

Abstract
Mitochondrial nitric oxide synthase (mtNOS), its cellular NOS isoform, and the effects of mitochondrially produced NO on bioenergetics have been controversial since mtNOS was first proposed in 1995. Here we functionally demonstrate the presence of a NOS in cardiac mitochondria. This was accomplished by direct porphyrinic microsensor measurement of Ca2+-dependent NO production in individual mitochondria isolated from wild-type mouse hearts. This NO production could be inhibited by NOS antagonists or protonophore collapse of the mitochondrial membrane potential. The similarity of mtNOS to the neuronal isoform was deduced by the absence of NO production in the mitochondria of knockout mice for the neuronal, but not the endothelial or inducible, isoforms. The effects of mitochondrially produced NO on bioenergetics were studied in intact cardiomyocytes isolated from dystrophin-deficient (mdx) mice. mdx cardiomyocytes are also deficient in cellular endothelial NOS, but overexpress mtNOS, which allowed us to study the mitochondrial enzyme in intact cells free of its cytosolic counterpart. In these cardiomyocytes, which produce NO beat-to-beat, inhibition of mtNOS increased myocyte shortening by approximately one-fourth. Beat-to-beat NO production and altered shortening by NOS inhibition were not observed in wild-type cells. A plausible mechanism for the reversible NO inhibition of contractility in these cells involves the reaction of NO with cytochrome c oxidase. This suggests a modulatory role for NO in oxidative phosphorylation and, in turn, myocardial contractility.
Keywords: cardiomyocytes‖cytochrome oxidase‖oxidative phosphorylation‖ respiration‖chronoamperometry
The existence of a nitric oxide synthase (NOS) that is localized in the mitochondria (mtNOS) was originally described in a series of immunohistochemical studies published between 1995 (1, 2) and 1996 (3). Because all of the known NOS isoforms are encoded by nuclear DNA and NOS is not encoded by mitochondrial DNA, this finding implied that one of the recognized NOS isoforms was targeted to the mitochondria after protein synthesis in the cytosol. In these early studies, it was reported that the endothelial NOS (eNOS) isoform was localized to the inner mitochondrial membrane in all tissues that were tested, which included brain, kidney, liver, and skeletal and cardiac muscle. Between 1997 and 1998, more in-depth studies using a variety of NO detection techniques with isolated rat liver mitochondria (4), submitochondrial particles (SMPs; ref. 4), and purified NOS enzyme (5–7) added further support for the existence of a mtNOS. However, these studies were unable to determine whether the enzyme was novel or related to the neuronal (nNOS), inducible (iNOS), or eNOS isoforms. In the last three years, some laboratories have extended their studies to the investigation of the functional implications of mtNOS (8–11), while others have used a NO-sensitive dye to stain the mitochondria in intact cells and demonstrate the presence of NO within these organelles (12). However, though this fluorescence technique showed that there is NO within the mitochondria, it could not definitively establish whether the NO reacting with the dye was produced in the mitochondria or whether it was derived from cellular sources. Likewise, in those studies using NOS antibodies to label mtNOS in intact cells, it could be argued that there was crossreactivity with other proteins or problems in interpretation because of low levels of NOS and high levels of background reactivity. In the studies using isolated mitochondria, SMPs, or purified NOS enzyme, it could similarly be argued that the reported mtNOS was merely one of the cellular isoforms that contaminated the different preparations. Thus, despite a number of positive reports by different laboratories, skepticism remains regarding the existence of a mtNOS. In this report we attempt to address this important controversy by demonstrating the real-time production (by using porphyrinic microsensors) and inhibition (by using NOS antagonists) of NO production in individual isolated mitochondria.
Materials and Methods
Purest available grades of all reagents were obtained from Aldrich/Sigma unless otherwise specified.
Isolation of Mitochondria and Cardiomyocytes.
Tightly coupled mitochondria were isolated from 8- to 12-week-old wild-type, knockout (nNOS−/−, iNOS−/−, or eNOS−/−) or mdx (dystrophin-deficient) mouse hearts as described for the rat heart (13), with modifications permitting the use of one heart per isolation and eliminating the need for collagenase and the Ca2+ required for collagenase activity. Briefly, after deep anesthesia with pentobarbital sodium (50 mg/kg i.p.), the thoracic cavity of a mouse was opened and the heart was cannulated in situ. The organ was then retrogradely perfused through the aorta for 5 min to remove the blood with a mitochondrial solution containing 300 mM sucrose, 5 mM Tris, 5 mM KH2PO4, 0.2 mM succinate, 0.2 mM EDTA, and 0.1% BSA, adjusted to pH 7.4 with 100 mM KOH. The heart (≈0.15 g) was then rapidly excised and minced in two passes at 90° to each other by using a McIlwain motorized tissue chopper (Brinkmann) set to chop at a 150-μm interval. The minced tissue was placed in 10 ml of mitochondrial solution and homogenized by six passes with a motorized Teflon pestle (0.01-mm wall clearance) turning at 75 rpm. The homogenate was spun at 1,000 × g for 10 min. The supernatant was then spun at 10,000 × g for 20 min to obtain a second pellet containing the mitochondria. This pellet was resuspended in 1 ml of mitochondrial solution, and 500 μl of the suspension was placed in a gas-tight vessel containing a Clark-type oxygen microelectrode (MI-730/OM-4; Microelectrodes, Londonderry, NH) to measure the state 3 (glutamate + malate + ADP added) and state 4 (glutamate + malate added) respiratory rates. The respiratory control ratio (RCR), a measure of the “tightness of coupling” between electron transport and oxidative phosphorylation, was determined from the ratio of state 3 to state 4 rates of respiration. Although a RCR of >6 may be considered acceptable (13), we typically obtain ratios of 8–12 in our mitochondrial preparations. If the mitochondria proved to be tightly coupled, the remainder of the suspension was used for NO measurements. Tightly coupled mitochondria could be reliably maintained for 2–3 h. For the mitochondria, all of the isolation steps were carried out at 4°C and all of the NO measurements were performed at room temperature.
Noncontracting Ca2+-tolerant cardiomyocytes were prepared from 8- to 12-week-old wild-type or mdx mice. Briefly, after deep anesthesia with pentobarbital sodium (50 mg/kg i.p.), the thoracic cavity was opened. The heart was cannulated at the aorta in situ and then rapidly excised and retrogradely perfused (120 mmHg; 1 mmHg = 133 Pa) in a modified Langendorff preparation. The heart was perfused for 5 min with cardiomyocyte solution containing 144 mM NaCl, 5.4 mM KCl, 0.4 mM NaH2PO4, 10 mM Hepes, 1 mM MgCl2, and 1.8 mM CaCl2, pH 7.4. The heart was then perfused for 5 min with Ca2+-free cardiomyocyte solution additionally containing 0.1 mM EGTA, 5 mM creatine, and 10 mM taurine. Finally the heart was perfused for 15 min with Ca2+-free cardiomyocyte solution without EGTA but additionally containing 1.5 mg/ml collagenase, 0.5 mg/ml type XIV bacterial (Streptomyces griseus) protease, and 0.01 mg/ml elastase. After perfusion, the atria and right ventricle were removed and discarded, and the remaining left ventricle was minced and incubated, with stirring, in the collected enzyme mix additionally containing 10 mg/ml BSA. Aliquots (10 ml) of the stirring enzyme mix containing suspended dissociated cells were centrifuged at 1,000 × g for 5 min. The supernatant was returned to the stirring enzyme mix, and the pellet was resuspended in medium 199 (Sigma). The cycle was repeated until the tissue was completely dissociated. The cells in medium 199 were used immediately and could be reliably maintained for 4–6 h. For intact cardiomyocytes, all of the isolation steps and NO measurements were performed at 37°C.
A suspension of tightly coupled mitochondria or Ca2+-tolerant cardiomyocytes was gently applied, dropwise, to collagen-coated dishes and allowed to settle out and attach. The dish with organelles or cells was placed in a temperature-regulated holder mounted on the stage of an inverted microscope (Olympus IX 70). Mitochondrial or cardiomyocyte solution entered the chamber from one end (0.1–1.0 ml/min) and drained out the other. Drugs were added to the bath along with the perfusate or were locally applied to an organelle or cell by means of a remote-controlled positive displacement nanoejector (Drummond Scientific, Broomall, PA). The nanoejector was mounted on a remote controlled micromanipulator (1-μm resolution) that allows the glass tip of the ejector to be positioned upstream and proximal to an organelle or cell.
NO and NO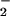 Microsensor Preparation and Data Acquisition.
Microsensor Preparation and Data Acquisition.
Porphyrinic microsensors (tip diameter, 0.1 μm; NO detection limit, 1 nM; response time, 1 ms) were prepared from carbon strands (one fiber, 5-μm diameter each; BP–AMOCO, Alpharetta, GA) as described (14). The 5-μm fiber was thermally sharpened to a 0.1-μm tip diameter. Monomeric tetrakis(3-methoxy-4-hydroxyphenyl)porphyrinnickel(II) (TMHPPNi; Frontier Scientific, Logan, UT) was dissolved in 0.1 M NaOH and deposited as a polymeric film on the carbon fiber by using cyclic voltammetry (−0.2 to +1.0 V; 20 cycles; 283 EG & G Potentiostat; Princeton Applied Research, Oak Ridge, TN). The cation exchanger, Nafion (Sigma), was applied to microsensors to be used for NO measurements by dipping in a 1% ethanolic solution. The microsensors were characterized by differential-pulse voltammetry to determine the oxidation potential of NO and NO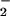 . Chronoamperometry was performed at a constant potential 50 mV more positive than the oxidation potential to determine the sensitivity and detection limit. High purity (>99.99%) NO and NO
. Chronoamperometry was performed at a constant potential 50 mV more positive than the oxidation potential to determine the sensitivity and detection limit. High purity (>99.99%) NO and NO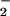 standards were prepared for accurate calibration as described (15). A three-electrode system that consisted of the working electrode (microsensor), a saturated calomel reference electrode, and a platinum counter electrode was used. The porphyrinic microsensor was mounted on an ultramicromanipulator (0.2-μm resolution), enabling the tip to be placed onto the cell surface for NO measurements, or to gently pierce the membrane for NO
standards were prepared for accurate calibration as described (15). A three-electrode system that consisted of the working electrode (microsensor), a saturated calomel reference electrode, and a platinum counter electrode was used. The porphyrinic microsensor was mounted on an ultramicromanipulator (0.2-μm resolution), enabling the tip to be placed onto the cell surface for NO measurements, or to gently pierce the membrane for NO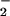 determinations. The currents generated by the oxidation of NO or NO
determinations. The currents generated by the oxidation of NO or NO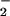 at the porphyrinic interface were amplified, converted to voltages, and digitized for real-time viewing, analysis, and storage.
at the porphyrinic interface were amplified, converted to voltages, and digitized for real-time viewing, analysis, and storage.
Video-Detected Cardiomyocyte Shortening as a Measure of Contractility.
The percent shortening of electrically stimulated cardiomyocytes was used as a determinant of myocardial force generation. Signals were recorded by using a Video Edge Motion Detector (VED105; Crescent Electronics, Sandy, UT) and charge-coupled device (CCD) camera system (FTM800; Philips Electronics). A given cell was viewed on a monitor and two raster points were locked on either end of the long axis of the cell. The temporal resolution of the system is 4.2 ms.
Measurement of Sarcoplasmic Ca2+ Levels in Isolated Cardiomyocytes.
Cells were loaded with the membrane permeant fluorescent Ca2+ indicator rhod-2-am (1 μM; Molecular Probes). To minimize incomplete de-esterification and trapping of rhod-2 in organelles, cardiomyocytes were loaded at 37°C with 1 μM dye. To limit dye penetration to the sarcoplasm, a 10- to 30-min incubation period was used. In order that the optical path of the inverted microscope might be used for simultaneous video-detected cell shortening, fluorescence signals were recorded with a light guide (250-μm diameter; Dolan–Jenner Industries, Lawrence, MA) positioned 0.2 mm above the cells. One branch of the guide focuses excitation light [λex(max) = 553 nm] on the cell surface; the second branch transmits fluorescent emission [λem(max) = 576 nm] through a cutoff filter (570 nm) to a photodiode (EG & G Instruments). Illumination was provided by a 300-W xenon arc lamp (EG & G Optoelectronics). Photodiode currents were amplified, converted to voltages, and then directed to an accessory channel of the EG & G potentiostat for digitization and real-time viewing on a monitor simultaneously with NO and NO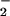 traces.
traces.
Results and Discussion
The functional demonstration of mtNOS has until now been based on the following approaches: (i) the conversion of oxyhemoglobin (or oxymyoglobin) to methemoglobin (or metmyoglobin) in SMPs of rat liver, brain, and thymus cells (16–18); or (ii) the conversion of l-[H3]arginine to l-[H3]citrulline in isolated intact rat liver mitochondria (16, 19). Although both of these NO measurement techniques are sensitive to NOS antagonists, which assures that any measured NO is produced by NOS, the studies cannot rule out the possibility that the NOS itself is a contaminant of cellular origin that was isolated with the SMPs or with the mitochondria. For example, there is a report of nNOS being localized to the sarcoplasmic reticulum in cardiomyocytes (20). Thus, any microsomal contamination in a SMP or mitochondrial preparation may bring along with it the nNOS isoform. Alternatively, the NO fluorescent probe, 4,5-diaminofluorescein diacetate (DAF-2/DA) has been used to stain the mitochondria in intact cells (12) and demonstrate the presence of NO within these organelles. However, this technique cannot definitively identify the source of NO, which could be from within the mitochondria or from the cytosol.
Mitochondrial purity was assessed as the ratio of mitochondrial protein, determined spectrophotometrically as cytochrome c oxidase content, to total protein (13). For example, this varied from ≈40% to 90% in the particular case of mitochondria from wild-type cells, whereas the variation in NO production by individual mitochondria was 28 ± 9 nM (n = 8, four different preparations), arguing very strongly against the measured activity being caused by nonmitochondrial contaminants. In addition, direct electrode measurements performed on the supernatant from the last step of the mitochondrial purification procedure showed an absence of NOS activity. Nevertheless, all mitochondrial preparations are, to some extent, contaminated with other organelles. Isolation procedures based on Percoll gradients, either as a single step (21) or in addition to differential centrifugation (22), still contain 10% or more other materials. The use of heart tissue in the present experiments leads to mitochondrial preparations that are less contaminated with microsomal fractions than if, say, liver were the starting tissue. Moreover, pelleting of mitochondria at ≈10,000 × g (rather than the ≈100,000 × g of many published procedures) ensures that the significantly smaller microsomes mostly remain dispersed throughout the supernatant. As mouse cardiomyocytes are ≈37.5% mitochondria as a percentage of cell volume and only ≈1% sarcoplasmic reticulum (23), heart preparations exhibit a higher ratio of mitochondria to contaminating organelles than preparations starting from other tissues. In mouse skeletal muscle for example, the mitochondria constitute only ≈2% of the cell volume. Nevertheless, the key feature of the present study is that we report measurements from individual mitochondria to overcome the problem of background contamination. Fig. 1 is a digital image taken by using phase-contrast optics to show a typical mitochondrial preparation where the 2- to 3-μm egg-shaped structures are the mitochondria, which always swell during isolation. In contrast, microsomes are <0.5 μm in diameter and are not resolved where they are present. In all of our active preparations, 80–90% of the individual mitochondria tested (6–12 per each of 14 preparation) produced NO in the same way. None of the individual mitochondria in preparations that were uncoupled either by aging or by application of protonophores produced NO. The notion that the remarkably reproducible production of NO by only coupled mitochondria could be due to low levels of contaminants that would have to be similarly associated with 80–90% of the individual mitochondria tested appears unlikely in the extreme. The additional observation that the results are quantitatively invariant between active preparations further argues against the measured effects being caused by contaminants.
Figure 1.

Photomicrograph of a mitochondrial preparation. This image (×1,200), which was acquired by using phase-contrast optics, shows a tightly coupled (respiratory control ratio = 10) mitochondrial preparation with a microsensor positioned next to one of the organelles. To-scale representations of an erythrocyte and a cardiomyocyte are superimposed on the image to show their relative sizes in comparison to the organelles, demonstrating the feasibility of measuring NO production from a single mitochondrion. Isolated cardiac mitochondria are ≈2–3 μm in length, somewhat larger than their counterparts in fixed cells (≈0.75–1.5 μm).
Withdrawing the microsensor tip (in 1-μm increments) from a mitochondrion generating NO, enabled us to determine that mitochondrially produced NO diffused ≤10 μm in our experimental setup (8 ± 2 μm; n = 24). Thus, by sparsely plating to provide a >10-μm zone around the mitochondrion from which we were recording, we were assured that the NO signals detected were derived from a single mitochondrion. Initially, with a microsensor positioned next to a mitochondrion, there was no detectable basal NO production. However, the addition of Ca2+ (10 μM) by itself evoked sustained NO production (Fig. 2, trace B). Supplementation of the mitochondria with l-arginine (1 mM) or tetrahydrobiopterin (10 μM) did not increase NO formation. This result implies that the mitochondria contain all of the substrate and cofactors necessary for NOS activity except for Ca2+. The fact that this NO production is inhibitable by the NOS antagonists l-NMA (100 μM; Fig. 2, trace A) or 7-nitroindazole (50 μM; not shown) demonstrates that it is being produced by a NOS. Moreover, the finding that this NO production is inhibited by blocking Ca2+ uptake through the electrogenic uniporter with ruthenium red (RR, 1 μM; Fig. 2 trace C) or by collapsing the membrane potential with a protonophore [carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), 100 nM; or carbonyl cyanide m-chlorophenylhydrazone, 100 nM; not shown] demonstrates that the measured NO is coming from mtNOS and not a cellular NOS contaminating the preparation.
Figure 2.

Electrochemical demonstration of NO production by isolated mitochondria. A porphyrinic microsensor was used to measure NO production by individual tightly coupled cardiac mitochondria. The organelles were in aerobic Ca2+-free mitochondrial solution without supplemental l-arginine or cofactors, under which conditions there was no detectable NO production. When exogenous Ca2+ (10 μM) was added to the bath, it evoked a rapid production of NO (28 ± 9 nM; n = 8; traces A and B) that was inhibited by the addition of the NOS antagonists, NG-monomethyl-l-arginine (l-NMA, 100 μM; trace A) or 7-nitroindazole (50 μM; not shown). The Ca2+-dependent production of NO was also inhibited by pretreatment with ruthenium red (RR, 1 μM; trace C), a blocker of the electrogenic uniporter that is responsible for Ca2+ uptake by mitochondria.
The Ca2+-evoked production of NO could be blocked with the nonselective NOS antagonist, l-NMA (100 μM; Fig. 2 trace A). However, the blockade of NO production could be achieved with a lower dose of the nNOS selective antagonist, 7-nitroindazole (50 μM; not shown), suggesting that mtNOS may be related to the nNOS isoform. This similarity of mtNOS to the neuronal isoform was confirmed by the absence of NO production in mitochondria isolated from the hearts of knockout mice for the neuronal, but not the endothelial or inducible NOS isoforms (Fig. 3). There are at least three nNOS alternative-splice variants: nNOSα, nNOSβ, and nNOSγ (24, 25). Only the nNOSα variant is knocked out in nNOS−/− mouse, which is accomplished by deleting exon2 near the N terminus. This exon contains the PDZ (postsynaptic disks-large zoe-1) motif responsible for tethering nNOS to membranes (26–28). Both the eNOS−/− and iNOS−/− mice have their respective NOS gene entirely knocked out, neither of which has been shown as of yet to contain a PDZ motif. Because mtNOS is reported to be membrane bound (1–3), it is not surprising that this isoform is related to nNOSα, as the other neuronal splice variants lack the PDZ-binding domain.
Figure 3.

Identification of the nNOS isoform in cardiac mitochondria. The identification of the isoform of cardiac mtNOS was deduced by using mitochondria isolated from the hearts of knockout mice for the neuronal (nNOS−/−), inducible (iNOS−/−), and endothelial (eNOS−/−) isoforms. Only the mitochondria isolated from the hearts of nNOS−/− mice failed to produce NO (n = 5).
To determine the effects of mitochondrially produced NO on bioenergetics, we used intact cardiomyocytes from dystrophin-deficient (mdx) mice. We have recently shown by a combination of immunohistochemistry and activity measurements (L.A.B., S. C. Froehner, P.R.C., and A.J.K., unpublished data) that mdx cardiomyocytes are deficient in cellular caveolae NOS (eNOS), but overexpress mtNOS. This provided the unique opportunity to study mtNOS in intact cells in the absence of the cellular isoform. Norepinephrine (NE) evoked NO production by caveolar-associated eNOS (Fig. 4A Left) is abolished (Fig. 4B Left) or greatly decreased (by ≈83%, not shown) in mdx cardiomyocytes. However, a new beat-to-beat production of NO that we propose is coming from mtNOS, is readily observed in mdx cells in response to electrical stimulation (Fig. 4B Right). Although we cannot unequivocally state that beat-to-beat NO production is entirely absent in the case of the wild-type cardiomyocytes, it is certainly at or below our detection limit (1 nM NO; Fig. 4A Right). We further hypothesize that the observed increase in mtNOS activity represents an attempt by the mdx cell to compensate for the low eNOS activity in caveolae.
Figure 4.

Beat-to-beat NO production in an mdx cardiomyocyte. Norepinephrine (NE; 1 μM) evoked NO (726 ± 260 nM; n = 7; A Left) production in a quiescent wild-type cardiomyocyte. Pacing (voltage = 2 × threshold; duration = 10 ms; f = 2 Hz) the cell did not evoke detectable NO formation (A Right). In mdx cardiomyocytes, however, NE (1 μM) evoked a diminished NO response (126 ± 90 nM; n = 7; not shown) or none at all (n = 5; B Left). All paced mdx cells released NO (141 ± 94 nM; n = 12; B Right) beat-to-beat.
Although contractility (video-detected shortening) is decreased by about 50% in mdx cardiomyocytes compared with wild-type cells, inhibition (l-NMA; 100 μM) of beat-to-beat NO production recovered about half of this lost contractility (Fig. 5A). On the other hand, the inhibition of NO production in wild-type cardiomyocytes had no observable effects on contractility. Abolition of beat-to-beat NO production by collapsing the mitochondrial membrane potential with a protonophore uncoupler (FCCP, 100 nM; Fig. 5B) further supports our hypothesis that this NO production is of mitochondrial origin. These observations also suggest that prolonged elevation of cellular Ca2+, a hallmark of dystrophin-deficient cells, activates mtNOS resulting in local NO production that can transiently inhibit mitochondrial ATP production and, in turn, contractility. In mdx cells, where the absence of dystrophin results in a weakening of the sarcolemmal membrane, this decrease in contractility may be a protective mechanism to guard against myocardial damage.
Figure 5.

Demonstration of the effects of mitochondrially produced NO on contractility. Administration of NOS antagonists, l-NMA (100 μM; n = 7; A) or 7-nitroindazole (50 μM; n = 6; not shown) effectively inhibited electrically stimulated beat-to-beat NO production in mdx cardiomyocytes and NE (1 μM) evoked NO release in mdx and wild-type cells (n = 7; not shown). An alternative way to block Ca2+-dependent NO production was by inhibiting the electrogenic Ca2+ uniporter. This was rapidly and reversibly accomplished by collapsing the membrane potential with the protonophore uncouplers, carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP, 100 nM; n = 5; B) or carbonyl cyanide m-chlorophenylhydrazone (CCCP, 100 nM; n = 4; not shown). Although contractility in mdx cardiomyocytes was approximately half that of normal cells, inhibition of NO production with l-NMA increased contractility by approximately one-fourth in mdx cells (Left; n = 8), but had no effect on normal cells.
A single electrical pulse applied to an mdx cardiomyocyte resulted in a transient increase of cytoplasmic Ca2+ that immediately preceded an NO burst (Fig. 6), indicating that the observed production of NO was Ca2+ dependent. Interestingly, microsensor measurements also revealed that the NO produced is almost completely converted to nitrite (NO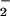 ; Fig. 6, traces B) rather than nitrate (NO
; Fig. 6, traces B) rather than nitrate (NO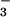 ) or a mixture of the two stable anions. The near-quantitative (≈85%) conversion of NO to NO
) or a mixture of the two stable anions. The near-quantitative (≈85%) conversion of NO to NO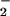 was unexpected, as the metabolic turnover of “excess” nonmitochondrial NO has been widely attributed to the reaction with oxymyoglobin (Mb) in red muscle and oxyhemoglobin in the blood producing NO
was unexpected, as the metabolic turnover of “excess” nonmitochondrial NO has been widely attributed to the reaction with oxymyoglobin (Mb) in red muscle and oxyhemoglobin in the blood producing NO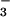 (29, 30):
(29, 30):
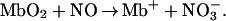 |
Thus, NO conversion to NO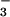 may be of importance in the bloodstream, whereas it appears to be of less significance in cardiomyocytes overexpressing mtNOS. Moreover, this argument can probably be extended to most other cell types that contain little or no Mb.
may be of importance in the bloodstream, whereas it appears to be of less significance in cardiomyocytes overexpressing mtNOS. Moreover, this argument can probably be extended to most other cell types that contain little or no Mb.
Figure 6.

Simultaneous measurement of Ca2+, NO, and NO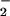 in an mdx cardiomyocyte. A single electrical stimulus (voltage = 2 × threshold; duration = 10 ms) from a bipolar microelectrode (area = 50 μm2) evoked a rise in cytoplasmic Ca2+ (411 ± 57; nM; trace A) followed by the production of NO (149 ± 97 nM; trace B) 10–20 ms thereafter. The onset of NO production, in turn, was followed, in 25–50 ms, by an increase in intracellular NO
in an mdx cardiomyocyte. A single electrical stimulus (voltage = 2 × threshold; duration = 10 ms) from a bipolar microelectrode (area = 50 μm2) evoked a rise in cytoplasmic Ca2+ (411 ± 57; nM; trace A) followed by the production of NO (149 ± 97 nM; trace B) 10–20 ms thereafter. The onset of NO production, in turn, was followed, in 25–50 ms, by an increase in intracellular NO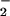 (112 ± 31 nM; n = 6; B). Although the NO transient rapidly peaked and returned to baseline, the NO
(112 ± 31 nM; n = 6; B). Although the NO transient rapidly peaked and returned to baseline, the NO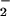 signal rose to a plateau and gradually declined (decline not shown). There was no detectable NO production in quiescent cells. Electrical stimulation evoked Ca2+ transients but did not evoke detectable NO production in wild-type cells (not shown).
signal rose to a plateau and gradually declined (decline not shown). There was no detectable NO production in quiescent cells. Electrical stimulation evoked Ca2+ transients but did not evoke detectable NO production in wild-type cells (not shown).
There is, so far as we are aware, no well-established biochemical mechanism by which NO could be near-quantitatively converted to NO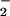 as we have observed (Fig. 6 traces B). Beckman et al. (31) have convincingly argued that, at physiological NO levels, uncatalyzed reactions in aqueous solutions are too slow to be relevant. Recently, reactions of cytochrome c oxidase with both peroxynitrite (32) and NO (33) have been shown to result in nitrite production. Accordingly, we propose the following, plausible pathway, involving a peroxynitrite intermediate in a cytochrome c oxidase reaction cycle, to account for the observed conversion of NO to nitrite (Fig. 7). Initially, NO may bind to one of the metal ions at the active site of cytochrome c oxidase (i.e., the binuclear pair). Next, O2 could enter the active site, which is able to accommodate at least two exogenous diatomic ligands (34), where it reacts with the other metal ion. After reduction of O2 to superoxide ion (O
as we have observed (Fig. 6 traces B). Beckman et al. (31) have convincingly argued that, at physiological NO levels, uncatalyzed reactions in aqueous solutions are too slow to be relevant. Recently, reactions of cytochrome c oxidase with both peroxynitrite (32) and NO (33) have been shown to result in nitrite production. Accordingly, we propose the following, plausible pathway, involving a peroxynitrite intermediate in a cytochrome c oxidase reaction cycle, to account for the observed conversion of NO to nitrite (Fig. 7). Initially, NO may bind to one of the metal ions at the active site of cytochrome c oxidase (i.e., the binuclear pair). Next, O2 could enter the active site, which is able to accommodate at least two exogenous diatomic ligands (34), where it reacts with the other metal ion. After reduction of O2 to superoxide ion (O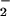 ), NO can then combine with O
), NO can then combine with O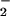 to form ONO
to form ONO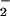 in a reaction that is known to be very rapid (35, 36). The subsequent conversion of ONO
in a reaction that is known to be very rapid (35, 36). The subsequent conversion of ONO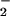 to NNO
to NNO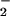 in a two-electron reduction at the active site is also a known reaction (32). After the transfer of electrons from the other components of the electron transport chain, the proposed cycle of reactions could be repeated. If the proposed NO-driven process is slower than the familiar four-electron reduction of O2 to H2O, then it follows that the reaction of NO at the enzyme active site is inhibitory (37–41). Therefore, the presence of NO will ultimately lead to decreased ATP production and contractility. Of course, the ideas encompassed by Fig. 7 do not exclude the possibility that reactivation of cytochrome c oxidase might simply involve dissociation of active-site bound NO, or some other process, under hypoxic conditions.
in a two-electron reduction at the active site is also a known reaction (32). After the transfer of electrons from the other components of the electron transport chain, the proposed cycle of reactions could be repeated. If the proposed NO-driven process is slower than the familiar four-electron reduction of O2 to H2O, then it follows that the reaction of NO at the enzyme active site is inhibitory (37–41). Therefore, the presence of NO will ultimately lead to decreased ATP production and contractility. Of course, the ideas encompassed by Fig. 7 do not exclude the possibility that reactivation of cytochrome c oxidase might simply involve dissociation of active-site bound NO, or some other process, under hypoxic conditions.
Figure 7.

Alternate reactions catalyzed by cytochrome c oxidase. (A) The normal (accepted) reaction cycle involving the four-electron reduction of dioxygen to water. (B) Proposed three-electron reduction of dioxygen and NO to nitrite ion and water, possibly via a peroxynitrite intermediate. For NO to be inhibitory, the rate-determining step in B must be slower than in A.
The short half-life (<6 s) of NO and the presence of caveolar eNOS have heretofore precluded the verification and characterization of mtNOS. We have overcome these limitations by using microsensors to directly measure NO production in isolated mitochondria and intact cardiomyocytes deficient in cellular caveolar NOS. The use of mitochondria isolated from the hearts of knockout mice for the nNOS, iNOS, and eNOS isoforms has demonstrated the absence of mtNOS in the mitochondria of nNOS−/− cells. The finding that the majority of intracellular NO is converted to NO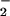 is contrary to the prevailing idea concerning NO metabolism and is, therefore, of some importance. Further experiments are needed to explore the hypothesis that inhibitory reactions at the cytochrome c oxidase active site are responsible for our observed NO
is contrary to the prevailing idea concerning NO metabolism and is, therefore, of some importance. Further experiments are needed to explore the hypothesis that inhibitory reactions at the cytochrome c oxidase active site are responsible for our observed NO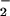 production.
production.
The mdx mouse, whose skeletal muscle is deficient in nNOS activity and whose cardiac muscle is deficient in eNOS activity, is the animal model of choice for Duchenne muscular dystrophy (DMD). In skeletal muscle, nNOS is expressed and localized to sarcolemma caveolae by the dystrophin–glycoprotein complex (26, 42). Dystrophin tethers NOS to the C terminus of dystrophin through the PDZ motif of the linker protein, syntrophin, and a corresponding PDZ motif on nNOS. Dystrophin also serves as an F-actin anchor point for the cytoskeleton through the N terminus. In human DMD and the mdx mouse, mutations in the X-linked dystrophin gene result in the absence of dystrophin and nNOS from the sarcolemma of skeletal muscle (42–46).
In cardiac muscle, eNOS is expressed and localized to sarcolemma caveolae, but lacks a corresponding PDZ domain. Therefore, a direct association of cardiac eNOS with dystrophin has not yet been conclusively demonstrated. However, in mdx cardiac muscle, our findings suggest a similar loss of NOS from the sarcolemma, and demonstrate inactivation of caveolae eNOS. Consequently, the mdx mouse provides a useful experimental model for studying other potential NOS isoforms that may be present at low levels in wild-type cells. Because NO produced by eNOS in the sarcoplasm is important for maintaining low resting Ca2+ by decreasing its influx across the cell membrane (47, 48) and by enhancing its reuptake into cellular stores (49), inactivation of caveolae eNOS may contribute to the elevation in sarcolemmal Ca2+ seen in mdx mouse cardiomyocytes (50, 51).
Although the muscular dystrophies are most readily recognized for their skeletal muscle involvement, DMD patients also have a cardiomyopathy. In Becker muscular dystrophy, which is allelic to DMD, the cardiomyopathy may be severe and may be the principal manifestation of dystrophin deficiency. Because the absence of dystrophin from the sarcolemma may weaken the skeletal and cardiac muscle membrane alike, making them leaky to Ca2+ and more susceptible to stretch-induced damage, it is interesting to speculate that the role of mitochondrially produced NO in the pathologic myocardium may be to limit myocardial contractility and thus protect the heart. Inhibition of ATP production and contractility would, however, eventually lead to cardiac hypertrophy and, eventually, heart failure as observed in patients with both DMD and Becker muscular dystrophy. Because mtNOS is present also in wild-type cardiomyocytes, it may serve a similar role under physiological conditions (in which mitochondrially produced NO could protect the healthy myocardium from overwork by limiting ATP production during acute Ca2+ overload) that may accompany strenuous exercise or other stressors. Thus, our direct measurements of mitochondrially produced NO production may offer important new insights into cardiac mtNOS and the modulatory role of NO in the normal and pathologically compromised heart.
Acknowledgments
This research is supported by National Institutes of Health Grants HL 57985 (to A.J.K.), HL 61411 (to J.P.), and DK 54824 (to L.A.B.), American Heart Association Grant AHA-PA 9808156U (to P.R.C.), and a grant from the Muscular Distrophy Association.
Abbreviations
- NOS
nitric oxide synthase
- iNOS
inducible NOS
- eNOS
endothelial NOS
- mtNOS
mitochondrial NO synthase
- nNOS
neuronal NOS
- DMD
Duchenne muscular dystrophy
- NE
norepinephrine
- SMP
submitochondrial particle
- PDZ motif
postsynaptic disks-large zoe-1 motif
- FCCP
carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- l-NMA
NG-monomethyl-l-arginine
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Kobzik L, Stringer B, Balligand J L, Reid M B, Stamler J S. Biochem Biophys Res Commun. 1995;211:375–381. doi: 10.1006/bbrc.1995.1824. [DOI] [PubMed] [Google Scholar]
- 2.Bates T E, Loesch A, Burnstock G, Clark J B. Biochem Biophys Res Commun. 1995;213:896–900. doi: 10.1006/bbrc.1995.2213. [DOI] [PubMed] [Google Scholar]
- 3.Bates T E, Loesch A, Burnstock G, Clark J B. Biochem Biophys Res Commun. 1996;218:40–44. doi: 10.1006/bbrc.1996.0008. [DOI] [PubMed] [Google Scholar]
- 4.Ghafourifar P, Richter C. FEBS Lett. 1997;418:291–296. doi: 10.1016/s0014-5793(97)01397-5. [DOI] [PubMed] [Google Scholar]
- 5.Tatoyan A, Giulivi C. J Biol Chem. 1998;273:11044–11048. doi: 10.1074/jbc.273.18.11044. [DOI] [PubMed] [Google Scholar]
- 6.Giulivi C, Poderoso J J, Boveris A. J Biol Chem. 1998;273:11038–11043. doi: 10.1074/jbc.273.18.11038. [DOI] [PubMed] [Google Scholar]
- 7.Giulivi C. Biochem J. 1998;332:673–679. doi: 10.1042/bj3320673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghafourifar P, Richter C. Biol Chem. 1999;380:1025–1028. doi: 10.1515/BC.1999.127. [DOI] [PubMed] [Google Scholar]
- 9.Ghafourifar P, Schenk U, Klein S D, Richter C. J Biol Chem. 1999;274:31185–31188. doi: 10.1074/jbc.274.44.31185. [DOI] [PubMed] [Google Scholar]
- 10.Richter C, Ghafourifar P. Biochem Soc Sym. 1999;66:27–31. doi: 10.1042/bss0660027. [DOI] [PubMed] [Google Scholar]
- 11.Bringold U, Ghafourifar P, Richter C. Free Radical Biol Med. 2000;29:343–348. doi: 10.1016/s0891-5849(00)00318-x. [DOI] [PubMed] [Google Scholar]
- 12.López-Figueroa M O, Caamaño C, Morano M I, Rønn L C, Akil H, Watson S J. Biochem Biophys Res Commun. 2000;272:129–133. doi: 10.1006/bbrc.2000.2748. [DOI] [PubMed] [Google Scholar]
- 13.Darley-Usmar V, Rickwood D, Wilson M T. Mitochondria: A Practical Approach. Oxford: IRL; 1987. pp. 1–16. [Google Scholar]
- 14.Kanai A J, Mesaros S, Finkel M S, Oddis C V, Birder L A, Malinski T. Am J Physiol. 1997;273:C1371–C1377. doi: 10.1152/ajpcell.1997.273.4.C1371. [DOI] [PubMed] [Google Scholar]
- 15.Kanai A J, Strauss H C, Truskey G A, Crews A L, Grunfeld S, Malinski T. Circ Res. 1995;77:284–293. doi: 10.1161/01.res.77.2.284. [DOI] [PubMed] [Google Scholar]
- 16.Busse R, Mulsch A. FEBS Lett. 1990;265:133–136. doi: 10.1016/0014-5793(90)80902-u. [DOI] [PubMed] [Google Scholar]
- 17.Cavalié A, Allen T J A, Trautwein W. Pflügers Arch. 1991;419:433–443. doi: 10.1007/BF00370785. [DOI] [PubMed] [Google Scholar]
- 18.Gardner P R, Costantino G, Szabo C, Salzman A L. J Biol Chem. 1997;272:25071–25076. doi: 10.1074/jbc.272.40.25071. [DOI] [PubMed] [Google Scholar]
- 19.Gardiner S M, Compton A M, Bennett T, Palmer R M J, Moncada S. Br J Pharmacol. 1990;101:10–12. doi: 10.1111/j.1476-5381.1990.tb12079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu K Y, Huso D L, Dawson T M, Bredt D S, Becker L C. Proc Natl Acad Sci USA. 1999;96:657–662. doi: 10.1073/pnas.96.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reinhart P H, Taylor W M, Bygrave R L. Biochem J. 1982;204:731–735. doi: 10.1042/bj2040731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Susin S A, Larochette N, Geuskens M, Kroemer G. Methods Enzymol. 2000;322:205–208. doi: 10.1016/s0076-6879(00)22020-x. [DOI] [PubMed] [Google Scholar]
- 23.Sommer J R, Johnson E A. In: The Heart. Berne R M, Sperelakis N, Geiger S R, editors. Bethesda: Am. Physiol. Soc.; 1979. pp. 113–186. [Google Scholar]
- 24.Saur D, Paehge H, Schusdziarra V, Allescher H-D. Gastroenterology. 2000;118:849–858. doi: 10.1016/s0016-5085(00)70171-5. [DOI] [PubMed] [Google Scholar]
- 25.Putzke J, Seidel B, Huang P L, Wolin M S. Mol Brain Res. 2000;85:13–23. doi: 10.1016/s0169-328x(00)00220-5. [DOI] [PubMed] [Google Scholar]
- 26.Brenman J E, Chao D S, Gee S H, McGee A W, Craven S E, Santillano D R, Wu Z, Huang F, Xia H, Peters M F, et al. Cell. 1996;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- 27.Stricker N L, Christopherson K S, Yi B A, Schatz P J, Raab R W, Dawes G, Bassett D E, Jr, Bredt D S, Li M. Nat Biotechnol. 1997;15:336–342. doi: 10.1038/nbt0497-336. [DOI] [PubMed] [Google Scholar]
- 28.Craven S E, Bredt D S. Cell. 1998;93:495–498. doi: 10.1016/s0092-8674(00)81179-4. [DOI] [PubMed] [Google Scholar]
- 29.Wilson M T, Torres J, Cooper C E, Sharpe M A. Biochem Soc Trans. 1997;25:905–909. doi: 10.1042/bst0250905. [DOI] [PubMed] [Google Scholar]
- 30.Groves G T. Curr Opin Chem Biol. 1999;3:226–235. doi: 10.1016/S1367-5931(99)80036-2. [DOI] [PubMed] [Google Scholar]
- 31.Beckman J S. In: Nitric Oxide: Principles and Actions. Lancaster J Jr, editor. San Diego: Academic; 1996. pp. 1–82. [Google Scholar]
- 32.Pearce L L, Pitt B R, Peterson J. J Biol Chem. 1999;274:35763–35767. doi: 10.1074/jbc.274.50.35763. [DOI] [PubMed] [Google Scholar]
- 33.Torres J, Sharpe M A, Rosquist A, Cooper C E, Wilson M T. FEBS Lett. 2000;475:263–266. doi: 10.1016/s0014-5793(00)01682-3. [DOI] [PubMed] [Google Scholar]
- 34.Hill B C, Brittain T, Eglinton D G, Gadsby P M A, Greenwood C, Nicholls P, Peterson J, Thomson A J, Woon T C. Biochem J. 1983;215:57–66. doi: 10.1042/bj2150057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beckmann J S, Koppenol W H. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 36.Huie R E, Padmaja S. Free Radical Res Commun. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- 37.Torres J, Darley-Usmar V, Wilson M T. Biochem J. 1995;312:169–173. doi: 10.1042/bj3120169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown G C. FEBS Lett. 1995;369:136–139. doi: 10.1016/0014-5793(95)00763-y. [DOI] [PubMed] [Google Scholar]
- 39.Borutaite V, Brown G C. Biochem J. 1996;315:295–299. doi: 10.1042/bj3150295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brooks P S, Bolanos J P, Heales S J. FEBS Lett. 1999;446:261–263. doi: 10.1016/s0014-5793(99)00217-3. [DOI] [PubMed] [Google Scholar]
- 41.Sarti P, Lendaro E, Ippoliti R, Benedetti P A, Brunori M. FASEB J. 1999;13:191–197. doi: 10.1096/fasebj.13.1.191. [DOI] [PubMed] [Google Scholar]
- 42.Brenman J E, Chao D S, Xia H, Aldape K, Bredt D S. Cell. 1995;82:743–752. doi: 10.1016/0092-8674(95)90471-9. [DOI] [PubMed] [Google Scholar]
- 43.Chao D S, Gorospe J R, Brenman J E, Rafael J A, Peters M F, Froehner S C, Hoffman E P, Chamberlain J S, Bredt D S. J Exp Med. 1996;184:609–618. doi: 10.1084/jem.184.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bredt D S. Proc Soc Exp Biol Med. 1996;211:41–48. doi: 10.3181/00379727-211-43950f. [DOI] [PubMed] [Google Scholar]
- 45.Grozdanovic Z, Gosztonyi G, Gossrau R. Acta Histochem. 1996;98:61–69. doi: 10.1016/S0065-1281(96)80051-1. [DOI] [PubMed] [Google Scholar]
- 46.Hoffman E P, Brown R H, Kunkel L M. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 47.Méry P F, Pavoine C, Belhassen L, Pecker F, Fischmeister R. J Biol Chem. 1993;268:26286–26295. [PubMed] [Google Scholar]
- 48.Campbell D L, Stamler J S, Strauss H C. J Gen Physiol. 1996;108:277–293. doi: 10.1085/jgp.108.4.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stoyanovsky D, Murphy T, Anno P R, Kim Y M, Salama G. Cell Calcium. 1997;21:19–29. doi: 10.1016/s0143-4160(97)90093-2. [DOI] [PubMed] [Google Scholar]
- 50.Turner P R, Westwood T, Regen C M, Steinhardt R A. Nature (London) 1988;335:735–738. doi: 10.1038/335735a0. [DOI] [PubMed] [Google Scholar]
- 51.Hope F W, Turner P R, Denetclaw W F, Jr, Reddy P, Steinhardt R A. Am J Physiol. 1996;271:C1325–C1339. doi: 10.1152/ajpcell.1996.271.4.C1325. [DOI] [PubMed] [Google Scholar]