2 | 5 | 3.64 (0.42) |
1.0 | 5 | 23.53 (1.54) |
1.0 | 5 | 2.59 (0.32) |
1.0 | ||
| 10 | 5 | 3.28 (0.41) |
0.9 | 5 | 21.54 (1.49) |
0.9 | 5 | 2.65 (0.31) |
1.0 | ||
| 20 | 6 | 4.14 (0.37) |
1.1 | 6 | 25.50 (1.36) |
1.1 | 6 | 2.39 (0.29) |
0.9 | ||
| 50 | 5 | 3.61 (0.40) |
1.0 | 5 | 28.27 (1.48) |
1.2 | 5 | 2.50 (0.31) |
1.0 | ||
| 7 | EE | 0 | 6 | 3.83 (0.26) |
6 | 25.68 (1.85) |
6 | 2.67 (0.20) |
|||
| 1 | 6 | 2.41** (0.26) |
0.6 | 6 | 16.00** (1.85) |
0.6 | 6 | 0.94*** (0.20) |
0.4 | ||
| 2.5 | 6 | 2.09*** (0.26) |
0.5 | 6 | 17.49* (1.85) |
0.7 | 6 | 0.96*** (0.20) |
0.4 | ||
| 5 | 5 | 2.04*** (0.28) |
0.5 | 5 | 16.80* (2.02) |
0.7 | 5 | 0.92*** (0.22) |
0.3 | ||
| 10 | 6 | 1.48*** (0.26) |
0.4 | 6 | 13.91*** (1.85) |
0.5 | 6 | 0.90*** (0.20) |
0.3 | ||
| 7 | PB | 0 | 6 | 4.50 (3.31) |
6 | 28.22 (10.41) |
6 | 3.24 (2.13) |
|||
| 600 | 6 | 27.44** (3.31) |
6.1 | 6 | 86.01** (10.41) |
3.0 | 6 | 16.44** (2.13) |
5.1 | ||
| 7 | PRED | 0 | 6 | 4.30 (3.63) |
6 | 25.45 (1.05) |
6 | 3.04 (1.46) |
|||
| 1.25 | 5 | 8.20 (4.07) |
1.9 | 5 | 21.77 (1.17) |
0.9 | 5 | 2.87 (1.64) |
0.9 | ||
| 2.5 | 6 | 4.14 (3.63) |
1.0 | 6 | 20.05** (1.05) |
0.8 | 6 | 3.45 (1.46) |
1.1 | ||
| 5 | 6 | 5.38 (3.63) |
1.3 | 6 | 16.09*** (1.05) |
0.6 | 6 | 2.63 (1.46) |
0.9 | ||
| 25 | 6 | 42.74*** (4.02) |
9.9 | 6 | 11.67*** (1.16) |
0.5 | 6 | 12.89*** (1.62) |
4.2 | ||
| 28 | A4 | 0 | 4 | 4.17 (0.55) |
4 | 22.63 (3.96) |
4 | 2.82 (0.47) |
|||
| 50 | 6 | 4.53 (0.44) |
1.1 | 6 | 32.64 (3.18) |
1.4 | 6 | 2.32 (0.37) |
0.8 | ||
| 28 | EE | 0 | 6 | 4.33 (0.58) |
6 | 25.50 (1.67) |
6 | 2.57 (0.12) |
|||
| 10 | 8 | 4.17 (0.53) |
1.0 | 8 | 17.31** (1.53) |
0.7 | 8 | 1.03*** (0.11) |
0.4 | ||
| 28 | PB | 0 | 6 | 3.79 (2.84) |
6 | 26.87 (4.04) |
6 | 2.57 (1.62) |
|||
| 600 | 7 | 58.31*** (2.70) |
15.4 | 7 | 108.03*** (3.84) |
4.0 | 7 | 28.63*** (1.54) |
11.1 | ||
| 28 | PRED | 0 | 6 | 4.04 (2.18) |
6 | 25.20 (1.32) |
6 | 2.33 (1.88) |
|||
| 25 | 7 | 66.39*** (2.07) |
16.4 | 7 | 18.98** (1.25) |
0.8 | 7 | 30.99*** (1.79) |
13.3 | ||
Asterisks indicate significant difference at
p < 0.05,
p < 0.01,
p < 0.001 compared to control group.
BROD: Benzyloxyresorufin O-debenzylase; PROD: Pentyloxyresorufin O-depentylase; EROD: Ethoxyresorufin O-deethylase
Dose is the gavage dose in mg chemical/kg body weight for A4, dietary concentration (ppm in diet) for EE and PRED, and ppm in drinking water for PB.
Global gene expression.
PCA showed no clear separation in global gene expression between the control and A4 dose groups at 7 or 28 days (Figure 2A), in contrast to EE and PRED (Supplemental Figure 3A-C). Microarray analysis did not identify any genes to be significantly altered by A4 at any dose at 7 or 28 days (Figure 2B), while EE and PRED significantly altered up to 3070 and 671 genes in a dose-dependent manner (Supplemental Table 4). A4 effects on known target genes of androgens in the mouse liver (Clodfelter et al., 2006; Oshida et al., 2016a) were also measured by qPCR. Out of the 11 genes measured, Hsd3b5 and Cyp2c29 both expected to increase in expression after androgen exposure exhibited decreased expression at 7 or 28 days, respectively (Supplemental Figure 4). These minor changes inconsistently observed across the treatment times including a 90-day time point (discussed below) likely are not due to a direct effect of A4 (discussed in legend of Supplemental Figure 4). One-dimensional clustering of DEGs showed a consistent direction of change across dose groups for EE and PRED at 7 and 28 days (Figure 2C). The pattern of expression was remarkably consistent across all doses and time points for EE, despite the fact that at 28 days, the 10 mg/kg/day and the 25 mg/kg/day doses for EE and PRED, respectively, exhibited differences in indicators of toxicity (terminal body weights and ALT). The extent of the overlap in the regulated genes by the two compounds was examined at 7 days (highest dose) or 28 days (Supplemental Figure 5). Out of the 205 genes that were regulated at 7 days by both EE and PRED, 167 (81%) had concordant regulation (i.e., genes were regulated in the same direction). Similarly, at 28 days, EE and PRED commonly regulated 335 genes, of which 296 (88%) had concordant direction of change. Genes regulated by EE significantly overlapped with those regulated by an ERα agonist in an ERα-dependent manner and genes regulated by PRED overlapped with those regulated by a GR agonist in a GR-dependent manner (Supplemental Figure 6). In summary, these results indicate that prototypical agonists of ER and GR result in the modulation of an overlapping set of genes. The biological basis for regulation and the impact on liver effects of the overlapping genes remains to be determined.
Figure 2. Steroid effects on global gene expression at 7 and 28 days.
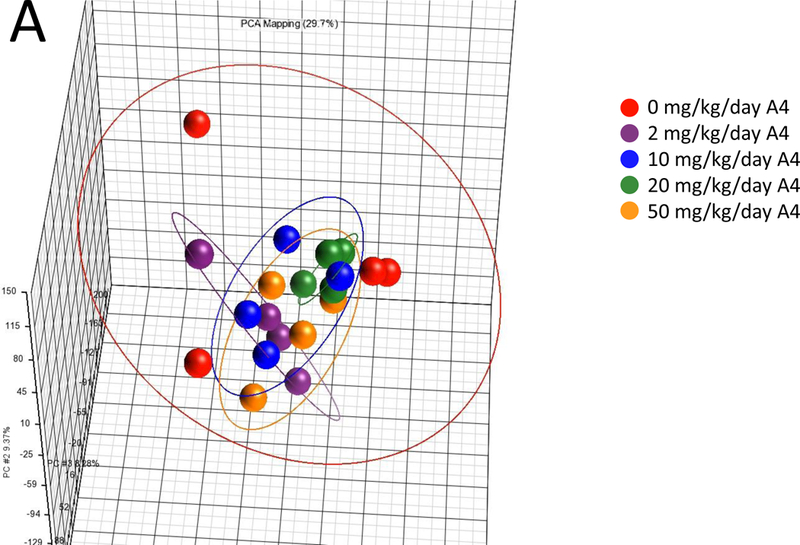
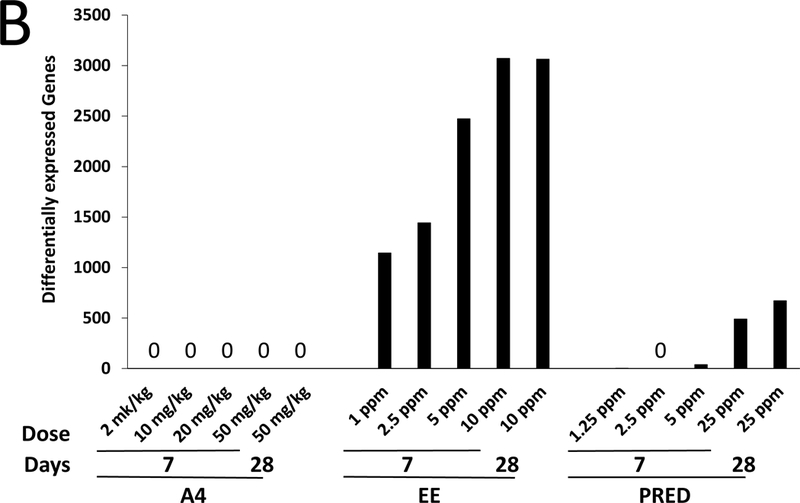
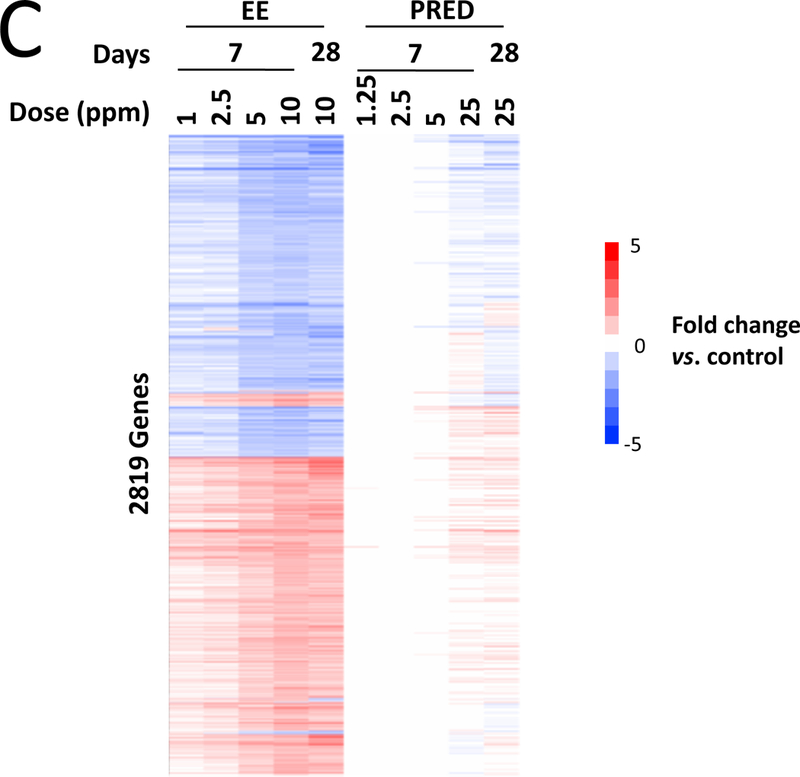
A. Principal component (PC) plot of microarray profiles for A4 samples at 7 days. B. Number of significant differentially expressed genes in each dose group. C. One-dimensional hierarchical clustering analysis of all significantly altered genes. (Clades are not shown.)
Genomic effects of A4 at 90 days.
Given the lack of any A4 effects at 7 or 28 days, we used targeted RNA sequencing to examine A4 responses in FFPE liver samples from male and female mice treated for 90 days with three doses of A4 (NTP study TR-560). All samples passed standard TempO-Seq quality control metrics, including a signal-to-noise ratio cutoff of 20:1, greater than 80% of reads mapped, and average reads per probe greater than 250. PCA showed a clear separation of control males and control females (Figure 3A). Genes significantly altered between control males and females were highly similar in expression differences to male vs. female comparisons from other studies examining liver gene expression in control mouse liver (Figure 3B). There was no obvious separation between the control and A4 groups for either sex. Only a small number of genes were altered in male mice (8 genes, or 0.26% of the total, at any dose, and only 1 gene, Fabp5, at all 3 doses). In contrast, there were greater numbers of genes altered in female mice at the low and high dose groups (Figure 3C). The complete list of genes altered by A4 at 90 days is found in Supplemental Table 5.
Figure 3. Gene expression analysis of the archival NTP liver samples from mice treated with A4 for 90 days.
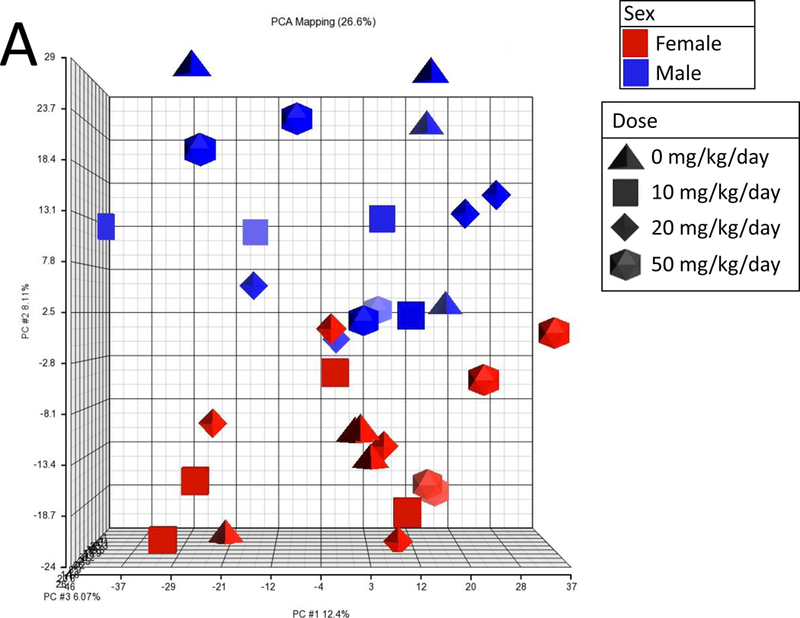
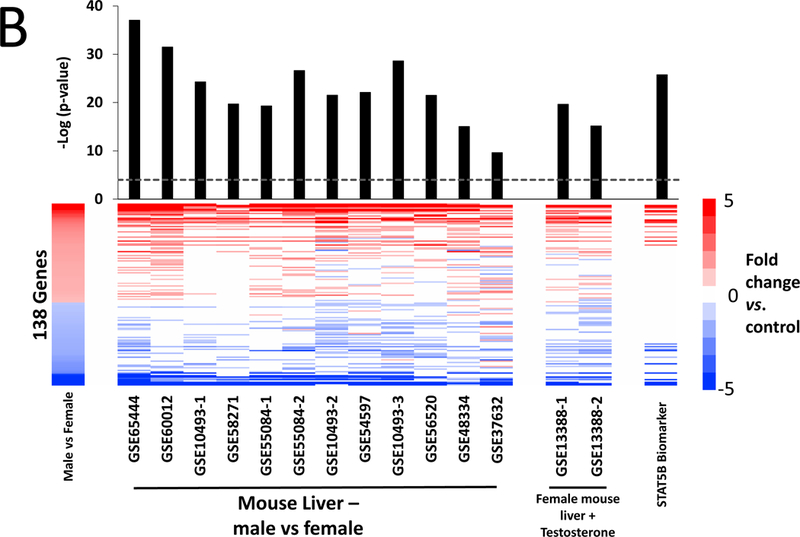
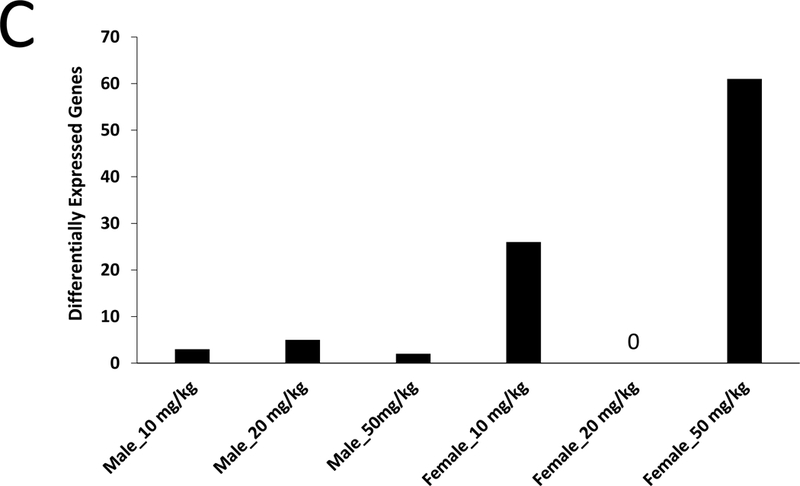
Sections of FFPE liver from male and female mice treated by gavage with 0, 10, 20, and 50 mg/kg A4 for 90 days were used to examine the expression of 3099 genes as described in the Methods. A. Principle component analysis of TempO-Seq profiles. B. Genes that were differentially expressed between control males and control females were identified as described in the Methods. The genes were compared to male vs. female comparisons from wild-type adult mice fed normal diets. The GEO number associated with each of the studies is indicated. Comparisons were made using the Running Fisher’s test in the NextBio environment as described in the Methods. The –Log(p-value) of each comparison is shown; positive values represent positive correlations. C. Number of significantly altered genes in each dose group.
The genes regulated in females at the high dose of A4 were subjected to an analysis in IPA (Supplemental Table 6). The top two canonical pathways were “Complement System” and “Acute Phase Response Signaling” and included a number of overlapping genes that were increased in expression (C1ra, C1s, C5, C8a, C9, Hp, Serpina3k). Other top 5 most significant canonical pathways included “Melatonin Degradation I”, “LPS/IL-1 Mediated Inhibition of RXR Function”, and “Nicotine Degradation II” due to changes in a number of phase I and II metabolism genes. The upstream activator analysis hypothesized that the transcription factor interferon regulatory factor 7 (IRF7) was activated due to modulation of target genes (C5, Ifi44, Ifitm3, Igtp, Irgm2). While it appears that A4 may be regulating genes which overlap with an inflammatory response, there is no evidence from histopathology in the present study or in the NTP bioassay for an inflammatory component in A4-induced liver tumors.
Marker genes for AhR, CAR, or PPARα were examined as alternate pathways of xenobiotic-induced liver cancer. Out of the 3099 genes examined, there were 18, 26, and 14 genes from the respective AhR, CAR, and PPARα gene expression biomarkers used to accurately predict receptor activation in previous studies (Oshida et al., 2015a-c). These genes included targets for AhR (Cyp1a1), CAR (Cyp2b10), or PPARα (Pdk4) activation. None of the genes in the AhR or CAR biomarkers were altered by A4, while only Hsd17b12, which is induced by PPARα activation, was increased in expression by A4 (data not shown). Of note, testosterone treatment of female mice for three weeks (from GEO study GSE13388) also did not activate AhR, CAR, or PPARα based on lack of responsiveness of target genes (data not shown). The gene expression profiles from EE and PRED groups did not significantly correlate with AhR, CAR, and PPARα biomarkers (p-values > 1E-4) (Supplemental Figure 7).
A4 masculinized the gene expression pattern in female mice.
We predicted that the gene expression pattern after A4 exposure in females from the 90-day study would resemble not only sexually dimorphic differences (males vs. females), but also the genomic profile induced by testosterone in female mice in reference datasets. For this analysis, we used the A4 profile observed at the high dose in females from the NTP study and compared the expression of these DEGs to male vs. female comparisons. Twelve male vs. female comparisons from 9 studies were randomly selected from the mouse liver compendium described in Oshida et al. (2016a). All male vs. female comparisons exhibited highly significant positive correlation to the A4 comparison (p-values = 6.4E-6 to 4.2E-14) (Figure 4A, left). The positive correlations reflected both increased expression of male-predominant genes and decreased expression of female-predominate genes by A4. Male-predominant genes included a cluster of major urinary protein (Mup) genes (Mup3, Mup12, Mup15, and Gm2083 (LOC100048885)), which are expressed in a sexually dimorphic (and often androgen-dependent) manner, with male mice expressing 2- to 8-fold higher protein levels than female mice (Cheetham et al., 2009). As testosterone treatment of female mice masculinizes the pattern of gene expression in the liver (Oshida et al., 2016a), we compared the A4-regulated genes with two testosterone vs. control comparisons in which female mice were treated twice weekly with 0.9 mg testosterone (GSE13388). There was significant positive correlation between A4 and testosterone in both comparisons (p-values = 1.5E-7 and 1.4E-8) (Figure 4A, middle).
Figure 4. A4 effects on sexually dimorphic genes.
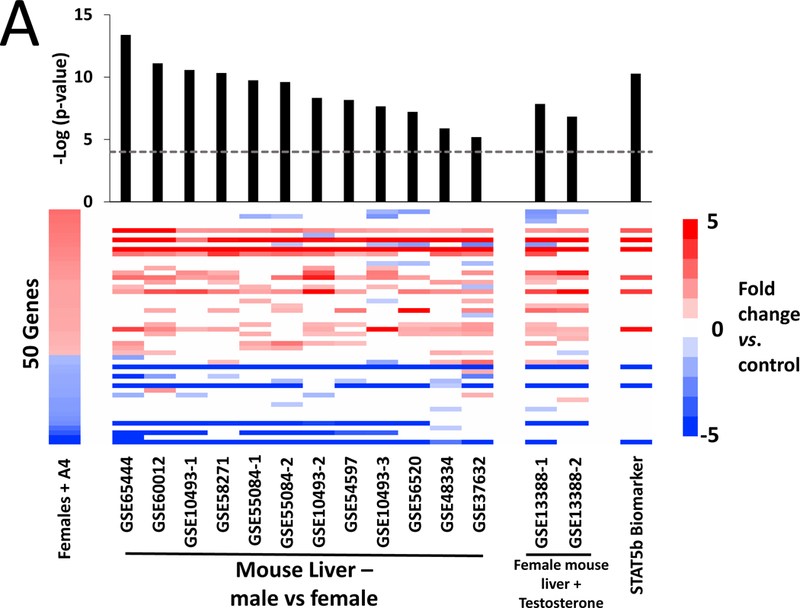
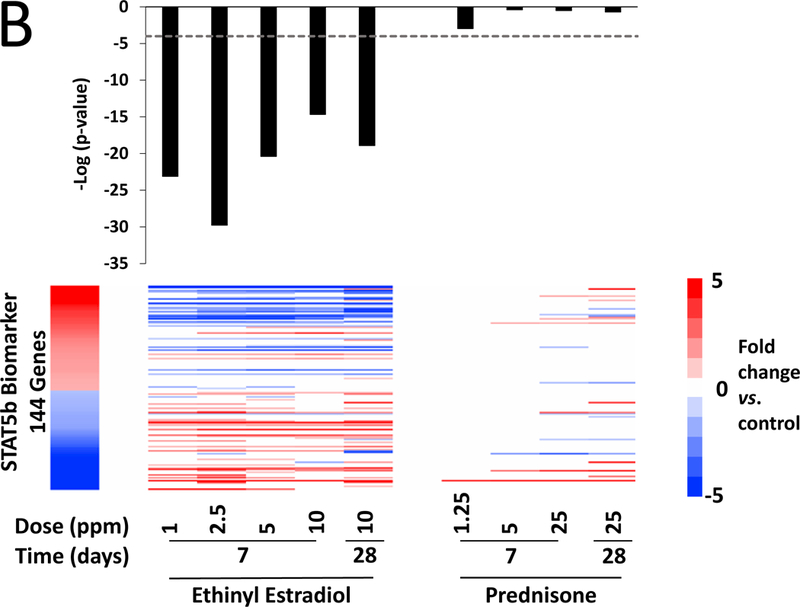
A. Significance of correlations between the A4 high dose group (left) and reference datasets for male vs. female mice, female mice treated with testosterone, and the STAT5b biomarker. Fifty genes from the 60 that were altered in the high dose group were also significantly altered in at least one of the comparisons. The male vs. female comparisons were from wild-type adult mice on standard diets. The GEO number associated with each study is indicated. Female mice were treated with testosterone (0.9 mg) or sesame oil (vehicle) twice a week for three weeks. Gene expression in the liver was analyzed either directly after treatment (GSE13388–1) or after three weeks of hormone withdrawal (GSE13388–2). The STAT5b biomarker was described previously (Oshida et al., 2016a,b). All of the indicated comparisons were compared to the A4 bioset using the Running Fisher’s test. The –Log(p-value)s of the comparisons are shown. B. Significance of the correlations between the genes altered by EE and PRED and the STAT5b biomarker. The –Log(p-value)s with negative values represent pairwise comparisons with negative correlation.
Differences in sex-dependent gene expression in the rodent liver are regulated by the transcription factor STAT5b, which is activated by the male-specific pulsatile pattern of growth hormone release from the pituitary (described in Waxman and O’Connor, 2006). To determine if the A4-regulated genes were dependent on STAT5b, the A4 comparison was compared to the STAT5b gene expression biomarker consisting of genes that are differentially expressed between males and females and dependent on STAT5b for sex differences. This biomarker has been previously found to accurately predict STAT5b activation or suppression in a mouse liver gene expression compendium (Oshida et al., 2016a,b). The p-value of the positive correlation was 5.2E-11 (Figure 4A, right). There was also highly significant negative correlation between the STAT5b biomarker and all of the EE treatment groups, consistent with suppression of the male-specific STAT5b activation pattern and feminization in male mice by estrogen treatment (discussed in Oshida et al., 2016a,b) (Figure 4B). There were no consistent correlations between PRED comparisons and the STAT5b biomarker. Overall, these results indicate that A4 exerts effects on expression of sexually dimorphic genes that are dependent on STAT5b.
DISCUSSION
Androgens induce liver cancer in mice and likely contribute to the higher risk of liver cancer in men compared to women (Zhang et al., 2015). Mechanistic drivers and early biomarkers for these effects are not well defined. In this study, we evaluated short-term effects of the androgen A4 in reference to other steroid hormones and investigated potential growth factor pathways mediated by androgenic signaling. Treatment of male mice with A4 did not induce key events for common chemical-induced modes of action (MOA) of liver cancer (Holsapple et al., 2009). A4 did not increase liver cell hypertrophy, cytotoxicity, or proliferation, alter enzymatic markers of xenobiotic receptor activation, or result in significant transcriptional changes at 7 or 28 days. Further analysis of dose-matched archival samples from the 90-day NTP study of A4 showed no consistent changes in genes regulated by AhR, CAR, or PPARα, which are common mitogenic targets of liver carcinogens. There were modest effects of A4 on expression of sexually dimorphic genes in female mice that are dependent on STAT5b and suppressed by estrogen. These findings indicate that liver cancer induced by A4 does not occur through the typical targets of liver carcinogens or through direct AR-mediated mitogenesis but rather by subtle long-term effects on male-predominant growth factor pathways that may be coordinated by AR or STAT5b.
The lack of short-term morphologic effects is unusual among liver carcinogens. A previous analysis of NTP studies by Allen et al. (2004) showed that 93% (25/27) of mouse liver carcinogens (genotoxic or not) exhibited increased liver weight and/or hepatocyte hypertrophy, cytomegaly, or necrosis in the corresponding pre-chronic 90-day studies. For A4, the absence of histopathological effects at 7 or 28 days is consistent with prior mouse NTP studies at 14 and 90 days, which found no effects in either sex across the same dose range (NTP 2010). A4 also had no effects on liver cell proliferation, in contrast to PB, the positive mitogenic control. This latter finding is supported by the NTP 14-day studies, which found no effect of A4 on liver cell proliferation markers as assessed by enzymatic assays in male and female mice. Most known non-genotoxic chemical carcinogens induce cancer through mitogenic effects, either directly or secondary to cytotoxicity (Wood et al., 2015), so the clear lack of a short-term proliferative effect here is notable. A4 and possibly other androgens may induce liver cancer by a MOA that requires an interaction with age-related factors. At least in male mice, A4 may maintain activation of androgen-dependent genes that normally decline with age (Oshida et al., 2016a).
Given that A4 is a direct precursor of testosterone and an AR agonist, we hypothesized that A4 increases liver cancer incidence through an androgen-dependent mechanism that may also underlie the greater susceptibility of males to liver cancer. Liver cancer incidence and mortality is higher in men than women in almost all populations examined (Ferlay et al., 2010), and in the U.S., the incidence is approximately three-fold higher in men compared to women (Altekruse et al., 2009). Sex hormones are thought to play a key role in this male-predominant pattern, although the molecular pathways are largely unknown. Male mice have higher incidences of both spontaneous and chemically-induced liver tumors compared to female mice (Smith et al., 1973; Toh, 1981; Vesselinovitch and Mihailovich, 1967), and castrated male mice have a lower incidence of spontaneous and chemically-induced liver cancer compared to intact mice (Bugni et al., 2001; Kemp et al., 1989a; Poole and Drinkwater, 1996; Toh, 1981; Vesselinovitch and Mihailovich, 1967). Conversely, ovariectomized female mice develop greater numbers of liver tumors compared to intact female mice (Bugni et al., 2001; Poole and Drinkwater, 1996; Vesselinovitch and Mihailovich, 1967; Yamamoto et al., 1991). In the A4 carcinogenicity studies, female mice also exhibited a more statistically significant liver tumor induction than male mice (NTP 2010) (Supplemental Figure 8). These studies collectively support the idea that androgens enhance and ovarian hormones suppress liver carcinogenesis.
If A4 is acting through AR-mediated pathways, genomic responses should be consistent with sexually dimorphic patterns. After 90 days of A4 exposure, gene expression changes were seen primarily in female mice, which had a larger set of genes at the highest dose tested (a total of 60 genes or 1.9% of the total) compared to male mice. The genes altered by the high dose of A4 in female mice were found to have significant positive correlations to 12 male vs. female comparisons across 9 different microarray studies. The A4-regulated gene set also exhibited significant positive correlation to the genes altered by a 3-week testosterone treatment in the livers of female mice. For these comparisons, the alterations included both increased expression of male-predominant genes as well as decreased expression of female-predominant genes. These comparisons support the idea that A4 is inducing a more male-like gene expression pattern.
A central candidate for regulating A4-induced genes is STAT5b, a master regulator of sexually dimorphic gene expression in the liver controlled by the male-specific pulsatile pattern of growth hormone secretion from the pituitary (Clodfelter et al., 2006, Zhang et al., 2012). We determined if the genes regulated by A4 overlapped with those regulated by STAT5b. Expression of A4-regulated genes was compared to a biomarker that accurately predicts STAT5b activation or suppression (Oshida et al., 2016a,b). This comparison showed significant positive correlation (p-value = 5.2E-11), providing strong support that A4 is activating STAT5b. In contrast, there were highly significant negative correlations between the STAT5b biomarker and the EE comparisons (p-value range = 4.8E-14 – 5.9E-29), consistent with the idea that estrogens suppress the growth hormone pulsatile pattern and, secondarily, STAT5b activation (Waxman and O’Connor, 2006). These results demonstrate that A4 causes masculinization of gene expression, most likely through STAT5b.
Previous studies have identified a role for both STAT5b and AR in liver carcinogenesis. STAT5b signaling was examined in three mouse strains by assessing liver cancer in the presence (wild-type) or absence of STAT5b (Stat5b-null mice) following diethylnitrosamine (DEN) exposure (Oberly et al., 2014). Stat5b-null mice on the C57BL/6J background developed more DEN-induced liver tumors than wild-type mice, while loss of STAT5b had no effect on the BALB/cJ background, and led to resistance to liver cancer on the C3H/HeJ background. It should be noted that the strain used in the NTP study was B6C3F1, a cross between female C57BL/6 and male C3H mice. Thus, STAT5b appears to act either as an oncogene or a tumor suppressor depending on the background strain. AR is another candidate target of A4 carcinogenesis. By comparing effects between wild-type and hepatocyte-specific AR-null mice, AR has been shown to be required for DEN- and hepatitis B virus-mediated hepatocellular carcinoma (Ma et al., 2008; Wu et al., 2010). In males, studies using mice lacking a functional AR (testicular feminization (Tfm) mice) have also revealed the importance of AR in promotion of liver tumors, as DEN treatment resulted in >25-fold more tumors in wild-type male mice than their Tfm/Y counterparts (Kemp et al., 1989b). Further studies are required to determine if STAT5b and AR are involved in A4-induced liver cancer, particularly as mice age and endogenous androgens decline (Oshida et al., 2016a).
Increased cellular proliferation is a central key event in the MOA for many non-genotoxic chemical carcinogens (Wood et al., 2015). In our study, exposure to EE led to increases in hepatocyte proliferation at 7 and 28 days. This finding is notable given that four of the five chronic bioassays of EE resulted in no significant increases in liver tumors in mice, and the one statistically significant increase has been considered equivocal because of the relatively low spontaneous liver tumor incidence and the small increase over controls in the treated animals (Schuppler and Gunzel, 1979). There are several differences in study design that might explain these apparent discrepancies. While we used B6C3F1 mice, CF-LP, and BDH-SPF strains were used in the chronic bioassays. We cannot rule out that B6C3F1 mice exhibit different short- and long-term responses to EE compared to the strains used in the bioassays or that B6C3F1 mice might be susceptible to increases in liver tumorigenesis resulting from EE. In addition, our cell proliferation results contrast with three studies that showed suppression of liver foci growth by EE in initiation-promotion assays (Lee et al., 1989; Standeven et al., 1994; Moser et al., 1996). None of these studies measured cell proliferation at time points that overlapped with our 28-day window. One study found that there was an approximate 2-fold (not significantly significant) increase in cell proliferation in the non-foci hepatocytes in female B6C3F1 mice upon treatment with EE for 16 weeks compared to non-foci hepatocytes from untreated mice (Moser et al., 1996). In a similar study from the same laboratory using B6C3F1 mice, there were increases in liver to body weights of ~70% upon EE exposure (Standeven et al., 1994), indicating that the liver increased in size due to hypertrophy and/or hyperplasia. The behavior of EE may be similar to that of peroxisome proliferators in which short-term exposures transiently induce cell proliferation but act to promote and induce cell proliferation in only a subset of foci, in this case basophilic foci, while apparently inhibiting growth in eosinophilic foci (Cattley and Popp, 1989; Marsman and Popp, 1994). It cannot be ruled out that EE under the conditions that increase acute cell proliferation might increase liver tumor induction only in a subset of foci that were not observed in the initiation-promotion studies.
One difference between our study and that of the NTP 2010 study is the composition of the diets. Diet is an important factor in the growth, body weight, survival, and development of age-related diseases in rodent chronic studies (Rao and Crockett, 2003). In the present study, AIN-93G was used, while in the NTP study, the NTP-2000 diet was used. It is possible that differences in diets could account for differences in responses to A4. However, we feel that this is unlikely based on the uniform lack of responses after short-term exposures in both studies. The fact that we did not see any morphologic effects in our 7 and 28 day studies is completely consistent with the original NTP study in which short-term effects on liver histopathology were not observed in male or female mice at 14 or 90 days. The results of the gene expression analysis of the FFPE livers from the 90-day NTP study were also consistent with the lack of histopathological effects in those same livers. We observed changes in a relatively low number of genes (~60 out of 3099 examined). Thus, our results are consistent with the NTP study and argue that differences in diet do not contribute to differences in responses to A4.
In summary, the short-term molecular effects of A4 exposure are not consistent with classical mitogenic or cytotoxic pathways of liver carcinogenicity. After 90 days of treatment, we identified genomic effects of A4 similar to sexually dimorphic patterns between males and females. We propose that A4 is acting via growth factor pathways that drive age-dependent increased susceptibility of males to liver cancer through an as yet unidentified mechanism. STAT5b activation in the livers of chemically-treated mice could be used as an indicator of such risk.
Supplementary Material
Acknowledgements
The authors would like to thank Tanya Moore, Brenda Edwards, Tony McDonald, Gail Nelson, Delores Kay Rigsbee, Kim Howell, Debbie Crawford, Beth Padnos, Judy Richards, Gleta Carswell, David Ross and Carlton Jones for technical assistance, including in-life portions of the study; Anna Fisher and Beena Vallanat of the NHEERL Genomic Research Cores Unit for microarray data generation; staff at EPL for histology, histopathology, and immunohistochemical support; staff at BioSpyder for technical assistance in carrying out the TempO-Seq assay; and Jennifer Fostel and Isabel Lee for archiving the microarray files in GEO. We also thank Drs. Norman Drinkwater and David Waxman for critical review of this manuscript. Funding for this study came from the U.S. EPA Office of Research and Development.
Disclaimer
The research described in this article has been reviewed by the U.S. EPA and approved for publication. Approval does not signify that the contents necessarily reflect the views or the policies of the Agency. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
References
- Allen DG, Pearse G, Haseman JK, and Maronpot RR. (2004). Prediction of rodent carcinogenesis: an evaluation of prechronic liver lesions as forecasters of liver tumors in NTP carcinogenicity studies. Toxicol Pathol 32(4), 393–401. [DOI] [PubMed] [Google Scholar]
- Altekruse SF, McGlynn KA, and Reichman ME. (2009). Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol 27(9), 1485–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blystone CR, Elmore SA, Witt KL, Malarkey DR, and Foster PM. (2011). Toxicity and Carcinogenicity of Androstenedione in F344/N Rats and B6C3F1 Mice. Food Chem Toxicol 49(9), 2116–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boobis AR, Cohen SM, Dellarco V, McGregor D, Meek ME, Vickers C, Willcocks D, and Farland W. (2006) IPCS framework for analyzing the relevance of a cancer mode of action for humans. Crit Rev Toxicol 36(10), 781–92. [DOI] [PubMed] [Google Scholar]
- Bugni JM, Poole TM, and Drinkwater NR. (2001). The little mutation suppresses DEN-induced hepatocarcinogenesis in mice and abrogates genetic and hormonal modulation of susceptibility. Carcinogenesis 22, 1853–62. [DOI] [PubMed] [Google Scholar]
- Burke MD, and Mayer Rt. (1974). Ethoxyresorufin: direct fluorimetric assay of amicrosomal dealkylation which is preferentially inducible by 3-methylcholanthrene. Drug Metab Dispos 2(6), 583–8. [PubMed] [Google Scholar]
- Burke MD, Thompson S, Elcombe CR, Halpert J, Haparanta T, and Mayer RT. (1985). Ethoxy-, pentoxy- and benzyloxyphenoxazones and homologues: a series of substrates to distinguish between different induced cytochromes P-450. Biochem Pharmacol 34(18), 3337–45. [DOI] [PubMed] [Google Scholar]
- Cain DW and Cidlowski JA. (2015). Specificity and sensitivity of glucocorticoid signaling in health and disease. Best Pract Res Clin Endocrinol Metab 29(4), 545–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattley RC, Popp JA. Differences between the promoting activities of the peroxisome proliferator WY-14,643 and phenobarbital in rat liver. Cancer Res. 1989. June 15;49(12):3246–51. [PubMed] [Google Scholar]
- Cheetham SA, Smith AL, Armstrong SD, Beynon RJ, and Hurst JL. (2009). Limited variation in the major urinary proteins of laboratory mice. Physiol Behav 96(2), 253–61. [DOI] [PubMed] [Google Scholar]
- Clodfelter KH, Holloway MG, Hodor P, Park SH, Ray WJ, and Waxman DJ. (2006). Sex-dependent liver gene expression is extensive and largely dependent upon signal transducer and activator of transcription 5b (STAT5b): STAT5b-dependent activation of male genes and repression of female genes revealed by microarray analysis. Mol Endocrinol 20(6), 1333–51. [DOI] [PubMed] [Google Scholar]
- Cohen SM 2004. Human carcinogenic risk evaluation: an alternative approach to the two-year rodent bioassay. Toxicol Sci 80, 225–229. [DOI] [PubMed] [Google Scholar]
- Crissman JW, Goodman DG, Hildebrandt PK, Maronpot RR, Prater DA, Riley JH, Seaman WJ, and Thake DC. (2004). Best practices guideline: Toxicologic pathology. Toxicol Pathol 32, 126–31. [DOI] [PubMed] [Google Scholar]
- Davey RA and Grossmann M. (2016). Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin Biochem Rev 37(1), 3–15. [PMC free article] [PubMed] [Google Scholar]
- De Maria N, Manno M, and Villa E. (2002). Sex hormones and liver cancer. Mol Cell Endocrinol 193(1–2), 59–63. [DOI] [PubMed] [Google Scholar]
- Deb S, Pandey M, Adomat H, and Tomlinson Guns ES. (2012). Cytochrome P450 3A-mediated microsomal biotransformation of 1α, 25-dihydroxyvitamin D3 in mouse and human liver: drug-related induction and inhibition of catabolism. Drug Metab Dispos 40(5), 907–18. [DOI] [PubMed] [Google Scholar]
- Delić D, Grosser C, Dkhil M, Al-Quraishy S, and Wunderlich F. (2010). Testosterone-induced upregulation of miRNAs in the female mouse liver. Steroids 75(12), 998–1004. [DOI] [PubMed] [Google Scholar]
- Dillberger JE, Cronin NS, and Carr GJ. (1992). Prednisone is not a mouse carcinogen. Toxicol Pathol 20(1), 18–26. [DOI] [PubMed] [Google Scholar]
- Dodge T and Hoagland MF. (2011). The use of anabolic androgenic steroids and polypharmacy: a review of the literature. Drug Alcohol Depend 114(2–3), 100–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EPA (U.S. Environmental Protection Agency). Guidelines for Carcinogen Risk Assessment. EPA/630/P-03/001F. Risk Assessment Forum, Washington, DC, March 2005. Available at: https://www.epa.gov/risk/guidelines-carcinogen-risk-assessment. Accessed April 13, 2016. [Google Scholar]
- Ferlay J, Shin H, Bray F, Forman D, Mathers C, and Parkin DM. (2010). Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127(12), 2893–917. [DOI] [PubMed] [Google Scholar]
- Fernandez E, Gallus S, Bosetti C, Franceschi S, Negri E, and La Vecchia C. (2003) Hormone replacement therapy and cancer risk: a systematic analysis from a network of case-control studies. Int J Cancer 105(3), 408–12. [DOI] [PubMed] [Google Scholar]
- Frijters R, Fleuren W, Toonen EJ, Tuckermann JP, Reichardt HM, van der Maaden H, van Elsas A, van Lierop MJ, Dokter W, de Vlieg J and Alkema W. (2010). Prednisolone-induced differential gene expression in mouse liver carrying wild type or a dimerization-defective glucocorticoid receptor. BMC Genomics 11, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannitrapani L, Soresi M, La Spada E, Cervello M, D’Alessandro N, and Montalto G. (2006). Sex hormones and risk of liver tumor. Ann N Y Acad Sci 1089, 228–36. [DOI] [PubMed] [Google Scholar]
- Guengerich FP. (1988). Oxidation of 17 alpha-ethynylestradiol by human liver cytochrome P-450. Mol Pharmacol 33(5), 500–8. [PubMed] [Google Scholar]
- Guengerich FP, Dannan GA, Wright ST, Martin MV, and Kaminsky LS. (1982). Purification and characterization of microsomal cytochrome P-450s. Xenobiotica 12(11), 701–16. [DOI] [PubMed] [Google Scholar]
- Hagemeyer CE, Burck C, Schwab R, Knoth R, and Meyer RP. (2010). 7-Benzyloxyresorufin-O-dealkylase activity as a marker for measuring cytochrome P450 CYP3A induction in mouse liver. Anal Biochem 398(1), 104–11. [DOI] [PubMed] [Google Scholar]
- Hall AP, Elcombe CR, Foster JR, Harada T, Kaufmann W, Knippel A, Küttler K, Malarkey DE, Maronpot RR, Nishikawa A, Nolte T, Schulte A, Strauss V, and York MJ. (2012). Liver hypertrophy: a review of adaptive (adverse and non-adverse) changes--conclusions from the 3rd International ESTP Expert Workshop. Toxicol Pathol 40, 971–94. [DOI] [PubMed] [Google Scholar]
- Holsapple MP, Pitot HC, Cohen SM, Boobis AR, Klaunig JE, Pastoor T, Dellarco VL, and Dragan YP. (2006). Mode of action MOA in relevance of rodent liver tumors to human cancer risk. Toxicol Sci 89(1), 51–6. Erratum in: Toxicol Sci 90(2), 596. [DOI] [PubMed] [Google Scholar]
- IARC. (2007). Combined estrogen-progesterone contraceptives and combined estrogen-progestogen menopausal therapy. IARC Monogr Eval Carcinog Risks Hum 91, 1–528. [PMC free article] [PubMed] [Google Scholar]
- Kemp CJ and Drinkwater NR. (1989a). Genetic variation in liver tumor susceptibility, plasma testosterone levels, and androgen receptor binding in six inbred strains of mice. Cancer Res 49, 5044–47. [PubMed] [Google Scholar]
- Kemp CJ, Leary CN, and Drinkwater NR. (1989b). Promotion of murine hepatocarcinogenesis by testosterone is androgen receptor-dependent but not cell autonomous. Proc Natl Acad Sci USA 86, 7505–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent UM, Mills DE, Rajnarayanan RV, Alworth WL, and Hollenberg PF (2002). Effect of 17-α-ethynylestradiol on activities of cytochrome P450 2B (P450 2B) enzymes: characterization of inactivation of P450s 2B1 and 2B6 and identification of metabolites. J Pharmacol Exp Ther 300(2), 549–58. [DOI] [PubMed] [Google Scholar]
- Kupershmidt I, Su QJ, Grewal A, Sundaresh S, Halperin I, Flynn J, Shekar M, Wang H, Park J, Cui W, Wall GD, Wisotzkey R, Alag S, Akhtari S, and Ronaghi M. (2010). Ontology-based meta-analysis of global collections of high-throughput public data. PLoS One 5, e13066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake AD, Wood CE, Bhat VS, Chorley BN, Carswell GK, Sey YM, Kenyon EM, Padnos B, Moore TM, Tennant AH, Schmid JE, George BJ, Ross DG, Hughes MF, Corton JC, Simmons JE, McQueen CA, and Hester SD. (2016). Dose and Effect Thresholds for Early Key Events in a PPARα-Mediated Mode of Action. Toxicol Sci 149(2), 312–25. [DOI] [PubMed] [Google Scholar]
- Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, and Kliewer SA. (1998). The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Invest 102(5), 1016–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GH, Nomura K, and Kitagawa T. (1989). Comparative study of diethylnitrosamine-initiated two-stage hepatocarcinogenesis in C3, C57BL and BALB mice promoted by various hepatopromoters. Carcinogenesis 10(12), 2227–30. [DOI] [PubMed] [Google Scholar]
- Li Z, Tuteja G, Schug J, Kaestner KH. (2012). Foxa1 and Foxa2 are essential for sexual dimorphism in liver cancer. Cell 148(1–2),72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ and Schmittgen TD. (2001). Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2 to the negative delta delta CT Method. Methods 25(4), 402–8. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12), 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijten M, Olthof ED, Hakkert BC, Rorije E, van der Laan JW, Woutersen RA, van Benthem J. An integrative test strategy for cancer hazard identification. Crit Rev Toxicol 2016. August;46(7):615–39. [DOI] [PubMed] [Google Scholar]
- Ma Q, and Lu AYH. (2007). CYP1A induction and human risk assessment: an evolving tale of in vitro and in vivo studies. Drug Metab Dispos 34(7), 1009–16. [DOI] [PubMed] [Google Scholar]
- Ma WL, Hsu CL, Wu MH, Wu CT, Wu CC, Lai JJ, Jou YS, Chen CW, Yeh S, and Chang C. (2008). Androgen receptor is a new potential therapeutic target for the treatment of hepatocellular carcinoma. Gastroenterology 135(3), 947–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma WL, Lai HC, Yeh S, Cai X, and Chang C. (2014). Androgen receptor roles in hepatocellular carcinoma, fatty liver, cirrhosis and hepatitis. Endocr Relat Cancer 21(3), R165–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsman DS, Popp JA. Biological potential of basophilic hepatocellular foci and hepatic adenoma induced by the peroxisome proliferator, Wy-14,643. Carcinogenesis. 1994. January;15(1):111–7. [DOI] [PubMed] [Google Scholar]
- Matthews LC, Berry AA, Morgan DJ, Poolman TM, Bauer K, Kramer F, Spiller DG, Richardson RV, Chapman KE, Farrow SN, Norman MR, Williamson AJ, Whetton AD, Taylor SS, Tuckermann JP, White MR, and Ray DW. (2015). Glucocorticoid receptor regulates accurate chromosome segregation and is associated with malignancy. Proc Natl Acad Sci USA 112(17), 5479–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser GJ, Wolf DC, Harden R, Standeven AM, Mills J, Jirtle RL, and Goldsworthy TL. (1996). Cell proliferation and regulation of negative growth factors in mouse liver foci. Carcinogenesis 17(9), 1835–40. [DOI] [PubMed] [Google Scholar]
- Mueller KM, Kornfeld JW, Friedbichler K, Blaas L, Egger G, Esterbauer H, Hasselblatt P, Schlederer M, Haindl S, Wagner KU, Engblom D, Haemmerle G, Kratky D, Sexl V, Kenner L, Kozlov AV, Terracciano L, Zechner R, Schuetz G, Casanova E, Pospisilik JA, Heim MH, and Moriggl R. (2011). Impairment of hepatic growth hormone and glucocorticoid receptor signaling causes steatosis and hepatocellular carcinoma in mice. Hepatology 54(4), 1398–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council (NRC), Guide for the Care and Use of Laboratory Animals. 8th Edition National Academies Press, Washington, DC: 2011 [Google Scholar]
- National Toxicology Program. (1999). NTP Toxicology and Carcinogenesis Studies of Oxymetholone (CAS NO. 434–07-1) in F344/N Rats and Toxicology Studies of Oxymetholone in B6C3F1 Mice (Gavage Studies). Natl Toxicol Program Tech Rep Ser 485, 1–233. [PubMed] [Google Scholar]
- National Toxicology Program. (2010). Toxicology and carcinogenesis study of androstenedione (CAS No. 63–05-8) in F344/N rats and B6C3F1 mice (gavage studies). National Toxicology Program Tech Rep 560, 1–196. [PubMed] [Google Scholar]
- Oberley CC, Bilger A, and Drinkwater NR. (2014). Genetic background determines if Stat5b suppresses or enhances murine hepatocarcinogenesis. Mol Carcinog 54(10), 959–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshida K, Vasani N, Thomas RS, Applegate D, Gonzalez FJ, Aleksunes LM, Klaassen CD, and Corton JC. (2015a). Screening a mouse liver gene expression compendium identifies modulators of the aryl hydrocarbon receptor (AhR). Toxicology 336, 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshida K, Vasani N, Jones C, Moore T, Hester S, Nesnow S, Auerbach S, Geter DR, Aleksunes LM, Thomas RS, Applegate D, Klaassen CD, and Corton JC. (2015b). Identification of chemical modulators of the constitutive activated receptor (CAR) in a gene expression compendium. Nucl Recept Signal 13, e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshida K, Vasani N, Thomas RS, Applegate D, Rosen M, Abbott B, Lau C, Guo G, Aleksunes LM, Klaassen C, and Corton JC. (2015c). Identification of modulators of the nuclear receptor peroxisome proliferator-activated receptor α (PPARα) in a mouse liver gene expression compendium. PLoS One 10(2), e0112655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshida K, Waxman DJ, and Corton JC. (2016a). Chemical and Hormonal Effects on STAT5b-Dependent Sexual Dimorphism of the Liver Transcriptome. PLoS One 11(3), e0150284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshida K, Vasani N, Waxman DJ, and Corton JC. (2016b). Disruption of STAT5b-Regulated Sexual Dimorphism of the Liver Transcriptome by Diverse Factors Is a Common Event. PLoS One 11(3), e0148308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedram A, Razandi M, O’Mahony F, Harvey H, Harvey BJ, and Levin ER. (2013). Estrogen reduces lipid content in the liver exclusively from membrane receptor signaling. Sci Signal 6(276), ra36. [DOI] [PubMed] [Google Scholar]
- Pichard L, Fabre I, Daujat M, Domergue J, Joyeux H, and Maurel P. (1992). Effect of corticosteroids on the expression of cytochromes P450 and on cyclosporin A oxidase activity in primary cultures of human hepatocytes. Mol Pharmacol 41(6), 1047–55. [PubMed] [Google Scholar]
- Poole TM and Drinkwater NR. (1996). Strain dependent effects of sex hormones on hepatocarcinogenesis in mice. Carcinogenesis 17, 191–6. [DOI] [PubMed] [Google Scholar]
- Rao GN, Crockett PW. Effect of diet and housing on growth, body weight, survival and tumor incidences of B6C3F1 mice in chronic studies. Toxicol Pathol. 2003. Mar-Apr;31(2):243–50. [DOI] [PubMed] [Google Scholar]
- Reznik-Schuller H (1979) Carcinogenic effects of diethylstilbestrol in male Syrian golden hamsters and European hamsters. J Natl Cancer Inst.62, 1083–8. [PubMed] [Google Scholar]
- Schuppler J and Günzel P. (1979). Liver tumors and steroid hormones in rats and mice. Arch Toxicol Suppl 2, 181–95. [DOI] [PubMed] [Google Scholar]
- Shepard C, Finelli L, and Alter M. (2005). Global epidemiology of hepatitis C virus infection. Lancet Infect Dis 5(9), 558–67. [DOI] [PubMed] [Google Scholar]
- Shi L, Feng Y, Lin H, Ma R, and Cai X. (2014). Role of estrogen in hepatocellular carcinoma: is inflammation the key? J Transl Med 12, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon TW, Simons SS Jr, Preston RJ, Boobis AR, Cohen SM, Doerrer NG, Fenner-Crisp PA, McMullin TS, McQueen CA, Rowlands JC; RISK21 Dose-Response Subteam. The use of mode of action information in risk assessment: quantitative key events/dose-response framework for modeling the dose-response for key events. Crit Rev Toxicol 2014. August;44 Suppl 3:17–43. [DOI] [PubMed] [Google Scholar]
- Smith GS, Walford RL, and Mickey MR. (1973). Lifespan and incidence of cancer and other diseases in selected long-lived inbred mice and their F1 hybrids. J Natl Cancer Inst 50, 1195–1213. [DOI] [PubMed] [Google Scholar]
- Standeven AM, Wolf DC, and Goldsworthy TL. (1994). Interactive effects of unleaded gasoline and estrogen on liver tumor promotion in female B6C3F1 mice. Cancer Res 54(5), 1198–204. [PubMed] [Google Scholar]
- Thigpen JE, Setchell KD, Ahlmark KB, Locklear J, Spahr T, Caviness GF, Goelz MF, Haseman JK, Newbold RR, Forsythe DB. (1999). Phytoestrogen content of purified, open- and closed-formula laboratory animal diets. Lab Anim Sci.49(5):530–6. [PubMed] [Google Scholar]
- Thigpen JE, Setchell KD, Kissling GE, Locklear J, Caviness GV, Whiteside T, Belckher SM, Brown NM, Colling BJ, Lih FB, Tmer KB, Padilla-Banks E, Camacho L, Adsit FG, Grant M. (2013) The estrogenic content of rodent diets, bedding, cages, and water bottles and its effect on bisphenol A studies. J Am Assoc Lab Anim Sci 42(2):131–41. [PMC free article] [PubMed] [Google Scholar]
- Thoolen B, Maronpot RR, Harada T, Nyska A, Rousseaux C, Nolte T, Malarkey DE, Kaufmann W, Küttler K, Deschl U, Nakae D, Gregson R, Vinlove MP, Brix AE, Singh B, Belpoggi F, and Ward JM. (2010). Proliferative and nonproliferative lesions of the rat and mouse hepatobiliary system. Toxicol Pathol 38, 5S–81S. [DOI] [PubMed] [Google Scholar]
- Toh YC. (1981). Effect of neonatal castration on liver tumor induction by N-2-fluorenylacetamide in suckling BALB/c mice. Carcinogenesis 2, 1219–21. [DOI] [PubMed] [Google Scholar]
- Tyl RW, Myers CB, Marr MC, Castillo NP, Veselica MM, Joiner RL, Dimond SS, Van Miller JP, Stropp GD, Waechter JM Jr., and Hentges SG (2008). One-generation reproductive toxicity study of dietary 17β-estradiol (E2; CAS No. 50–28-2) in CD-1® (Swiss) mice. Reprod. Toxicol 25(2), 144–60. [DOI] [PubMed] [Google Scholar]
- Vesselinovitch SD and Mihailovich N. (1967). The effect of gonadectomy on the development of hepatomas induced by urethan. Cancer Res 27, 1788–91. [PubMed] [Google Scholar]
- Villeneuve DL, Crump D, Garcia-Reyero N, Hecker M, Hutchinson TH, LaLone CA, Landesmann B, Lettieri T, Munn S, Nepelska M, Ottinger MA, Vergauwen L, and Whelan M. (2014). Adverse outcome pathway development II: best practices. Toxicol Sci 142(2), 321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve DL, Crump D, Garcia-Reyero N, Hecker M, Hutchinson TH, LaLone CA, Landesmann B, Lettieri T, Munn S, Nepelska M, Ottinger MA, Vergauwen L, and Whelan M. (2014). Adverse outcome pathway (AOP) development I: strategies and principles. Toxicol Sci 142(2), 312–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman DJ and O’Connor C. (2006). Growth hormone regulation of sex-dependent liver gene expression. Mol Endocrinol 20(11), 2613–29. [DOI] [PubMed] [Google Scholar]
- Wilsnack R, Wilsnack S, Kristjanson A, Vogeltanz-Holm N, and Gmel G. Gender and alcohol consumption: Patterns from the multinational GENACIS project. Addiction 104(9), 1487–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood CE, Hukkanen RR, Sura R, Jacobson-Kram D, Nolte T, Odin M, and Cohen SM. (2015). Scientific and Regulatory Policy Committee (SRPC) Review: Interpretation and Use of Cell Proliferation Data in Cancer Risk Assessment. Toxicol Pathol 43(6), 760–75. [DOI] [PubMed] [Google Scholar]
- Wu MH, Ma WL, Hsu CL, Chen YL, Ou JH, Ryan CK, Hung YC, Yeh S, and Chang C. (2010). Androgen receptor promotes hepatitis B virus-induced hepatocarcinogenesis through modulation of hepatitis B virus RNA transcription. Sci Transl Med 2(32), 32ra35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto R, Iishi H, Tatsuta M, Tsuji M, and Terada N. (1991). Roles of ovaries and testes in hepatocellular tumorigenesis induced in mice by 3′-methyl-4-dimethylaminoazobenzene. Int J Cancer 49, 83–8. [DOI] [PubMed] [Google Scholar]
- Yoshinari K, Sueyoshi T, Moore R, and Negishi M. (2001). Nuclear receptor CAR as a regulatory factor for the sexually dimorphic induction of CYP2B1 gene by phenobarbital in rat livers. Mol Pharmacol 59(2), 278–84. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Laz EV, Waxman DJ. (2012). Dynamic, sex-differential STAT5 and BCL6 binding to sex-biased, growth hormone-regulated genes in adult mouse liver. Mol Cell Biol 32(4), 880–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ren JS, Shi JF, Li N, Wang YT, Qu C, Zhang Y, and Dai M. (2015). International trends in primary liver cancer incidence from 1973 to 2007. BMC Cancer 15, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.