

Abstract
Serum amyloid A (SAA) proteins were isolated and named over 50 years ago. They are small (104 amino acids) and have a striking relationship to the acute phase response with serum levels rising as much as 1000-fold in 24 hours. SAA proteins are encoded in a family of closely-related genes and have been remarkably conserved throughout vertebrate evolution. Amino-terminal fragments of SAA can form highly organized, insoluble fibrils that accumulate in “secondary” amyloid disease. Despite their evolutionary preservation and dynamic synthesis pattern SAA proteins have lacked well-defined physiologic roles. However, considering an array of many, often unrelated, reports now permits a more coordinated perspective. Protein studies have elucidated basic SAA structure and fibril formation. Appreciating SAA’s lipophilicity helps relate it to lipid transport and metabolism as well as atherosclerosis. SAA’s function as a cytokine-like protein has become recognized in cell-cell communication as well as feedback in inflammatory, immunologic, neoplastic and protective pathways. SAA likely has a critical role in control and possibly propagation of the primordial acute phase response. Appreciating the many cellular and molecular interactions for SAA suggests possibilities for improved understanding of pathophysiology as well as treatment and disease prevention.
Keywords: Serum amyloid A, SAA, inflammation, amyloidosis, acute phase response (APR), arthritis, apolipoprotein, liver, cytokine, lipopolysaccharide (LPS), myeloid-derived suppressor cells (MDSC), atherosclerosis
Background
Homeostasis is essential for most biological systems. Both primordial and adaptable mechanisms exist for reestablishing this important state following perturbations. Among the former, the so-called “acute-phase response” (APR) is prominent. The APR comprises a stereotyped set of physiologic changes (Kushner 1982; Gabay and Kushner 1999; Yoo and Desiderio 2003) that occur as a consequence of inflammation, infection, trauma and other events. The APR involves many physiologic responses – fever, hormonal changes, metabolic alterations. Changes in serum protein levels during the APR are particularly remarkable. Among these, altered serum levels of C-reactive protein (CRP) and serum amyloid A (SAA) are the most notable. Proteins and genes of the SAA family are particularly prominent in the APR. This review emphasizes human SAA genes and proteins but their close relationships to those in murine and other species means that relevant studies from other systems also will be considered when informative.
Both CRP and SAA are present, but at generally quite low levels, in the blood of healthy individuals (SAA is normally found at 20-50 mcg/ml). However, their levels can rise as much as 1000-fold 24 hours after APR onset, largely reflecting de novo synthesis in the liver. Consistent with the APR definition, blood levels of both CRP and SAA fall rapidly as the stereotyped APR pattern resolves.
CRP is an endogenous human protein that specifically reacts with the type C polysaccharides of Streptococcus pneumoniae (Tillett and Francis 1930; Macleod and Avery 1941). Its level also was found to rise after other acute infections. Thus, it has been considered to participate in primordial, endogenous, host-defense against a commonly encountered bacterial pathogen. The serum CRP level is widely used as a non-specific but clinically useful marker for inflammation of many types.
By contrast, the biology of SAA protein(s) has been less clear; both the name and discovery have been more indirect. The term “amyloid” originally referred to material found in plants and presumed to be carbohydrate (Gk “amylon” = starch). “Amyloidosis” was used by 19th century pathologists to describe apparently amorphous, infiltrative histopathologic changes in kidney/liver/heart often found in postmortem examinations using light microscopy. Although the term implied that the infiltrating material was carbohydrate, later studies showed that “amyloid” changes comprised protein deposits (Hass 1942) and electron microscopy revealed arrays of microfibrils (Cohen and Calkins 1959). These deposits shared a characteristic birefringence when stained with planar dyes such as Congo red or thioflavin T and this tinctorial property remains the basis for current histopathologic diagnosis. Clinical data implied that the proteins constituting “amyloid” might differ because the deposits were encountered in very different pathologic contexts; many of these have subsequently been defined. For example, amyloid deposits in multiple myeloma comprise fibrillar arrays of fragments of immunoglobulin light chains. Thus, identifying the constituent protein(s) in amyloid deposits is important. In addition to immunoglobulin-derived polypeptides, Benditt and Eriksen (1961) distinguished a protein, referred to as “amyloid of unknown origin” (AUO), isolated by gel electrophoresis in cases of so-called “secondary” amyloidosis (i.e. those associated with chronic or recurrent inflammatory conditions). Subsequent studies revealed a distinct amino acid composition, including absence of cysteine and threonine and an amino terminal sequence of R-S-F-F-S (Benditt et al. 1971). Levin et al. (1972) presented the first complete sequence of a 76 aa protein found in secondary amyloid deposits. The fibril-derived proteins isolated by different laboratories differed slightly in length from 68 – 76 aa but shared common N-terminal residues (Benditt et al. 1971; Levin et al. 1972; Ein et al. 1972a, b). These were referred to as “AA” (i.e. “amyloid A”). Antibodies prepared against these proteins identified a small (104 aa) serum protein that was initially presumed and later shown to be their precursor (Linke et al. 1975; Meek et al. 1986; Prelli et al. 1987; Husby and Natvig 1974; Rosenthal et al. 1976). Because this was the first non-immunoglobulin serum protein identified as a precursor of amyloid disease deposits, it was named “serum amyloid A” (SAA). Subsequently, it has been shown that SAA is a prominent constituent of APR proteins arising from various stimuli.
The SAA sequence (see Fig. 1a) is remarkably conserved throughout the mammalian radiation as well as in birds and other animals (see Fig. 1b and (Uhlar et al. 1994; Jensen et al. 1997)). In humans, multiple, apparently minor, variants have been identified. Figure 2 (Faulkes et al. 1994; Sipe 1999; Sun and Ye 2016) shows reported sequences. Unfortunately, many past reports did not distinguish among SAA gene and/or protein family members. Unless specified originally, the APR serum species (SAA1 and/or 2) should be considered relevant; other family members, when identified, will be noted in this review.
Fig. 1.

a Consensus amino acid sequence for human SAA (SAA1 shown although variants are recognized as well – see Fig. 2) b Comparison of SAA sequences in different organisms. Conserved residues noted in boxes. (USCS Genome Browser [GRCh38/hg38] Assembly)
Fig. 2.

Reported amino acid variants for SAA1 and SAA2 in humans. Underlined regions appear invariant.
The APR profile of SAA protein levels in blood is similar to that described for CRP, rising as much as 1000-fold within the initial 24 hours and then returning to very low levels as the event resolves. Although proteolysis of SAA yields the N-terminal fragment(s) found in the microfibrils in secondary amyloid deposits the responsible protease(s) has(ve) not yet been unequivocally identified. SAA is poorly soluble in aqueous solutions; in blood it is partitioned into high density lipoproteins (HDL (Benditt and Eriksen 1977; Benditt et al. 1979)). It functions as an apolipoprotein in this regard, possibly changing the character of the HDL particles (see below and (Benditt et al. 1979; Hoffman and Benditt 1982; Cabana et al. 1989; Kisilevsky and Subrahmanyan 1992)). Aside from its apolipoprotein character, no function was initially identified for SAA although its APR association implied that it might be related to primordial host defense or inflammation, similar to the situation for CRP.
SAA proteins
Although SAA proteins are small and their sequences and polymorphisms have been carefully catalogued details of their 3-dimensional structure(s) have been elusive. A barrier to fully characterizing these proteins has been their generally poor solubility. The previously recognized tendency of SAA 1, 2 and 4 to associate with HDL lipoproteins (they have been referred to as apolipoproteins) is consistent with this poor aqueous solubility although hydrophobic residues are not strikingly prominent in their primary sequences. Thus, despite their relatively small size, it has been difficult to achieve SAA protein concentrations suitable for NMR study and/or crystallization. Based on earlier algorithms, Turnell et al. (1986) proposed a model with two α-helices and a β-sheet region. Stevens (2004) proposed a theoretical structure based on possible homology to the N-terminal domain of arthropod hemocyanins with a predominant helical bundle and a disordered C-terminal region. A soluble fusion protein between SAA and staphylococcal nuclease (Meeker and Sack 1998) showed only α-helical domains by CD. Only a single surface tryptophan residue was identified by iodide quenching.
Lu et al. (2014) determined SAA monomer structure by multiwavelength anomalous dispersion to 2.2 Å. As predicted from earlier CD studies (Meeker and Sack 1998), the monomer comprises four α helices in a cone-shaped array (see Fig. 3a). The C-terminal residues are wrapped around the bundle. Further study of His6-tagged SAA1 (which could be purified without denaturation) showed a hexamer formed by two identical trimers (consistent with earlier studies in solution (Wang et al. 2002)). Interestingly, Wang et al. (2005, 2011) earlier had indicated that a less stable octamer resolved to a stable hexamer. Based on this structure, potential sites for binding of both HDL and glycosaminoglycans were predicted to overlap near residues R-1, R-62 and H-71, consistent with a structural basis for competitive binding. This is also consistent with earlier mutation studies (Patel et al. 1996) as well as the observation that SAA binding to HDL could be disrupted by heparan sulfate (Noborn et al. 2012). The models show that helix h4 contains the site where cleavage usually occurs prior to amyloid fibril formation.
Fig. 3.

a Monomeric three-dimensional structure of SAA1.1 (Lu et al. (2014), Copyright [2014], National Academy of Sciences, used by permission). Note 4 α-helices 1 (aa 1-27), 2 (aa 32-47), 3 (aa 50-69), 4 (aa 73-88) and C-terminal tail (aa 89-104). b Proposed model for SAA interaction on HDL surface showing potential binding sites to receptors and other molecules. (Frame and Gursky (2016), Copyright John Wiley and Sons, 2016, used by permission)
Frame and Gursky (see Fig. 3b) noted that helices 1 and 3 have both hydrophobic and hydrophilic faces. The latter interact with helices 2 and 4 while the hydrophobic domains could bind to the lipid surface of HDL. According to this model, SAA could serve as a “hub” – binding HDL on one side while interactions with glycosaminoglycans, cystatin C, retinoic acid and cell receptors could occur on other domains specifically involving the C-terminus. Binding to each set of domains likely would influence packing of the entire structure due to allosteric changes and the flexible linkage between the N-terminal and C-terminal regions which is most prominent between h3 and h4 helices.
A model for murine SAA3 (where the N-terminal helix has a different primary sequence) presented by Derebe et al. (2014) resembled the structure in Fig. 3 but had differences and associated as a tetramer. The core of the tetramer was predicted to be the site for retinal binding. There was no evidence for octamer formation (cf (Wang et al. 2011)).
SAA fibrils
As noted earlier, electron microscopy showed that amyloid deposits are fibrillary with relatively similar electron microscopic dimensions. (Upon review, the rabbit tissue deposits earlier (Cohen and Calkins 1959) were likely to comprise SAA-derived polypeptides.) X-ray diffraction patterns of dried (Eanes and Glenner 1968) and native (Glenner et al. 1971) amyloid fibrils showed reflections consistent with a cross-β fibrillar model as proposed by Pauling and Corey (1951). Later studies of amyloid fibrils derived from various human sources (including AA fibrils) revealed common synchrotron X-ray diffraction patterns with intense meridional reflections from 4.6 to 4.84 Å (Sunde et al. 1997). Subsequent studies have confirmed the common feature of β sheets parallel to the fibril axis; the sheets themselves are generally about 10 Å apart (Eisenberg and Jucker 2012; Knowles et al. 2014). In their review of amyloid structures Riek and Eisenberg (2016) emphasized that the interacting domain(s) forming intermolecular β sheets and characteristic cross-β structures may be limited to specific domains of the constituent monomers rather than the entire monomer (they did not have data for AA fibrils).
Different SAA isoforms appear to influence fibril formation, Takase et al. (2014) described fibril forms from SAA1.1 as “long and curly” while those formed from SAA1.3 were “thin and straight.” Srinivasan et al. (2013) showed that SAA1.1 first formed oligomers followed by a lag phase of several days to form fibrils while SAA2.2 formed fibrils quickly. Interestingly, Mori et al. (2014) noted that CE/J mice with a Q30L substitution in both SAA1 and SAA2 are resistant to amyloidosis, presumably implicating a critical domain.
Finding a common unit of β sheet structure has suggested that at least some fibrils in nature may grow via “seeding” from other fibrils or precursors; this was shown by Yan et al. (2007) for AA and apoprotein A-11 fibrils in mice. Lundmark et al. (2005) reported that at least some proteins in nature can “seed” AA fibril formation in mice. Westermark et al. (2010) observed similarities to prion transmission and emphasized nucleation as central to the rate of fibril formation. Such a mechanism could underlie apparent transmission of AA amyloidosis in captive animals (Ranlov 1967).
As noted above, initial sequencing revealed a polypeptide of ≈76 aa common to most AA fibrils (Benditt et al. 1971; Levin et al. 1972; Ein et al. 1972a, b). This sequence has been reported frequently although the N-terminal arginine often is missing. The structure proposed by Lu et al. (see Fig. 3a (Lu et al. 2014)) features an exposed, non-helical region between aa 69-73, potentially making that and adjacent regions more susceptible to proteolysis. A 1978 study (Lavie et al. 1978) showed that human blood monocytes from at least some individuals could degrade SAA into a stable 76 aa species using an outer membrane-associated serine protease; whether this observation can be generalized is unknown. To date, the C-terminal ≈28 aa species has not been detected.
Later studies have found that polypeptides of different lengths can be associated with AA-derived fibrils in different pathologic states. The 76 residue species is commonly found in renal glomeruli. However, Westermark et al. (1989) found that about 10% of patients affected with amyloidosis have AA deposits in blood vessels and the renal medulla with relatively little glomerular involvement. Proteinuria was not prominent in these individuals and AA fragments of 45-50 aa were found (all beginning at serine – residue 2). A species of 94 aa was noted in another sample. Thus, while the ≈76 residue fragment is likely the most common constituent of pathologic deposits both longer and shorter species also can be found. Based on accumulating data for other types of amyloid fibrils (see (Riek and Eisenberg 2016)), it is likely that the critical β sheet region for AA fibril formation is relatively short. It is thus possible either that residues at the C-terminal regions are removed after fibril formation (and that this process may differ depending on the site(s) of deposition) or that early cleavage of the parent SAA protein leads to monomers differing in length which, themselves, are structurally predisposed to become parts of different types of fibrils. SAA 1 and 2 have isoelectric points ≈5.6 with a more basic N-terminal region and a relative acidic C-terminal domain. Thus, cleavage can alter the pI of the resulting fragment and, possibly, the tissue affinity (Westermark 2005).
In order to form fibrils, SAA cannot be constrained by being bound to HDL. Li et al. (2005) showed that transgenic mice overexpressing human heparanase (which led to shortening of heparin sulfate chains) did not have AA fibril deposition in kidney and liver following the established protocol of AEF and AgNO3 injection. As noted above (Noborn et al. 2012) heparin sulfate disrupted SAA binding to HDL. Digre et al. (2016) showed interaction of heparin with both SAA and apoA1 in HDL and that SAA and apoA1 were likely within 25Å on the HDL surface (a distance likely to be too short for the products of heparanase treatment (Westermark 2005) to be effective). These observations are consistent with dependence on both the degree of sulfation as well as the length of the sulfated domains on the effectiveness of fragmented glycosaminoglycans (Takase et al. 2016) to release SAA.
The process of SAA fibril formation has an obligate intracellular stage and Jayaraman et al. (2017) evaluated SAA’s fate in murine lysosomes. Detailed spectroscopic features were related to environmental pH. In the pH range of 3.5-4.5 SAA1 forms oligomers that are remarkably stable and resistant to proteolysis. This pH-dependent structural transition for lipid-free SAA led to oligomers with 5% α-helix, 50+/- 5% random coil and the remainder β-structure. Such oligomers convert from the α-helix described above (Lu et al. 2014) to β-sheets within lysosomes (and, experimentally, other lipid vesicles in which pH can be adjusted) and then disrupt the enclosing membranes and, ultimately, the organelles and cells themselves. Susceptibility of SAA to at least some proteolysis persisted at pH 4.3 (consistent with the possible involvement of cathepsin B (Rὄcken et al. 2005)).
Claus et al. (2017) used a sensitive FRET assay to confirm that HDL binding helped maintain α-helical conformation and blocked fibril formation. Internalization into lysosomes, freeing SAA from HDL, was essential for fibril development. Furthermore, a sufficient time within the acidic lysosomal environment was required, consistent with relatively slow structural reorganization of the monomer.
Figure 4 presents a scheme for AA amyloid fibril formation consistent with these data; it emphasizes the obligate cellular (and intralysosomal) pathway. Beginning with clathrin-mediated endocytosis SAA progresses through lysosomes (with structural reorganization at pH 4.3 with or without cathepsin B proteolysis). SAA oligomers are formed that disrupt membranes leading to cell toxicity with lysis. Finally, extracellular growth of the deposits and pathologically recognizable amyloid fibrils occurs (cellular debris also is present).
Fig. 4.

Proposed scheme for SAA fibril formation. a HDL-bound SAA dissociates from HDL. b {S}AA enters cell via clathrin-coated pits to reside in low pH lysosomal environment. c {S}AA monomers undergo structural rearrangement(s) within lysosome. d AA oligomers form within cells. e Lysis of lysosomal and cellular membranes leads to extracellular oligomers and debris from necrotic cells. f AA oligomers extend into fibrils based on β-pleated sheet domain interactions and become visible as tissue “amyloid” deposits with congo red binding. (modified after Claus et al. (2017), Copyright John Wiley and Sons, 2017, used by permission). As described in the text, cleavage of SAA occurs during this process, generally yielding a 76 aa N-terminal fragment. Cleavage may involve a serine protease on the cell surface (Lavie et al. 1978) but the precise site at which this occurs remains unestablished and the undefined nature of the intracellular species in the early stages (a-c) is indicated by {S}AA. By stage (d) the AA (post-cleavage) species likely predominates
While the most prominent constituent of pathologic secondary amyloid fibrils is the SAA-derived “AA” polypeptide other molecules are present as well. Heparan- and dermatan-sulfated glycosaminoglycans and proteoglycans are non-covalently associated (Pepys 2001). In addition, all pathogenic amyloid fibrils contain the plasma glycoprotein serum amyloid P (SAP), a member of the pentraxin protein family (CRP also is a member of this family). SAP contains a calcium-dependent ligand binding site that recognizes an as yet unidentified feature apparently common to all types of amyloid fibrils (Pepys 2001).
SAA genes and molecular biology
Morrow et al. (1981) showed that the level of murine hepatic mRNA for SAA rose 500-fold after inducing the APR by intraperitoneal administration of bacterial lipopolysaccharide (LPS) and that SAA synthesis could rise to comprise 2.5% of total mouse hepatic protein synthesis. This permitted cloning of mRNA for murine SAA by Lowell et al. (1986a). All of the genes mapped to murine chromosome 7 (Taylor and Rowe 1984). Two members of this gene family (Saa1, 2) show 96% homology over their entire length; a third gene (Saa3) is similar in general structure but with distinct sequence differences. In the mouse Saa1, Saa2 and Saa3 are all acute phase genes. The murine SAA gene family comprises a 42 kb cluster of 5 members (Butler and Whitehead 1996); one (Saa5) is a pseudogene.
The striking evolutionary conservation of SAA protein sequences permitted using murine SAA cDNA clones to isolate their human genomic counterparts (Sack 1983). The human SAA gene family is clustered within a 160 kb region of chromosome 11p15.1 (Kluve-Beckerman et al. 1986; Sack et al. 1989), a region analogous to the chromosome 7 region in the mouse (Taylor and Rowe 1984). Human Saa1 and Saa2 are acute phase genes.
Humans and mice both contain another gene family member (Saa4) mapping within the cluster (Steel et al. 1993a; DeBeer et al. 1996; Uhlar and Whitehead 1999a). The corresponding protein, SAA4, also found as an apoliprotein of HDL, is synthesized constitutively (i.e. it is not induced in the APR) and in both murine and human liver and includes an insertion of 8 aa between residues 69 and 70 of Saa1 and Saa2. Figure 5a presents a map of the human Saa gene family. All genes share the same organization of 4 exons and 3 introns (e.g. Fig. 5b). An 18 aa signal sequence is present in the initial transcript but is removed in the serum proteins.
Fig. 5.

a Organization of the four members of the human Saa gene family on chromosome 11p. b Exon/intron structure of human SAA1 gene (a common pattern for all Saa gene family members)
Despite the remarkable conservation of much of their sequences there is considerable short- and long-range variation among human Saa genes and proteins (see Fig. 2), their relationship(s) to the APR and their site(s) of transcription/translation. Saa1 and Saa2 are the genes for the classic APR serum proteins in humans and mice. The liver, a major source of APR serum proteins, initially was considered to be the sole site of SAA 1/2 synthesis. However, SAA 1/2 proteins are now known to be synthesized in many other tissues including macrophages, kidney, lung, adipocytes, and mammary gland (Urieli-Shoval et al. 1998; Sack et al. 2018). Transcription also has been found in synovial cells, brain and mammary gland (Sack and Zink 1992; Tucker and Sack 2001; Larson et al. 2003a).
As noted above, the Saa5 locus is a pseudogene in the mouse. However, the status of the human Saa3 locus has been confusing. The first reported sequence of a genomic clone (Sack and Talbot 1989) predicted a 104 aa transcribed protein differing from the APR serum proteins due to changes in the N-terminal region. The predicted sequence was similar to that of an SAA-like protein produced by rabbit macrophages following phorbol ester treatment and that functioned as an autocrine inducer of collagenase (Brinckerhoff et al. 1989). Later, partial sequencing of a clone from another human library (Kluve-Beckerman et al. 1991) found an early stop codon suggesting that this was a non-translated pseudogene. More recently, Larson et al. (2003a) detected an RT-PCR product in human mammary epithelial cells stimulated with prolactin or LPS and corresponding to SAA3. Their data predicted an mRNA encoding a protein with an N-terminal sequence matching 29/31 residues of the initial (Sack and Talbot 1989) report. However, their nucleic acid sequence also included an additional T residue at their nt 204 changing the reading frame and, hence, the aa sequence beyond residue 31 with a stop at residue 42 (after the 18 aa signal sequence). In addition, RACE 3’ extension predicted a long 3’ UTR of 406 nt (due to the presumably “early” stop codon generated by the altered reading frame) while the earlier report (Sack and Talbot 1989) predicted only a 145 nt 3’ UTR. Both predicted the same polyadenylation signal (AAUAAAA). The position of the termination codon would lead to a transcript likely susceptible to nonsense-mediated decay (Nagy and Maquat 1998); the corresponding short protein has not been found. GenBank data, based on multiple species, show this “early” stop codon in human, chimp and bonobo genes reflecting a frameshift due to a single “A” nucleotide in the genomic sequence (following codon 30 of the putatively secreted protein (Sack and Talbot 1989)); in these organisms Saa3 is thus considered to be a pseudogene. In other mammals the sequence is compatible with transcription and translation of a full-length 104 aa protein whose site(s) of translation may be limited to mammary epithelium (Larson et al. 2003a) and adipose tissue (Benditt and Meek 1989; Lin et al. 2001). Thus, the human Saa3 locus is transcribed but is not a source for a 104 aa protein (with presumably the same situation in chimps and bonobos). This distinction is important because Saa3 is an authentic gene in other mammals (e.g. mice (Benditt and Meek 1989), see also below).
Read-through transcription, presumably due to alternative splicing, between human Saa genes has been reported. As shown in Fig. 5, Saa2, Saa3, and Saa4 have the same transcriptional orientation. A low-level transcript connecting Saa2 (exon 3) with Saa4 (exon 2) has been identified but the presumptive 208 aa protein has not been found. The distance between these exons is relatively short – 10 kb. By contrast, Tomita et al. (2015) reported read-through transcripts connecting exon 3 of Saa2 with exon 1 of Saa3 in several human cell lines. The C-terminus of the resulting protein is at aa 42 of the potentially secreted protein, consistent with the frameshift and early stop codon in Saa3 described above. Such a read-through implies a long distance (≈130 kb) for splicing which appears to be rare (Nacu et al. 2011; Hiratsuka et al. 2008).
Control of SAA protein synthesis and serum levels
Control of Saa gene transcription varies with the individual gene. In addition, details differ in different organisms and different cells and tissues. (This review will emphasize humans and mice although important work also has been done using rat and rabbit systems.) Many studies have emphasized human Saa1 and Saa2 genes (those sharing APR expression profiles). As noted above, the APR can be induced with a wide array of stimuli including tumor necrosis factor (TNF), Interleukin-1β (IL-1β), Interleukin-6 (IL-6) and Interferon-γ (IFN-γ) (Edbrooke et al. 1989; Betts et al. 1993; Edbrooke et al. 1991; Woo et al. 1987; Edbrooke and Woo 1989). These likely are released from macrophages during the APR. In their earlier review, Uhlar and Whitehead (1999a) described different responses in different cell types and conditions. More recently, de Buck et al. (2016) reviewed SAA production in cells of hepatic origin. In some systems, but not all, the temporal order of exposure to stimuli is important. An earlier map of 5’ sites in Saa genes (see Fig. 6) indicates some regions incriminated in transcriptional control (Uhlar and Whitehead 1999a; Edbrooke and Woo 1989) but all details have not yet been resolved. IL-1β induces a factor that binds to the NF-κB site, displacing an apparently constitutive repressive nuclear factor that recognizes portions of the adjacent NF-κB and C/EBP sites (Brasier et al. 1990).
Fig. 6.

Promoter maps for mammalian SAA genes showing regions for transcription-factor binding. Transcription begins at the arrow (+1). Sites are identified for NF-κB, C/EBP, AP-2, SAS (binds SAF and SEF-1) and YY1. SEF is a factor identified in the mouse distal response element. (Uhlar and Whitehead (1999a), Copyright John Wiley and Sons, 2011, used by permission)
Vitamin A is required for Saa gene expression in mice (see de Buck et al. 2016). Derebe et al. (2014) showed reduced Saa1 and Saa2 transcription in the intestines of mice fed a vitamin A-deficient diet. This was confirmed by real-time qPCR and immunofluorescence. Adding retinol to the epithelial surface of small intestinal explants and of liver after intraperitoneal introduction of retinoic acid led to elevated Saa1 expression. Using HepG2 cells in the presence of both IL-1β and IL-6 adding retinol or retinoic acid enhanced Saa1 expression further, consistent with a dietary requirement for vitamin A under normal circumstances. They proposed a model for SAA containing a binding site for retinol (see also below).
IL-6 is most effective early in APR but the combined activity of all factors gives the highest level of transcription (Steel et al. 1993b; Uhlar and Whitehead 1999b). Ray & Ray (1996) found 3 homologous pyrimidine-rich octanucleotide sequence motifs (CACCGTCA, CACCCACA, CAGCCCCC) in the -280 - -224 nt region in the mouse that specifically reacted with an “SAA activating factor” (SAF). Mutations in these motifs reduced induction by IL-6 in non-hepatic cells. SAF is a 477 aa zinc-finger protein that becomes phosphorylated during inflammation (Ray et al. 2004a) and a mouse transgenic for SAF-1 production developed prominent amyloid deposition (Ray et al. 2006). IL-6 is known to bind to gp130 leading to the activation of the STAT3 pathway. Interestingly, although Saa1 lacks a STAT3 consensus sequence a JAK inhibitor reduced SAA1 transcription by ≈30% but did not eliminate it, thus implicating the STAT pathway in SAA1 transcription (Hagihara et al. 2005).
Mice deficient in IL-6 can produce at least a partial APR, depending on the stimulus (Kopf et al. 1994). Cytokines other than IL-6 also share the gp130 receptor and may compensate for IL-6 deficiency (Murakami et al. 1993). A signaling complex with two gp130 receptors is phosphorylated on an intracellular domain, in turn activating STAT3 and/or MAPK signaling. Hagihara et al. (2004) found that an antibody to the IL-6 receptor blocked the APR whereas an IL-1 receptor antagonist or anti-TNF antibody only partially inhibited it. Using a hepatocyte-specific deletion of gp130 in mice, Sander et al. (2010) showed that APR protein synthesis was minimized, implicating this receptor and its associated intracellular pathway(s) in both SAA and CRP synthesis. RelA also is essential. Induction of murine APR hepatic Saa1 was inhibited by RNAi that knocked down STAT3 or RelA, thus incriminating both pathways (Quinton et al. 2009, 2012).
Glucocorticoids bind to glucocorticoid receptors that recognize glucocorticoid-responsive elements (GRE). A GRE is found at nt -208- -194 of human Saa1 but not Saa2 (Thorn and Whitehead 2002). This difference may explain why adding dexamethasone preferentially stimulates Saa1 in acute phase stimulation (overcoming a slight bias toward Saa2 stimulation – for instance in HepG2 cells – (Thorn and Whitehead 2003)). Using a reporter assay in human aortic smooth muscle cells Kumon et al. (2002) found that SAA1 synthesis responded to dexamethasone but not to other acute phase stimuli. Glucocorticoids bind to their cellular receptor leading to GRE upregulation of IL-6 (Ray et al. 1995) and hepatic responsiveness to TNF and IL-1. However, they also can interact with NF-κB subunits in the cytoplasm and increase transcription of IκBα, a cytoplasmic inhibitor of NF-κB. Hence glucocorticoids ultimately inhibit cytokine effects and dampen the APR (Ray et al. 1995; Scheinman et al. 1995a, b; Auphan et al. 1995; Kerppola et al. 1993; Knudsen et al. 1987).
Two chromosomal regions affecting SAA blood levels were identified in a genome-wide association study (Marzi et al. 2010). The first region, 11p15.5-p13, contains the human SAA gene family and the general transcription factor 2 H1, consistent with the map location of the structural gene sequences. The second region implicated (1p31) contains the leptin receptor gene, possibly implicating leptin with APR proteins and SAA biology.
A single nucleotide polymorphism (SNP) in the 5’ flanking region of the human Saa1 gene (-13 C/T) affects transcription with the -13T allele having greater activity (Moriguchi et al. 2005). These workers suggested that this SNP may affect susceptibility to secondary amyloid disease but this has not been confirmed in clinical studies.
Despite impressive levels of SAA1 and SAA2 mRNA seen in APR cells in vitro (e.g. human Hep3B), translation, although clearly increased, is not proportionally higher (Jiang et al. 1995a). In addition, the peak rate of SAA mRNA translation can be 10-fold lower than the rate of mRNA synthesis (Steel et al. 1993b). Thus, post-transcriptional regulation likely contributes to blood levels of APR SAA1 and SAA2 proteins. These features are in contrast to those of human Saa4 where transcription and translation are constitutive. As noted above, Saa3 is transcribed in murine adipocytes (Benditt and Meek 1989) but less prominently in murine liver. It is also transcribed in rabbit macrophages and other inflammatory cells. As noted earlier, human Saa3 is transcribed, although apparently not translated, in mammary epithelial cells. In bone, Saa1 is expressed while both Saa1 and Saa2 are expressed in osteoblast-like cells (Koracevic et al. 2008).
The steady-state level of SAA mRNA is not only regulated by transcription. As shown earlier (Lowell et al. 1986b), mRNA stabilization also is important in maintaining the level. Rienhoff & Groudine (1988) implicated sequences 3’ to the transcription initiation region in determining the half-life of murine SAA3 mRNA. Jiang et al. (1995a, b) reported that SAA mRNA levels in Hep3B cells following IL-6 or IL-1β stimulation could be higher than transcriptional rates suggested.
At least some factors controlling SAA mRNA in human and murine cells include: defined binding sites for C/EBP and NF-κB, STAT3 octanucleotide sites responsive to IL-6 and the -13 C/T SNP. Implicating leptin in recent mapping studies widens the scope of APR control and, likely, interactions. Human mammary gland epithelial cells transcribe SAA3 mRNA in response to prolactin or LPS (Larson et al. 2003a). However, as discussed above, the mRNA contains an early stop codon and no protein product has been detected.
Less is known about control of murine Saa3 expression. In mice, a 350 bp promoter fragment can control expression after conditioned medium exposure. The critical region is a 42 bp “distal response element” (DRE) containing 3 functional sections. IL-1, the major cytokine in conditioned medium, is largely responsible for Saa3 induction but adding IL-6 has a synergistic effect. Integrity of the 3 DRE elements is essential (Huang and Liao 1999). Later studies identified the CCAAT/enhancer-binding protein (C/EBP) and “SAA3 enhancer factor” (SEF). NF-κB and SEF function synergistically at the Saa3 promoter (Bing et al. 2000).
SAA and APR control (proposed model)
The ≈1000-fold elevations of serum APR protein levels after 24-36 hours and their subsequent rapid decline imply impressive regulation and feedback. All constituents of this presumptive “loop” have not been characterized fully but murine model systems have been particularly valuable. Pattern recognition receptors on cell surfaces (PAMPS – for bacterial sepsis and DAMPS - damage-associated molecular patterns (Lamkanfi and Dixit 2014)) can be recognized by Toll-like receptors on innate immune cells such as macrophages (of the proinflammatory or M1 phenotype) leading to NF-κB activation, T-cell priming and inflammatory cytokine secretion. Absence of residential T cells can be lethal in such situations (Kim et al. 2007). In particular, cytokine IL-6 stimulates hepatocytes via gp130/STAT3 to secrete APR proteins – SAA and CRP. (cxcl1, also known as KC, also is stimulated but less strikingly.) Rapid parallel mobilization of myeloid-derived suppressor cells (MDSC – GR-1+/ CD11b+) through MyD88 also is prominent in the mouse (Sander et al. 2010; Delano et al. 2007). MDSC are susceptible to apoptosis induction by TNF but in mice their survival (in spleen) is enhanced by both SAA and KC (a chemoattractant for MDSC and granulocytes (Delano et al. 2007; Newton and Dixit 2012)). These mobilized MDSC can then mediate feedback inhibition of proinflammatory (M1) macrophage features as well as conversion to pro-resolving (historically referred to as M2) features (more frequently seen in later stages of inflammation, associated with resolution of inflammation and tissue repair) and dampen the entire APR. Sun et al. (2015) showed that the conversion of macrophages from proinflammatory (M1) to proresolving (M2) phenotype requires MyD88 and interferon regulatory factor (IRF) 4 in addition to SAA. (Fig. 7 presents a hypothetical cycle including at least some participants).
Fig. 7.
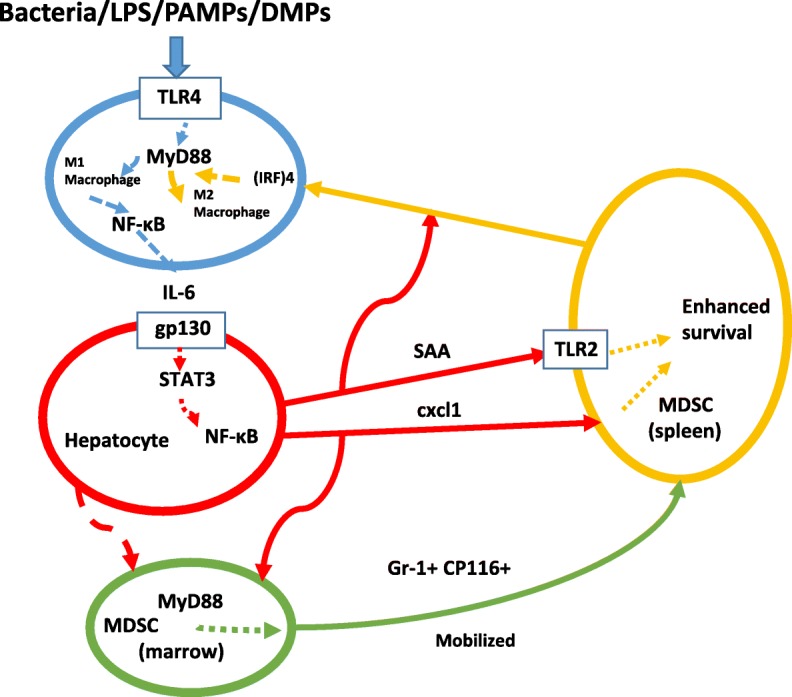
Proposed relationships between cytokines, chemokines and participating cells in APR consistent with current data. Note 3 distinct cell types: 1) macrophages, 2) hepatocytes, 3) myeloid-derived suppressor cells [MDSC]. MDSC can be mobilized from bone marrow and undergo further differentiation in spleen and elsewhere
This model has been tested in mice with a hepatocyte-specific gp130 deletion (gp103Δhepa) in which SAA and KC production are not stimulated. In these mice, APR due to CLP (cecal ligation and puncture) protocol for polymicrobial sepsis or LPS can be ≈90% lethal. Normal mice subjected to the CLP showed only ≈25% lethality by the third day. In normal mice, intraperitoneal injection of murine monoclonal antibodies against aa 33-43 of SAA (a well-conserved region – see Figs. 1 and 2) led to 90% lethality at the same time point. Treating the gp130 deletion mice with recombinant SAA1 substantially prevented the enhanced lethality due to CLP (survival was restored to that of normal mice when KC was added as well (Linke et al. 2017)). CRP was not effective.
As summarized by de Buck et al. (2016) SAA itself can induce a wide array of both cytokines and chemokines. Interrelationships between these have not yet been clarified. Nevertheless, SAA and possibly other APR constituents (not all identified but including chemokine CXCL1 {called KC in the mouse}) participate in a counterregulatory scheme for acute inflammation, critically enhancing its control and down-regulation by prolonging survival of at least one form of MDSC. Other type(s) of MDSC otherwise are susceptible to apoptosis induction by TNF (and possibly other cytokines (Newton and Dixit 2012; Veglia et al. 2018)). A feedback loop is thus established for interaction(s) of cells and soluble molecules as APR mediators. This complex process has been rigorously conserved in evolution. Absent or defective function of any of the cellular or molecular participants can lead to prolongation of the APR inflammatory response and likely lethality. Linke et al. (2017) proposed that SAA might be therapeutically useful in managing sepsis and other APR-like events.
As described above, chronic inflammation (even at relatively low intensity) can be associated with persistently elevated SAA levels and, ultimately, “secondary” amyloidosis (e.g. (McGlennen et al. 1986; Chae et al. 2009; Lane et al. 2017; Kuroda et al. 2017)). Participation of SAA in this innate host defense system is consistent with its evolutionary stability. Additional interactions and likely functions for SAA can thus be viewed from this important perspective.
SAA pathophysiology
While the proposed essential role of SAA in APR regulation (presented above) is compatible with many aspects of its biology, the nature of and basis for other SAA function(s) have remained poorly defined. As noted above, SAA proteins were isolated and described based on their association with the APR and their accumulation in amyloid deposits of “secondary” amyloid disease but no specific function was initially suggested based on these features. The rather small size of SAA proteins makes enzymatic activity unlikely (although this has not been rigorously evaluated). Several observations offer a perspective on possible function(s). Pathophysiologic implications for processes occurring later in life could have developed based on SAA protein chemistry and receptor interactions. Differences in structure and transcriptional patterns of different SAA gene family members suggest a wide range of possible activities.
SAA binding and receptor(s) (see Table 1)
Table 1.
SAA receptor interactions
| Receptor | Function | References |
|---|---|---|
| SR-B1 | Cholesterol efflux, clears SAA | Tomita et al. (2015); He et al. (2003, 2006); Ye and Sun (2017); van der Westhuyzen et al. (2005); Cai et al. (2005) |
| FPRL1 | Stimulates MMP-9 | Badolato et al. (1994, 1995); Gao and Murphy (1993); Fiore et al. (1994); Su et al. (1999); Patel et al. (1998); Furlaneto and Campa (2000); Dufton et al. (2010); Ji et al. (2015) |
| FPR2 | Phagocyte migration; antiapoptotic | de Buck et al. (2016); Gao and Murphy (1993); Furlaneto and Campa (2000); Ye and Sun (2017); Chen et al. (2014) |
| TLR2 | Induce IRZF7, IRF4-jmjd3; M2 markers (IL-33, IL-10, IL-1m) | de Buck et al. (2016); Ray et al. (2006); He et al. (2006, 2007); Cheng et al. (2008); Ji et al. (2015); Ye and Sun (2017); Rosenthal et al. (1976); Chen and Moller (2010) |
| TLR4 | Activate NF-κB, AP-1→NOS | Hiratsuka et al. (2008); de Buck et al. (2016); Sandri et al. (2008); Deguchi et al. (2013); Li et al. (2015); Ye and Sun (2017); Poltorak et al. (1998) |
| RAGE | Activate NF-κB, induce cytokines | Li et al. (2015); Yan et al. (2000); Rὄcken et al. (2003); Kennel et al. (2016); Ye and Sun (2017) |
| Bacteria | Opsonize Gram negative species | Hari-Dass et al. (2005); Shah et al. (2006); Ye and Sun (2017) |
SAA1 binds to the outer membrane protein A (OmpA, identified by MALDI-ToF) of E. coli and, presumably, its homologues in other Gram-negative bacteria (Hari-Dass et al. 2005). An E. coli strain specifically lacking OmpA showed no SAA binding. Shah et al. (2006) found that SAA1 can function as an opsonin increasing uptake of these bacteria by PMNs. Gram-positive organisms (e.g. Streptococcus pneumoniae and Staphylococcus aureus) did not bind SAA. (Recall that CRP does bind to Streptococcus pneumoniae (Tillett and Francis 1930; Macleod and Avery 1941).) As noted earlier (Derebe et al. 2014) SAA1 can bind retinol and can provide it during an infection – mice deficient in both SAA1 and SAA2 have increased bacterial numbers in spleen and liver following acute infection. Eckhardt et al. (2010) found that SAA1/2 derived from mouse colonic epithelial cells (where it apparently was produced constitutively) reduced E. coli growth in cultures. These observations are consistent with the idea of SAA being part of a “primordial” host defense and innate recognition system, particularly for Gram-negative bacteria (cf CRP as cited above).
It is important to note that E. coli and other Gram-negative bacteria are themselves capable of generating “amyloid” fibrils, known as “curli” (van Gerven et al. 2009). These β-sheet containing fibrils are synthesized through a specific pathway involving intracellular assembly, chaperoning and ultimately secretion to the extracellular surface where they become part of biofilms that can protect the organisms. Although biofilms themselves can contribute to bacterial stability and survival it is notable that curli fibrils themselves can be a pathogen-associated molecular pattern (PAMP) and interact with TLR2 receptors on macrophages to induce inflammatory responses (e.g. Nos2 and IL-8 expression) (Tükel et al. 2009). Curli-associated pathophysiology appears important in vivo (especially via biofilm constituents) where PAMP recognition can lead to an inflammatory response (see Fig. 7). This curli pathway differs from the “opsin” model described above for host protection by SAA. It is likely, but not yet established, that both approaches can provide host defense against at least some Gram-negative bacteria, possibly at different times and locations (e.g. nascent bacterial infection vs established biofilms). Whether SAA is directly associated with curli fibril production is unknown.
In addition to opsonin activity noted above SAA physiology including possible role(s) in mediating inflammation and its relation to lipids (details below) implies specific cell surface receptor(s). Delineating such interaction(s) has developed slowly (and likely remains incomplete). A series of studies used a recombinant SAA (rhSAA PeproTech). Badolato et al. found that this recombinant induced apparent chemotactic activity with migration, adhesion and infiltration of monocytes and PMN cells as well as intracellular calcium mobilization. These effects could be blocked by pretreating the cells with pertussis toxin (Badolato et al. 1994, 1995) and they proposed that SAA could interact with a conventional seven-membrane-spanning G-protein-coupled receptor (Gao and Murphy 1993; Fiore et al. 1994). Subsequent studies showed that rhSAA could interact with a homolog of the phagocyte receptor for the bacterial chemotactic polypeptide N-formyl-methionyl-leucyl-phenylalanine (fMLP). This homolog (FPRL1) could interact with high levels of fMLP to mobilize calcium, bind with the eicosanoid lipoxin A4 (LXA4) with high affinity and increase arachidonic acid production. Su et al. (1999) distinguished rhSAA binding to FPRL1 (rather than FPR) and showed that this interaction could induce calcium mobilization and migration in HEK cells transgenic for FPRL1. rhSAA also bound to human phagocytes and FPRL1-transfected 293 cells at concentrations achievable during inflammation.
SAA-induced cytokine release was shown by Patel et al. (1998). Furlaneto and Campa (2000) exposed human blood neutrophils to recombinant human SAA and found 75-400 fold release of cytokines TNF-α, IL-1β and IL-8. This effect was not seen when either normal or acute phase HDL (from febrile patients) was used, suggesting an effect of lipid-free SAA. He et al. (2003) confirmed this and showed that overexpression of FPRL1 (also called FPRL1/LXA4R, LXA4 and ALX) in HeLa cells increased NF-κB and IL-8 luciferase reporter gene expression after SAA exposure and that proximal events in the pathway included calcium mobilization, MAPkinase (ERK1/2 and p38), and NF-κB activation. The effect also was blocked by pertussis toxin (consistent with earlier observations implicating a G-protein-coupled receptor) as well as an antibody against the N-terminal domain of FPRL1. The SAA effect was significantly reduced in the presence of LXA4 which interacts with the FPRL1 receptor.
In 2007, El Kebir et al. (2007) showed that SAA extends the lifespan of PMN cells by suppressing apoptosis – it stimulates phosphorylation of the proapoptotic protein BAD by activating ERK and Akt preventing mitochondrial dysfunction and caspase-3 activation. Importantly, they showed that 15-epi-LXA4 triggered by aspirin overrides this effect, suppressing ERK and Akt signals. These observations are consistent with SAA’s prolongation of PMN participation in inflammation and antagonism by 15-epi-LXA4. In mice with deletion of Fpr2 (murine FprL1/FPR2 homologue) macrophages and PMNs failed to respond to SAA-mediated chemotaxis (as noted earlier (Su et al. 1999; Patel et al. 1998)).
Tomita et al. (2015) reported binding of human SAA3 (only the short, 42 aa N-terminal species containing the TFLK region – see above) to CHO cells expressing human LDL receptor leading to ERK phosphorylation and MAP kinase signaling. The level of stimulation was similar to that produced by oxidized LDL. Binding of the same SAA3 recombinant to HeLa cells was enhanced by expression of CD36, a phagocyte class B scavenger receptor. Using recombinant SAA (combining SAA1 and SAA2 isotypes) Baranova et al. had previously shown that CD36 overexpression in HEK293 cells led to JNK- and ERK1/2- mediated proinflammatory cytokine (IL-8) signaling (Baranova et al. 2005, 2010).
O’Hara et al. (2004) associated SAA and FMLPL1 gene expression in synovial tissue by noting matrix metalloproteinase (MMP) expression in individuals with inflammatory arthritis (see also below).
Dufton et al. (2010) created mice with a deletion of FPRL1 (also called FPR-2) and examined ischemia-reperfusion and carageenan-induced paw edema. Interestingly, phosphorylation of ERK in response to SAA was not diminished in the fpr-/- mice consistent with SAA interaction with other receptors as well.
Relating SAA to lipid transport and cholesterol mobilization also involves receptor interaction(s). This is discussed in the section on lipid biology below. Importantly, Marsche et al. (2007) emphasized considering the lipidation status of SAA in determining its cholesterol acceptance. Cholesterol efflux from cells involves the ABCA1 and/or SR-BI pathways.
Toll-like receptors also have been implicated. He et al. (2006, 2007; Cheng et al. 2008) showed that SAA can induce IL-12, IL-23, G-CSF and granulocytosis. The latter two effects could be inhibited by anti-TLR2 antibody. HeLa cells expressing TLR2 had a good response to SAA that was blocked by Toll/IL-1 receptor/resistance deletion mutants of TLR1, TLR2 and TLR6. Tlr2-/-mouse macrophages had reduced SAA-induced gene expression compared with wild type cells. TLR2 was implicated as a responsive receptor using transgenic HeLa cells expressing TLR2. A solid-phase binding assay showed SAA interaction with the ectodomain of TLR2. Ji et al. (2015) noted that T-cell mediated hepatitis was accelerated by SAA1 acting through TLR2. Similar results were not seen in parallel studies using TLR4 (He et al. 2007). The SAA effect was reduced using macrophages from tlr2-/- mice.
TLR4 also was implicated in SAA interactions (Sandri et al. 2008). Treating murine peritoneal macrophages with SAA induced NO production; this depended upon ERK1/2 and p38 MAPKs. However, this effect was lost when the cells were derived from C3H/HeJ or C57BL/10ScCr (lacking a functional TLR4 receptor). The intracellular pathway appeared to be independent of MyD88. Hiratsuka et al. (2008) reported that murine SAA3 bound more strongly to TLR4 in lung endothelial cells and macrophages than to other leucine-rich repeat family members. SAA3 stimulated NF-κB signaling via TLR4, enhancing metastasis. Other studies (Deguchi et al. 2013) indicate that murine SAA3 binds TLR4/MD-2 to activate the MyD88-dependent pathway. Li et al. (2015) showed that some recombinant human SAA species required TLR4 and RAGE to stimulate expression of double-stranded RNA-activated protein kinase R and release of serum high mobility group 1 box (HMG1) release from murine macrophages.
It is important to note that many studies of SAA have used recombinant protein(s) produced in E. coli. (e.g. rhSAA 300-13 and 300-53 PeproTech). Simple aa changes in at least some recombinant molecules may affect their physiology (although they also may aid solubility). Bacterial endotoxin or LPS, which may stimulate TLR4, may be present in such preparations but the manufacturer claims that the level is <0.1ng/mcg SAA protein. The endotoxin level found in amebocyte preparations was even lower (<0.05 ng/mcg SAA protein). Thus, contamination was very low, if not absent, and experiments uniformly controlled for this. (e.g. (Sun et al. 2015; Badolato et al. 1994, 1995)).
The receptor for advanced glycation end products (RAGE) in a soluble form can bind to murine AA amyloid (Yan et al. 2000). Heterogeneity in glycation may influence AA fibril formation and/or deposition (Rὄcken et al. 2003). A synthetic form of RAGE (VC1) which has a highly basic surface can specifically bind to murine AA deposits (Kennel et al. 2016).
These observations support cytokine-like interaction of SAA with several cell-surface receptors. ALX has been studied most thoroughly but TLR2 and TLR4 also appear to be important in at least some aspects of the inflammatory response. Controversy remains regarding induction by SAA which was not shown by Kim et al. (2013). Nevertheless, other more recent studies are consistent with interactions which, however, differ with the study system(s) used. Ye and Sun (2017) reviewed SAA/receptor interactions.
SAA and lipids
Early studies showed SAA association with lipids, particularly HDL (Benditt and Eriksen 1977; Benditt et al. 1979). Predicted amphipathic features for SAA monomers implicated regions for lipid binding (Meeker and Sack 1998; Lu et al. 2014; Frame and Gursky 2016). Kisilevsky & Manley (2012) estimated that 95% of circulating SAA is in the HDL fraction (consistent with (Benditt et al. 1979) showing only 5-6% of SAA reactivity at the top of a density gradient). Using an egg phosphatidyl choline monolayer they also showed high affinity of SAA for phospholipids, similar to that of apo A-1. As noted above, the SAA monomer is poorly soluble in aqueous environments, justifying use of a fusion protein (SAA and Staphylococcal nuclease) for biophysical characterization (Meeker and Sack 1998). Early reports, however, noted that a “small proportion” of SAA can be found in a lipid-free form in human plasma (van der Westhuyzen et al. 1986; Coetzee et al. 1986). Relationships between SAA, HDL and cholesterol not only vary temporally but also depending upon the environment. In particular, SAA is involved with cholesterol metabolism in both normal physiologic and inflammatory conditions. Interpreting all available studies is complicated by hydrophobic partitioning of SAA monomers.
The physiologic relevance of studies using delipidated SAA has been questioned (Ye and Sun 2017). However, it can be estimated that an equilibrium must exist such that ~50 ng/ml of SAA (presumably monomeric) free of HDL and, possibly, free of lipids, should exist under baseline conditions and that this concentration could rise to ~50 mcg/ml in APR (as noted but not quantified in (van der Westhuyzen et al. 1986; Coetzee et al. 1986)). Yamada et al. (2009) used surface plasmon resonance to estimate binding of human SAA isotypes and HDL. Their data showed KD values for recombinant SAA family members - 1.1, 1.3 and 1.5 - of 1.4 X 10-5, 1.8 X 10-5, and 3.7 X 10-6, respectively. They also estimated the concentration of lipid-free SAA as a fraction of total SAA in patients with rheumatoid arthritis (in whom the serum concentrations were elevated) as 10-40 mcg/ml, consistent with the estimate above. In mice deficient for ApoA-1 and ApoE some recombinant SAA was found in a “lipid-poor fraction” of serum as well as in VLDL and LDL fractions although this does not correspond to the in vivo situation (Cabana et al. 2004). Thus, while lipophilicity of SAA and, particularly, its partitioning into HDL, accounts for ~95% of the protein in serum, a small lipid-free fraction may exist in equilibrium with lipids although details of this fraction remain elusive. Even in quiescent cells in culture a low level of lipid turnover and remodeling is present. This is likely modified qualitatively and quantitatively during the dynamic changes accompanying the APR as well as in other inflammatory conditions. We will consider both acute and chronic conditions. The following studies emphasize the steady state.
Tam et al. (2002) loaded murine macrophages with 3H cholesterol, cholesterol oleate or oleate alone and exposed them to HDL (50 mcg/ml) from either normal or APR mice. The latter showed reduced oleate to cholesterol ester conversion (i.e. reduced ACAT activity) and an increased release of labeled cholesterol (enhanced CEH activity). Trypsinizing HDL (eliminating both apoA-1 and SAA) eliminated this activity with SAA2.1 specifically being essential for this effect. The same effect could be produced using apoprotein-containing liposomes.
Delipidated SAA at 10 mcg/ml promoted cholesterol and phospholipid efflux from HeLa cells, an effect attributed to the ABCA1 transporter (Stonik et al. 2004; Qian et al. 2017). However, even when the ABCA1 transporter was inhibited (in fixed cells or in fibroblasts from individuals with Tangier disease where the transporter is defective (Rust et al. 1999)) reduced efflux was seen. By contrast, similar exposure to apoA-1 could not mediate efflux in cells defective for ABCA1 (Tangier disease or fixed cells), implying that delipidated SAA might use an alternative (undefined) pathway to mediate lipid release. In Chinese hamster ovary (CHO) and Hep G2 cells both delipidated SAA (at both 10 and 30 mcg/ml) and APR murine HDL (containing SAA) increased cholesterol efflux 2-fold (over murine baseline HDL alone) (van der Westhuyzen et al. 2005). Using BLT-1 (an inhibitor of scavenger receptor B1 {SR-B1}), intracellular cholesterol release induced by delipidated SAA was reduced (but not eliminated). When added together, SAA and HDL synergistically increased efflux. SAA induced cholesterol efflux in an apparently ABCA1- and SR-B1- (i.e. Tangier and BLT-1-treated cells, respectively) independent manner, suggesting 3 efflux pathways.
SAA binding to SR-B1 inhibits HDL binding (Cai et al. 2005). It also permits SAA uptake and cleavage within macrophages where degradation – leading to AA fragments - may occur (Kluve-Beckerman et al. 2002) (recall Fig. 4).
Labeled delipidated SAA (at 1.25 and 10 mcg/ml) bound to HeLa cells transfected with CLA-1 (human orthologue of SR-B1). Unlabeled SAA and helical polypeptides L-37pA and D-37pA could compete for this binding but an L3D-37pA polypeptide (containing 3 D-amino acid substitutions interrupting the helical structure) did not compete. Confocal microscopy colocalized transferrin and SAA in the endocytic recycling compartment. HDL competed with delipidated SAA for CLA-1 binding. Exposure of CLA-1 expressing HeLa cells to SAA and HDL led to IL-8 secretion as well as activation of MAPKs (He et al. 2003).
Treating HEK293 cells stably expressing human ABCA1 with delipidated SAA (10 mcg/ml) led to HDL particles with higher density, larger diameter and slower electrophoretic mobility than those following apoA-1 treatment. Increasing ABCA1 protein by liver retinoic acid receptor (RXR) agonists increased lipid release (Abe-Dohmae et al. 2006).
In HEK-293 cells ABCA1 mediated cholesterol release to lipid-free SAA (10 mcg/ml) (He et al. 2007) However, overexpressing SR-B1 in HEK-293 cells did not lead to cholesterol release in response to lipid-free or lipid-poor SAA (in contrast to (Abe-Dohmae et al. 2006)). These SR-B1 transfected cells transferred cholesterol only to lipid-phosphatidyl choline-associated SAA. By comparison with CHO and HepG2 cells the cell type studied was at least partially responsible for the differences because HEL-293 and HEK-293 cells transfected with SR-B1 showed minimal cholesterol efflux in response to lipid-free and lipid-poor SAA while efflux was found in CHO cells and fibroblasts. Thus, both the cell type and SAA lipidation status must be considered.
Hu et al. (2008) used the model of LPS-induced APR inflammation in mice to examine intact mouse hepatocytes as well as those with a knockout of the ABCA1 transporter. They concluded that ABCA1 is required to generate HDL within hepatocytes using either SAA or apoA-1. Both delipidated SAA and apoA-1 interacted with ABCA1 causing release of cellular lipids. In ABCA1 knockout mice, no HDL was found in the plasma following LPS stimulus and SAA secretion was reduced (compared with wild-type) and largely found in the low-density fraction. SAA was increased within the liver, however, consistent with the APR pattern. Adding either lipid-free SAA or apo A-1 to fibroblasts derived from both types of mice caused release of cellular lipids from wild-type but not ABCA1-knockout cells. Getz and Reardon (2008) suggested that apoA-1 may mobilize cholesterol and phospholipids from the inner to the outer leaflets of the cell membrane in preparation for exovesiculation to form at least primordial (possibly discoidal) HDL in the presence of amphipathic protein domains. Such nascent HDL particles may require LCAT to convert them to spherical HDL.
Mice with deletions of SAA 1.1 and SAA 2.1 showed no effect on HDL levels in the LPS-induced APR. However, HDL particle size increased due to increases in surface lipid rather than proteins (deBeer et al. 2010). The murine SAA3 gene remained intact in these mice, however, and, as reviewed earlier, has considerable sequence conservation especially in the C-terminal region.
Reconciling, at the molecular level, all studies relating SAA to HDL cholesterol is difficult. The studies have used cells from humans, mice and hamsters (in addition different to cell types, e.g. CHO, HepG2, fibroblasts, HEK293, HeLa). Furthermore, quantitation of cholesterol flux has been difficult to compare among the systems. An additional complexity is that the precise molecular status of the SAA used in “lipid-free” and “lipid-poor” studies is not clear. In this regard, Kinkley et al. (2006) suggested that “lipid-free SAA” may be aggregated even at low levels but this may be part of an equilibrium situation as discussed above. An additional complication (introduced above) has been the relation(s) of the studies to the APR with its attendant cytokine changes (for example, the sequelae of the LPS response) in intact organisms. Ye and Sun (2017) also noted that some recombinant SAA proteins used in reported studies contain aa changes (including N-terminal methionine and D61N and H72R). Moreover, Chen et al. (2014) reported differences among SAA isotypes and specific receptors. Specifically, SAA1.1 and SAA2.2 were similar in FPR2 activation and ERK phosphorylation while all tested species gave similar results using a TLR2-dependent luciferase reporter. They also explored the possibility that LPS contamination of recombinant SAA (made in E. coli) could affect signaling – using macrophages from Tlr3-deletion mice (Poltorak et al. 1998) they found that SAA1.3 induced higher levels of TNFα and IL-1 receptor antagonist transcription. In addition, SAA1.5 promoted the highest IL-10 expression while being less active in IL-1β induction. Thus, the SAA isoform(s) present may differ in receptor selectivity.
Several reasonable summary conclusions can be formulated. 1) The ABCA1 transporter is involved in cholesterol efflux (Stonik et al. 2004; Qian et al. 2017). 2) SAA promotes an increase in cholesterol ester hydrolase, leading to increased free intracellular cholesterol. 3) SAA reduces activity of macrophage ACAT. 4) HDL containing SAA is targeted to the macrophage within which it can be loaded with free cholesterol (increased by steps #2 and 3) for transport. 5) Although the scavenger receptor SR-B1 may be involved in SAA recognition/uptake it is not the only mediator. 6) The in vitro data implicate multiple pathways. 7) Different SAA isoform(s) likely contribute to different downstream activities (e.g. Fig. 2) but most studies have not distinguished the species in molecular detail and commercially available SAA proteins often have small aa changes that can assist with their solubility.
Based on the structural data for SAA monomers (Meeker and Sack 1998; Lu et al. 2014) and the model proposed by Frame and Gursky (2016) (Fig. 3a, b) the potential for hydrophilic domain exposure after HDL binding has suggested regions responsible for fibronectin and laminin binding (aa 39-41 and 29-33, respectively). Residues toward the C-terminus (including aa 70-104) include regions implicated in binding to heparan sulfate (aa 83-102 (Ancsin and Kisilevsky 1999)), cystatin C (11 86-104 (Spodzieja et al. 2013)) and receptors LOX 1 and CD36 (Tomita et al. 2015). Such interactions could permit SAA-mediated binding and possibly internalization into cells with these receptors for these species. Other contents of HDL also could be internalized through this mediation (e.g. fat-soluble vitamins). These same structural considerations could help explain the C-terminal region’s stimulation of cholesterol ester hydrolase while the N-terminal region reduces ACAT and LCAT (Kisilevsky and Tam 2003; Hosoai et al. 1999) thus changing cholesterol from esterified to nonesterified species and increasing transport from cells to HDL.
SAA and cholesterol salvage
The most prominent changes in serum SAA levels (both 1 and 2, often not distinguished in past reports) occur in the APR as reviewed above and transport and partitioning of SAA under these conditions has been studied extensively. Inflammation, both acute (including the APR) and chronic is central to SAA pathophysiology. Plasma HDL apoproteins change during the APR. Specifically, the fraction of apoA1 in HDL falls while that of SAA rises (Ye and Sun 2017; Kisilevsky and Manley 2012). In addition, HDL3 (denser fraction) preferentially associates with SAA and becomes physically altered with an increase in relative size while remaining a polydisperse mixture. The fractional apoprotein composition of HDL (particularly HDL3) can shift such that SAA comprises as much as 80% of the total with an SAA:apoA1 ratio of nearly 10:1 (van der Westhuyzen et al. 1986). Such changes are consistent with SAA’s lipophilicity as well as its altered synthesis (and relative concentration) during the APR.
Kisilevsky, Manley and coworkers (Kisilevsky and Manley 2012; Kisilevsky and Tam 2003) noted that the evolutionary conservation of SAA and its relation to the APR (reviewed earlier) strongly support a central role for this family of proteins in host survival. They defend the notion that SAA mobilizes cholesterol from macrophages at sites of acute injuries, infections, etc. Under such circumstances cellular debris accumulates in macrophages which then release cytokines that induce the APR (e.g. IL-1, IL-6, TNF). Subsequent SAA synthesis and release alter HDL composition as described above as well as the affinity of the reorganized HDL for lipid-laden macrophages. Such a role would be consistent with early survival and evolutionary maintenance. Hosoai et al. (1999) emphasized that SAA alone did not lead to reduced HDL cholesterol among the lipoproteins.
Despite this role in “survival” (which, along with APR control, could have been valuable early in life and selected for evolutionarily), it remains important to also consider role(s) of SAA proteins in physiologic processes that, at least in humans, have accompanied longevity as well as more chronic biologic and medical situations that presumably did not initially contribute to evolutionary APR preservation. At least in part, these considerations help explain multiple, often confusing, observations and possibly additional roles for SAA proteins.
SAA and atherosclerosis
The intimate relationship between cytokines and SAA synthesis and release as well as the SAA/HDL/cholesterol relationships discussed above have implicated SAA proteins in atherosclerosis. Clearly, this is a process complicating longevity and would not necessarily have been related to evolutionary conservation of these proteins and their genes (cf Getz and Reardon 2008; Kisilevsky and Manley 2012). Rather it has developed more recently, largely in older individuals. In this context the notion of “APR” has less relevance and a chronic role is likely more important.
Ross (1993, 1999) reviewed evidence for inflammation as a basic feature of atherosclerosis. His work (as well as that of many others) emphasized important features. Particularly notable are: 1) endothelial dysfunction, 2) chronicity, 3) intimate relationship to serum lipids, particularly LDL, 4) participation of monocytes/macrophages and T-cells (less frequently PMN cells), 5) cytokines, and 6) local tissue remodeling. These processes have been reviewed extensively and will not be presented in detail here.
In addition to their APR changes, both CRP and SAA levels are correlated in chronic inflammatory conditions (Ridker et al. 2000; Jousilahti et al. 2001). As early as 1994, Meek et al. (1994) detected SAA mRNA in atherosclerotic lesions in human cells. O’Brien et al. (2005) detected both SAA and lipoproteins in murine vascular disease lesions. Chiba et al. (2011) showed that HDL containing SAA bound to proteoglycans from vascular endothelium leading to lipoprotein retention within the vessel wall. Zhang et al. (2017) found that SAA could induce a change in the phenotype of vascular smooth muscle cells from a quiescent and contractile form to a synthetic, proliferative form and that the latter could be important in development and propagation of atherosclerosis. Using adenovirus expressing SAA in Apoe-/- and Rag-/- mice, Thompson et al. (2015) found prominent aortic and brachiocephalic vascular lesions containing high numbers of macrophages. They also noted that this effect was particularly prominent early after SAA induction. Local production of SAA – especially within the lesion(s) was important.
IL-1α induced SAA synthesis in aortic smooth muscle cells (O'Brien and Chait 2006). Kumon et al. (1997) distinguished transcriptional regulators of SAA in human aortic smooth muscle cells where C/EBP is prominent (as compared with HepG2 cells). Wilson et al. (2008) documented SAA stimulation of vascular proteoglycan synthesis. SAA also stimulated synthesis of phospholipase A2, apparently by transcriptional control (Sullivan et al. 2010), and the association of SAA with HDL blocked this effect consistent with local direct activity of SAA on lipid metabolism. Wroblewski et al. (2011) noted that the combination of SAA and endothelial lipase was associated with reduced HDL cholesterol formation – a situation that could accompany chronic inflammation. Association studies using SAA gene SNPs have suggested relationships to carotid artery intimal media thickness, HDL level and cardiovascular disease (Carty et al. 2009) but these have not yet been confirmed in large populations. Interestingly, apoprotein E-deficient mice also deficient in SAA1.1 and SAA2.1 continue to develop atherosclerotic lesions (De Beer et al. 2014).
Mice with a hepatocyte-specific knock-out of gp130 (IL-6 signal transducer) and with an atherosclerosis-prone genetic background show less aortic atherosclerosis and decreased macrophages in plaques. Humans with variants in IL6ST (the human gp130 homologue) show significant association with coronary artery disease, particularly involving ostia of coronary arteries (Luchtefeld et al. 2007).
Despite the evidence (above) for APR participation of SAA in cholesterol transport, recent studies have suggested that SAA may impair removal of cholesterol from some peripheral sites. Combined observations (McGillicuddy et al. 2009; Annema et al. 2010; Feingold and Grunfeld 2010) imply that even in the absence of an inflammatory response (e.g. SAA overproduction mediated by a viral vector – see (Meek et al. 1994)) SAA can reduce HDL-based cholesterol removal. As reviewed by Getz and Krishack (2016) and King et al. (2011) the underlying effect may be modification of HDL structure and/or transport function. The same authors propose the interactions as shown in Fig. 8.
Fig. 8.

Proposed sites for participation of SAA in atherosclerosis. (King et al. (2011), Copyright Lippincott Williams & Wilkins, 2011, used by permission.)
Although there have not been as many direct clinical studies relating SAA and vascular disease, multiple reports have been devoted to the “other” acute phase reactant – CRP, measured by a high sensitivity (hs) assay – as a “biomarker” for atherosclerosis (e.g. (Ridker et al. 2002)). Historically, CRP levels have been determined more routinely in the clinic than those of SAA so there are large data sets supporting the correlation. Although some reports (e.g. (Danesh et al. 2004)) have shown that CRP levels are less well-correlated than other common risk factors, the notion of an underlying inflammatory process with associated CRP elevation has appeared valid (Ridker et al. 2000; O'Brien and Chait 2006). Reducing cholesterol levels with statin treatment alone is associated with reduced inflammatory marker levels (Ridker et al. 2005). Even in asymptomatic individuals with normal levels of LDL but elevated CRP levels statin treatment reduced the CRP level (although the LDL levels fell as well, possibly contributing to the outcome) (Ridker et al. 2008). Interestingly, earlier studies (Choi et al. 2002; Suissa et al. 2006) had reported fewer cardiovascular deaths among patients with rheumatoid arthritis treated with methotrexate and other disease-modifying agents, consistent with the hypothesis that lowering inflammation itself might reduce clinical events. Because IL-1β precedes CRP in the inflammatory cascade (Ridker et al. 2017) these and other observations led to the recent CANTOS trial to evaluate the effect(s) of reducing vascular inflammation on cardiovascular events using Canakinumab, a human monoclonal antibody against IL-1β. Including over 10,000 patients with high CRP levels who had had a previous myocardial infarction and were studied over 4 years, this trial showed a significantly lower rate of recurrent cardiovascular events as well as reduced CRP levels (Ridker et al. 2017). This important study has provided a “proof-of-concept” for linking inflammation and clinical atherosclerosis (Ibañez and Fuster 2017).
SAA and cancer
Elevated levels of SAA in blood were detected relatively early in the largely phenomenologic studies of blood protein changes in cancer (Rosenthal and Sullivan 1979). SAA has been evaluated as a possible serum biomarker for many tumors including ovarian (Moshkovskii et al. 2005; Edgell et al. 2010), lung (Sung et al. 2011), renal (Sung et al. 2011; Tolson et al. 2004; Paret et al. 2010; Wood et al. 2010; Cocco et al. 2010), endometrial (Cocco et al. 2010), uterine (Cocco et al. 2009), and melanoma (Findeisen et al. 2009). These correlative studies were interesting but generally were not introduced as clinical markers. More recently, proteomics has substantially enlarged the scope of reports.
Finding local SAA production within tumor tissues added a new dimension to biologic relationship(s). Colorectal (Gutfeld et al. 2006), ovarian (Urieli-Shoval et al. 2010) and uterine (Cocco et al. 2009, 2010) tumor tissues showed SAA production. Murine SAA3 is prominently expressed in cancer-associated fibroblasts in pancreatic ductal adenocarcinoma (Djurec et al. 2018) where, in addition to SAA detection as a biomarker, the cells themselves could stimulate tumor cell growth. Prominent SAA expression on melanoma cells correlated with immunosuppressive neutrophils (surface markers CD11b+CD15+) within the tumor as well as IL-10 (De Santo et al. 2010). Thus, tumor (or contiguous stromal) production of SAA may be directly related to downregulation of antitumor immunity.
Further evidence linking SAA and tumor behavior has been shown in several systems. Hansen et al. (2015) related SAA 1 (and A3) in mice and SAA1 in humans to the S100 protein S100A4, a marker of metastasis and inflammation. SAA 1 and 3 and transcriptional targets of S100A4 in mice via at TLR4/NF-κB pathway. Their data establish SAA as “effectors for the metastasis-promoting functions of S100A4.”
Knebel et al. (2017) found that SAA1 was both expressed and secreted by human glioblastoma cells. SAA production was associated with tumor associated macrophages of the M2 type. Such an association would be consistent with a role in promoting angiogenesis and metastasis. Thus, SAA could have a feedback role in reducing anti-tumor effects of macrophages by inducing transition to the M2 type of functions.
While using SAA as a biomarker may have clinical utility as a “biomarker”, a larger question relates to direct SAA participation in tumor biology. As noted above, SAA interaction(s) as well as the function(s) and phenotype(s) of local macrophages could be central to this question and directly related to understanding tumor immunity. This extremely important and complex topic is beyond the scope of this current review.
SAA and tissue remodeling - matrix metalloproteinases and collagenase
Tissue remodeling in response to inflammation, trauma or other damage is, at least in part, mediated by enzymatic degradation of proteins in the extracellular matrix (ECM). Matrix metalloproteinases (MMP) are central to this process and the neutral proteinase collagenase has been examined closely. In a series of studies, Brinckerhoff et al. evaluated collagenase mRNA levels in rabbit synovial fibroblasts in response to inducers including phorbol myristate acetate (PMA) and monosodium urate (Brinckerhoff et al. 1982). Collagenase production was regulated by both inhibitory and stimulatory proteins released by these cells (Brinckerhoff et al. 1985; Brinckerhoff and Mitchell 1988). N-terminal sequencing of the secreted proteins identified β2-microglobulin and a protein with homology to SAA (Brinckerhoff et al. 1989). Adding either of these proteins to synovial cells led to collagenase release, consistent with an autocrine pathway that could both provoke and prolong inflammatory remodeling. The “SAA-like” protein sequence they identified differed from SAA1/2 but was quite similar to murine SAA3 (as well as human SAA3 - originally designated as GSAA1 – (Larson et al. 2003a)) and, as will be noted below in more detail, this SAA species uniquely contains the TFLK aa sequence near its N-terminus. Antibody raised against this protein could abolish this induction (Mitchell et al. 1991).
Collagenase and stromelysin both could degrade rabbit SAA3 and human SAA, consistent with a feedback mechanism controlling autocrine (or paracrine) effects (Mitchell et al. 1993). SAA cleavage occurred in the aa 50-57 region (conserved between both species) leaving an identifiable N-terminal fragment. (C-terminal residues were not identified and may have been degraded further.) Further studies confirmed that MMPs could cleave SAA (and SAA-amyloid fibril proteins) in the same aa 50-57 region and also identified other cleavage sites toward the N-terminus (Stix et al. 2001). In the rabbit, SAA3 functioned as an intermediate in the IL-1α stimulation pathway (Strissel et al. 1997). SAA also stimulated release of MMPs from human synovial fibroblasts (Migita et al. 1998). IL-1 and IL-6 induction of SAA synthesis and release in the same cells required both factors Sp1 and SAF (see also above) acting on promoter elements between nt -254 and -226 (Ilay et al. 1999).
Vallon et al. (2001) detected local SAA gene expression in rheumatoid arthritis tissues and showed that it induced MMP transcription. Synovial tissue from patients with inflammatory arthritis showed local expression of both SAA and formyl peptide receptor-like (FPLR1) genes as well as MMP production (O'Hara et al. 2004). SAA transcription also was found in inflammatory synovium from caprine retroviral arthritis by in situ hybridization (Sack and Zink 1992). Mullan et al. (2006) using synoviocytes from individuals with rheumatoid arthritis as well as human microvascular endothelial cells found that SAA induced expression of intracellular adhesion molecule (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1) and MMP 1. SAA induced IκBα degradation and NF-κB translation in these cells, implicating this pathway. These observations are consistent with a role for SAA in prolonging local inflammation and tissue degradation by inducing at least one component of the tissue remodeling pathway in both rheumatologic and atherosclerotic tissues. It is important to note that all of these studies did not specifically identify the SAA species (or sequence) used although detailed review shows that SAA1 and 2 were likely to be the species involved; later studies indicate that this may be important. Although the C-terminal regions of most SAA family members are quite similar (and likely cross-hybridize – see above) the N-terminal regions have multiple differences, including the characteristic TFLK sequence near the N-terminal end (see below).
Rosiglitazone (a thiazolidinedione) decreased MMP-9, SAA and TNF levels, consistent with common control site(s) for inflammation in individuals with diabetes (Marx et al. 2003; Ghanim et al. 2006). Human synovial fibroblasts contained mRNA for both SAA and formyl peptide receptor-like-1 (Dufton et al. 2010; Marsche et al. 2007) and exogenous SAA could induce MMP-1 and 3 in these cells. In mice transgenic for SAF-1, infection with Borelia burgdorferi led to inflammatory arthritis as well as MMP-1 induction (Ray et al. 2004b). In human THP1 cells SAA induced MMP9 synthesis through FPRL1 (Lee et al. 2005). MMP9 transcription responds to SAF-1 and AP1 whose binding sites are separated by only 14 nt in the upstream control region (Ray et al. 2005). SAA 1.1 was more susceptible to degradation by MMP1 than SAA1.5, correlating with a polymorphism at aa 57 (van der Hilst et al. 2008).
SAA and pulmonary physiology and disease - sarcoidosis
Lung changes in chronic obstructive pulmonary disease (COPD) are associated with inflammation and tissue destruction. Serologic markers correlating with acute exacerbations have been sought - CRP levels are related to functional decline in affected patients (Donaldson et al. 2005; Man et al. 2006).
Using SELDI-ToF proteomics, Bozinovski et al. (2008) identified SAA in sera of individuals with acute, glucocorticoid-refractory exacerbations of COPD and quantified the changes by ELISA. SAA levels were better correlated with clinical status than those of CRP. Subsequently, they studied bronchoalveolar lavage fluid (BALF) where there was good correlation between levels of SAA, IL-8 and neutrophil elastase (Bozinovski et al. 2012). Their data implicated alveolar macrophages as a potential source of SAA in BALF. There was no evidence for AA fibril deposition in affected tissues. Using human lung epithelial cells with and without the FPRL1 receptor showed that the receptor was required in order for SAA to induce significant amounts of the monocyte chemoattractant protein-1 (MCP-1), GMN-CSF and IL-8. This effect was antagonized by LXA4 and 15-epi-LXA4. Interestingly, despite their recognized effects in reducing other inflammatory reactions, glucocorticosteroids increased SAA production. Glucocorticosteroids can increase FPRL1 expression in leukocytes (Sawmynaden and Perretti 2006) and, as noted above, SAA1 has a glucocorticoid-responsive element (Thorn and Whitehead 2002, 2003). These observations were consistent with the lack of general efficacy of glucocorticosteroids in treating COPD exacerbations. They also revealed potential antagonism for LXA4 derivatives.
SAA has been implicated in the pathophysiology of sarcoidosis, a chronic, systemic, inflammatory disorder characterized by formation of noncaseating granulomata (Chen and Moller 2010). Local production of TH-1 cytokines and TNF have been recognized. Using a proteomics-based approach, Moller et al. (Song et al. 2005; Chen et al. 2010) identified IgG responses to the catalase-peroxidase protein from M. tuberculosis (mKatG) in >50% of affected individuals. BALF from sarcoidosis patients showed significantly higher levels of SAA. Tissue staining localized SAA to macrophages and multinucleated giant cells and was compatible with the notion that SAA is regulated by local CD3 T cells. SAA was prominent in areas of fibrosis. Studying the human macrophage-like cell line THP-1 they showed SAA stimulation of NF-κB that could be inhibited by preincubation with anti-TLR2 antibodies. A competitive binding ligand (Pam3CSK4) had a similar effect. They noted that polymorphisms in both RAGE and TLR2 have been associated with sarcoidosis. Interaction of SAA with multiple receptors (as recounted above and Table 1) is consistent with involvement of multiple pathways in regulating the granulomatous changes typical of sarcoidosis. They proposed that SAA is central to a specific (possibly unique) pathway for “epithelioid granulomatous inflammation in association with polarized Th1 responses to specific mycobacterial antigens.”
Cytokine-like function, intestinal physiology
Host defense against intestinal pathogens has revealed an important SAA role. Ivanov et al. (2009) reported specific, IL-17-secreting, Th17 cell accumulation in the lamina propria of the murine intestine as a specific consequence of non-invasive segmental filamentous bacteria (sfb) adhesion. Sfb adhesion to the intestinal epithelial cells (not simply colonization) led to SAA1 transcription as the most prominent upregulated species. Adding SAA to cocultures of dendritic cells and naïve CD4+T cells led to a concentration-dependent induction of Th17 cell differentiation (including cytokines and RORγt).
Atarashi et al. (2015) proposed that actin reorganization in the epithelial cells secondary to sfb adhesion led to elevated C/EBPδ expression which they proposed interacted with 2 DNase hypersensitive sites 3’ of the SAA1 gene. C/EBPδ levels were upregulated in sfb-colonized mice where they emphasized the need for direct contact between the cells and the sfb.
Sano et al. (2015) emphasized that IL17A production was greatest in the ileum. They identified type 3 innate lymphoid cells (ILC3) which, following sfb adhesion, secreted IL22. This response then led to SAA production by the epithelial cells through a STAT-dependent mechanism. The SAA effect on Th17 cells appeared to resemble cytokine activation. Binding of retinol derivatives to SAA (as suggested by Derebe et al. (2014)) could provide another role for SAA in maintaining intestinal barrier integrity. Pedicord and Mucida (2015) emphasized that these studies implicate “local environmental cues in licensing effector cytokine production.” Gury-BenAri et al. (2015) distinguished transcriptional differences between antigen-presenting cells that inhibit microbiota-directed T cell responses from those that produce IL11 and promote T cell responses (although SAA transcription was not reported). Mao et al. (2018) emphasized the role of ILC3 cells in maturation of gut commensalism.
Studying mice, Reigstad et al. (2009) showed that gut bacteria and possibly other bacterial products were associated with SAA3 expression in both adipose and colonic tissue. The Toll-like receptor/MyD88/NF-κB signal was at least partially responsible for the colon response. Gardiner et al. (2009) showed that using 10 or 42 aa N-terminal polypeptides from SAA3 in vitro to pretreat HT29 cells reduced adherence of enteropathogenic E. coli by 72%. However, using either polypeptide to pretreat mice did not reduce Salmonella invasion, consistent with likely participation of other components in vivo.
Maternal/fetal health
Preterm labor and sepsis
Serum SAA levels have been evaluated as markers for preterm labor. In mice, using a model of intrauterine LPS injection, Yang et al. (2009) found that SAA2 levels were increased in animals with preterm delivery. In women with unexplained recurrent early pregnancy loss Ibrahim et al. (2017) found significant elevations of SAA level (p<0.001). They identified SAA as “an independent indicator of primary unexplained recurrent early pregnancy loss after adjusting for maternal age and gestational age.” Sandri et al. (2014) showed that SAA at moderate levels enhances placentation via a TLR-4 receptor and that increasing SAA levels can disturb it. Mithal et al. (2017) found that elevated cord blood levels of SAA, CRP and haptoglobin levels were each associated with early onset neonatal sepsis in preterm (29.7 weeks) infants.
Mammary-derived SAA
McDonald et al. found SAA-reactivity in colostrum from cow, horse and sheep (McDonald et al. 2001) using a monoclonal antibody ELISA (McDonald et al. 1991). Using both batch purification and affinity chromatography they isolated substantial quantities of the protein (relatively little was found in mature milk) and sequencing revealed a sequence corresponding to the sequence reported earlier in rabbit (Brinckerhoff et al. 1989) as well as that predicted for human SAA 3 (Sack and Talbot 1989). The N-terminal region of this protein differed from that for the serum protein and there was no elevation in serum SAA levels in the animals. All isolates contained a 4 aa TFLK sequence near the N-terminus.
Adding either a 10 aa polypeptide containing the TFLK sequence or the 4 aa TFLK peptide alone to HT29 human intestinal epithelial cells led to intestinal MUC3 production (Larson et al. 2003b). A polypeptide corresponding to the human SAA3 gene sequence (GWLTFLKAAG) produced the same effect and led to a 73% decrease in adherence of enteropathogenic E. coli to the cultured cells (Mack et al. 2003) (later confirmed by Gardiner et al. (2009) as noted above).
Prolactin or LPS stimulation of a bovine mammary cell line (CLR-10274) revealed only SAA3 expression by RT-PCR (other SAA species were not transcribed) implying tissue-specific control (Larson et al. 2005a). Adding lipoteichoic acid from S. aureus to bovine mammary gland cells confirmed stimulation of SAA synthesis consistent with the potential use of SAA3 as a biomarker for mastitis (Weber et al. 2005). Detailed analysis of the bovine SAA3 gene structure identified an enhancer region between nt -2571 and -2338 but also a silencer region between nt -2338 and -1003. The minimal fragment retaining responsiveness was 352 nt and 53 nt of the untranslated region of exon 1 also enhanced expression (Larson et al. 2005b). LPS remained the most effective stimulant.
As discussed earlier, following LPS or prolactin stimulation, human mammary gland epithelial cell lines MCF-7 and T47-D showed SAA3 transcripts and RACE revealed a 655 bp cDNA sequence including an open reading frame corresponding to a 42 aa protein with truncation in the C-terminal region due an early stop codon. The corresponding short protein was not detected ((Larson et al. 2003a), see above). SAA3 was not found in human colostrum. An earlier report suggested that SAA3 was present (based on ELISA activity alone (Knee et al. 2015)). However, further study showed that human colostrum contains only a full-length, 12 kD protein shown by MS/tof sequencing to be SAA1.1; no other reactive species were detected (Sack et al. 2018). Finding SAA1.1 in human colostrum implies evolutionary pressure to change expression of at least SAA1.1 in human mammary cells. SAA1.1 does not contain the TFLK aa sequence (found in mice and other mammals e.g. (McDonald et al. 2001)) and whether it also contributes to intestinal mucin secretion is not yet known. Nevertheless, finding this switch in expression to another gene family member is consistent with possible evolutionary selection for a molecule with a likely important role in host defense.
Conclusions
Since its discovery, SAA has been an enigma. Nevertheless, its impressive evolutionary conservation as well as its dynamic synthesis control have always implied a consequential position in biology. SAA’s participation in the primordial survival physiology of the APR is consistent with both the evolutionary preservation and stability of SAA genes and proteins and with their cytokine and chemokine relationships (see Fig. 7). SAA helps link the complex network of cells (including macrophages, hepatocytes, MDSC) and proteins mediating inflammation.
In humans, clinical aspects of SAA protein pathobiology largely present later in life and thus may be appear to be “side effects” of generic APR preservation. For example, the 3-dimensional and amphipathic features of the SAA monomer help explain complex lipid interactions including HDL formation and cholesterol transport (details unresolved). Combining lipid associations with inflammatory cells and proteins is central to development of atherosclerosis. Stimulation (possibly via feedback loop(s)) of proteinase secretion contributes to chronic tissue injury in arthritis as well as pulmonary damage in sarcoidosis. Mediating signals reflecting the intestinal bacterial environment can modulate host immune defense(s) and possibly the gut flora itself. SAA’s presence in colostrum, stimulating neonatal intestinal mucin production, is consistent with SAA’s preservation as a “pro-survival” molecule. Other aspects of maternal-fetal health are characterized by SAA’s value as a biomarker for both prematurity and mastitis. Not surprisingly, myriad gene and cell surface changes can implicate SAA as a biomarker in malignancy. However, likely even more important is the relationship of SAA to immunologic pathways involving macrophage biology that are related to both metastasis and treatment including immunotherapy.
Finally, the propensity of fragment(s) of SAA monomers to form highly ordered amyloid fibrils explains many pathophysiologic features of “secondary” amyloidosis where chronic inflammation (or periodic – e.g. familial Mediterranean fever) overwhelms degradation that otherwise would occur under baseline conditions. It is ironic that “Serum Amyloid A” - the name acquired for this molecule soon after its description - (and now applied to the entire gene and protein family) is based on this property which now appears to be an epiphenomenon based on its unique protein structure and inherent propensity to form fibrils rather than its basic, well preserved physiologic function(s).
Convergence of multiple disciplines has begun to clarify the multifaceted relationships of SAA proteins and helps explain their remarkable evolutionary preservation. In addition to many valuable descriptive aspects of SAA biology, possibilities for prevention and specific, molecular-based treatments are emerging.
Acknowledgements
The author is grateful for collegial support and encouragement from Drs. Thomas McDonald and Natasha Zachara.
Funding
Support for these studies was provided by generous gifts to GS from: Stuart Bainum, Mary Nell and George A. Berry, Ethelbert Cooper, Gloria and Edward Felsenthal, Ruth Frey, Shirley Griffin, Ruth and George Harms, Yancey and David Hillegas, Diantha Johnson, Bassem Kudsi, Steven Lazinsky, Judy Lewent and Mark Shapiro, Terrence O’Donnell, Jay Ripley, Israel Vainboim, The Foundation for Greater Good, the Health Network Foundation, and The Met Life Foundation.
Availability of data and materials
All data are available through cited literature
Abbreviations
- ABCA
ATP-binding cassette (transporter)
- ACAT
Acyl-CoA:cholesterol acyl transferase
- APR
Acute phase response
- BALF
Broncho-alveolar lavage fluid
- C/EBP
CCAAT/enhancer-binding protein
- CD
Circular dichroism
- CEH
Cholesterol ester hydrolase
- CHO
Chinese hamster cell (line)
- COPD
chronic obstructive pulmonary disease
- CRP
C-reactive protein
- DNA
Deoxyribonucleic acid
- ELISA
Enzyme-linked immunosorbent assay
- fMLP
Formylmethionine-leucyl-phenylalanine
- FPRL
Formyl peptide receptor-like
- gp130
Glycoprotein130 – class of cytokine receptor
- HDL
High-density lipoprotein
- HepG2
Cell line
- ICAM
Intercellular adhesion molecule
- IFN-
Interferon-
- IL-
Interleukin-
- Kb
Kilobase
- LCAT
Lecithin/cholesterol acyl transferase
- LPS
Lipopolysaccharide
- MAPK
Mitogen-activated protein kinase
- MDSC
Myeloid-derived suppressor cell
- MMP
Matrix metalloproteinase
- mRNA
Messenger RNA
- NMR
Nuclear magnetic resonance
- PAMPS
Pathogen-associated molecular patterns
- PMA
Phorbol myristic acid
- PMN
Polymorphonuclear leukocyte
- RACE
Rapid amplification of cDNA ends (technique)
- RAGE
Advanced glycation end product receptor
- RNA
Ribonucleic acid
- RT-PCR
Reverse transcriptase-polymerase chain reaction
- SAA
serum amyloid A
- SAF
SAA activating factor
- SAP
Serum amyloid P
- sfp
Segmented filamentous bacteria
- SNP
Single nucleotide polymorphism
- SR-B1
Scavenger receptor-= B1
- TLR
Toll-like receptor
- TNF
Tumor necrosis factor
- VCAM
Vascular cell adhesion molecule
Authors’ contributions
The author is solely responsible for the entire review. The author read and approved the final manuscript.
Author’s information
The author received his M.D. and Ph.D. degrees from Johns Hopkins University where he also completed training in Internal Medicine and Medical Genetics. His research has emphasized genetic disorders, amyloid diseases and diagnostic problems in internal medicine. He is a member of the International Society for Amyloidosis, AAAS, ASHG and a founding fellow of ACMG.
Ethics approval
Human subjects were not directly involved in preparing this review
Consent for publication
No individual personal data are involved in this review
Competing interests
The author declares that he has no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Abe-Dohmae S, Kato KH, Kumon Y, Hu W, Ishigami H, Iwamoto N, Okazaki M, Wu C-A, Tsujita M, Ueda K, Yokoyama S. Serum amyloid A generates high density lipoprotein with cellular lipid in an ABCA1- or ABCA7-dependent manner. J Lipid Res. 2006;47:1542–1550. doi: 10.1194/jlr.M600145-JLR200. [DOI] [PubMed] [Google Scholar]
- Ancsin JB, Kisilevsky R. The heparin/heparin sulfate-binding site on apo-serum amyloid A. Implications for the therapeutic intervention of amyloidosis. J Bioi Chem. 1999;274:7172–7181. doi: 10.1074/jbc.274.11.7172. [DOI] [PubMed] [Google Scholar]
- Annema W, Nijstad N, Tὄlle M, deBeer JF, Buijs RVC, Heeringa P, van der Giet M, UJF T. Myeloperoxidase and serum amyloid A contribute to impaired in vivo reverse cholesterol transport during the acute phase response but not group IIA secretory phospholipase A2. J Lipid Res. 2010;51:743–754. doi: 10.1194/jlr.M000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, Suda W, Imaoka A, Steoyama H, Nagamori T, Ishikawa E, Shima T, Hara T, Kado S, Jinnohara T, Ohno H, Kondo T, Toyooka K, Watanabe E, Yokoyama S, Tokoro S, Mori H, Noguchi Y, Morita H, Ivanov II, Sugiyama T, Nunez G, Camp JG, Hattori M, Umesaki Y, Honda K. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell. 2015;163:357–380. doi: 10.1016/j.cell.2015.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: Inhibition of NF-κB activity through induction of IκB synthesis. Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- Badolato R, Johnson JA, Wang JM, McVicar D, Xu LL, Oppenheim JJ, Kelvin OJ. Serum amyloid A induces calcium mobilization and chemotaxis of human monocytes by activating a pertussis toxin-sensitive signaling pathway. J Immunol. 1995;155:4004–4010. [PubMed] [Google Scholar]
- Badolato R, Wang JM, Murphy WJ, Lloyd AR, Michiel OF, Bausserman LL, Kelvin OJ, Oppenheim JJ. Serum amyloid A is a chemoattractant: induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J Exp Med. 1994;180:203–209. doi: 10.1084/jem.180.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranova IN, Bocharov AV, Vishnyakova TG, Kurlander R, Chen Z, Fu D, Arias IM, Csako G, Patterson AP, Eggerman TL. CD36 is a novel serum amyloid A receptor mediating SAA binding and SAA-induced signaling in human and rodent cells. J Biol Chem. 2010;285:8492–8506. doi: 10.1074/jbc.M109.007526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranova IN, Vishnyakova TG, Bocharov AV, Kurlander R, Chen Z, Kimelman ML, Remaley AT, Csako G, Thomas F, Eggerman TL, Patterson AP. Serum amyloid A binding to CLA-l (CD36 and LlMPII analogous-1) mediates serum amyloid A protein-induced activation of ERK1/2 and p38 mitogen-activated protein kinases. J Biol Chem. 2005;280:8031–8040. doi: 10.1074/jbc.M405009200. [DOI] [PubMed] [Google Scholar]
- Benditt EP, Eriksen N. Amyloid II. Starch gel electrophoresis of some proteins extracted from amyloid. Arch Path. 1961;78:326–330. [PubMed] [Google Scholar]
- Benditt EP, Eriksen N. Amyloid protein SAA is associated with high density lipoprotein from human serum. Proc Natl Acad Sci USA. 1977;74:4025–4028. doi: 10.1073/pnas.74.9.4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benditt EP, Eriksen N, Hanson RH. Amyloid protein SAA is an apoprotein of mouse plasma high density lipoprotein. Proc Natl Acad Sci USA. 1979;76:4092–4096. doi: 10.1073/pnas.76.8.4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benditt EP, Eriksen N, Hermonson MA, Ericsson LH. The major proteins of human and monkey amyloid substance: Common properties including unusual N-terminal amino acid sequences. FEBS ltrs. 1971;19:169–173. doi: 10.1016/0014-5793(71)80506-9. [DOI] [PubMed] [Google Scholar]
- Benditt EP, Meek RL. Expression of the third member of the serum amyloid A gene family in mouse adipocytes. J Exp Med. 1989;169:1841–1846. doi: 10.1084/jem.169.5.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts JC, Cheshire JK, Akira S, Kishimoto T, Woo P. The role of NF-κB and NF-IL6 transactivating factors in the synergistic activation of human serum amyloid A gene expression by interleukin-1 and interleukin-6. J Biol Chem. 1993;268:25624–25631. [PubMed] [Google Scholar]
- Bing Z, Huang JH, Liao WS. NFkappa B interacts with serum amyloid A3 enhancer factor to synergistically activate mouse serum amyloid A3 gene transcription. J Biol Chem. 2000;275:31616–31623. doi: 10.1074/jbc.M005378200. [DOI] [PubMed] [Google Scholar]
- Bozinovski S, Hutchinson A, Thompson M, MacGregor L, Black J, Giannakis E, Karlsson A-S, Silvestrini R, Smallwood D, Vlahos R, Irving LB, Anderson GP. Serum amyloid A is a biomarker of acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177:269–278. doi: 10.1164/rccm.200705-678OC. [DOI] [PubMed] [Google Scholar]
- Bozinovski S, Uddin M, Vlahos R, Thompson M, McQualter JL, Merritt A-S, Wark PAB, Hutchinson A, Irving IB, Levy BD, Anderson GP. Serum amyloid A opposes lipoxin A4 to mediate glucocorticoid refractory lung inflammation in chronic obstructive pulmonary disease. Proc Natl Acad Sci USA. 2012;109:935–940. doi: 10.1073/pnas.1109382109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasier AR, Ron D, Tate JE, Habener JF. A family of constitutive C/EBP-like DNA binding proteins attenuate the IL-1α induced, NF-κB mediated trans-activation of the angiotensinogen gene acute-phase response element. EMBO J. 1990;9:3933–3944. doi: 10.1002/j.1460-2075.1990.tb07614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinckerhoff CE, Benoit MC, Culp WJ. Autoregulation of collagenase production by a protein synthesized and secreted by synovial fibroblasts: Cellular mechanism for control of collagen degradation. Proc Natl Acad Sci USA. 1985;82:1916–1920. doi: 10.1073/pnas.82.7.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinckerhoff CE, Gross RH, Nigase H, Sheldon L, Jackson RC, Harris ED., Jr Increased level of translatable collagenase messenger ribonucleic acid in rabbit synovial fibroblasts treated with phorbol myristate acetate or crystals of monosodium urate monohydrate. Biochem. 1982;21:2674–2679. doi: 10.1021/bi00540a015. [DOI] [PubMed] [Google Scholar]
- Brinckerhoff CE, Mitchell TI. Autocrine control of collagenase synthesis by synovial fibroblasts. J Cell Phys. 1988;136:72–80. doi: 10.1002/jcp.1041360109. [DOI] [PubMed] [Google Scholar]
- Brinckerhoff CE, Mitchell TJ, Karmilowicz MJ, Kluve-Beckerman B, Benson MD. Autocrine induction of collagenase by serum amyloid A-like and β2-microglobulin-like proteins. Science. 1989;243:655–657. doi: 10.1126/science.2536953. [DOI] [PubMed] [Google Scholar]
- Butler A, Whitehead AS. Mapping of the mouse serum amyloid A gene cluster by long-range polymerase chain reaction. Immunogenet. 1996;44:468–474. doi: 10.1007/BF02602809. [DOI] [PubMed] [Google Scholar]
- Cabana VG, Feng N, Reardon CA, Lukens J, Webb NR, de Beer FC, Getz GS. Influence of apoA-1 and apoE on the formation of serum amyloidosis A-containing lipoproteins in vivo and in vitro. J Lipid Res. 2004;45:317–325. doi: 10.1194/jlr.M300414-JLR200. [DOI] [PubMed] [Google Scholar]
- Cabana VG, Siegel JN, Sabesin SM. Effects of the acute phase response on the concentration and density distribution of plasma lipids and apolipoproteins. J Lipid Res. 1989;30:39–49. [PubMed] [Google Scholar]
- Cai L, deBeer MC, deBeer FC, van der Westhuysen DR. Serum amyloid A is a ligand for scavenger receptor class B type I and inhibits high density lipoprotein binding and selective lipid uptake. J Biol Chem. 2005;280:2954–2961. doi: 10.1074/jbc.M411555200. [DOI] [PubMed] [Google Scholar]
- Carty CL, Heagerty P, Heckbert SR, Enquobahrie OS, Jarvik G, Davis S, Tracy RP, Reiner AP. Association of genetic variation in serum amylold-A with cardiovascular disease and interactions with IL6, IL1RN, IL1beta and TNF genes in the Cardiovascular Health Study. J Atheroscler Thromb. 2009;16:419–430. doi: 10.5551/jat.No968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae JJ, Aksentijevich I, Kastner DL. Advances in the understanding of familial Mediterranean fever and possibilities for targeted therapy. Br J Haematol. 2009;146:467–478. doi: 10.1111/j.1365-2141.2009.07733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ES, Moller DR. Sarcoidosis – scientific progress and clinical challenges. Nat Rev Rheumatol. 2010;7:457–467. doi: 10.1038/nrrheum.2011.93. [DOI] [PubMed] [Google Scholar]
- Chen ES, Song Z, Willett MH, Heine S, Yung RC, Liu MC, Groshong SD, Zhang Y, Tuder RM, Moller DR. Serum amyloid A regulates granulomatous inflammation in sarcoidosis through Toll-like receptor-2. Am J Respir Crit Care Med. 2010;181:360–373. doi: 10.1164/rccm.200905-0696OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Zhou H, Cheng N, Qian F, Ye RD. Serum amyloid A1 isoforms display different efficacy at Toll-like receptor 2 and formyl peptide receptor 2. Immunobiol. 2014;219:916–923. doi: 10.1016/j.imbio.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng N, He R, Tian J, Ye PP, Ye RD. Cutting Edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J Immunol. 2008;181:22–26. doi: 10.4049/jimmunol.181.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba T, Chany MY, Wang S, Wight TN, McMillen TS, Oram JF, Vaisar T, Heinecke JW, De Beer FC, De Beer MC, Chait A. Serum amyloid A facilitates the binding of high-density lipoprotein from mice injected with lipopolysaccharide to vascular proteoglycans. Arterioscler Thromb Vasc Biol. 2011;31:1326–1332. doi: 10.1161/ATVBAHA.111.226159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HK, Hernán MA, Seeger JD, Robins JM, Wolfe F. Methotrexate and mortality in patients with rheumatoid arthritis: a prospective study. Lancet. 2002;359:1173–1177. doi: 10.1016/S0140-6736(02)08213-2. [DOI] [PubMed] [Google Scholar]
- Claus S, Meinhardt K, Aumüller T, Puscalau-Girtu I, Linder J, Haupt C, Walther P, Syrovets T, Simmer T, Fӓndrich M. Cellular mechanism of fibril formation from serum amyloid a1 protein. EMBO Rep. 2017;18:1352–1366. doi: 10.15252/embr.201643411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocco E, Bellone S, El-Sahwi K, Cargnelutti M, Buza N, Tavassoli FA, Schwartz PE, Rutherford TJ, Pecorelli S, Santin AD. Serum Amyloid A – A novel biomarker for endometrial cancer. Cancer. 2010;116:843–851. doi: 10.1002/cncr.24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocco E, Bellone S, El-Sahwi K, Cargnelutti M, Casagrande F, Buza N, Tavassoli FA, Siegel ER, Visintin I, Ratner E, Silasi D-A, Azodi M, Schwartz PE, Rutherford TJ, Pecorelli S, Santin AD. Serum amyloid A (SAA): A novel biomarker for uterine serous papillary cancer. Br J Cancer. 2009;101:335–341. doi: 10.1038/sj.bjc.6605129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee GA, Strachan AF, van der Westhuyzen DR, Hoppe HC, Jeenah MS, deBeer FC. Serum amyloid A-containing human high density lipoprotein 3. Density, size and apolipoprotein composition. J Biol Chem. 1986;261:9644–9651. [PubMed] [Google Scholar]
- Cohen AS, Calkins E. Electron microscopic observations on a fibrous component in amyloid of diverse origins. Nature. 1959;183:1202–1203. doi: 10.1038/1831202a0. [DOI] [PubMed] [Google Scholar]
- Danesh J, Wheeler JG, Hirschfield GM, Eda S, Eiriksdottir G, Rumley A, Lowe GDO, Pepys MB, Gudnason V. C-reactive protein and other Circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med. 2004;350:1387–1397. doi: 10.1056/NEJMoa032804. [DOI] [PubMed] [Google Scholar]
- De Beer MC, Wroblewski JM, Noffsinger VP, Rateri DL, Howatt DA, Balakrishnan A, Ji A, Shridas P, Thompson JC, van der Westhuyzen DR, Tannock LR, Daugherty A, Webb NR, De Beer FC. Deficiency of endogenous acute phase serum amyloid A does not affect atherosclerotic lesions in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Bioi. 2014;34:255–261. doi: 10.1161/ATVBAHA.113.302247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Buck M, Gouwy M, Wang JM, Van Snick J, Proost P, Struyf S, Van Damme J. The cytokine-serum amyloid A-chemokine network. Cytokine Growth Factor Rev. 2016;30:55–69. doi: 10.1016/j.cytogfr.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santo C, Arscott R, Booth S, Karydis I, Jones M, Asher R, Salio M, Middleton M, Cerundolo V. Invariant NKT cells modulate the suppressive activity of IL-10-secreting neutrophils differentiated with serum amyloid A. Nat Immunol. 2010;11:1039–1046. doi: 10.1038/ni.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBeer MC, DeBeer FC, Gerardot CJ, Cecil DR, Webb NR, Goodson ML, Kindy MS. Structure of the mouse Saa4 gene and its linkage to the Serum Amyloid A Gene Family. Genomics. 1996;34:139–142. doi: 10.1006/geno.1996.0253. [DOI] [PubMed] [Google Scholar]
- deBeer MC, Webb NR, Wroblewski JM, Noffsinger VP, Rateri DL, Ji A, van der Westhuyzen DR, deBeer FC. Impact of serum amyloid A on high density lipoprotein composition and levels. J Lipid Res. 2010;51:3117–3125. doi: 10.1194/jlr.M005413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deguchi A, Tomita T, Omori T, Komatsu A, Ohto U, Takahashi S, Tanimura N, Akashi-Takamura S, Miyake K, Maru Y. Serum amyloid A3 binds MD-2 to activate p38 and NF-κB pathways in a MyD88-dependent manner. J Immunol. 2013;191:1856–1864. doi: 10.4049/jimmunol.1201996. [DOI] [PubMed] [Google Scholar]
- Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, O’Malley KA, Wynn JL, Amtonenko S, Al-Quran SZ, Swan R, Chung C-S, Atkinson MA, Ramphal R, Gabrilovich DI, Reeves WH, Ayala A, Phillips J, LaFace D, Heyworth PG, Clare-Salzer M, Moldawer LL. MyD88-dependent expansion of an immature GR-1+CDllb+ population induces T cell suppression and h2 polarization in sepsis. J Exp Med. 2007;204:1463–1474. doi: 10.1084/jem.20062602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derebe MG, Ziatkov CM, Gattu S, Ruhn KA, Vaishnava S, Diehl GE, MacMillan JB, Williams NS, Hooper LV. Serum amyloid A is a retinol binding protein that transports retinol during bacterial infection. 2014. p. 03206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digre A, Nan J, Frank M, Li J-P. Heparin interactions with apoA1 and SAA in inflammation-associated HDL. Biochem Biophys Res Comm. 2016;474:309–314. doi: 10.1016/j.bbrc.2016.04.092. [DOI] [PubMed] [Google Scholar]
- Djurec M, Graňa O, Lee A, Troulé K, Espinet E, Cabras L, Navas C, Blasco MT, Martin-Diaz L, Burdiel M, Li J, Liu Z, Vallespinós M, Sanchez-Bueno F, Sprick MR, Trumpp A, Sainz BJR, Al-Shahrour F, Rabadan R, Guerra C, Barbacid M. SAA3 is a key mediator of the protumorigenic properties of cancer-associated fibroblasts in pancreatic tumors. Proc Natl Acad Sci USA. 2018;115:E1147–E1156. doi: 10.1073/pnas.1717802115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson GC, Seegmungal TA, Patel S, Bhowmik A, Wilkinson TM, Hurst JR, Maccallum PK, Wedzicha JA. Airway and systemic inflammation and decline in lung function in patients with COPD. Chest. 2005;128:1995–2004. doi: 10.1378/chest.128.4.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufton N, Hannon R, Brancaleone V, Dalli J, Patel HB, Gray M, D'Acquisto F, Buckingham JC, Perretti M, Flower RJ. Anti-inflammatory role of the murine formyl-peptide receptor 2: Ligand-specific effects on leukocyte responses and experimental inflammation. J Immunol. 2010;184:2611–2619. doi: 10.4049/jimmunol.0903526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eanes ED, Glenner GG. X-ray diffraction studies on amyloid filaments. J Histochem Cytochem. 1968;16:673–677. doi: 10.1177/16.11.673. [DOI] [PubMed] [Google Scholar]
- Eckhardt ERM, Witta J, Zhong J, Arsenescu R, Arsenescu V, Wang Y, Ghoshal S, deBeer MC, de Beer FC, de Villiers WJS. Intestinal epithelial serum amyloid A modulates bacterial growth in vitro and proinflammatory responses in mouse experimental colitis. BMC Gastroenterology. 2010;10:133. doi: 10.1186/1471-230X-10-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edbrooke MR, Burt DW, Cheshire JK, Woo P. Identification of cis-acting sequences responsible for phorbol ester induction of human serum amyloid A gene expression via a nuclear factor κB-like transcription factor. Mol Cell Biol. 1989;9:1908–1916. doi: 10.1128/MCB.9.5.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edbrooke MR, Foldi J, Cheshire JK, Li F, Faulkes DJ, Woo P. Constitutive and NF-κB-like proteins in the regulation of the serum amyloid A gene by interleukin-1. Cytokine. 1991;3:380–388. doi: 10.1016/1043-4666(91)90041-B. [DOI] [PubMed] [Google Scholar]
- Edbrooke MR, Woo P. Regulation of human SAA gene expression by cytokines. In: Pepys M, editor. Acute Phase Proteins in the Acute Phase Response. London: Springer-Verlag; 1989. pp. 21–27. [Google Scholar]
- Edgell T, Martin-Roussety G, Barker G, Auteliano DJ, Allen D, Grant P, Rice GE. Phase II biomarker trial of a multimarker diagnostic for ovarian cancer. J Cancer Res Clin Oncol. 2010;136:1079–1088. doi: 10.1007/s00432-009-0755-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ein D, Kimura S, Glenner GG. An amyloid fibril protein of unknown origin: Partial amino acid sequence analysis. Biochem Biophys Res Commun. 1972;46:498–500. doi: 10.1016/S0006-291X(72)80166-9. [DOI] [PubMed] [Google Scholar]
- Ein D, Kimura S, Terry WD, Magnotta J, Glenner GG. Amino acid sequence of an amyloid fibril protein of unknown origin. J Bioi Chem. 1972;247:5653–5655. [PubMed] [Google Scholar]
- Eisenberg D, Jucker M. The amyloid state of proteins in human diseases. Cell. 2012;148:1188–1203. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Kebir D, Jozsef L, Khreiss T, Pan W, Petasis NA, Serhan CN, Filep JG. Aspirin-triggered lipoxins override the apoptosis-delaying action of serum amyloid A in human neutrophils: A novel mechanism for resolution of inflammation. J ImmunoI. 2007;179:616–622. doi: 10.4049/jimmunol.179.1.616. [DOI] [PubMed] [Google Scholar]
- Faulkes DJ, Betts JC, Woo P. Characterization of five human serum amyloid A1 alleles. Amyloid. 1994;1:255–262. doi: 10.3109/13506129409146117. [DOI] [Google Scholar]
- Feingold KR, Grunfeld C. The acute phase response inhibits reverse cholesterol transport. J Lipid Res. 2010;51:682–684. doi: 10.1194/jlr.E005454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findeisen P, Zapatka M, Peccerella T, Matzk H, Neumaier M, Schadendorf D, Ugure S. Serum Amyloid A as a prognostic marker in melanoma identified by proteomic profiling. J Clin Onc. 2009;27:2199–2208. doi: 10.1200/JCO.2008.18.0554. [DOI] [PubMed] [Google Scholar]
- Fiore S, Maddox JF, Perez HD, Serhan CN. Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. J Exp Med. 1994;180:253–260. doi: 10.1084/jem.180.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame NM, Gursky O. Structure of serum amyloid A suggests a mechanism for selective lipoprotein binding and functions: SAA as a hub in macromolecular interaction networks. FEBS Lett. 2016;590:866–879. doi: 10.1002/1873-3468.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlaneto CJ, Campa A. A novel function of serum amyloid A: a potent stimulus for the release of tumor necrosis factor-α, interleukin-lβ, and interleukin-8 by human blood neutrophil. Biochem Biophys Res Commun. 2000;268:405–408. doi: 10.1006/bbrc.2000.2143. [DOI] [PubMed] [Google Scholar]
- Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448–454. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- Gao JL, Murphy PM. Species and subtype variants of the N-formyl peptide chemotactic receptor reveal multiple important functional domains. J Biol Chem. 1993;268:25395–25401. [PubMed] [Google Scholar]
- Gardiner GE, O'Flaherty S, Casey PG, Weber A, McDonald TL, Cronin M, Hill C, Ross RP, Gahan CGM, Shanahan F. Evaluation of colostrum-derived human mammary-associated serum amyloid A3 (M-SAA3) protein and peptide derivatives for the prevention of enteric infection: in vitro and in murine models of intestinal disease. FEMS Immunol Med Microbiol. 2009;55:404–413. doi: 10.1111/j.1574-695X.2009.00539.x. [DOI] [PubMed] [Google Scholar]
- Getz GA, Reardon CA. SAA, HDL biogenesis, and inflammation. J Lipid Res. 2008;49:269–270. doi: 10.1194/jlr.E700012-JLR200. [DOI] [PubMed] [Google Scholar]
- Getz GS, Krishack PA, Reardon CA. Serum amyloid A and atherosclerosis. Curr Opin Lipidol. 2016;27:531–535. doi: 10.1097/MOL.0000000000000331. [DOI] [PubMed] [Google Scholar]
- Ghanim H, Dhindsa S, Aljada A, Chaudhuri A, Viswanathan P, Dandona P. Low-dose rosiglitazone exerts an anti-inflammatory effect with an increase in adiponectin independently of free fatty acid fall and insulin sensitization in obese type 2 diabetics. J Clin Endocrinol Metab. 2006;91:3553–3558. doi: 10.1210/jc.2005-2609. [DOI] [PubMed] [Google Scholar]
- Glenner GG, Page D, Isersky C, Harada M, Cuatrecasas P, Eanes ED, RA DL, Bladen HA, Keiser HR. Murine amyloid fibril protein: Isolation, purification and characterization. J Histochem Cytochem. 1971;19:16–28. doi: 10.1177/19.1.16. [DOI] [PubMed] [Google Scholar]
- Gury-BenAri M, Thaiss CA, Serafini N, Winter DR, Giladi A, Lara-Astiaso D, Levy M, Salame TM, Weiner A, David E, Shapiro H, Dori-Bachhash M, Pevsner-Fischer M, Lorenzo-Vivas E, Keren-Shaul H, Paul F, Harmelin A, Ebery G, Itzkovitz S, Tanay A, DiSanto JP, Elinav E, Amit I. The spectrum and regulatory landscape of intestinal innate lymphoid cells are shaped by the microbiome. Cell. 2015;155:1231–1246. doi: 10.1016/j.cell.2016.07.043. [DOI] [PubMed] [Google Scholar]
- Gutfeld O, Prus D, Ackerman Z, Dishon S, Linke RP, Levin M, Urieli-Shoval S. Expression of Serum Amyloid A, in normal, dysplastic, and neoplastic human colonic mucosa: Implication for a role in colonic tumorigenesis. J Histochem Cytochem. 2006;54:63–73. doi: 10.1369/jhc.5A6645.2005. [DOI] [PubMed] [Google Scholar]
- Hagihara K, Nishikawa T, Isobe T, Song J, Sugamata Y, Yoshizaki K. IL-6 plays a critical role in the synergistic induction of human serum amyloid A (SAA) gene when stimulated with proinflammatory cytokines as analyzed with an SAA isoform real-time quantitative RT-PCR assay system. Biochem Biophys Res Commun. 2004;314:363–369. doi: 10.1016/j.bbrc.2003.12.096. [DOI] [PubMed] [Google Scholar]
- Hagihara K, Nishikawa T, Sugamata Y, Song J, Isobe T, Taga T, Yoshizaki K. Essential role of STAT3 in cytokine-driven NF-κB-mediated serum amyloid A gene expression. Genes Cells. 2005;10:1051–1063. doi: 10.1111/j.1365-2443.2005.00900.x. [DOI] [PubMed] [Google Scholar]
- Hansen MT, Forst B, Cremers N, Quagliata L, Ambartsumian N, Grum-Schwensen B, Klinghὄfer J, Abdul-Al A, Herrmann P, Osterland M, Stein U, Nielsen GH, Scherer PE, Lukanidin E, Sleeman JP, Grigorian M. A link between inflammation and metastasis: serum amyloid A1 and A3 induce metastasis, and are targets of metastasis-inducing S100A4. Oncogene. 2015;34:424–435. doi: 10.1038/onc.2013.568. [DOI] [PubMed] [Google Scholar]
- Hari-Dass R, Shah C, Meyer OJ, Raynes JG. Serum amyloid A protein binds to outer membrane protein A of gram-negative bacteria. J Biol Chem. 2005;280:18562–18567. doi: 10.1074/jbc.M500490200. [DOI] [PubMed] [Google Scholar]
- Hass G. Studies of amyloid II. The isolation of a polysaccharide from amyloid-bearing tissues. 1942. [Google Scholar]
- He R, Sang H, Ye RD. Serum amyloid A induces IL-8 secretion through a G protein-coupled receptor, FPRL1/LXA4R. Blood. 2003;101:1572–1581. doi: 10.1182/blood-2002-05-1431. [DOI] [PubMed] [Google Scholar]
- He R, Shepard LW, Chen J, Pan ZK, Ye RD. Serum amyloid A is an endogenous ligand that differentially induces IL-12 and IL-23. J Immunol. 2006;177:4072–4079. doi: 10.4049/jimmunol.177.6.4072. [DOI] [PubMed] [Google Scholar]
- He R, Zhou J, Hanson C, Chen J, Ye RD. The acute-phase protein serum amyloid A induces G-CSF expression and granulocytosis. J Leukoc Biol Suppl. 2007;44-45:abs 96. [Google Scholar]
- Hiratsuka S, Watanabe A, Sakurai Y, Akashi-Takamura S, Ishibashi S, Miyake K, Shibuya M, Akira S, Aburatani H, Maru Y. The S100A8- serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nature Cell Biol. 2008;10:1349–1355. doi: 10.1038/ncb1794. [DOI] [PubMed] [Google Scholar]
- Hoffman JS, Benditt EP. Changes in high density lipoprotein content following endotoxin administration in the mouse. J Bioi Chem. 1982;257:10510–10517. [PubMed] [Google Scholar]
- Hosoai H, Webb NR, Glick JM, Tietge UJF, Purdom MS, de Beer FC, Rader DJ. Expression of serum amyloid A protein in the absence of the acute phase response does not reduce HDL cholesterol or apoA-levels in human apoA-1 transgenic mice. J Lip Res. 1999;40:648–653. [PubMed] [Google Scholar]
- Hu W, Abe-Dohmae S, Tsujita M, Iwamoto N, Ogikubo O, Otsuka T, Kumon Y, Yokoyama S. Biogenesis of HDL by SAA is dependent on ABCA1 in the liver in vivo. J Lipid Res. 2008;49:386–393. doi: 10.1194/jlr.M700402-JLR200. [DOI] [PubMed] [Google Scholar]
- Huang JH, Liao WS. Synergistic induction of mouse serum amyloid A3 promoter by the inflammatory mediators IL-1 and IL-6. I Interferon Cytokine Res. 1999;12:1403–1411. doi: 10.1089/107999099312867. [DOI] [PubMed] [Google Scholar]
- Husby G, Natvig JB. A serum component related to nonimmunoglobulin amyloid protein AS, a possible precursor of the fibrils. J Clin Invest. 1974;53:1054–1061. doi: 10.1172/JCI107642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibañez B, Fuster V. CANTOS A gigantic proof-of-concept trial. Cir Res. 2017;121:1320–1322. doi: 10.1161/CIRCRESAHA.117.312200. [DOI] [PubMed] [Google Scholar]
- Ibrahim MI, Ramy AR, Abdelhamid AS, Ellaithy MI, Omar A, Harara RM, Fathy H, Abolouz AS. Maternal serum amyloid A level as a novel marker of primary unexplained recurrent early pregnancy loss. Int J Gynecol Obstet. 2017;136:298–303. doi: 10.1002/ijgo.12076. [DOI] [PubMed] [Google Scholar]
- Ilay A, Schatten H, Ray BK. Activation of Sp1 and its functional co-operation with serum amyloid A-activating sequence binding factor in synoviocyte cells trigger synergistic action of interleukin-1 and interleukin-6 in serum amyloid A gene expression. J Biol Chem. 1999;274:4300–4308. doi: 10.1074/jbc.274.7.4300. [DOI] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umcsaki Y, Honda K, Littman DR. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman S, Gantz DL, Haupt C, Gursky O. Serum amyloid A forms stable oligomers that disrupt vesicles at lysosomal pH and contribute to the pathogenesis of reactive amyloidosis. Proc Natl Acad Sci USA. 2017;114:E6507–E6515. doi: 10.1073/pnas.1707120114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen LE, Hiney MP, Shields DC, Uhlar CM, Lindsay AJ, Whitehead AS. Acute phase proteins in salmonids: evolutionary analyses and acute phase response. J ImmunoI. 1997;158:384–392. [PubMed] [Google Scholar]
- Ji YR, Kim HJ, Bae KB, Lee S, Kim MO, Ryoo ZY. Hepatic serum amyloid A1 aggravates T Cell-mediated hepatitis by inducing chemokines via Toll-like receptor 2 in mice. J Biol Chem. 2015;290:12804–12811. doi: 10.1074/jbc.M114.635763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S-L, Lozanski G, Samols D, Kushner I. Induction of human serum amyloid A in Hep3B cells by IL-6 and IL-1β involves both transcriptional and post-transcriptional mechanisms. J ImmunoI. 1995;154:825–831. [PubMed] [Google Scholar]
- Jiang S-L, Samols D, Rzewnicki D, Macintyre SS, Greber I, Sipe J, Kushner I. Kinetic modeling and mathematical analysis indicate that acute phase gene expression in Hep 3B cells is regulated by both transcriptional and posttranscriptional mechanisms. J Clin Inv. 1995;95:1253–1261. doi: 10.1172/JCI117775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jousilahti P, Salomaa V, Rasi V, Vahtera E, Palosuo T. The association of C-reactive protein, serum amyloid a and fibrinogen with prevalent coronary heart disease - baseline findings of the PAIS project. Atherosclerosis. 2001;156:451–456. doi: 10.1016/S0021-9150(00)00681-X. [DOI] [PubMed] [Google Scholar]
- Kennel SJ, Williams A, Stuckey A, Richey T, Wooliver C, Chazin W, Stern DA, Martin EB, Wall JS. The pattern recognition reagents RAGE VC1 and peptide p5 share common binding sites and exhibit specific reactivity with AA amyloid in mice. Amyloid. 2016;23:8–16. doi: 10.3109/13506129.2015.1112782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerppola TK, Luk D, Curran T. Fos is a preferential target of glucocorticoid receptor inhibition of AP-1 activity in vitro. Mol Cell Biol. 1993;13:3782–2791. doi: 10.1128/MCB.13.6.3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KD, Zhao J, Auh S, Yang X, Du P, Tang H, Fu Y-X. Adaptive immune cells temper initial innate responses. Nat Med. 2007;13:1248–1252. doi: 10.1038/nm1207-1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MH, de Beer MC, Wroblewski JM, Webb NR, de Beer FC. SAA does not induce cytokine production in physiological conditions. Cytokine. 2013;61:506–512. doi: 10.1016/j.cyto.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King VL, Thompson J, Tannock LR. Serum amyloid A in atherosclerosis. Curr Opin Lipidol. 2011;22:302–307. doi: 10.1097/MOL.0b013e3283488c39. [DOI] [PubMed] [Google Scholar]
- Kinkley SM, Bagshaw WL, Tam SP, Kisilevsky R. The path of murine serum amyloid A through peritoneal macrophages. Amyloid. 2006;13:123–134. doi: 10.1080/13506120600877201. [DOI] [PubMed] [Google Scholar]
- Kisilevsky R, Manley PN. Acute-phase amyloid A: Perspectives on its physiological and pathological roles. Amyloid. 2012;19:5–14. doi: 10.3109/13506129.2011.654294. [DOI] [PubMed] [Google Scholar]
- Kisilevsky R, Subrahmanyan L. Serum amyloid A changes high density lipoprotein's cellular affinity. Lab Inv. 1992;66:778–785. [PubMed] [Google Scholar]
- Kisilevsky R, Tam SP. Macrophage cholesterol efflux and the active domains of serum amyloid A 2.1. J Lipid Res. 2003;44:2257–2269. doi: 10.1194/jlr.M300133-JLR200. [DOI] [PubMed] [Google Scholar]
- Kluve-Beckerman B, Drumm ML, Benson MD. Nonexpression of the human serum amyloid A three (SAA3) gene. DNA Cell Biol. 1991;10:651–661. doi: 10.1089/dna.1991.10.651. [DOI] [PubMed] [Google Scholar]
- Kluve-Beckerman B, Manaloor JJ, Liepnieks JJ. A pulse-chase study tracking the conversion of macroendocytosed serum amyloid A into extracellular amyloid. Arth Rheum. 2002;46:1905–1913. doi: 10.1002/art.10335. [DOI] [PubMed] [Google Scholar]
- Kluve-Beckerman B, Naylor SL, Marshall A, Gardner JC, Shows TB, Benson MD. Localization of human SAA gene(s) to chromosome 11 and detection of DNA polymorphisms. Biochem Biophys Res Commun. 1986;137:1196–1204. doi: 10.1016/0006-291X(86)90352-9. [DOI] [PubMed] [Google Scholar]
- Knebel FH, Uno M, Galatro TF, Bellé LP, Oba-Shinbjo SM, Marie SKN, Campa A. Serum amyloid A1 is upregulated in human glioblastoma. J Neurooncol. 2017;132:383–391. doi: 10.1007/s11060-017-2386-z. [DOI] [PubMed] [Google Scholar]
- Knee O, Gupta A, Curley A, Charnok-Jones DS, GCS S, Belteki G. The acute-phase protein SAA3 is present in the preterm human colostrum and breast milk. Arch. Dis. Child. Fetal Neonatal Ed. 2015;100:F369–F371. doi: 10.1136/archdischild-2014-307795. [DOI] [PubMed] [Google Scholar]
- Knowles TPJ, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol. 2014;15:384–396. doi: 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- Knudsen PJ, Dinarello CA, Strom TB. Glucocorticoids inhibit transcriptional and post-transcriptional expression of interleukin 1 in U937 cells. J Immunol. 1987;139:4129–4134. [PubMed] [Google Scholar]
- Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, Zinkernagel R, Bluethmann H, Kὄhler G. Impaired immune and acute-phase responses in Interleukin-6-deficient mice. Nature. 1994;368:339–342. doi: 10.1038/368339a0. [DOI] [PubMed] [Google Scholar]
- Koracevic A, Hammer A, Stadelmeyer E, Windischhofer W, Sundl R, Ray A, Schweighofer N, Friedl G, Windhager R, Sattler W, Malle W. Expression of serum amyloid A transcripts in human bone tissues, differentiated osteoblast-like stem cells and human osteosarcoma cell lines. J Cell Biochem. 2008;103:994–1004. doi: 10.1002/jcb.21472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumon Y, Sipe JD, Brinckerhoff CE, Schreiber BM. Regulation of extrahepatic apolipoprotein Serum amyloid A gene expression by interleukin-1α alone. Scand J Immunol. 1997;46:284–291. doi: 10.1046/j.1365-3083.1997.d01-128.x. [DOI] [PubMed] [Google Scholar]
- Kumon Y, Suehiro T, Faulkes DJ, Hosakawa T, Ikeda Y, Woo P, Sipe JD, Hashimoto K. Transcriptional regulation of serum amyloid Al gene expression in human aortic smooth muscle cells involves CCAAT/enhancer binding proteins (C/EBP) and is distinct from HepG2 cells. Scand J Immunol. 2002;56:504–511. doi: 10.1046/j.1365-3083.2002.01169.x. [DOI] [PubMed] [Google Scholar]
- Kuroda T, Tanabe N, Hasegawa E, Wakamatsu A, Nozawa Y, Sato H, Nakatsue T, Wada Y, Ito Y, Imai N, Ueno M, Nakano M, Narita I. Significant association between renal function and area of amyloid deposition in kidney biopsy specimens in both AA amyloidosis associated with rheumatoid arthritis and AL amyloidosis. Amyloid. 2017;24:123–130. doi: 10.1080/13506129.2017.1338565. [DOI] [PubMed] [Google Scholar]
- Kushner I. The phenomenon of the acute phase response. Ann NY Acad Sci. 1982;39:39–48. doi: 10.1111/j.1749-6632.1982.tb22124.x. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM. Mechanisms and function of inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- Lane T, Pinney JH, Gilbertson JA, Hutt DF, Rowczenio DM, Mahmood A, Sachchithanantham S, Fontana M, Youngstein T, Quarta CC, Wechalekar AD, Gillmore JD, Hawkins PN, Lachmann HJ. Changing epidemiology of AA amyloidosis: clinical observations over 25 years at a single national referral center. Amyloid. 2017;24:162–166. doi: 10.1080/13506129.2017.1342235. [DOI] [PubMed] [Google Scholar]
- Larson MA, Weber A, McDonald TL. Bovine serum amyloid A3 gene structure and promoter analysis: Induced transcriptional expression by bacterial components and the hormone prolactin. Gene. 2005;380:104–110. doi: 10.1016/j.gene.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Larson MA, Weber A, Weber AT, McDonald TL. Differential expression and secretion of bovine serum amyloid A3 (SAA3) by mammary epithelial cells stimulated with prolactin or lipopolysaccharide. Vet Immunol lmmunopath. 2005;107:255–254. doi: 10.1016/j.vetimm.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Larson MA, Wei SH, Weber A, Mack DR, McDonald TL. Human serum amyloid A3 peptide enhances intestinal MUC3 expression and inhibits EPEC adherence. Biochem Biophys Res Comm. 2003;300:531–540. doi: 10.1016/S0006-291X(02)02901-7. [DOI] [PubMed] [Google Scholar]
- Larson MA, Wei SH, Weber A, Weber AT, McDonald TL. Induction of human mammary-associated serum amyloid A3 expression by prolactin or lipopolysaccharide. Biochem Biophys Res Comm. 2003;301:1030–1037. doi: 10.1016/S0006-291X(03)00045-7. [DOI] [PubMed] [Google Scholar]
- Lavie G, Zucker-Franklin D, Franklin EC. Degradation of serum amyloid A protein by surface-associated enzymes of human blood monocytes. J Exp Med. 1978;148:1020–1031. doi: 10.1084/jem.148.4.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Kim MK, Park KS, Bae YH, Yun J, Park JL, Kwak JY, Bae YS. Serum amyloid A stimulates matrix-metalloproteinase-9 upregulation via formyl peptide receptor like-1 mediated signaling in human monocytic cells. Biochem Biophys Res Commun. 2005;330:989–998. doi: 10.1016/j.bbrc.2005.03.069. [DOI] [PubMed] [Google Scholar]
- Levin M, Franklin EC, Frangione B, Pras M. The amino acid sequence of a major nonimmunoglobulin component of some amyloid fibrils. J Clin Inv. 1972;51:2773–2776. doi: 10.1172/JCI107098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J-P, Galvis MLE, Gong F, Zhang X, Zcharia E, Metzger S, Vlodavsky I, Kisilevsky R, Lindahl U. In vivo fragmentation of heparin sulfate by heparanase overexpression renders mice resistant to amyloid protein A amyloidosis. Proc Natl Acad Sci USA. 2005;102:6473–6477. doi: 10.1073/pnas.0502287102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Zhu S, Li J, D’Amore J, D’Angelo J, Yang H, Wang P, Tracey KJ, Wang H. Serum amyloid A stimulates PKR expression and HMGB1 release possibly through TLR4/RAGE receptors. Mol Med. 2015;21:515–525. doi: 10.2119/molmed.2015.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Rajala MW, Berger JP, Moller DE, Barzsilai N, Scherer OE. Hyperglycemia-induced production of acute phase reactants in adipose tissue. J Biol Chem. 2001;276:42077–42083. doi: 10.1074/jbc.M107101200. [DOI] [PubMed] [Google Scholar]
- Linke RP, Meinel A, Chalcroft JP, Urieli-Shoval S. Serum amyloid A (SAA) treatment enhances the recovery of aggravated polymicrobial sepsis in mice, whereas blocking SAA’s invariant peptide results in early death. Amyloid. 2017;24(S1):149–150. doi: 10.1080/13506129.2017.1295950. [DOI] [PubMed] [Google Scholar]
- Linke RP, Sipe JD, Pollock PS, Ignaczak TF, Glenner GG. Isolation of a low-molecular-weight serum component antigenically related to an amyloid fibril protein of unknown origin. Proc Natl Acad Sci USA. 1975;72:1473–1476. doi: 10.1073/pnas.72.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowell CA, Potter DA, Stearman RS, Morrow JF. Structure of the murine serum amyloid A gene family: Gene conversion. J Biol Chem. 1986;261:8442–8452. [PubMed] [Google Scholar]
- Lowell CA, Stearman RS, Morrow JF. Transcriptional regulation of serum amyloid A gene expression. J Biol Chem. 1986;261:8453–8461. [PubMed] [Google Scholar]
- Lu J, Yu Y, Zhu I, Cheng Y, Sun PD. Structural mechanism of serum amyloid A-mediated inflammatory amyloidosis. Proc Natl Acad Sci USA. 2014;111:5189–5194. doi: 10.1073/pnas.1322357111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchtefeld M, Schunkert H, Stoll M, Selle T, Lorier R, Grote K, Sagebiel C, Jagavelu K, Tietge UJF, Assmus U, Streetz K, Hengstenberg C, Fischer M, Mayer B, Maresso K, El Mokhtari NE, Schreiber S, Műller W, Bavendiek U, Grothusen C, Drexler H, Trautwein C, Broeckel U, Schieffer B. Signal transducer of inflammation gp130 modulates atherosclerosis in mice and man. J Exp Med. 2007;204:1935–1944. doi: 10.1084/jem.20070120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundmark K, Westermark GT, Olsen A, Westermark P. Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: cross-seeding as a disease mechanism. Proc Natl Acad Sci USA. 2005;102:6098–6102. doi: 10.1073/pnas.0501814102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack DR, McDonald TL, Larson MA, Wei S, Weber A. The conserved TFLK motif of mammary-associated serum amyloid A3 is responsible for up-regulation of intestinal MUC3 mucin expression In Vitro. Ped Res. 2003;53:137–114. doi: 10.1203/00006450-200301000-00023. [DOI] [PubMed] [Google Scholar]
- Macleod CM, Avery OT. The occurrence during acute infections of a protein not normally present in the blood. J Exp Med. 1941;73:183–190. doi: 10.1084/jem.73.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man SF, Connett JE, Anthonisen NR, Wise RA, Tashkin DP, Sin DD. C-reactive protein and mortality in mild to moderate chronic obstructive pulmonary disease. Thorax. 2006;61:849–853. doi: 10.1136/thx.2006.059808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao K, Baptista AP, Tamaoutounour S, Zhuang L, Bouladoux N, Martins AJ, Huang Y, Gerner MY, Belkaid Y, Germain RN. Innate and adaptive lymphocytes sequentially shape the gut microbiota and lipid metabolism. Nature. 2018;554:255–259. doi: 10.1038/nature25437. [DOI] [PubMed] [Google Scholar]
- Marsche G, Frank S, Raynes JG, Kozarsky KF, Sattler W, Malle E. The lipidation status of acute-phase protein serum amyloid A determines cholesterol mobilization via scavenger receptor class B, type 1. Biochem J. 2007;402:117–124. doi: 10.1042/BJ20061406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx N, Froehlich J, Siam L, Ittner J, Wierse G, Schmidt A, Scharnagl H, Hombach V, Koenig W. Antidiabetic PPARy-Activator Rosiglitazone reduces MMP-9 serum levels in Type 2 diabetic patients with coronary artery disease. Arterioscler Thromb Vasc Biol. 2003;23:283–288. doi: 10.1161/01.ATV.0000054195.35121.5E. [DOI] [PubMed] [Google Scholar]
- Marzi C, Albrecht E, Hysi PG, Lagou V, Waldenberger M, Tonjes A, Prokopenko J, Heim K, Blackburn H, Ried JS, Kleber ME, Mangino M, Thorand B, Peters A, Hammond CJ, Graliert HO, Boehm B, Kovacs P, Geistlinger L, Prokisch H, Winkelmann BR, Spector TO, Wichmann H-E, Stumvoll M, Soranzo N, Marz W, Koenig W, Illig T, Gieger C. Genome-wide association study identifies two novel regions at 11p15.5-p13 and 1p31 with major impact on acute-phase serum amyloid A. PLOS Genet. 2010;6:1–8. doi: 10.1371/journal.pgen.1001213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald TL, Larson MA, Mack DR, Weber A. Elevated extrahepatic expression and secretion of mammary-associated serum amyloid A3 (M-SAA3) into colostrum. Vet Immunol lmmunopath. 2001;83:203–211. doi: 10.1016/S0165-2427(01)00380-4. [DOI] [PubMed] [Google Scholar]
- McDonald TL, Weber A, Smith JW. A monoclonal antibody sandwich immunoassay for serum amyloid A (SAA) protein. J Immunol Meth. 1991;144:149–155. doi: 10.1016/0022-1759(91)90081-P. [DOI] [PubMed] [Google Scholar]
- McGillicuddy FC, de la Llera MM, Hinkle CC, Joshi MR, Chiquone EH, Billheimer JT, Rothblat GH, Reilly MP. Inflammation impairs reverse cholesterol transport in vivo. Circ. 2009;119:1135–1145. doi: 10.1161/CIRCULATIONAHA.108.810721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlennen RC, Burke BA, Dehner LP. Systemic amyloidosis complicating cystic fibrosis: A retrospective pathologic study. Arch Pathol Lab Med. 1986;110:879–884. [PubMed] [Google Scholar]
- Meek RL, Hoffman JS, Benditt EP. Amyloidogenesis - One serum amyloid A isotype is selectively removed from the circulation. J Exp Med. 1986;163:499–510. doi: 10.1084/jem.163.3.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek RL, Urieli-Shoval S, Benditt EP. Expression of apolipoprotein serum amyloid A mRNA in human atherosclerotic lesions and cultured vascular cells: implications for serum amyloid A function. Proc Natl Acad Sci USA. 1994;91:3186–3190. doi: 10.1073/pnas.91.8.3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeker AK, Sack GH., Jr A fusion protein between Serum Amyloid A and Staphylococcal Nuclease - Synthesis, purification, and structural studies. Proteins. 1998;30:381–387. doi: 10.1002/(SICI)1097-0134(19980301)30:4<381::AID-PROT5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Migita K, Kawabe Y, Tominaga M, Riguchi TO, Aoyagi T, Eguchi K. Serum amyloid A protein induces production of matrix metalloproteinases by human synovial fibroblasts. Lab Invest. 1998;78:535–539. [PubMed] [Google Scholar]
- Mitchell TI, Coon CI, Brinckerhoff CE. Serum amyloid A (SAA3) produced by rabbit synovial fibroblasts treated with phorbol esters or Interleukin 1 induces synthesis of collagenase and is neutralized with specific antiserum. J Clin Invest. 1991;87:1177–1185. doi: 10.1172/JCI115116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell TI, Jeffrey JJ, Palmiter RD, Brinckerhoff CE. The acute phase reactant serum amyloid A (SAA3) is a novel substrate for degradation by the metalloproteinases collagenase and stromolysin. Biochim Biophys Acta. 1993;1156:245–254. doi: 10.1016/0304-4165(93)90038-A. [DOI] [PubMed] [Google Scholar]
- Mithal LB, Palac HL, Yogev R, Ernst LM, Mestan KK. Cord blood acute phase reactants predict early onset neonatal sepsis in preterm infants. PLOS one. 2017; 10.1371/journal.pone.0168677. [DOI] [PMC free article] [PubMed]
- Mori M, Tiang G, Higuchi K. AA amyloidosis-resistant CE/J mice have Saal and Saa2 genes that encode an identical SAA isoform. Amyloid. 2014;21:1–8. doi: 10.3109/13506129.2013.852529. [DOI] [PubMed] [Google Scholar]
- Moriguchi M, Kaneko H, Terai C, Koseki Y, Kajiyama H, Inada S, Kitamura Y, Kamatani N. Relative transcriptional activities of SAA1 promoters polymorphic at position -13 (T/C): Potential association between increased transcription and amyloidosis. Amyloid. 2005;12:26–32. doi: 10.1080/13506120500032394. [DOI] [PubMed] [Google Scholar]
- Morrow JF, Stearman RS, Peltzman CG, Potter DA. Induction of hepatic synthesis of serum amyloid A protein and actin. Proc Natl Acad Sci USA. 1981;78:4718–4722. doi: 10.1073/pnas.78.8.4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moshkovskii SA, Serebryakova MV, Kuteykin-Teplyakov KB, Tikhonova OV, Goufman EI, Zgoda VG, Taranets IN, Makarov OV, Archakov AI. Ovarian cancer marker of 11.7 kDa detected by proteomics is a serum amyloid A1. Proteomics. 2005;5:3790–3797. doi: 10.1002/pmic.200401205. [DOI] [PubMed] [Google Scholar]
- Mullan RN, Bresnihan B, Golden-Mason L, Markham T, O'Hara R, FitzGerald O, Veale DJ, Fearon U. Acute-phase serum amyloid A stimulation of angiogenesis, leukocyte recruitment, and matrix degradation in rheumatoid arthritis through an NF-κB-dependent signal transduction pathway. Arthritis Rheum. 2006;54:105–114. doi: 10.1002/art.21518. [DOI] [PubMed] [Google Scholar]
- Murakami M, Hibi M, Nakagawa N, Nakagawa T, Yasukawa K, Yamanishi K, Taga T, Kishimoto T. IL-6-induced homodimerization of gp130 and associated activation of a tyrosine kinase. Science. 1993;260:1808–1810. doi: 10.1126/science.8511589. [DOI] [PubMed] [Google Scholar]
- Nacu S, Yuan W, Kan Z, Bhatt D, Rivers CS, Stinson J, Peters BA, Modrusan Z, Jung K, Seshagiri S, Wu TD. Deep RNA sequencing analysis of readthrough gene fusions in human prostate adenocarcinoma and reference samples. BMC Med Genom. 2011;4:11. doi: 10.1186/1755-8794-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. TIBS. 1998;23:198–199. doi: 10.1016/s0968-0004(98)01208-0. [DOI] [PubMed] [Google Scholar]
- Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspec Biol. 2012;4:1–19. doi: 10.1101/cshperspect.a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noborn F, Ancsin JB, Ubhayasekera W, Kisilevsky R, Li J-P. Heparan sulfate dissociates serum amyloid A (SAA) from acute-phase lipoprotein, promoting SAA aggregation. J Biol Chem. 2012;287:25669–25677. doi: 10.1074/jbc.M112.363895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien KD, Chait A. Serum amyloid A: The “Other” inflammatory protein. Curr Atherosclerosis Rep. 2006;8:62–68. doi: 10.1007/s11883-006-0066-0. [DOI] [PubMed] [Google Scholar]
- O'Brien KD, McDonald TO, Kunjathoor V, Eng KL, Knopp EA, Lewis K, Lopez R, Kirk EA, Chait A, Wight TN, deBeer FC, LeBoeuf RC. Serum amyloid A and lipoprotein retention in murine models of atherosclerosis. Arterioscler Thromb Vasc Bioi. 2005;25:785–790. doi: 10.1161/01.ATV.0000158383.65277.2b. [DOI] [PubMed] [Google Scholar]
- O'Hara R, Murphy EP, Whitehead AS, FitzGerald O, Bresnihan B. Local expression of the serum amyloid A and formyl peptide receptor-like 1 genes in synovial tissue is associated with matrix metalloproteinase production in patients with inflammatory arthritis. Arthritis Rheum. 2004;50:1788–1799. doi: 10.1002/art.20301. [DOI] [PubMed] [Google Scholar]
- Paret C, Schὄn Z, Szponar A, Kovacs G. Inflammatory protein serum amyloid A1 marks a subset of conventional renal cell carcinomas with fatal outcome. Eur Urol. 2010;57:859–866. doi: 10.1016/j.eururo.2009.08.014. [DOI] [PubMed] [Google Scholar]
- Patel H, Bramall J, Waters H, DeBeer MC, Woo P. Expression of recombinant human serum amyloid A in mammalian cells and demonstration of the region necessary for high-density lipoprotein binding and amyloid fibril formation by site-directed mutagenesis. Biochem J. 1996;318:1041–1049. doi: 10.1042/bj3181041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel H, Fellowes R, Coade S, Woo P. Human serum amyloid A has cytokine-like properties. Scand J ImmunoI. 1998;48:410–418. doi: 10.1046/j.1365-3083.1998.00394.x. [DOI] [PubMed] [Google Scholar]
- Pauling L, Corey RB. Configurations of polypeptide chains with favored orientation around single bonds: Two new pleated sheets. Proc Natl Acad Sci USA. 1951;37:729–740. doi: 10.1073/pnas.37.11.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedicord VA, Mucida D. “A sledgehammer breaks glass but forges steel”: Bacteria adhesion shapes gut immunity. Cell. 2015;153:273–274. doi: 10.1016/j.cell.2015.09.040. [DOI] [PubMed] [Google Scholar]
- Pepys MB. Pathogenesis, diagnosis and treatment of systemic amyloidosis. Phil Trans R Soc Lond B. 2001;356:203–211. doi: 10.1098/rstb.2000.0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu M-Y, van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/Hej and C57BL/10ScCr mice: mutations in the Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Prelli F, Pras M, Frangione B. Degradation and deposition of amyloid AA fibrils are tissue specific. Biochem. 1987;26:8251–8256. doi: 10.1021/bi00399a035. [DOI] [PubMed] [Google Scholar]
- Qian H, Zhao X, Cao P, Lei J, Van N, Gong X. Structure of the human lipid exporter ABCA1. Cell. 2017;169:1228–1239. doi: 10.1016/j.cell.2017.05.020. [DOI] [PubMed] [Google Scholar]
- Quinton U, Blahna MT, Jones MR, Allen E, Ferrari JD, Hilliard KL, Zhang X, Sabharwal V, Algal H, Akira S, Schmid RM, Pelton SI, Spira A, Mizgerd JP. Hepatocyte-specific mutation of both NF-κB RelA and STAT3 abrogates the acute phase response in mice. J Clin Inv. 2012;122:1758–1763. doi: 10.1172/JCI59408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinton U, Jones MR, Robson BE, Mizgerd JP. Mechanisms of the hepatic acute-phase response during bacterial pneumonia. Infect Immun. 2009;77:2417–2426. doi: 10.1128/IAI.01300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranlov P. The adoptive transfer of experimental mouse amyloidosis by intravenous injections of spleen cell extracts from casein treated syngenic donor mice. Acta Path Microbiol Scand. 1967;70:321–335. doi: 10.1111/j.1699-0463.1967.tb01300.x. [DOI] [PubMed] [Google Scholar]
- Ray A, Bal BS, Ray BK. Transcriptional induction of matrix metalloproteinase-9 in the chondrocyte and synoviocyte cells is regulated via a novel mechanism: evidence for functional cooperation between serum amyloid A-activating factor-1 and AP-l. J ImmunoI. 2005;175:4039–4048. doi: 10.4049/jimmunol.175.6.4039. [DOI] [PubMed] [Google Scholar]
- Ray A, Kumar D, Ray P, Ray BK. Transcriptional activity of serum amyloid A-activating factor-1 is regulated by distinct functional modules. J Biol Chem. 2004;279:54637–54646. doi: 10.1074/jbc.M411830200. [DOI] [PubMed] [Google Scholar]
- Ray A, Kumar D, Shakya A, Brown CR, Cook JL, Ray BK. Serum amyloid A-activating factor (SAF-1) transgenic mice are prone to develop a severe form of inflammation-induced arthritis. J Immunol. 2004;173:4684–4691. doi: 10.4049/jimmunol.173.7.4684. [DOI] [PubMed] [Google Scholar]
- Ray A, Ray BK. A novel cis-acting element is essential for cytokine-mediated transcriptional induction of the serum amyloid A gene in nonhepatic cells. Mol Cell Biol. 1996;16:1584–1594. doi: 10.1128/MCB.16.4.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Shakya A, Kumar D, Benson MD, Ray BK. Inflammation-responsive transcription factor SAF-1 activity is linked to the development of amyloid A amyloidosis. J Immunol. 2006;177:2601–2609. doi: 10.4049/jimmunol.177.4.2601. [DOI] [PubMed] [Google Scholar]
- Ray A, Shang DH, Siegel MD, Ray P. Regulation of interleukin-6 gene expression by steroids. Ann N Y Acad Sci. 1995;762:79–87. doi: 10.1111/j.1749-6632.1995.tb32316.x. [DOI] [PubMed] [Google Scholar]
- Reigstad CS, Lundén GO, Felin J, Bӓckhed F. Regulation of Serum Amyloid A3 (SAA3) in mouse colonic epithelium and adipose tissue by the intestinal microbiota. PLOS one. 2009; 10.1371/journal.pone.0005842. [DOI] [PMC free article] [PubMed]
- Ridker PM, Cannon CP, Morrow D, Rifai N, Rose LM, McCabe CH, Pfeffer MA, Braunwald E. C-reactive protein levels and outcomes after statin therapy. New Engl J Med. 2005;351:20–28. doi: 10.1056/NEJMoa042378. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Danielson E, FAH F, Genest J, AMJr G, JJP K, Koenig W, Libby P, Lorenzatti AJ, JG MF, Bordestgaard BG, Shepherd J, Willerson JT, Glynn RJ, for the JUPITER Study Group Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. New Engl J Med. 2008;359:2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau F, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, for the CANTOS trial group Antiinflammatory therapy with Canakinumab for atherosclerotic disease. New Engl J Med. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000;342:836–843. doi: 10.1056/NEJM200003233421202. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 2002;347:1557–1565. doi: 10.1056/NEJMoa021993. [DOI] [PubMed] [Google Scholar]
- Riek R, Eisenberg DS. The activities of amyloids from a structural perspective. Nature. 2016;539:227–235. doi: 10.1038/nature20416. [DOI] [PubMed] [Google Scholar]
- Rienhoff HY, Jr, Groudine M. Regulation of Amyloid A gene expression in cultured cells. Mol Cell BioI. 1988;8:3710–3716. doi: 10.1128/MCB.8.9.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal CJ, Franklin EC, Frangione B, Greenspan J. Isolation and partial characterization of SAA-an amyloid-related protein from human serum. J ImmunoI. 1976;116:1415–1418. [PubMed] [Google Scholar]
- Rosenthal CJ, Sullivan LM. Serum amyloid A to monitor cancer dissemination. Ann Int Med. 1979;91:383–390. doi: 10.7326/0003-4819-91-3-383. [DOI] [PubMed] [Google Scholar]
- Ross R. Atherosclerosis: a defense mechanism gone awry. Am J Path. 1993;143:987–1002. [PMC free article] [PubMed] [Google Scholar]
- Ross R. Atherosclerosis - An inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- Rust S, Rosier M, Funke H, Real J, Amoura Z, Piette J-C, Deleuze J-F, Brewer HB, Duverger N, Denèfle P, Assmann G. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat Genet. 1999;22:352–355. doi: 10.1038/11921. [DOI] [PubMed] [Google Scholar]
- Rὄcken C, Kientsch-Engel SM, Stix B, Stubenrach K, Weigle B, Bűhling F, Schwan M, Saeger W. Advanced glycation end products and receptor for advanced glycation end products in AA amyloidosis. Amer J Path. 2003;162:1213–1220. doi: 10.1016/S0002-9440(10)63917-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rὄcken C, Menard R, Bühling F, Vὄckler S, Raynes J, Stix B, Krüger S, Roessner A, Kӓhne T. Proteolysis of serum amyloid A and AA amyloid proteins by cysteine proteases: cathepsin B generates AA amyloid proteins and cathepsin L may prevent their formation. Ann Rheum Dis. 2005;64:808–815. doi: 10.1136/ard.2004.030429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack GH., Jr Molecular cloning of human genes for serum amyloid A. Gene. 1983;21:19–24. doi: 10.1016/0378-1119(83)90143-9. [DOI] [PubMed] [Google Scholar]
- Sack GH, Jr, Talbot CC., Jr The human serum amyloid A (SAA)-encoding gene GSAA1: nucleotide sequence and possible autocrine collagenase-inducer function. Gene. 1989;84:509–515. doi: 10.1016/0378-1119(89)90528-3. [DOI] [PubMed] [Google Scholar]
- Sack GH, Jr, Talbot CC, Jr, Seuanez H, O'Brien SJ. Molecular analysis of the human serum amyloid A (SAA) gene family. Scand J Immunol. 1989;29:113–119. doi: 10.1111/j.1365-3083.1989.tb01105.x. [DOI] [PubMed] [Google Scholar]
- Sack GH, Jr, Zachara N, Rosenblum N, Talbot CC, Jr, Kreimer S, Cole R, TJ MD. Serum amyloid A1 (SAA1) protein in human colostrum. FEBS Open Bio. 2018;8:1–7. doi: 10.1002/2211-5463.12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack GH, Jr, Zink MC. Serum amyloid A (SAA) gene expression in synovial cells in retroviral arthritis. Am J Path. 1992;141:525–529. [PMC free article] [PubMed] [Google Scholar]
- Sander LE, Sackett SD, Dierssen Y, Beraza N, Linke RP, Müller M, Blander JM, Tacke F, Trautwein C. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J Exp Med. 2010;207:1453–1464. doi: 10.1084/jem.20091474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri S, Borbely AU, Fernandes I, deOliveira EM, Knebel FH, Ruano R, Zugaib M, Filippin-Monteiro F, Bevilaqua E, Campa A. Serum Amyloid A in the Placenta and its Role in Trophoblast Invasion. PLOSone. 2014; 10.1371/journal.pone.0090881. [DOI] [PMC free article] [PubMed]
- Sandri S, Rodriguez D, Gomes E, Monteiro HP, Russo M, Campa A. Is serum amyloid A an endogenous TLR4 agonist? J Leukoc Bioi. 2008;83:1174–1180. doi: 10.1189/jlb.0407203. [DOI] [PubMed] [Google Scholar]
- Sano T, Huang W, Hall JA, Yang Y, Chen A, Gavzy SJ, Lee J-Y, Ziel JW, Miraldi ER, Domingos AI, Bonneau R, Littman DR. An IL-23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell. 2015;163:381–393. doi: 10.1016/j.cell.2015.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawmynaden P, Perretti M. Glucocortocoid upregulation of the annexin-A1 receptor in leukocytes. Biochem Biophys Res Commun. 2006;349:1351–1355. doi: 10.1016/j.bbrc.2006.08.179. [DOI] [PubMed] [Google Scholar]
- Scheinman RI, Cogswell PC, Lofquist AK, Baldwin AS., Jr Role of transcriptional activation of IκBα in mediation of immunosuppression by glucocorticoids. Science. 1995;270:283–286. doi: 10.1126/science.270.5234.283. [DOI] [PubMed] [Google Scholar]
- Scheinman RI, Gualberto A, Jewell CM, Cidlowski JA, Baldwin AS., Jr Characterization of mechanisms involved in transrepression of NF-kappaB by activated glucocorticoid receptor. Mol Cell Biol. 1995;15:943–953. doi: 10.1128/MCB.15.2.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah C, Hari-Dass R, Raynes JG. Serum amyloid A is an innate immune opsonin for Gram-negative bacteria. Blood. 2006;108:1751–1757. doi: 10.1182/blood-2005-11-011932. [DOI] [PubMed] [Google Scholar]
- Sipe J. Revised nomenclature for serum amyloid A (SAA). Nomenclature Committee of the International Society of Amyloidosis. Part 1. Amyloid. 1999;6:67–70. doi: 10.3109/13506129908993291. [DOI] [PubMed] [Google Scholar]
- Song Z, Marsilli L, Greenlee BM, Chen ES, Silver RF, Askin FB, Teirstein AS, Zhang Y, Cotter RJ, Moller DR. Mycobacterial catalase-peroxidase is a tissue antigen and target of the adaptive immune response in systemic sarcoidosis. J Exp Med. 2005;201:755–767. doi: 10.1084/jem.20040429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spodzieja M, Rafalik M, Szymańska A, Kolodziejczyk AS, Czaplewska P. Interaction of serum amyloid A with human cystatin C - assessment of amino acid residues crucial for hCC-SAA formation (part II) J Mol Recognit. 2013;26:415–425. doi: 10.1002/jmr.2283. [DOI] [PubMed] [Google Scholar]
- Srinivasan S, Patke S, Wang Y, Ye Z, Litt J, Srivastava SK, Lopez MM, Kurouski D, Lednev IK, Kane RS, Colon W. Pathogenic Serum Amyloid A 1.1 shows a long oligomer-rich fibrillation lag phase contrary to the highly amyloidogenic non-pathogenic SAA2.2. J Biol Chem. 2013;288:2744–2755. doi: 10.1074/jbc.M112.394155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel DM, Roger JT, DeBeer MC, DeBeer FC, Whitehead AS. Biosynthesis of human acute-phase serum amyloid A protein (A-SAA) in vitro: the roles or mRNA accumulation, poly (A) tail shortening and translational efficiency. Biochem J. 1993;291:701–707. doi: 10.1042/bj2910701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel DM, Sellar GC, Uhlar CM, Simon S, deBeer FC, Whitehead AS. A constitutively expressed serum amyloid A protein gene is closely linked to, and shares structural similarities with, an acute phase serum amyloid A protein gene. Genomics. 1993;16:447–454. doi: 10.1006/geno.1993.1209. [DOI] [PubMed] [Google Scholar]
- Stevens JF. Hypothetical structure of human serum amyloid A protein. Amyloid. 2004;11:71–80. doi: 10.1080/13506120412331272296. [DOI] [PubMed] [Google Scholar]
- Stix B, Kähne T, Sletten K, Raynes J, Roessner A, Rὄcken C. Proteolysis of AA amyloid fibril proteins by matrix metalioproteinases-1,- 2, and -3. Am J PathoI. 2001;159:561–570. doi: 10.1016/S0002-9440(10)61727-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stonik JS, Remaley AT, Demosky SJ, Neufeld EB, Bocharov A, Brewer HB. Serum amyloid A promotes ABCA1-dependent and ABCA1- independent lipid efflux from cells. Biochem Biophys Res Comm. 2004;321:936–941. doi: 10.1016/j.bbrc.2004.07.052. [DOI] [PubMed] [Google Scholar]
- Strissel KJ, Girard MT, West-Mays J-A, Rinehart WB, Cook JR, Brinckerhoff CE, Fini ME. Role of serum amyloid A as an intermediate in the IL-1 and PMA-stimulated signaling pathways regulating expression of rabbit fibroblast collagenase. Exp Cell Res. 1997;237:275–287. doi: 10.1006/excr.1997.3783. [DOI] [PubMed] [Google Scholar]
- Su SB, Gong W, Gao J-L, Shen W, Murphy PM, Oppenheim JJ, Wang JM. A seven-transmembrane, G protein-coupled receptor, FPRLl, mediates the chemotactic activity of serum amyloid A for human phagocytic cells. J Exp Med. 1999;189:395–402. doi: 10.1084/jem.189.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suissa S, Bernatsky S, Hudson M. Antirheumatic drug use and the risk of acute myocardial infarction. Arth Rheum. 2006;55:531–536. doi: 10.1002/art.22094. [DOI] [PubMed] [Google Scholar]
- Sullivan CP, Seidl SE, Rich CB, Raymondjean M, Schreiber BM. Secretory phospholipase A2 , Group IIA is a novel serum amyloid A target gene. J Biol Chem. 2010;285:565–575. doi: 10.1074/jbc.M109.070565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Ye RD. Serum amyloid AI: Structure, function and gene polymorphism. Gene. 2016;583:48–57. doi: 10.1016/j.gene.2016.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Zhou H, Zhu Z, Yan Q, Wang L, Liang Q, Ye RD. Ex Vivo and In Vitro effect of serum amyloid A in the induction of macrophage M2 markers and efferocytosis of apoptotic neutrophils. J Immunol. 2015;194:4891–4900. doi: 10.4049/jimmunol.1402164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CCF. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J Mol Bioi. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- Sung H-J, Ahn J-M, Yoon Y-H, Rhim ZT-Y, Park C-S, Park J-Y, Lee S-Y, Kim E-W, Cho J-Y. Identification and validation of SAA as a potential lung cancer biomarker and its involvement in metastatic pathogenesis of lung cancer. J Proteome Res. 2011;10:1383–1395. doi: 10.1021/pr101154j. [DOI] [PubMed] [Google Scholar]
- Takase H, Tanaka M, Miyagawa S, Yamada T, Mukai T. Effect of amino acid variations in the central region of human serum amyloid A on the amyloidogenic properties. Biochem Biophys Res Comm. 2014;444:92–97. doi: 10.1016/j.bbrc.2014.01.029. [DOI] [PubMed] [Google Scholar]
- Takase H, Tanaka M, Yamamoto A, Watanabe S, Takahashi S, Nadanaka S, Kitagawa H, Yamada T, Mukai T. Structural requirements of glycosaminoglycans for facilitating amyloid fibril formation of human serum amyloid A. Amyloid. 2016;23(2):67–75. doi: 10.3109/13506129.2016.1168292. [DOI] [PubMed] [Google Scholar]
- Tam SP, Flexman A, Hulme J, Kisilevsky R. Promoting export of macrophage cholesterol: the physiological role of a major acute phase protein, serum amyloid A 2.1. J Lipid Res. 2002;43:1410–1420. doi: 10.1194/jlr.M100388-JLR200. [DOI] [PubMed] [Google Scholar]
- Taylor BA, Rowe L. Genes for serum amyloid A proteins map to chromosome 7 in the mouse. Mol Gen Genet. 1984;195:491–499. doi: 10.1007/BF00341452. [DOI] [PubMed] [Google Scholar]
- Thompson JC, Jayne C, Thompson J, Wilson PG, Yoder MH, Webb N, Tannock LR. A brief elevation of serum amyloid A is sufficient to increase atherosclerosis. J Lipid Res. 2015;56:286–293. doi: 10.1194/jlr.M054015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorn CF, Whitehead AS. Differential glucocorticoid enhancement of the cytokine-driven transcriptional activation of the human acute phase serum amyloid genes, SAA1 and SAA2. J ImmunoI. 2002;169:399–405. doi: 10.4049/jimmunol.169.1.399. [DOI] [PubMed] [Google Scholar]
- Thorn CF, Whitehead AS. Tissue-specific regulation of the human acute-phase serum amyloid A genes, SAA1 and SAA2, by glucocorticoids in hepatic and epithelial cells. Eur J Immunol. 2003;33:2630–2639. doi: 10.1002/eji.200323985. [DOI] [PubMed] [Google Scholar]
- Tillett WS, Francis T., Jr Serological reactions in pneumonia with a non-protein somatic fraction of pneumococcus. J Exp Med. 1930;52:561–571. doi: 10.1084/jem.52.4.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolson J, Bogumil R, Brunst E, Beck H, Elsner R, Humeny A, Kratzin H, Deeg M, Kuczyk M, Mueller GA, Mueller CA, Flad T. Serum protein profiling by SELDI mass spectrometry: detection of multiple variants of serum amyloid alpha in renal cancer patients. Lab Invest. 2004;84:845–856. doi: 10.1038/labinvest.3700097. [DOI] [PubMed] [Google Scholar]
- Tomita T, Ieguchi K, Sawamura T, Maru Y. Human serum amyloid A3 (SAA3) protein, expressed as a fusion protein with SAA2, binds the oxidized low density lipoprotein receptor. PLOS one. 2015; 10.1371/journal.pone.0118835. [DOI] [PMC free article] [PubMed]
- Tucker PC, Sack GH., Jr Expression of serum amyloid A (SAA) genes in mouse brain - unprecedented response to inflammatory mediators. FASEB J. 2001;15:2241–2246. doi: 10.1096/fj.01-0133com. [DOI] [PubMed] [Google Scholar]
- Tükel C, Wilson RP, Nishimori JH, Pezeshki M, Chromy BA, Bӓumler AJ. Responses to amyloids of microbial and host origin are mediated through Toll-like receptor 2. Cell Host and Microbe. 2009;6:45–53. doi: 10.1016/j.chom.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnell W, Sarra R, Glover ID, Baum JO, Caspi O, Baltz ML, Pepys MB. Secondary structure prediction of human SAA1. Presumptive identification of calcium and lipid binding sites. Mol Biol Med. 1986;3:387–407. [PubMed] [Google Scholar]
- Uhlar CM, Burgess CJ, Sharp PM, Whitehead AS. Evolution of the serum amyloid A (SAA) protein superfamily. Genomics. 1994;19:228–235. doi: 10.1006/geno.1994.1052. [DOI] [PubMed] [Google Scholar]
- Uhlar CM, Whitehead AS. Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem. 1999;265:S01–523. doi: 10.1046/j.1432-1327.1999.00657.x. [DOI] [PubMed] [Google Scholar]
- Uhlar CM, Whitehead AS. The kinetics and magnitude of the synergistic activation of the serum amyloid A promoter by IL-1β and IL-6 is determined by the order of cytokine addition. Scand J ImmunoI. 1999;49:399–404. doi: 10.1046/j.1365-3083.1999.00515.x. [DOI] [PubMed] [Google Scholar]
- Urieli-Shoval S, Cohen P, Eisenberg S, Matzner Y. Widespread expression of serum amyloid A in histologically normal human tissues: Predominant localization to the epithelium. J Histochem Cytochem. 1998;46:1377–1384. doi: 10.1177/002215549804601206. [DOI] [PubMed] [Google Scholar]
- Urieli-Shoval S, Finci-Yeheskel Z, Dishon S, Galinsky D, Linke RP, Ariel I, Levin M, Ben-Shachar I, Prus D. Expression of Serum Amyloid A in human ovarian epithelial tumors: Implication for a role in ovarian tumorigenesis. J Histochem Cytochem. 2010;58:1015–1023. doi: 10.1369/jhc.2010.956821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallon R, Freuler F, Desta-Tesdu N, Robeva A, Dawson J, Wenner P, Engelhardt P, Boes L, Schnyder J, Tschopp C, Urfer R, Baumann G. Serum amyloid A (apoSAA) expression is up-regulated in rheumatoid arthritis and induces transcription of matrix metalloproteinases. J Immunol. 2001;166:2801–2807. doi: 10.4049/jimmunol.166.4.2801. [DOI] [PubMed] [Google Scholar]
- van der Hilst JC, Yamada T, Op den Camp TJ, van der Meer JW, Drenth JP, Simon A. Increased susceptibility of serum amyloid A 1.1 to degradation by MMP-1: potential explanation for higher risk of type AA amyloidosis. Rheumatol (Oxford) 2008;47:1651–1654. doi: 10.1093/rheumatology/ken371. [DOI] [PubMed] [Google Scholar]
- van der Westhuyzen DR, Cai L, deBeer MC, de Beer FC. Serum amyloid A promotes cholesterol efflux mediated by scavenger receptor B-1. J Biol Chem. 2005;280:35890–35895. doi: 10.1074/jbc.M505685200. [DOI] [PubMed] [Google Scholar]
- van der Westhuyzen DR, Coetzee GA, deBeer FC. Serum amyloid A protein in plasma: Characteristics of acute phase HDL. In: Marrink J, van Rijswijk MH, editors. Amyloidosis. Dordrecht: Martinus Nijhoff; 1986. pp. 115–125. [Google Scholar]
- van Gerven N, Klein RD, Hultgren SJ, Remaut H. Bacterial amyloid formation:Structural insights into curli biogenesis. Trends in Microbiol. 2009;23:693–706. doi: 10.1016/j.tim.2015.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19:108–119. doi: 10.1038/s41590-017-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Lashuel HA, Colon W. From hexamer to amyloid: Marginal stability of apolipoprotein SAA2.2 leads to in vitro fibril formation at physiological temperature. Amyloid. 2005;12:139–148. doi: 10.1080/13506120500223084. [DOI] [PubMed] [Google Scholar]
- Wang L, Lashuel HA, Walz T, Colon W. Murine apolipoprotein serum amyloid A in solution forms a hexamer containing a central channel. Proc Natl Acad Sci USA. 2002;99:15947–15952. doi: 10.1073/pnas.252508399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Srinivasan S, Ye Z, Aguilera J, Lopez MM, Colon W. Serum amyloid A 2.2 refolds into a octameric oligomer that slowly converts to a more stable hexamer. Biochem Biophys Res Commun. 2011;407:725–729. doi: 10.1016/j.bbrc.2011.03.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A, Weber AT, McDonald TL, Larson MA. Staphylococcus aureus lipotechoic acid induces differential expression of bovine serum amyloid A3 (SAA3) by mammary epithelial cells: Implications for early diagnosis of mastitis. Vet Immunol lmmunopath. 2005;109:79–83. doi: 10.1016/j.vetimm.2005.07.023. [DOI] [PubMed] [Google Scholar]
- Westermark GT, Sletten K, Westermark P. Massive vascular AA-Amyloidosis: A histologically and biochemically distinctive subtype of reactive systemic amyloidosis. Scand J Immunol. 1989;30:605–613. doi: 10.1111/j.1365-3083.1989.tb02468.x. [DOI] [PubMed] [Google Scholar]
- Westermark GT, Westermark P. Prion-like aggregates: infectious agents in human disease. Trends Mol Med. 2010;16:501–507. doi: 10.1016/j.molmed.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Westermark P. Aspects on human amyloid forms and their fibril polypeptides. FEBS J. 2005;272:5942–5949. doi: 10.1111/j.1742-4658.2005.05024.x. [DOI] [PubMed] [Google Scholar]
- Wilson PG, Thompson JC, Webb NR, deBeer FC, King VL, Tannock LR. Serum Amyloid A, but not C-reactive protein, stimulates vascular proteoglycan synthesis in a pro-atherogenic manner. Am J Pathol. 2008;173:1902–1920. doi: 10.2353/ajpath.2008.080201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P, Sipe J, Dinarello CA, Colten HR. Structure of a human serum amyloid A gene and modulation of its expression in transfected L cells. J Biol Chem. 1987;262:15790–15795. [PubMed] [Google Scholar]
- Wood SL, Rogers M, Cairns DA, Paul A, Thompson D, Vasudev NS, Selby PJ, Banks RE. Association of serum amyloid A protein and peptide fragments with prognosis in renal cancer. Br J Cancer. 2010;103:101–111. doi: 10.1038/sj.bjc.6605720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wroblewski JM, Jahangiri A, Ji A, deBeer FC, van der Westhuyzen DR, Webb NR. Nascent HDL formation by hepatocytes is reduced by the concerted action of serum amyloid A and endothelial lipase. J Lipid Res. 2011;52:2255–2261. doi: 10.1194/jlr.M017681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Sato J, Okuda Y. Differential affinity of serum amyloid A1 isotypes for high-density lipoprotein. Amyloid. 2009;16:196–200. doi: 10.3109/13506120903421546. [DOI] [PubMed] [Google Scholar]
- Yan J, Fu X, Ge F, Zhang B, Yao J, Zhang H, Qian J, Tomozawa H, Naiki H, Sawashita J, Mori M, Higuchi K. Cross-seeding and cross-competition in mouse apolipoprotein A-ll amyloid fibrils and protein A amyloid fibrils. Amer J Path. 2007;171:172–180. doi: 10.2353/ajpath.2007.060576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SD, Zhu H, Zhu A, Golabek A, Du H, Roher A, Yu J, Soto C, Schmidt AM, Stern D, Kindy M. Receptor-dependent cell stress and amyloid accumulation in systemic amyloidosis. Nat Med. 2000;6:643–651. doi: 10.1038/76216. [DOI] [PubMed] [Google Scholar]
- Yang Q, Whitin JC, Ling XB, Nayak NR, Cohen HJ, Jin J, Schilling J, Yu TT-S, Madan A. Plasma biomarkers in a mouse model of preterm labor. Ped Res. 2009;66:11–16. doi: 10.1203/PDR.0b013e3181a207e3. [DOI] [PubMed] [Google Scholar]
- Ye RD, Sun L. Emerging functions of serum amyloid A in inflammation. J Leukocyte Biol. 2017;98:923–929. doi: 10.1189/jlb.3VMR0315-080R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo J-Y, Desiderio S. Innate and acquired immunity intersect in a global view of the acute-phase response. Proc Natl Acad Sci USA. 2003;100:1157–1162. doi: 10.1073/pnas.0336385100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen J, Wang S. Serum amyloid A induces a vascular smooth muscle cell phenotype switch through the p38 MAPK signaling pathway. 2017. p. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are available through cited literature