

Abstract
Background
Sex differences in idiopathic pulmonary fibrosis (IPF) suggest a protective role for estrogen (E2); however, mechanistic studies in animal models have produced mixed results. Reports using cell lines have investigated molecular interactions between transforming growth factor beta1 (TGF-β1) and estrogen receptor (ESR) pathways in breast, prostate, and skin cells, but no such interactions have been described in human lung cells. To address this gap in the literature, we investigated a role for E2 in modulating TGF-β1-induced signaling mechanisms and identified novel pathways impacted by estrogen in bronchial epithelial cells.
Methods
We investigated a role for E2 in modulating TGF-β1-induced epithelial to mesenchymal transition (EMT) in bronchial epithelial cells (BEAS-2Bs) and characterized the effect of TGF-β1 on ESR mRNA and protein expression in BEAS-2Bs. We also quantified mRNA expression of ESRs in lung tissue from individuals with IPF and identified potential downstream targets of E2 signaling in BEAS-2Bs using RNA-Seq and gene set enrichment analysis.
Results
E2 negligibly modulated TGF-β1-induced EMT; however, we report the novel observation that TGF-β1 repressed ESR expression, most notably estrogen receptor alpha (ESR1). Results of the RNA-Seq analysis showed that TGF-β1 and E2 inversely modulated the expression of several genes involved in processes such as extracellular matrix (ECM) turnover, airway smooth muscle cell contraction, and calcium flux regulation. We also report that E2 specifically modulated the expression of genes involved in chromatin remodeling pathways and that this regulation was absent in the presence of TGF-β1.
Conclusions
Collectively, these results suggest that E2 influences unexplored pathways that may be relevant to pulmonary disease and highlights potential roles for E2 in the lung that may contribute to sex-specific differences.
Electronic supplementary material
The online version of this article (10.1186/s12931-018-0861-5) contains supplementary material, which is available to authorized users.
Keywords: Transforming growth factor beta1, Estrogen, Estrogen receptor, Lung, Fibrosis
Background
Epidemiological studies have associated sex with idiopathic pulmonary fibrosis (IPF) where males are more negatively impacted and females have better survival rates [1–5]. In addition, a study by our group found a statistical interaction between sex and IPF severity based on gene expression in diseased lung tissue [6]. Based on these observations, several hypotheses suggest a role for estrogens and androgens.
Sex-based studies of pulmonary fibrosis in small animal models have produced mixed results. Several reports suggest that estrogen (E2) is protective and androgens exacerbate fibrotic responses. For example, bleomycin caused increased pulmonary fibrosis in male mice compared to females [7, 8], and a recent study found that it was the presence of the Y chromosome and not necessarily sex itself that predisposed the lung to increased bleomycin-induced fibrosis in male and female mice [9]. Conversely, a study in rats indicated that females exhibited increased pulmonary fibrosis in response to bleomycin compared to males [10]. Importantly, variable responses to bleomycin in mice may be due to differential activity of bleomycin hydrolase between male and females [11]. Conversely, E2 was protective in ovariectomized mice where a significant increase in total lung collagen content and airway subepithelial collagen deposition was observed in ovariectomized mice which was mitigated by E2 administration [12].
Current reports have not probed the molecular mechanisms impacted by hormones in the lung contributing to sex-based differences in fibrotic disease except one study which found increased TGF-β1 expression in rat fibroblasts exposed to E2 [10]. Regulation of TGF-β1 by E2 has been extensively characterized in other model systems and the effects appear to be contextual. For example, E2 inhibited TGF-β1 signaling in breast cancer cells by reducing the expression of activators of TGF-β1 [13] and by increasing degradation of SMAD proteins [14, 15]. Conversely, E2 increased TGF-β1 secretion in dermal fibroblasts [16], and TGF-β1 levels were reduced in the kidneys of diabetic female mice lacking estrogen receptor alpha (ESR1) compared to wildtype mice suggesting positive regulation of TGF-β1 by E2 [17].
E2 binds and activates several receptors including the nuclear transcription factors estrogen receptor alpha (ESR1), estrogen receptor beta (ESR2), and several variants thereof, and the putative membrane-bound G-protein coupled estrogen receptor 1 (GPER1). Studies aiming to decipher receptor-specific effects on TGF-β1 signaling have been limited to non-lung cells and tissues (i.e. breast). For example, Stope et al. found that ESR1 but not ESR2 inhibited TGF-β1 activation in breast cancer cells [18] while others found that both ESR1 and ESR2 suppressed TGF-β1 signaling by associating with and acting as a transcriptional corepressor for SMAD3 [19, 20]. Other studies showed a role for GPER1 in mediating E2-dependent reduction in SMAD protein activation in breast cancer cells [21] and TGF-β1-induced extracellular matrix (ECM) production in human and rat mesangial cells [22].
Given the preponderance of opposing actions of E2 on TGF-β1 in in vitro systems other than the lung and the epidemiological evidence suggesting a male sex-bias in IPF, we hypothesized that E2 would inhibit TGF-β1-induced signaling in lung epithelial cells. To test this hypothesis, we investigated the impact of E2 on TGF-β1-induced epithelial to mesenchymal transition (EMT), characterized the expression of ESRs in bronchial epithelial cells and lung tissue from individuals with IPF, and performed RNA-Seq analysis to identify targets of E2 in bronchial epithelial cells.
Methods
Chemicals
Recombinant human transforming growth factor beta1 (TGF-β1) was purchased from R&D Systems (> 97% purity, Catalog No. 240-B002, Minneapolis, MN) and 17β-Estradiol (E2) was purchased from Sigma (≥98% purity, Catalog No. E2758, Saint Louis, MO).
Human lung samples
The human lung tissue samples used in this study were a kind gift from Dr. Andrew Bryant. The explanted lung tissue was obtained from subjects undergoing lung transplant for IPF and from lungs rejected for transplant from normal controls per the National Institutes of Health Lung Tissue Research Consortium (protocol no. 14-99-0011). This study consisted of fifteen patients, eight with IPF (n = 8) and seven controls (n = 7) described in detail in Table 1. A diagnosis of IPF was determined based on ATS criteria [23, 24]. The protocol for collection of lung tissue samples and subsequent studies were approved by the institutional review board at Vanderbilt University and the University of Florida [25]. Immediately after lung biopsy or resection, a portion of the lung was removed and flash-frozen in liquid nitrogen or dry ice and stored at − 80 °C.
Table 1.
Patient demographic data
| Characteristics | Controls | Patients |
|---|---|---|
| Male/Female | 2/3 | 8/0 |
| Agea | 64 ± 14.78 | 61.5 ± 7.11 |
| Smoking Status | ||
| Never | 4 | 6 |
| Ever | 1 | 2 |
| FVC, % predicteda | 105.2 ± 7.79 | 42.9 ± 14.39 |
aMean ± SD
Cell culture
Human bronchial epithelial cells (BEAS-2Bs, CRL-9609™) were purchased from ATCC and cultured according to manufacturer’s specifications. STR analysis and mycoplasma contamination testing was not performed. Cells were cultured in bronchial epithelial growth medium (BEGM) consisting of bronchial epithelial basal medium (BEBM, Lonza CC-3171, Walkersville, MD) and the BEGM SingleQuot Kit Supplements & Growth Factors (Lonza CC-4175) but exchanging gentamicin for Penicillin-Streptomycin-Neomycin (PSN) Antibiotic Mixture (Gibco 15,640, ThermoFisher Scientific Inc., Waltham, MA). Cells were cultured in T75 Corning™ U-Shaped Cell Culture Flasks (Corning 430,641 U, Fisher Scientific Co LLC, Pittsburgh, PA) coated with a matrix (4.5 mL per 75 cm2) consisting of 0.01 mg/mL fibronectin (Akron AK8350, City, State), 0.03 mg/mL bovine collagen (Gibco A10644–01), and 0.01 mg/mL BSA (Fisher BP1605) in BEBM. Cells were sub-cultured up to 11 times before use in exposure studies. All exposures were performed in BEGM without the supplied bovine pituitary extract (BPE) aliquot because its composition is not defined. For the gene expression experiments, BEAS-2Bs were plated at 40,000 cells/mL on matrix-coated 12-well Nunc™ Cell-Culture-Treated Multidishes (ThermoFisher Scientific Inc.), allowed to adhere overnight, and subsequently exposed for 48 h to the indicated concentrations of TGF-β1 dissolved in 0.1% BSA (Fisher BP1605) and 4 mM HCl or 10 nM E2 dissolved in DMSO (Mediatech Inc. MT25950CQC). Doses of TGF-β1 and E2 were chosen based on efficacy in previous experiments [26–30]. For the protein expression experiments, BEAS-2Bs were plated at 60,000 cells/mL on matrix-coated 6-well Corning® Costar® cell culture plates (Costar 3516), allowed to adhere overnight, and subsequently exposed for 48 h to the indicated concentrations of TGF-β1 in BEBM without BPE. All chemical solvent concentrations were maintained below 0.1%.
Total RNA extraction and purification
Frozen whole lung samples were pulverized over liquid nitrogen using a mortar and pestle then mechanically disrupted in RNA STAT-60™ Reagent (Tel-Test, Inc. Cs-502, Friendswood, TX) using a handheld homogenizer, and cell lysates were collected in RNA STAT-60™ Reagent and vortexed to promote lysis. RNA was extracted per manufacturer’s specifications followed by overnight precipitation at − 20 °C using 100% molecular biology grade isopropanol (Fisher BioReagents™ BP26184) containing 0.067% v/v GlycoBlue™ Coprecipitant (Ambion® AM9515). Precipitated RNA was pelleted by centrifugation at 14000 x g for 45 min at 4 °C and purified by washing 2X with 75% molecular biology grade absolute ethanol (Fisher BioReagents™ BP28184). RNA pellets were reconstituted in 15 μL RNAsecure™ (Ambion® AM7010). RNA was quantified using a Synergy™ H1 plate reader (BioTek Instrument, Inc., Winooski, VT) and RNA integrity was analyzed using a Bioanalyzer 2100 instrument (Agilent Technologies, Santa Clara, CA).
qPCR
RNA (1–2 μg) was DNase-treated using the PerfeCTa DNase I Kit (Quanta BioSciences 95,150-01 k, VWR International LLC, Suwanee, GA) and subsequently reverse transcribed using the qScript™ cDNA Synthesis Kit (Quanta BioSciences 95,047). cDNA was diluted 1:20 in RNase-DNase free water. Each 10 μL qPCR reaction contained 1× SsoAdvanced™ Universal SYBR® Green Supermix (Bio-Rad 172–5270, Hercules, CA), 850 nM forward and reverse primers, and 3.3 μL of the cDNA dilution. Per MIQE guidelines [31], gene specific primers and cycling parameters are displayed in Table 2. ESR2 primers were purchased from Bio-Rad (Unique Assay ID: qHsaCID0013184). Each qPCR reaction was followed by melt curve analysis to verify primer specificity. Cq values were determined by regression method using the CFX Manager 2.1 software and quantified using the relative ΔΔCq method [32] or the ratio method [33] when indicated. In the case of no amplification, a Cq value of 40 was applied. For the cell culture experiments, target gene expression was normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression and to ribosomal protein S13 (RPS13) expression in the human lung tissue samples.
Table 2.
Primer information for qPCR
| Gene | Forward (5′-3′) | Reverse (5′-3′) | Protocol | Efficiency | Source |
|---|---|---|---|---|---|
| GAPDH | GAAGGTGAAGGTCGGAGTC | GAAGATGGTGATGGGATTTC | 95C 3m; 95C 10s, 60C 30s, ×40 | 93.9% | [92] |
| ESR1 | CCACCAACCAGTGCACCATT | GGTCTTTTCGTATCCCACCTTTC | 95C 3m; 95C 10s, 60C 30s, ×40 | 100.7% | [93] |
| ESR2 | Proprietary | Proprietary | 95C 3m; 95C 10s, 60C 30s, ×40 | 102.9% | Bio-Rad |
| GPER | GCTCCCTGCAAGCAGTCTTT | GAAGGTCTCCCCGAGAAAGC | 95C 3m; 95C 10s, 60C 30s, ×40 | 97.2% | [94] |
| SNAI1 | CCAGACCCACTCAGATGTCAA | GGACTCTTGGTGCTTGTGGA | 95C 3m; 95C 10s, 58C 30s, ×40 | 97.2% | [94] |
| CDH1 | GAAAGCGGCTGATACTGACC | CTCAGACTAGCAGCTTCGGA | 95C 3m; 95C 10s, 58C 30s, ×40 | 104.9% | [94] |
| ACTA2 | CATCATGCGTCTGGATCTGG | GGACAATCTCACGCTCAGCA | 95C 3m; 95C 10s, 60C 30s, ×40 | 94.8% | [95] |
| MMP2 | TGTGTTCTTTGCAGGGAATG | TCCAGAATTTGTCTCCAGCA | 95C 3m; 95C 10s, 58C 30s, ×40 | 93.6% | [96] |
| CTGF | AATGCTGCGAGGAGTGGGT | CGGCTCTAATCATAGTTGGGTCT | 95C 3m; 95C 10s, 60C 30s, ×40 | 96.4% | [97] |
| VIM | GCGTGAAATGGAAGAGAACT | GGTATCAACCAGAGGGAGTG | 95C 3m; 95C 10s, 56C 10s, 72C 30S, ×40 | 104.0% | [30] |
| MUC15 | CCATCGGCGACTTTATGACG | TCTTCACTTTCTGGCATGGCT | 95C 3m; 95C 10s, 60C 30s, ×40 | 92.1% | [94] |
Protein extraction and purification
After exposure, media was removed, and cells were washed 3X with ice-cold PBS and subsequently collected in 200 μL RIPA Lysis and Extraction Buffer (Pierce Biotechnology 89,900) containing Pierce™ Protease Inhibitor Tablets (Pierce Biotechnology 88,265) using a cell scraper. The lysates were passed 5X through a 25-gauge needle and incubated on ice for 30 min with intermittent mixing. Thereafter, the lysates were centrifuged at 14000 x g for 20 min at 4 °C, supernatants were removed, and total protein quantified using the Pierce BCA Protein Assay Kit (Pierce Biotechnology 23,225).
Western blot
For SDS-PAGE, total protein (15 μg) was diluted in Novex™ Tris-Glycine SDS Sample Buffer (2X) (Invitrogen LC2676) and loaded onto a Novex™ WedgeWell™ 4–12% Tris-Glycine Mini Gel (Invitrogen XP04120BOX). Electrophoresis was performed for 30 min at 225 V and electrophoresed proteins were subsequently transferred to nitrocellulose membranes (GVS Life Sciences EP4HY00010) by semi-dry transfer method under 15 V for 30 min. Blots were blocked with 5% dehydrated milk dissolved in TBST for 1 h at room temperature, then incubated with mouse monoclonal antibody specific for estrogen receptor α (anti-ESR1, Santa Cruz Biotechnology, Inc. SC-514857, Dallas, TX) diluted 1:100 in blocking solution overnight at 4 °C. Blots were washed 3X in TBST for 10 min and incubated with HRP-linked Rabbit anti-Mouse IgG (H + L) Secondary Antibody (Pierce 31,450, Rockford, IL) diluted 1: 4000 in TBST for 1 h at room temperature. Blots were washed 3X with TBST for 10 min at room temperature, then 1X with TBS at room temperature. Thereafter, blots were incubated with 1:1 solution of Clarity™ Western ECL Blotting Substrates (Bio-Rad 170–5060, Hercules, CA) and imaged using the auto-exposure option on a Bio-Rad ChemiDoc™ MP system (Bio-Rad 17,001,402). After probing with anti-ESR1, blots were incubated 2X for 10 min at room temperature in mild stripping buffer (0.1 M Glycine, 20 mM MgAcetate, 50 mM KCl, pH 2.2), then washed 3X with TBST for 5 min at room temperature. To verify antibody stripping, the blot was probed with HRP-linked secondary antibody and re-imaged as before. After verification of anti-ESR1 removal, the blot was incubated in blocking solution for 1 h at room temperature, then incubated with mouse monoclonal antibody specific for β-Actin (anti-β-Actin, Sigma A5441) diluted 1:5000 in blocking solution overnight at 4 °C. The blot was subsequently washed as before, incubated in HRP-linked Rabbit anti-Mouse IgG (H + L) Secondary Antibody (Pierce 31,450) diluted 1:5000 in TBST for 1 h at room temperature, and imaged as before. Densitometry was performed in ImageJ [34] using the Gel Analysis method outlined in the ImageJ documentation.
RNA-Seq library preparation and sequencing
RNA library construction was performed at the Interdisciplinary Center for Biotechnology Research (ICBR) Gene Expression Core, University of Florida (UF). RNA concentration was determined on Qubit® 2.0 Fluorometer (ThermoFisher/Invitrogen, Grand Island, NY), RNA quality was assessed using the Agilent 2100 Bioanalyzer. Total RNA with 28S/18S > 1 and RNA integrity number (RIN) ≥ 7 was used for RNA-seq library construction. The RINs of all total RNA samples were 9.7–10. 2 μL of 1:2000 diluted External RNA Controls Consortium (ERCC) Spike-In Mix (Ambion™ 4,456,740, ThermoFisher Scientific Inc.) was added to 100 ng of high-quality total RNA followed by mRNA isolation using NEBNext Poly(A) mRNA Magnetic Isolation module (New England Biolabs E7490, Ipswich, MA) and RNA library construction with NEBNext Ultra RNA Library Prep Kit for Illumina (New England Biolabs E7530) according to the manufacturer’s user guide. Specifically, 100 ng of total RNA together with 2 μL of 1:2000 diluted ERCC was added to extracted mRNA with 15 μL of NEBNext Magnetic Oligo d(T)25 and fragmented in NEBNext First Strand Synthesis Buffer by heating at 94 °C for 8 min, followed by first-strand cDNA synthesis using reverse transcriptase and random primers. Synthesis of ds cDNA was done using the 2nd strand master mix provided in the kit. The resulting double-stranded cDNA was end-repaired, dA-tailed and ligated with NEBNext adaptors. Finally, library was enriched by 13 cycles of amplification, and purified by Meg-Bind RxnPure Plus beads (Omega Biotek M1386, Norcross, GA). Bar-coded libraries were sized on the bioanalyzer, quantitated by QUBIT and qPCR (Kapa Biosystems KK4824, Wilmington, MA). Seventeen individual libraries were pooled at equal molar (20 nM).
Bar-coded cDNA was sequenced using the 2 × 100 configuration in 2 lanes of a HiSeq 3000 instrument (Illumina, San Diego, CA). The yield for the run was in the expected range, the quality was good with Q30 > 96.25%, and the pool was well-balanced (in terms of number of reads per samples).
Bioinformatics
Short reads were trimmed and filtered to remove low-quality reads using Trimmomatic version 0.36. Quality control was assessed using the FastQC tool, version 0.11.4. Short reads were aligned to the transcriptome using STAR version 2.5.2a. Transcript quantification and differential analysis was performed using RSEM version 1.2.31. Differential analysis was performed at the level of coding genes, all transcript, and all splicing isoforms. Coding genes, transcripts, and splicing isoforms were considered statistically significant if FDR-corrected p-value ≤5% and fold change > 1.5 in either direction. Clustering analysis was performed using the ‘gplots’ package in R version 3.3.2 (2016-10-31). Gene set enrichment analysis was performed using Pathway Studio® Version 11.4.0.8 operating on the ResNet Mammalian database (Elsevier). Statistically significant enrichment (p ≤ 0.05) of predefined gene sets was determined by Mann-Whitney U-test. The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus [35] and are accessible through GEO Series accession number GSE100574 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE100574).
Statistics
Normality of experimental data was determined by D’Agostino & Pearson omnibus normality test, Shapiro-Wilk normality test, or KS normality test using GraphPad Prism software (Version 5.01, GraphPad Software, Inc.). Data were determined to be normal by passing at least one normality test (p < 0.05). For qPCR and western blot analyses, statistically significant differences (p < 0.05) in mean fold changes between experimental groups were determined by one-way ANOVA followed by Newman-Keuls multiple comparison test or unpaired, two-tailed t test when indicated using GraphPad Prism software. If the data were not normal, statistically significant differences were determined by two-tailed Mann-Whitney U test.
Results
TGF-β1 induces changes consistent with EMT
To begin investigations, we optimized a well-characterized model of TGF-β1-induced EMT based on changes in gene expression. BEAS-2Bs were exposed to TGF-β1 (0.1, 1, and 5 ng/mL) for 48 h and mRNA expression of molecular markers for EMT were assayed by qPCR. Results revealed a dose-dependent response where exposure of cells for 48 h to 1 and 5 ng/mL TGF-β1 caused a significant reduction in expression of the epithelial cell type marker, E-cadherin (CDH1), compared to control cells (Fig. 1a). Conversely, exposure of cells to 1 and 5 ng/mL TGF-β1 caused a significant increase in expression of the mesenchymal cell type markers, Vimentin (VIM), Snail family transcriptional repressor 1 (SNAI1), N-cadherin (CDH2), and Fibronectin (FN1) compared to control cells (Fig. 1b, d-f). Exposure of cells to TGF-β1 did not affect expression of the myofibroblast cell type marker, alpha smooth muscle actin (ACTA2) at any tested doses (Fig. 1c).
Fig. 1.

TGF-β1 induces EMT-like changes in mRNA expression in BEAS-2B cells. (a-f) BEAS-2B cells were exposed to TGF-β1 (0.1, 1, and 5 ng/mL) for 48 h and mRNA expression of the epithelial cell type marker, E-cadherin (CDH1, a), the mesenchymal cell type markers Vimentin (VIM, b), Snail family transcriptional repressor 1 (SNAI1, d), Cadherin 2 (CDH2, e), and Fibronectin (FN1, f), and the myofibroblast cell type marker, Alpha smooth muscle actin (ACTA2) was measured by qPCR. Target gene expression was normalized to GAPDH mRNA expression and quantified as fold change to control using the relative ΔΔCq method. Data are mean ± SEM of three or four independent experiments. Different letters indicate statistically significant (p < 0.05) differences between groups as determined by one-way ANOVA and Newman-Keuls multiple comparison test
E2 does not significantly affect TGF-β1-induced EMT
A role for E2 in modulating TGF-β1-induced EMT was assessed by exposing BEAS-2B cells to 5 ng/mL TGF-β1 in the presence and absence of 10 nM E2 for 48 h. Thereafter, expression of EMT marker genes was assayed by qPCR. As expected, 5 ng/mL TGF-β1 caused a reduction (0.686-fold, p > 0.05) in CDH1 mRNA expression (Fig. 2a). However, the addition of 10 nM E2 did not significantly affect the TGF-β1-induced response or CDH1 expression individually (Fig. 2a). Exposure of cells to 5 ng/mL TGF-β1 caused a trend of increased mRNA expression of the mesenchymal markers, VIM (p > 0.05), SNAI1 (p > 0.05), CDH2, FN1 (Fig. 2b, d-f) and similar to CDH1, exposure of cells to 5 ng/mL TGF-β1 in the presence of 10 nM E2 did not result in a statistically significant difference from cells exposed to TGF-β1 individually (Fig. 2b, d-f). As before, there were no statistically significant differences in expression of ACTA2 compared to control cells in any exposure group, however there was a 1.74-fold trend of increased expression in the co-exposure group (Fig. 2c). Statistically significant differences could not be determined between groups for CDH2 and FN1 because these data were generated from only two independent experiments.
Fig. 2.

E2 does not significantly affect TGF-β1-induced EMT. BEAS-2B cells were exposed to 5 ng/mL TGF-β1 in the presence or absence of 10 nM E2 for 48 h. (a-f) Expression of CDH1 (a), VIM (b), ACTA2 (c), SNAI1 (d), CDH2 (e), and FN1 (f) mRNA was measured by qPCR. Target gene expression was normalized to GAPDH mRNA expression and quantified as fold change to control using the relative ΔΔCq method. Data are mean ± SEM of three or four (a-d) or two (e-f) independent experiments. Different letters indicate statistically significant (p < 0.05) differences between groups as determined by one-way ANOVA and Newman-Keuls multiple comparison test
TGF-β1 reduces estrogen receptor mRNA and protein expression
Next, we investigated whether estrogen receptor mRNA expression was was a target for TGF-β1. The baseline expression levels of estrogen receptor alpha (ESR1), estrogen receptor beta (ESR2), and g-protein coupled estrogen receptor (GPER1) in control cells were expressed as a ratio to ESR1 based on the method described by Pfaffl et al. [33] to determine relative baseline expression levels. The relative expression of each receptor subtype was GPER1 > ESR1 > ESR2 (Fig. 3a). Exposing BEAS-2Bs to increasing concentrations of TGF-β1 (0.1, 1, and 5 ng/mL) for 48 h caused a 1.81-, 3.11-, and 2.87-fold (p < 0.05) decrease in ESR1 mRNA expression compared to controls (Fig. 3b). Similar trends were observed for ESR2 mRNA expression compared to controls (Fig. 3c), and a 1.44, 1.72, and 1.78-fold (p < 0.05) decrease in GPER1 mRNA expression compared to controls (Fig. 3d) was observed for the three doses, respectively.
Fig. 3.
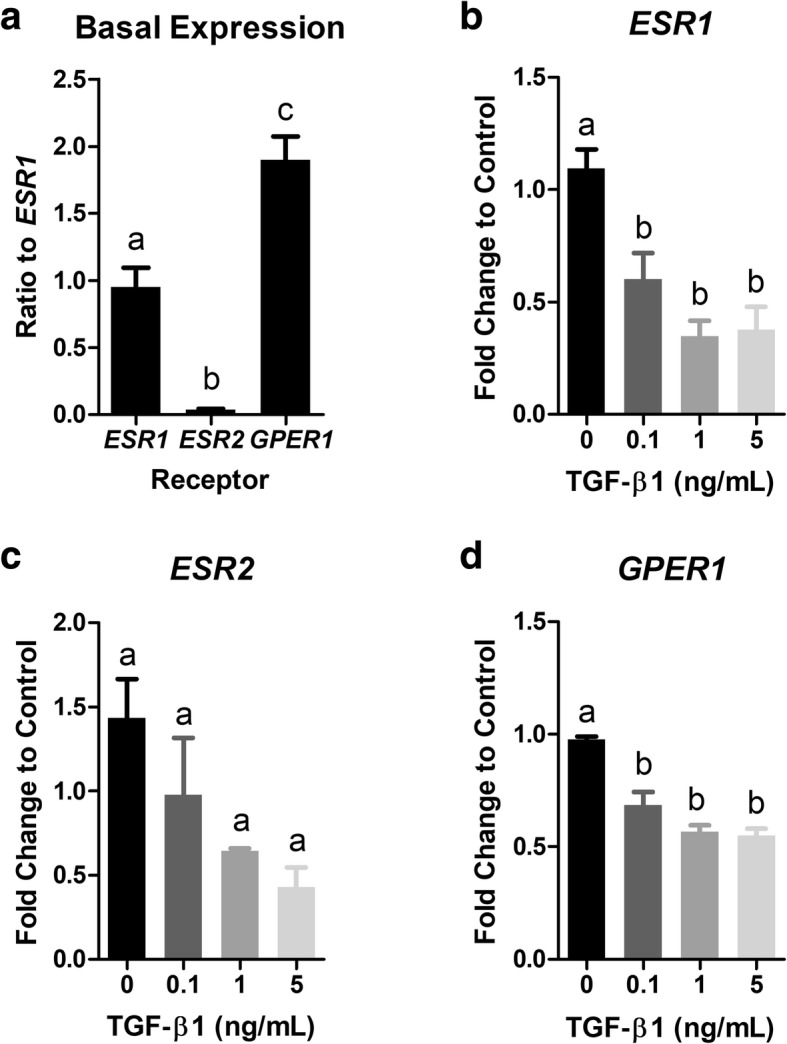
TGF-β1 down-regulates ESR1, ESR2, and GPER1 mRNA expression in BEAS-2Bs. (a) Relative expression of estrogen receptor subtypes in control cells was GPER1 > ESR1 > ESR2. ESR gene expression was normalized to GAPDH mRNA expression and calculated as a ratio to ESR1 mRNA expression. (b-d) BEAS-2B cells were exposed to TGF-β1 (0.1, 1, and 5 ng/mL) for 48 h and expression of ESR1 (n = 3; b), ESR2 (n = 2; c), and GPER1 (n = 3; d) mRNA was measured by qPCR. Target gene expression was normalized to GAPDH mRNA expression and quantified as fold change to control using the relative ΔΔCq method. Data are mean ± SEM and different letters indicate statistically significant (p < 0.05) differences between groups as determined by one-way ANOVA and Newman-Keuls multiple comparison test
BEAS-2Bs were exposed to TGF-β1 (0.1, 1, and 5 ng/mL) for 48 h and ESR1 was detected by western blot to determine if TGF-β1 reduced ESR1 protein levels (Fig. 4a). Signal intensity was quantified by densitometry using ImageJ (Fig. 4b). Similar to the mRNA results, TGF-β1 (0.1, 1, and 5 ng/mL) caused a 2.09-, 2.77-, and 3.76-fold significant decrease in ESR1 protein levels, respectively (Fig. 4b).
Fig. 4.
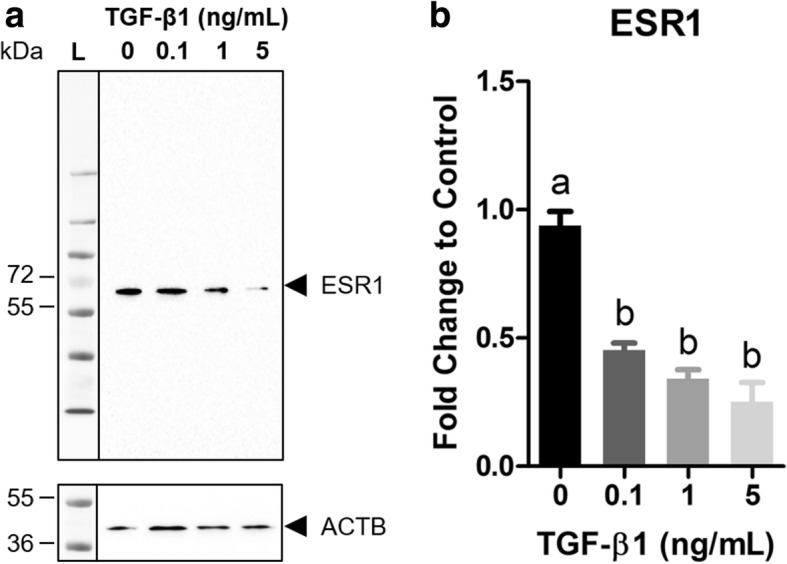
TGF-β1 down-regulates ESR1 protein expression. BEAS-2B cells were exposed to TGF-β1 (0.1, 1, and 5 ng/mL) for 48 h and ESR1 protein expression was measured by western blot followed by densitometric analysis in ImageJ. a Representative western blot. b Fold change ESR1 protein expression was normalized to Beta-actin (ACTB) and calculated as fold change to vehicle control (0 ng/mL TGF-β1). Data are mean ± SEM normalized arbitrary density units of duplicate measurements per blot of three independent experiments. Different letters indicate statistically significant (p < 0.05) differences between groups as determined by one-way ANOVA and Newman-Keuls multiple comparison test
Estrogen receptor mRNA expression is reduced in lungs of patients with IPF
We next questioned whether our in vitro results translated in vivo. To answer this, mRNA expression of ESR1, ESR2, and GPER1 was compared in lung tissue from healthy controls to individuals with end-stage IPF given that those with IPF tend to have higher TGF-β1 serum levels compared to healthy controls [36]. A qPCR analysis found that ESR1 and GPER1 mRNA expression was significantly reduced in the lungs of patients with end-stage IPF compared to healthy controls while there was a trend of reduced expression of ESR2 in the former group (Fig. 5a-c).
Fig. 5.

Estrogen receptor mRNA expression is reduced in lungs of patients with severe IPF compared to healthy control subjects. a-c Gene expression of ESR1 (a), ESR2 (b), and GPER1 (c) was measured in lung tissue from patients with IPF and healthy controls by qPCR. Target gene expression was normalized to GAPDH mRNA expression and quantified as fold change to control using the relative ΔΔCq method. Box plots represent 5–95% confidence intervals and asterisks (*) indicate statistically significant (p < 0.05) differences compared to controls as determined by two-tailed Mann-Whitney U test
TGF-β1 and E2 exhibit unique transcriptional profiles
RNA-Seq was performed to identify transcriptional targets of E2 in bronchial epithelial cells and cellular processes that may be affected by the observed down-regulation of ESR expression by TGF-β1. For this experiment, BEAS-2Bs were exposed to either vehicle control, 5 ng/mL TGF-β1 for 48 h, 10 nM E2 for 24 h, or pre-exposed to 5 ng/mL TGF-β1 for 24 h and subsequently co-exposed to both 5 ng/mL TGF-β1 and 10 nM E2 for 24 h (Fig. 6a). Differential expression analysis resulted in 2182 coding genes with FDR-corrected p-value ≤0.05 and Log2(Fold Change) > |0.6| compared to controls in the TGF-β1 group. The expression of 2119 coding genes was altered in the group co-exposed to TGF-β1 and E2, and 10 in the group exposed to E2. In sum, 379, 316, and 6 genes were specifically altered in the TGF-β1, TGF-β1 + E2, and E2 groups, respectively, while 1798 genes were differentially regulated in both the TGF-β1 and TGF-β1 + E2 groups, and 4 were differentially regulated in all groups (Fig. 6b). Many of the genes significantly up-regulated by TGF-β1, such as CTGF, MMP2 and VIM, are well-known targets of this pathway (Fig. 7, Additional file 1: Table S1). Other genes significantly up-regulated in all treatment groups included Sprouty RTK signaling antagonist 4 (SPRY4) and Dual specificity phosphatase 6 (DUSP6), and significantly down-regulated genes included Potassium voltage-gated channel subfamily Q member 1 (KCNQ1) and RAS protein activator like 1 (RASAL1) (Additional file 1: Tables S1-S2, Table 3). Genes that were specifically regulated by E2 included Retinol binding protein 7 [RBP7, Log2(Fold Change) = − 1.65] and Chloride intracellular channel 3 [CLIC3, Log2(Fold Change) = − 0.73] (Table 3). A hierarchical clustering analysis of genes differentially regulated in at least one exposure group showed that the expression profiles of the TGF-β1 and TGF-β1 + E2 group were more similar to each other than to the expression profile of E2 (Fig. 6c).
Fig. 6.

TGF-β1 and E2 exhibit distinct transcriptional profiles. a BEAS-2Bs were exposed to 5 ng/mL TGF-β1 and 10 nM E2 individually and in combination. Cells were acclimated for 24 h, then groups 2 and 3 were exposed to TGF-β1. After 24 h, groups 3 and 4 were exposed to E2, and all samples were collected 24 h thereafter. b Venn diagram highlighting distribution of differentially expressed genes [Log2(Fold Change) ≥ |0.6| and FDR-corrected p-value < 0.05] among the treatment groups. c Heat map showing the clustering and relative expression levels [Log2(Fold Change) compared to controls] of genes that were differentially expressed in at least one treatment group. Red coloring indicates up-regulation compared to controls and green coloring indicates down-regulation compared to controls, (T, TGF-β1; T + E, TGF-β1 + E2; E, E2)
Fig. 7.

Orthogonal validation of RNA-Seq data. a-d Expression of select genes was validated by qPCR; ESR1 (a), Connective tissue growth factor (CTGF, b), VIM (c), and Matrix metalloproteinase 2 (MMP2, d), in an identical and independent experiment. Bars represent expression [Log2(Fold Change)] of each gene in the RNA-Seq analysis, and black dots represent expression [Log2(Fold Change)] in each sample (n = 6) in the orthogonal experiment as determined by qPCR relative to vehicle control (DMSO). Target gene expression as measured by qPCR was normalized to GAPDH mRNA expression and quantified as fold change to control using the relative ΔΔCq method. Asterisks (*) indicate differential expression compared to controls [Log2(Fold Change) ≥ |0.6| and FDR-corrected p-value < 0.05] in the RNA-Seq analysis, and pound signs (#) indicate statistically significant (p < 0.05) differences compared to vehicle controls in the qPCR data as determined by one-way ANOVA and Newman-Keuls multiple comparison test
Table 3.
Genes differentially regulated by E2
| ENSEMBL Gene ID | Gene | Description | Log2(Fold Change) | p-value |
|---|---|---|---|---|
| ENSG00000258588 | TRIM6-TRIM34 | Tripartite Motif-Containing 6 And Tripartite Motif-Containing 34 | 5.36 | 1.33E-06 |
| ENSG00000256966 | RP11-613 M10.8 | AL513165.2 | 3.61 | 2.98E-02 |
| ENSG00000274944 | RP5-864 K19.6 | AL139260.3 | 2.93 | 1.34E-14 |
| ENSG00000255439 | RP11-196G11.1 | AC135050.2 | 1.43 | 9.12E-03 |
| ENSG00000187678 | SPRY4 | sprouty RTK signaling antagonist 4 | 0.76 | 3.20E-05 |
| ENSG00000139318 | DUSP6 | dual specificity phosphatase 6 | 0.75 | 2.25E-08 |
| ENSG00000169583 | CLIC3 | chloride intracellular channel 3 | −0.73 | 1.40E-06 |
| ENSG00000053918 | KCNQ1 | potassium voltage-gated channel subfamily Q member 1 | −0.86 | 4.87E-03 |
| ENSG00000111344 | RASAL1 | RAS protein activator like 1 | −1.34 | 1.79E-03 |
| ENSG00000162444 | RBP7 | retinol binding protein 7 | −1.65 | 3.08E-02 |
The expression of genes relevant to pulmonary fibrosis and this current work was validated by qPCR in an independent experiment (Fig. 7). As expected, exposure to TGF-β1 caused a significant reduction in ESR1 mRNA expression (Fig. 7a) and increased the expression of known targets of TGF-β1 such as Connective tissue growth factor (CTGF, Fig. 7b), VIM (Fig. 7c), and Matrix metalloproteinase 2 (MMP2, Fig. 7d). The presence of E2 in the co-exposure group did not have a clear effect on the expression of these genes compared to the TGF-β1 group.
TGF-β1 and E2 differentially regulate gene sets
Gene set enrichment analysis (GSEA) [37] was performed to identify statistical enrichment in the RNA-Seq data of curated pathways using Pathway Studio® Version 11.4.0.8 (Elsevier). The GSEA resulted in differential enrichment of biological function and disease pathways among the exposure groups. As expected, exposure to TGF-β1 resulted in enrichment of pathways such as ECM turnover and skin fibrosis (Table 4). Exposure to TGF-β1 also resulted in statistical enrichment of pathways including alveolar epithelial cell dysfunction, Ca2+ flux regulation, classical and alternative complement pathways, and neutrophil chemotaxis (Table 4). In most cases, similar enrichment and median changes were observed in the co-exposure group (TGF-β1 + E2) and the TGF-β1 only exposure group except for alveolar epithelial cell dysfunction which was not statistically enriched in the co-exposure group (Table 4). E2 also caused enrichment of classical and alternative complement pathways, airway smooth muscle cell contraction, Ca2+ flux regulation, and ECM turnover, however, the median change of the latter two pathways was inverse (down-regulated) compared to the median change observed in the TGF-β1 and co-exposure groups (Table 4). The ECM pathway is presented graphically to highlight the inverse regulation of genes in the pathway by TGF-β1 (Fig. 8, top) and E2 (Fig. 8, bottom). E2 specifically caused statistical enrichment of pathways including histone acetylation and phosphorylation pathways, nucleosome-remodeling factor (NURF) in chromatin remodeling, and vasodilation activation (Table 4).
Table 4.
Significantly enriched gene sets
| Name | TGF-β1 | TGF-β1 + E2 | E2 | ||||
|---|---|---|---|---|---|---|---|
| Pathway Type | Median Change | p-value | Median Change | p-value | Median Change | p-value | |
| Airway Smooth Muscle Cell Contraction | Diseases | 1.14 | 4.72E-04 | 1.10 | 4.34E-03 | −1.01 | 3.96E-02 |
| Alveolar Epithelial Cell Dysfunction | Diseases | 1.01 | 4.98E-02 | – | – | – | – |
| Ca2+ Flux Regulation | Biological Function | 1.02 | 2.12E-07 | 1.06 | 1.40E-05 | −1.00 | 5.48E-09 |
| Complement Alternative Pathway | Biological Function | −1.63 | 3.17E-04 | −1.49 | 2.64E-05 | −1.11 | 1.36E-03 |
| Complement Classical Pathway | Biological Function | −1.34 | 2.27E-04 | −1.49 | 3.09E-05 | −1.15 | 4.87E-04 |
| Extracellular Matrix Turnover | Biological Function | 1.55 | 2.26E-08 | 1.53 | 3.98E-07 | −1.04 | 6.86E-04 |
| Histone Acetylation | Biological Function | – | – | – | – | 1.04 | 3.68E-02 |
| Histone Phosphorylation | Biological Function | – | – | – | – | 1.05 | 2.99E-05 |
| Neutrophil Chemotaxis | Biological Function | 1.09 | 1.27E-03 | 1.07 | 7.25E-03 | – | – |
| NURF in Chromatin Remodeling | Biological Function | – | – | – | – | 1.06 | 2.48E-02 |
| Skin Fibrosis | Diseases | 1.07 | 2.15E-02 | 1.06 | 1.64E-02 | – | – |
| Vasodilation Activation | Biological Function | – | – | – | – | −1.00 | 3.34E-02 |
(−), not significantly enriched in specified exposure group
Fig. 8.

TGF-β1 and E2 cause differential regulation of genes involved in extracellular matrix turnover. A gene set enrichment analysis using Pathway Studio of genes identified by RNA-Seq revealed that exposure to 5 ng/mL TGF-β1 (top) and 10 nM E2 (bottom) caused statistically significant (p < 0.05) enrichment of the extracellular matrix turnover pathway. Gray boxes denote cellular processes involved in the extracellular matrix turnover pathway. Red proteins indicate up-regulation and blue proteins indicate down-regulation as determined by RNA-Seq
Discussion
This work was motivated by evidence suggesting hormones may influence gene regulation in the lung and contribute to sex differences in pulmonary diseases such as fibrosis. Using a well-established model lung epithelial cell line, we investigated the impact of E2 on TGF-β1-induced EMT. We report that although TGF-β1-induced EMT was not significantly affected by E2, this may be due to the novel observation that TGF-β1 repressed ESR expression, most notably ESR1. We extended this observation to identify novel targets of E2 by RNA-Seq that may be susceptible to TGF-β1-induced repression of ESRs such as chromatin remodeling processes and ECM turnover.
We first characterized the relative expression levels of the ESRs in our model cell line. We found that GPER1 was the most abundant followed by ESR1 while ESR2 was least expressed (Fig. 3a). Our results are similar to a study by Stabile et al. that found higher expression of ESR1 than ESR2 in human lung adenocarcinomas and squamous cell lung tumors although a difference between ESR1 and ESR2 expression was not evident in normal lung cells [38]. These results are in contrast to a study by Mollerup et al. that found that ESR2 was more abundantly expressed than ESR1 [39] and another study by Couse et al. that found greater expression of ESR2 in mouse lung [40]. The discrepancies may be a result of variable detection methods [38–40].
We probed a role for E2 in modulating TGF-β1-induced EMT because this process has been observed in fibrosis models although the relative contribution to fibrogenesis in humans is heavily debated [41–45]. TGF-β1 caused a significant reduction in expression of the epithelial cell type marker and a significant increase in expression of the mesenchymal cell type markers (Fig. 1b, d-f) similar to other studies [28–30]. Unlike the study by Doermer et al., we did not measure a significant increase in expression of ACTA2 mRNA (Fig. 1c) which may be a result of the duration of exposure as this particular marker tends to be more highly induced at later time-points (5 days) [29] and is indicative of further differentiation of fibroblasts into contractile myofibroblasts [46].
Using this model system, we determined whether E2 affected TGF-β1-induced EMT. A role for E2 in inhibiting EMT in humans was suggested in a study which found that reduced expression of ESR1 was associated with increased expression of genes involved in EMT in endometrial carcinoma samples [47]. Further, EMT is a target for sex hormones in cell types such as breast and prostate cancer cells where E2 signaling maintains an epithelial phenotype and suppresses EMT [48–51]. In our analysis, exposure to E2 did not significantly affect EMT marker gene expression individually nor did it impact the normal TGF-β1 response (Fig. 2a-f). Co-exposure to TGF-β1 and E2 resulted in a trend of increased expression of VIM and ACTA2 compared to TGF-β1 alone which is consistent with one study that reported E2 promoted reversible EMT-like transition and collective motility in breast cancer cells [52].
E2 may not affect TGF-β1-induced EMT due to direct actions of TGF-β1 on ESRs themselves. Interestingly, we found that exposure to TGF-β1 caused a dose-dependent and significant reduction in ESR1, ESR2, and GPER1 mRNA expression (Fig. 3b-c). We extended this to show that TGF-β1-induced repression of ESR1 persisted at the protein level (Fig. 4a-b). Other studies have shown that TGF-β1 reduces ESR1 mRNA expression [53, 54] and ESR1 protein expression [54, 55] in breast epithelial cancer cells and ESR2 protein expression in prostate cancer cells [51], however this is the first study to show that TGF-β1 reduced ESR2 and GPER1 mRNA expression and certainly the first to report any interaction between TGF-β1 and ESRs in bronchial epithelial cells. In support of this interaction occurring in vivo, we observed reduced expression of ESR1, ESR2, and GPER1 in lung tissue from patients with end-stage IPF compared to healthy control subjects (Fig. 5a-c). Although we do not have measurements of TGF-β1 in our samples, others have shown increased serum TGF-β1 levels in patients with IPF compared to healthy controls [36]. These results should be carefully interpreted given the small sample size and the absence of mechanistic information linking increased serum TGF-β1 levels to ESR1, ESR2, and GPER1 mRNA expression. Future studies should investigate which signaling mediators downstream of TGF-β1, e.g. SMADs, CTGF, or SNAI1, among others, are responsible for the observed repression.
Few studies to date have revealed a functional role for E2 in lung cells as measured by genes and pathways modulated downstream. Using RNA-Seq, we searched for enriched pathways in BEAS-2Bs exposed to E2 and TGF-β1 individually, and in combination, to both identify points of convergence of E2 and TGF-β1 signaling and to highlight novel E2 targets that may be susceptible to TGF-β1-induced repression of ESRs. Sequencing data indicated greater regulation of genes in response to TGF-β1 exposure in comparison to E2 exposure, perhaps consistent with the well-recognized strong pro-fibrotic response associated with TGF-β1. Although some genes were differentially regulated in the TGF-β1 or TGF-β1 + E2 exposure groups, most were shared suggesting that the presence of E2 had a minimal effect on the TGF-β1-induced transcriptome (Fig. 5b) potentially due to TGF-β1-induced repression of ESRs. We confirmed the expression of selected genes relevant to this work and/or known to be targets of TGF-β1 (Fig. 6, ESR1, VIM, CTGF, MMP2) which was consistent with our previous results and those reported in the literature [56–64].
Exposure to E2 did not induce as robust a transcriptional response in BEAS-2Bs compared to TGF-β1. However, we identified statistically significant regulation of 10 genes by E2 (Table 3) that have not been previously reported. Two genes that were specifically down-regulated by E2 included Chloride intracellular channel 3 (CLIC3) and Retinol binding protein 7 (RBP7) (Table 3). CLIC3 promotes migration and invasion of cancer cells by facilitating the functions of MT1-MMP (MMP14) [65, 66]. MT1-MMP is the most highly expressed MMP in IPF lungs [63] and may protect against PF by degrading collagen [67] and promoting lung repair [68]. Another study indicated that MT1-MMP promoted pulmonary fibrosis by activating latent TGF-β1 [69]. Our results suggest E2 may repress MT1-MMP function by downregulating CLIC3 mRNA expression. RBP7, also known as CRABP4, is a retinol binding protein thought to play an important role in retinol uptake, storage, and metabolism [70]. RBP7 has been shown to be up-regulated in IPF lung tissue [71] and in wound tissue in the normal chicken chorioallantoic wound model [72] although its role in fibrosis is unclear. RBP7 is positively regulated by E2 in breast cancer cells [73] and mouse mammary gland [74]. The discrepancy in regulation in our study may be a result of variable exposure dynamics as the study by Calvo et al. exposed mice to one dose of E2 and sacrificed the animals 3 h later [74] while we exposed cells in vitro for 24 h. Nonetheless, E2 appears to regulate RBP7 which may exhibit an unexplored effect on fibrogenic signaling.
As expected, exposure to TGF-β1 resulted in significant enrichment of the ECM turnover (Fig. 8a), alveolar epithelial cell dysfunction, and skin fibrosis pathways as determined by GSEA (Table 4). It is well known that TGF-β1 is involved in organization of the ECM [75] and neutrophil chemotaxis [76], and one of the prevailing hypotheses in IPF research is that it is a result of dysfunctional behavior of alveolar epithelial cells [77]. In this case, skin fibrosis serves as a surrogate for pulmonary fibrosis because the underlying mechanisms are similar and largely regulated by TGF-β1 [78], and pulmonary fibrosis does not exist as a curated, predefined pathway in Pathway Studio. In most cases, pathways enriched in the TGF-β1 individual exposure group were also enriched in the TGF-β1 + E2 co-exposure group, and the overall directionality as indicated by the median change was similar. This suggests that E2 has a limited effect on TGF-β1 once the pathways are in motion and/or was a result of TGF-β1-induced repression of ESRs thus mirroring the results seen at the gene level (Fig. 3b-c).
Interestingly, exposure to E2 resulted in specific enrichment of multiple pathways involved in epigenetic regulation of chromatin structure and organization including Histone Acetylation, Histone Phosphorylation, and NURF in Chromatin Remodeling (Table 4). While E2 has been shown to regulate histone acetylation in A549 cells [79], little is known about a role for E2 in transcriptionally regulating the expression of genes involved in chromatin remodeling in the lung. This is important because evidence for the importance of epigenetics and chromatin organization in lung disease is growing, particularly in the context of pulmonary fibrosis [80–85]. For example, histone deacetylases are involved in activation of lung fibroblasts to myofibroblasts [86] and accumulation of ECM components and EMT in the diabetic kidney [87]. Notably, exposure to TGF-β1 individually and in the presence of E2 did not result in enrichment of chromatin remodeling gene sets. This suggests that the absence of enrichment in the co-exposure group, despite the presence of E2, may be a result of TGF-β1-induced repression of ESR expression and not through direct regulation of genes by TGF-β1.
Similar to TGF-β1, exposure to E2 resulted in statistical enrichment of genes associated with ECM turnover, airway smooth muscle cell contraction, and calcium flux regulation pathways (Table 4). E2 is known to influence the ECM in the uterus and vaginal tissues [88], in osteoblasts [89], and in the skin [90]. Interestingly, the overall directionality of the pathway as indicated by the median change, was opposite (negative) compared to the directionality of the pathway in the TGF-β1 and TGF-β1 + E2 groups (positive, Table 4). This is consistent with a study that found that E2 inhibited TGF-β1-induced ECM production in human and rat mesangial cells through GPER1 activation [22]. Of note is the repression of MMP14 and MMP2 as another study showed that E2 decreased MMP2-, MMP13-, and MMP14-mediated tissue matrix destruction [91]. These results are consistent with the significant reduction of CLIC3 mRNA expression by E2 (Table 3) which is known to regulate MMP14 [65, 66]. Future studies should delineate the precise role of each ESR in regulating genes involved in ECM turnover.
Conclusions
In conclusion, we were not able to decipher an effect of E2 on TGF-β1-induced EMT, but we do report the novel observation that TGF-β1 inhibited ESR1, ESR2, and GPER1 mRNA expression and ESR1 protein expression in BEAS-2Bs. We also report that E2 specifically down-regulated the expression of CLIC3 and RBP7 which have been associated with pathogenic mechanisms of pulmonary fibrosis. We further highlight cellular pathways involved in chromatin remodeling as novel and specific targets of E2 in bronchial epithelial cells and opposing actions of TGF-β1 and E2 signaling on genes involved in ECM turnover. Although these data do not explicitly indicate a protective role for E2 in pulmonary fibrosis, these results suggest that E2 influences pathways relevant to pulmonary fibrosis and highlights potential roles for E2 in the lung that may contribute to sex-specific differences.
Additional file
Table S1. Genes differentially expressed (FDR-corrected p-value < 0.05) in TGFB1 group compared to vehicle control group. Table S2. Genes differentially expressed (FDR-corrected p-value < 0.05) in TGFB1 + E2 group compared to vehicle control group. (XLS 479 kb)
Acknowledgements
The authors would like to thank Yanping Zhang in the Gene Expression & Genotyping Core of the Interdisciplinary Center for Biotechnology Research (ICBR) for assistance with RNA-Seq library preparation, David Moraga of the NextGen DNA Sequencing core of UF ICBR for assistance with RNA sequencing, and Alberto Riva of the Bioinformatics core of UF ICBR for assistance with bioinformatics analysis of RNA-Seq data. The authors would also like to thank David Dreier and Amanda Buerger for assistance performing clustering analysis and creating heat maps.
Funding
This work was supported by funding from National Institutes of Health (R01HL114907 to T.S.A). Research reported in this publication was supported by the UF Clinical and Translational Science Institute, which is supported in part by the NIH National Center for Advancing Translational Sciences under award number UL1TR001427. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Availability of data and materials
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (39) and are accessible through GEO Series accession number GSE100574 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE100574).
Abbreviations
- ACTA2
Alpha smooth muscle actin
- ACTB
Beta-actin
- BEAS-2Bs
Bronchial epithelial cells
- CDH1
E-cadherin
- CDH2
N-cadherin
- CLIC3
Chloride intracellular channel 3
- CRABP4
Cellular retinoic acid-binding protein 4
- CTGF
Connective tissue growth factor
- DMSO
Dimethyl sulfoxide
- DUSP6
Dual specificity phosphatase 6
- E2
17β-Estradiol
- EMT
Epithelial to mesenchymal transition
- ERCC
External RNA controls consortium
- ESR
Estrogen receptor
- ESR1
Estrogen receptor alpha
- ESR2
Estrogen receptor beta
- FN1
Fibronectin
- FVC
Forced vital capacity
- FVC%
Percent predicted forced vital capacity
- GAPDH
Glyceraldehyde phosphate dehydrogenase
- GPER1
G-protein coupled estrogen receptor
- GSEA
Gene set enrichment analysis
- IPF
Idiopathic pulmonary fibrosis
- KCNQ1
Potassium voltage-gated channel subfamily Q member 1
- MIQE
Minimum information for publication of qPCR experiments
- MMP14
Matrix metalloproteinase 14
- MMP2
Matrix metalloproteinase 2
- MT1-MMP
Matrix metalloproteinase 14
- MT-MMP
Membrane-type matrix metalloproteinase
- NURF
Nucleosome remodeling factor
- PDGF
Platelet-derived growth factor
- RASAL1
RAS protein activator like 1
- RBP7
Retinol binding protein 7
- RPS13
Ribosomal protein S13
- SMAD3
SMAD family member 3
- SNAI1
Snail family transcriptional repressor 1
- SPRY4
Sprouty RTK signaling antagonist 4
- TGF-β1
Transforming growth factor beta1
- UIP
Usual interstitial pneumonia
- VIM
Vimentin
Authors’ contributions
LCS and TSA conceived the study and designed the experiments. LCS, SM, LR, KG, and SR performed the experiments. LCS and TSA interpreted the results. LCS drafted the manuscript and TSA edited. LCS, SM, LR, SR, AJB, and TSA approved the final version of the manuscript.
Ethics approval and consent to participate
The human lung tissue samples used in this study were a kind gift from Dr. Andrew Bryant. The deidentified, explanted lung tissue was obtained from subjects undergoing lung transplant for IPF and from lungs rejected for transplant from normal controls per the National Institutes of Health Lung Tissue Research Consortium (protocol no. 14-99-0011). The protocol for collection of lung tissue samples, and subsequent studies, were approved by the institutional review board at Vanderbilt University and the University of Florida (30).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
L. Cody Smith, Email: leycodysmith@gmail.com.
Santiago Moreno, Email: smorenoc@live.com.
Lauren Robertson, Email: laurenfay@ufl.edu.
Sarah Robinson, Email: sarah.robinson@phhp.ufl.edu.
Kristal Gant, Email: kgant@wisc.edu.
Andrew J. Bryant, Email: andrew.bryant@medicine.ufl.edu
Tara Sabo-Attwood, Phone: (352) 294-5293, Email: sabo@phhp.ufl.edu.
References
- 1.Han MK, Murray S, Fell CD, Flaherty KR, Toews GB, Myers J, Colby TV, Travis WD, Kazerooni EA, Gross BH, Martinez FJ. Sex differences in physiological progression of idiopathic pulmonary fibrosis. Eur Respir J. 2008;31:1183–1188. doi: 10.1183/09031936.00165207. [DOI] [PubMed] [Google Scholar]
- 2.Gribbin J, Hubbard RB, Le Jeune I, Smith CJ, West J, Tata LJ. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax. 2006;61:980–985. doi: 10.1136/thx.2006.062836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olson AL, Swigris JJ, Lezotte DC, Norris JM, Wilson CG, Brown KK. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med. 2007;176:277–284. doi: 10.1164/rccm.200701-044OC. [DOI] [PubMed] [Google Scholar]
- 4.Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174:810–816. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 5.Raghu G, Chen SY, Hou Q, Yeh WS, Collard HR. Incidence and prevalence of idiopathic pulmonary fibrosis in US adults 18–64 years old. Eur Respir J. 2016;48:179–186. doi: 10.1183/13993003.01653-2015. [DOI] [PubMed] [Google Scholar]
- 6.McGee SP, Zhang H, Karmaus W, Sabo-Attwood T. Influence of sex and disease severity on gene expression profiles in individuals with idiopathic pulmonary fibrosis. Int J Mol Epidemiol Genet. 2014;5:71–86. [PMC free article] [PubMed] [Google Scholar]
- 7.Redente EF, Jacobsen KM, Solomon JJ, Lara AR, Faubel S, Keith RC, Henson PM, Downey GP, Riches DW. Age and sex dimorphisms contribute to the severity of bleomycin-induced lung injury and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2011;301:L510–L518. doi: 10.1152/ajplung.00122.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voltz JW, Card JW, Carey MA, LM DG, Ferguson CD, Flake GP, Bonner JC, Korach KS, Zeldin DC. Male sex hormones exacerbate lung function impairment after bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2008;39:45–52. doi: 10.1165/rcmb.2007-0340OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valera IC, Kafaja S, Pham B, Fishbein M, Singh RR. Investigating mechanisms of sex bias in lung fibrosis using mice with sex chromosome complement. J Immunol. 2016;196:197–198. [Google Scholar]
- 10.Gharaee-Kermani M, Hatano K, Nozaki Y, Phan SH. Gender-based differences in bleomycin-induced pulmonary fibrosis. Am J Pathol. 2005;166:1593–1606. doi: 10.1016/S0002-9440(10)62470-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haston CK, Wang M, Dejournett RE, Zhou X, Ni D, Gu X, King TM, Weil MM, Newman RA, Amos CI, Travis EL. Bleomycin hydrolase and a genetic locus within the MHC affect risk for pulmonary fibrosis in mice. Hum Mol Gen. 2002;11:1855–1863. doi: 10.1093/hmg/11.16.1855. [DOI] [PubMed] [Google Scholar]
- 12.Lekgabe ED, Royce SG, Hewitson TD, MLK T, Zhao C, Moore XL, Tregear GW, Bathgate RA, Du XJ SCS. The effects of relaxin and estrogen deficiency on collagen deposition and hypertrophy of nonreproductive organs. Endocrinology. 2006;147:5575–5583. doi: 10.1210/en.2006-0533. [DOI] [PubMed] [Google Scholar]
- 13.Chang EC, Frasor J, Komm B, Katzenellenbogen BS. Impact of estrogen receptor β on gene networks regulated by estrogen receptor α in breast cancer cells. Endocrinology. 2006;147:4831–4842. doi: 10.1210/en.2006-0563. [DOI] [PubMed] [Google Scholar]
- 14.Ito I, Hanyu A, Wayama M, Goto N, Katsuno Y, Kawasaki S, Nakajima Y, Kajiro M, Komatsu Y, Fujimura A. Estrogen inhibits transforming growth factor β signaling by promoting Smad2/3 degradation. J Biol Chem. 2010;285:14747–14755. doi: 10.1074/jbc.M109.093039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goto N, Hiyoshi H, Ito I, Tsuchiya M, Nakajima Y, Yanagisawa J. Estrogen and antiestrogens alter breast cancer invasiveness by modulating the transforming growth factor-β signaling pathway. Cancer Sci. 2011;102:1501–1508. doi: 10.1111/j.1349-7006.2011.01977.x. [DOI] [PubMed] [Google Scholar]
- 16.Ashcroft GS, Dodsworth J, Van Boxtel E, Tarnuzzer RW, Horan MA, Schultz GS, Ferguson MWJ. Estrogen accelerates cutaneous wound healing associated with an increase in TGF-b1 levels. Nat Med. 1997;3:1209–1215. doi: 10.1038/nm1197-1209. [DOI] [PubMed] [Google Scholar]
- 17.Lovegrove AS, Sun J, Gould KA, Lubahn DB, Korach KS, Lane PH. Estrogen receptor α-mediated events promote sex-specific diabetic glomerular hypertrophy. Am J Physiol Renal Physiol. 2004;287:F586–F591. doi: 10.1152/ajprenal.00414.2003. [DOI] [PubMed] [Google Scholar]
- 18.Stope MB, Popp SL, Knabbe C, Buck MB. Estrogen receptor α attenuates transforming growth factor-β signaling in breast cancer cells independent from agonistic and antagonistic ligands. Breast Cancer Res Treat. 2010;120:357–367. doi: 10.1007/s10549-009-0393-2. [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto T, Saatcioglu F, Matsuda T. Cross-talk between bone morphogenic proteins and estrogen receptor signaling. Endocrinology. 2002;143:2635–2642. doi: 10.1210/endo.143.7.8877. [DOI] [PubMed] [Google Scholar]
- 20.Matsuda T, Yamamoto T, Muraguchi A, Saatcioglu F. Cross-talk between transforming growth factor-β and estrogen receptor signaling through Smad3. J Biol Chem. 2001;276:42908–42914. doi: 10.1074/jbc.M105316200. [DOI] [PubMed] [Google Scholar]
- 21.Kleuser B, Malek D, Gust R, Pertz HH, Potteck H. 17-β-estradiol inhibits transforming growth factor-β signaling and function in breast cancer cells via activation of extracellular signal-regulated kinase through the G protein-coupled receptor 30. Mol Pharmacol. 2008;74:1533–1543. doi: 10.1124/mol.108.046854. [DOI] [PubMed] [Google Scholar]
- 22.Li YC, Ding XS, Li HM, Zhang Y, Bao J. Role of G protein-coupled estrogen receptor 1 in modulating transforming growth factor-β stimulated mesangial cell extracellular matrix synthesis and migration. Mol Cell Endocrinol. 2014;391:50–59. doi: 10.1016/j.mce.2014.04.014. [DOI] [PubMed] [Google Scholar]
- 23.Society AT. Idiopathic pulmonary fibrosis, diagnosis and treatment. (International consensus statement) Am J Respir Crit Care Med. 2000;161:646–664. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- 24.Travis WD, King TE, Bateman ED, Lynch DA, Capron F, Center D, Colby TV, Cordier JF, DuBois RM, Galvin J. American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2002;165:277–304. doi: 10.1164/ajrccm.165.2.ats01. [DOI] [PubMed] [Google Scholar]
- 25.Bryant AJ, Carrick RP, McConaha ME, Jones BR, Shay SD, Moore CS, Blackwell TR, Gladson S, Penner NL, Burman A. Endothelial HIF signaling regulates pulmonary fibrosis-associated pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2016;310:L249–L262. doi: 10.1152/ajplung.00258.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith LC, Ralston-Hooper KJ, Ferguson PL, Sabo-Attwood T. The G protein-coupled estrogen receptor agonist G-1 inhibits nuclear estrogen receptor activity and stimulates novel phosphoproteomic signatures. Toxicol Sci. 2016;151:434–446. doi: 10.1093/toxsci/kfw057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith LC, Clark JC, Bisesi JH, Ferguson PL, Sabo-Attwood T. Differential recruitment of co-regulatory proteins to the human estrogen receptor 1 in response to xenoestrogens. Comp Biochem Physiol Part D Genomics Proteomics. 2016;19:159–173. doi: 10.1016/j.cbd.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamitani S, Yamauchi Y, Kawasaki S, Takami K, Takizawa H, Nagase T, Kohyama T. Simultaneous stimulation with TGF-β1 and TNF-α induces epithelial mesenchymal transition in bronchial epithelial cells. Int Arch Allergy Immunol. 2011;155:119–128. doi: 10.1159/000318854. [DOI] [PubMed] [Google Scholar]
- 29.Doerner AM, Zuraw BL. TGF-β1 induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells is enhanced by IL-1β but not abrogated by corticosteroids. Respir Res. 2009;10:100. doi: 10.1186/1465-9921-10-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hosper NA, van den Berg PP, de Rond S, Popa ER, Wilmer MJ, Masereeuw R, Bank RA. Epithelial-to-mesenchymal transition in fibrosis: collagen type I expression is highly upregulated after EMT, but does not contribute to collagen deposition. Exp Cell Res. 2013;319:3000–3009. doi: 10.1016/j.yexcr.2013.07.014. [DOI] [PubMed] [Google Scholar]
- 31.Taylor S, Wakem M, Dijkman G, Alsarraj M, Nguyen M. A practical approach to RT-qPCR—publishing data that conform to the MIQE guidelines. Methods. 2010;50:S1–S5. doi: 10.1016/j.ymeth.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 32.Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8:R19. doi: 10.1186/gb-2007-8-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pfaffl MW. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature Methods. 2012;9:671. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alhamad EH, Cal JG, Shakoor Z, Almogren A, AlBoukai AA. Cytokine gene polymorphisms and serum cytokine levels in patients with idiopathic pulmonary fibrosis. BMC Med Genet. 2013;14:66. doi: 10.1186/1471-2350-14-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stabile LP, Davis ALG, Gubish CT, Hopkins TM, Luketich JD, Christie N, Finkelstein S, Siegfried JM. Human non-small cell lung tumors and cells derived from normal lung express both estrogen receptor α and β and show biological responses to estrogen. Cancer Res. 2002;62:2141–2150. [PubMed] [Google Scholar]
- 39.Mollerup S, Jørgensen K, Berge G, Haugen A. Expression of estrogen receptors α and β in human lung tissue and cell lines. Lung Cancer. 2002;37:153–159. doi: 10.1016/S0169-5002(02)00039-9. [DOI] [PubMed] [Google Scholar]
- 40.Couse JF, Lindzey J, Grandien K, Gustafsson J, Korach KS. Tissue distribution and quantitative analysis of estrogen receptor-α (ERα) and estrogen receptor-β (ERβ) messenger ribonucleic acid in the wild-type and ERα-knockout mouse. Endocrinology. 1997;138:4613–4621. doi: 10.1210/endo.138.11.5496. [DOI] [PubMed] [Google Scholar]
- 41.Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willis BC, Borok Z. TGF-b-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. 2007;293:L525–L534. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- 43.Kage H, Borok Z. EMT and interstitial lung disease: a mysterious relationship. Curr Opin Pulm Med. 2012;18:517–523. doi: 10.1097/MCP.0b013e3283566721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bartis D, Mise N, Mahida RY, Eickelberg O, Thickett DR. Epithelial-mesenchymal transition in lung development and disease: does it exist and is it important? Thorax. 2014;69:760–765. doi: 10.1136/thoraxjnl-2013-204608. [DOI] [PubMed] [Google Scholar]
- 45.Kasai H, Allen JT, Mason RM, Kamimura T, Zhang Z. TGF-b1 induces human alveolar epithelial to mesenchymal cell transition (EMT) Respir Res. 2005;6:56. doi: 10.1186/1465-9921-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kis K, Liu X, Hagood JS. Myofibroblast differentiation and survival in fibrotic disease. Expert Rev Mol Med. 2011;13:e27. doi: 10.1017/S1462399411001967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wik E, Ræder MB, Krakstad C, Trovik J, Birkeland E, Hoivik EA, Mjos S, Werner HM, Mannelqvist M, Stefansson IM. Lack of estrogen receptor-α is associated with epithelial–mesenchymal transition and PI3K alterations in endometrial carcinoma. Clin Cancer Res. 2013;19:1094–1105. doi: 10.1158/1078-0432.CCR-12-3039. [DOI] [PubMed] [Google Scholar]
- 48.Dhasarathy A, Kajita M, Wade PA. The transcription factor snail mediates epithelial to mesenchymal transitions by repression of estrogen receptor-α. Mol Endocrinol. 2007;21:2907–2918. doi: 10.1210/me.2007-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guttilla IK, Adams BD, White BA. ERα, microRNAs, and the epithelial–mesenchymal transition in breast cancer. Trends Endocrinol Metab. 2012;23:73–82. doi: 10.1016/j.tem.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 50.Ye Y, Xiao Y, Wang W, Yearsley K, Gao J, Shetuni B, Barsky S. ERα signaling through slug regulates E-cadherin and EMT. Oncogene. 2010;29:1451–1462. doi: 10.1038/onc.2009.433. [DOI] [PubMed] [Google Scholar]
- 51.Mak P, Leav I, Pursell B, Bae D, Yang X, Taglienti CA, Gouvin LM, Sharma VM, Mercurio AM. ERβ impedes prostate cancer EMT by destabilizing HIF-1α and inhibiting VEGF-mediated snail nuclear localization: implications for Gleason grading. Cancer Cell. 2010;17:319–332. doi: 10.1016/j.ccr.2010.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Planas-Silva MD, Waltz PK. Estrogen promotes reversible epithelial-to-mesenchymal-like transition and collective motility in MCF-7 breast cancer cells. J Steroid Biochem Mol Biol. 2007;104:11–21. doi: 10.1016/j.jsbmb.2006.09.039. [DOI] [PubMed] [Google Scholar]
- 53.Fridriksdottir AJ, Kim J, Villadsen R, Klitgaard MC, Hopkinson BM, Petersen OW, Rønnov-Jessen L. Propagation of oestrogen receptor-positive and oestrogen-responsive normal human breast cells in culture. Nature communications. 2015;6:8786. doi: 10.1038/ncomms9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stoica A, Saceda M, Fakhro A, Solomon HB, Fenster BD, Martin MB. The role of transforming growth factor-β in the regulation of estrogen receptor expression in the MCF-7 breast cancer cell line. Endocrinology. 1997;138:1498–1505. doi: 10.1210/endo.138.4.5074. [DOI] [PubMed] [Google Scholar]
- 55.Petrel TA, Brueggemeier RW. Increased proteasome-dependent degradation of estrogen receptor-alpha by TGF-β1 in breast cancer cell lines. J Cell Biochem. 2003;88:181–190. doi: 10.1002/jcb.10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sonnylal S, Xu S, Jones H, Tam A, Sreeram VR, Ponticos M, Norman J, Agrawal P, Abraham D, de Crombrugghe B. Connective tissue growth factor causes EMT-like cell fate changes in vivo and in vitro. J Cell Sci. 2013;126:2164–2175. doi: 10.1242/jcs.111302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leask A, Abraham DJ. The role of connective tissue growth factor, a multifunctional matricellular protein, in fibroblast biology. Biochem Cell Biol. 2003;81:355–363. doi: 10.1139/o03-069. [DOI] [PubMed] [Google Scholar]
- 58.Mäkelä M, Larjava H, Pirilä E, Maisi P, Salo T, Sorsa T, Uitto V-J. Matrix metalloproteinase 2 (gelatinase A) is related to migration of keratinocytes. Exp Cell Res. 1999;251:67–78. doi: 10.1006/excr.1999.4564. [DOI] [PubMed] [Google Scholar]
- 59.Kim ES, Sohn YW, Moon A. TGF-β-induced transcriptional activation of MMP-2 is mediated by activating transcription factor (ATF) 2 in human breast epithelial cells. Cancer Lett. 2007;252:147–156. doi: 10.1016/j.canlet.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 60.Seomun Y, Kim J, Choun-Ki J. Overexpression of matrix metalloproteinase-2 mediates phenotypic transformation of lens epithelial cells. Biochem J. 2001;358:41–48. doi: 10.1042/bj3580041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kheradmand F, Rishi K, Werb Z. Signaling through the EGF receptor controls lung morphogenesis in part by regulating MT1-MMP-mediated activation of gelatinase A/MMP2. J Cell Sci. 2002;115:839–848. doi: 10.1242/jcs.115.4.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ravanti L, Kahari V. Matrix metalloproteinases in wound repair. Int J Mol Med. 2000;6:391–407. [PubMed] [Google Scholar]
- 63.García-Alvarez J, Ramirez R, Sampieri CL, Nuttall RK, Edwards DR, Selman M, Pardo A. Membrane type-matrix metalloproteinases in idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2006;23:13–21. [PubMed] [Google Scholar]
- 64.Craig VJ, Zhang L, Hagood JS, Owen CA. Matrix metalloproteinases as therapeutic targets for idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2015;53:585–600. doi: 10.1165/rcmb.2015-0020TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dozynkiewicz MA, Jamieson NB, MacPherson I, Grindlay J, van den Berghe PV, von Thun A, Morton JP, Gourley C, Timpson P, Nixon C. Rab25 and CLIC3 collaborate to promote integrin recycling from late endosomes/lysosomes and drive cancer progression. Dev Cell. 2012;22:131–145. doi: 10.1016/j.devcel.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Macpherson IR, Rainero E, Mitchell LE, Van Den Berghe PV, Speirs C, Dozynkiewicz MA, Chaudhary S, Kalna G, Edwards J, Timpson P. CLIC3 controls recycling of late endosomal MT1-MMP and dictates invasion and metastasis in breast cancer. J Cell Sci. 2014;127:3893–3901. doi: 10.1242/jcs.135947. [DOI] [PubMed] [Google Scholar]
- 67.Koenig GC, Rowe RG, Day SM, Sabeh F, Atkinson JJ, Cooke KR, Weiss SJ. MT1-MMP–dependent remodeling of cardiac extracellular matrix structure and function following myocardial infarction. Am J Pathol. 2012;180:1863–1878. doi: 10.1016/j.ajpath.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Karow M, Popp T, Egea V, Ries C, Jochum M, Neth P. Wnt signalling in mouse mesenchymal stem cells: impact on proliferation, invasion and MMP expression. J Cell Mol Med. 2009;13:2506–2520. doi: 10.1111/j.1582-4934.2008.00619.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL. The integrin αvβ8 mediates epithelial homeostasis through MT1-MMP–dependent activation of TGF-β1. J Cell Biol. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Folli C, Calderone V, Ramazzina I, Zanotti G, Berni R. Ligand binding and structural analysis of a human putative cellular retinol-binding protein. J Biol Chem. 2002;277:41970–41977. doi: 10.1074/jbc.M207124200. [DOI] [PubMed] [Google Scholar]
- 71.Molyneaux PL, Willis Owen SA, Cox MJ, James P, Cowman S, Loebinger M, Blanchard A, Edwards LM, Stock C, Daccord C. Host-microbial interactions in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2017;195(12):1640–1650. doi: 10.1164/rccm.201607-1408OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Soulet F, Kilarski WW, Antczak P, Herbert J, Bicknell R, Falciani F, Bikfalvi A. Gene signatures in wound tissue as evidenced by molecular profiling in the chick embryo model. BMC Genomics. 2010;11:495. doi: 10.1186/1471-2164-11-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kinyamu HK, Collins JB, Grissom SF, Hebbar PB, Archer TK. Genome wide transcriptional profiling in breast cancer cells reveals distinct changes in hormone receptor target genes and chromatin modifying enzymes after proteasome inhibition. Mol Carcinog. 2008;47:845–885. doi: 10.1002/mc.20440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Calvo E, Belleau P, Martel C, Labrie F. Specific transcriptional response of four blockers of estrogen receptors on estradiol-modulated genes in the mouse mammary gland. Breast Cancer Res Treat. 2012;134:625–647. doi: 10.1007/s10549-012-2104-7. [DOI] [PubMed] [Google Scholar]
- 75.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor β in human disease. New Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 76.Parekh T, Saxena B, Reibman J, Cronstein BN, Gold LI. Neutrophil chemotaxis in response to TGF-beta isoforms (TGF-beta 1, TGF-beta 2, TGF-beta 3) is mediated by fibronectin. J Immunol. 1994;152:2456–2466. [PubMed] [Google Scholar]
- 77.Fernandez IE, Eickelberg O. The impact of TGF-β on lung fibrosis: from targeting to biomarkers. Proc Am Thorac Soc. 2012;9:111–116. doi: 10.1513/pats.201203-023AW. [DOI] [PubMed] [Google Scholar]
- 78.Border WA, Noble NA. Transforming growth factor β in tissue fibrosis. New Engl J Med. 1994;331:1286–1292. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- 79.Lai J-C, Cheng Y-W, Chiou H-L, Wu M-F, Chen C-Y, Lee H. Gender difference in estrogen receptor alpha promoter hypermethylation and its prognostic value in non-small cell lung cancer. Int J Cancer. 2005;117:974–980. doi: 10.1002/ijc.21278. [DOI] [PubMed] [Google Scholar]
- 80.Coward WR, Saini G, Jenkins G. The pathogenesis of idiopathic pulmonary fibrosis. Ther Adv Respir Dis. 2010;4:367–388. doi: 10.1177/1753465810379801. [DOI] [PubMed] [Google Scholar]
- 81.Coward WR, Watts K, Feghali-Bostwick CA, Jenkins G, Pang L. Repression of IP-10 by interactions between histone deacetylation and hypermethylation in idiopathic pulmonary fibrosis. Mol Cell Biol. 2010;30:2874–2886. doi: 10.1128/MCB.01527-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Coward WR, Watts K, Feghali-Bostwick CA, Knox A, Pang L. Defective histone acetylation is responsible for the diminished expression of cyclooxygenase 2 in idiopathic pulmonary fibrosis. Mol Cell Biol. 2009;29:4325–4339. doi: 10.1128/MCB.01776-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rabinovich EI, Kapetanaki MG, Steinfeld I, Gibson KF, Pandit KV, Yu G, Yakhini Z, Kaminski N. Global methylation patterns in idiopathic pulmonary fibrosis. PLoS One. 2012;7:e33770. doi: 10.1371/journal.pone.0033770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sanders YY, Ambalavanan N, Halloran B, Zhang X, Liu H, Crossman DK, Bray M, Zhang K, Thannickal VJ, Hagood JS. Altered DNA methylation profile in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186:525–535. doi: 10.1164/rccm.201201-0077OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang IV, Schwartz DA. Epigenetics of idiopathic pulmonary fibrosis. Transl Res. 2014;165.1(2015):48–60. [DOI] [PMC free article] [PubMed]
- 86.Wang Z, Chen C, Finger S, Jung M, Schwarz H, Swanson N, Lareu R, Raghunath M. Suberoylanilide hydroxamic acid: a potential epigenetic therapeutic agent for lung fibrosis? Eur Respir J. 2009;34:145–155. doi: 10.1183/09031936.00084808. [DOI] [PubMed] [Google Scholar]
- 87.Noh H, Oh EY, Seo JY, Yu MR, Kim YO, Ha H, Lee HB. Histone deacetylase-2 is a key regulator of diabetes-and transforming growth factor-β1-induced renal injury. Am J Physiol Renal Physiol. 2009;297:F729–F739. doi: 10.1152/ajprenal.00086.2009. [DOI] [PubMed] [Google Scholar]
- 88.Cox DA, Helvering LM. Extracellular matrix integrity: a possible mechanism for differential clinical effects among selective estrogen receptor modulators and estrogens? Mol Cell Endocrinol. 2006;247:53–59. doi: 10.1016/j.mce.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 89.Komm BS, Terpening CM, Benz DJ, Graeme KA, Gallegos A. Estrogen binding, receptor mRNA, and biologic response in osteoblast-like osteosarcoma cells. Science. 1988;241:81. doi: 10.1126/science.3164526. [DOI] [PubMed] [Google Scholar]
- 90.Son ED, Lee JY, Lee S, Kim MS, Lee BG, Chang IS, Chung JH. Topical application of 17β-estradiol increases extracellular matrix protein synthesis by stimulating TGF-β signaling in aged human skin in vivo. J Invest Dermatol. 2005;124:1149–1161. doi: 10.1111/j.0022-202X.2005.23736.x. [DOI] [PubMed] [Google Scholar]
- 91.Pincus DJ, Kassira N, Gombosh M, Berho M, Glassberg M, Karl M, Elliot SJ, Thaller S. 17β-Estradiol modifies diabetic wound healing by decreasing matrix metalloproteinase activity. Wounds. 2010;22:171–178. [PubMed] [Google Scholar]
- 92.Beck LA, Tancowny B, Brummet ME, Asaki SY, Curry SL, Penno MB, Foster M, Bahl A, Stellato C. Functional analysis of the chemokine receptor CCR3 on airway epithelial cells. J Immunol. 2006;177:3344–3354. doi: 10.4049/jimmunol.177.5.3344. [DOI] [PubMed] [Google Scholar]
- 93.Spizzo R, Nicoloso M, Lupini L, Lu Y, Fogarty J, Rossi S, Zagatti B, Fabbri M, Veronese A, Liu X. miR-145 participates with TP53 in a death-promoting regulatory loop and targets estrogen receptor-α in human breast cancer cells. Cell Death Differ. 2010;17:246–254. doi: 10.1038/cdd.2009.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tanaka KI, Tanaka Y, Namba T, Azuma A, Mizushima T. Heat shock protein 70 protects against bleomycin-induced pulmonary fibrosis in mice. Biochem Pharmacol. 2010;80:920–931. doi: 10.1016/j.bcp.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 96.Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nagaraja T, Chen L, Balasubramanian A, Groopman JE, Ghoshal K, Jacob ST, Leask A, Brigstock DR, Anand AR, Ganju RK. Activation of the connective tissue growth factor (CTGF)-transforming growth factor β 1 (TGF-β 1) axis in hepatitis C virus-expressing hepatocytes. PLoS One. 2012;7:e46526. doi: 10.1371/journal.pone.0046526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genes differentially expressed (FDR-corrected p-value < 0.05) in TGFB1 group compared to vehicle control group. Table S2. Genes differentially expressed (FDR-corrected p-value < 0.05) in TGFB1 + E2 group compared to vehicle control group. (XLS 479 kb)
Data Availability Statement
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (39) and are accessible through GEO Series accession number GSE100574 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE100574).