

Abstract
Among the various proteins expressed by the periodontopathogen Aggregatibacter actinomycetemcomitans, two proteins play important roles for survival in the oral cavity. The autotransporter Aae facilitates the attachment of the pathogen to oral epithelial cells, which act as a reservoir, while the biofilm-degrading glycoside hydrolase dispersin B facilitates the movement of daughter cells from the mature biofilm to a new site. The objective of this study was to utilize the potential of these two proteins to control biofilms. To this end, we generated a hybrid construct between the Aae C-terminal translocating domain and dispersin B, and mobilized it into E. coli Rosetta (DE3) pLysS cells. Immunofluorescence analysis of the modified E. coli cells confirmed the presence of dispersin B on the surface. Further, the membrane localization of the displayed dispersin B was confirmed with western blot analysis. The integrity of the E. coli cells displaying the dispersin B was confirmed through FACS analysis. The hydrolytic activity of the surface-displayed dispersin B was confirmed by using 4-methylumbelliferyl-β-D-gluocopyranoside as the substrate. The detachment ability of the dispersin B surface-displaying E. coli cells was shown using Staphylococcus epidermidis and Actinobacillus pleuropneumoniae biofilms in a microtiter assay. We concluded that the Aae β-domain is sufficient to translocate foreign enzymes in the native folded form and that the method of Aae mediated translocation of surface displayed enzymes might be useful for control of biofilms.
Keywords: Autotransporter, surface display, biofilm, dispersin B, oral pathogen
INTRODUCTION
Autotransporters are a superfamily of virulence factors that are large in size and are secreted by Gram-negative bacteria to carry out diverse functions. They consist of a) an N terminal signal sequence that is responsible for crossing the inner membrane and is eventually cleaved off; b) a mature functional protein designated as the passenger domain; and c) a carboxy terminal β domain, which forms a β barrel that is anchored into the outer membrane (Jose et al., 1995). The C-terminal β barrel domain, necessary for translocating the passenger domain into the extracellular space, folds in the periplasm and inserts into the outer membrane as a barrel (~10 Å pore) via an energetically independent pathway (Henderson et al., 2000). Although some autotransporters require trimerization others are monomeric and possess the ability to translocate passenger domains (Cotter et al., 2005). The simplistic mechanism of autotransport provides a vehicle for exchanging a protein of interest to be secreted in place of the original passenger domain. There have been such reports utilizing the β domain’s ability to display proteins on the surface of cells for either delineating the mechanism of translocation (Skillman et al., 2005) or to assess the proper folding of the translocated proteins by testing their enzyme activity (Freeman et al., 1996; Lattemann et al., 2000; Van Gerven et al., 2009). For example, the surface display of various proteins using IgA protease in Neisseria and OMP-A and AIDA-I in Escherichia coli have already been well characterized as the anchoring protein for surface display (Freeman et al., 1996; Lattemann et al., 2000; Van Gerven et al., 2009).
The display of heterologous proteins on the surface of bacteria offers many practical applications, such as epitope presentation for vaccines, screening of peptide libraries, and whole cell biocatalysis (Klauser et al., 1990; Rutherford & Mourez, 2006). Although initial research was done using bacteriophages to display select foreign proteins, bacteria are now the preferred organisms (Jose & Meyer, 2007). This is due to bacteria’s fast replication time, which allows for more recombinant proteins to be made at high levels with little to no toxic effects (Rutherford & Mourez, 2006), as well as their size, which makes them easy to view under a microscope. In spite of these studies using surface display of enzymes, none have been performed to display an enzyme on the bacterial surface for the purpose of degradation and detachment of biofilms.
Biofilms are responsible for a myriad of medical problems including nosocomial infections of catheters and heart valves, as well as diseases like cystic fibrosis pneumonia (Costerton et al., 1995; Costerton et al., 1999). Biofilms are complex communities of bacteria that are encased in a sticky matrix that allows them to adhere to both biotic and abiotic surfaces. The self-synthesized, three-dimensional matrix (extracellular polysaccharide, EPS) participates in holding the bacterial cells together in a mass and firmly attaches the bacterial mass to the underlying surface (Costerton et al., 1999). The EPS matrix also protects the attached bacteria from antibiotics and the host antibodies, thus making them extremely difficult to eradicate. Almost 80% of pathogenic bacteria exist as biofilm communities and utilize a number of matrix substances for attachment and colonization. While the matrix is largely composed of EPS, it is often more complex and may be interacting with other macromolecules such as proteins and extracellular DNA (Izano et al., 2008; Izano et al., 2007). The EPS also facilitates in the aggregation and coaggregation of pathogenic as well as non-pathogenic bacteria in to multispecies biofilms.
Among the oral pathogens that are causative agents for periodontal diseases, Aggregatibacter actinomycetemcomitans has been shown to be involved in localized aggressive periodontitis (Slots et al., 1980). This Gram-negative bacterium possesses a secreted, non-cell associated matrix-degrading enzyme called dispersin B (DspB), which is capable of detaching biofilms (Kaplan et al., 2003a; Manuel et al., 2007). DspB belongs to the family 20 glycosyl hydrolase (β-hexosaminidase; EC 3.2.1.52), whose substrate is a linear homopolymer of N-acetyl-D-glucosamine residues in β(1,6)-linkages. The crystal structure of DspB in complex with glycerol and an acetate at the active site suggested that the active site architecture of DspB is similar to the family 20 β-hexosaminidases (Ramasubbu et al., 2005). Dispersin B has been shown to inhibit biofilm formation and/or promote biofilm detachment in different Gram-negative and Gram-positive bacteria: A. actinomycetemcomitans (Kaplan et al., 2003a); Actinobacillus pleuropneumoniae (Kaplan et al., 2004); E. coli, Pseudomonas fluorescens and Yersinia pestis (Itoh et al., 2005); Bordetella bronchiseptica (Parise et al., 2007); and Staphylococcus epidermidis (Kaplan et al., 2004). Recently, we demonstrated that transgenic tobacco plants expressing DspB are resistant to Pectobacterium carotovorum infection (Ragunath et al., 2011).
Since enzymes that degrade exopolysaccharide, such as DspB, are capable of causing the detachment of cells from biofilms (Johansen et al., 1997), the matrix-degrading enzymes are attractive anti-biofilm agents. Utilizing such agents against biofilm bacteria renders the bacteria more susceptible to the effect of antibiotics. Also, the targeted bacteria are not under selective pressure to develop resistance to enzyme mediated dispersal because these enzymes do not kill bacteria or inhibit their growth (Kaplan et al., 2003a).
A. actinomycetemcomitans possess a number of autotransporters that can be utilized for surface display as described above. One such protein is Aae (Rose et al., 2003), which is a 90-kDa surface-anchored protein. Aae is a member of the autotransporter family of bacterial proteins that is homologous to an epithelial cell adhesin (Hap) produced by Haemophilus influenzae (Hendrixson et al., 1997; Hendrixson & St Geme, 1998). This monomeric autotransporter, which has a truncated passenger domain, is primarily involved in the binding of A. actinomycetemcomitans to human buccal epithelial cells (Fine et al., 2005; Rose et al., 2003). In this study, we have used the translocating potential of the β-domain of Aae and the biofilm degrading potential of DspB to form a hybrid protein that is displayed on the surface of E. coli. While DspB is a secreted protein, displaying it on the surface of a bacterium is unlikely to enhance its catalytic activity; however, it is expected to retain its catalytic activity when it is exported in the folded conformation. It is noteworthy that autotransporter systems have been used in the surface display of enzymes such as lipase (Becker et al., 2005) or sorbitol dehydrogenase (Jose & von Schwichow, 2004), while retaining their corresponding catalytic activity. Our hypothesis is that Aae β-domain will translocate DspB in a correctly folded form. Our results show that the protein encoded by the hybrid construct was functional and able to detach S. epidermidis and A. pleuropneumoniae biofilms. Utilization of surface-displayed biofilm degrading enzymes provides a novel method to control biofilm infections independently or in combination with other applications (Jose, 2006).
METHODS
Bioinformatics
The C-terminus of the autotransporters is the region of highest homology among its close homologs (Rose et al., 2003) and presumably adopts a β-barrel used for translocation of the passenger domain. The goal for the hybrid generation was to identify the right size of the C-terminal domain such that only a few N-terminal amino acid(s) will exude out of the outer membrane. To select the appropriate β-domain length of Aae for our study we used in silico analysis. First, the amino acid sequence of the Aae protein (deduced from the genome of A. actinomycetemcomitans strain IDH781) was analyzed for protein homology detection and structure prediction using the HHpred server (Soding et al., 2005). Homologs of Aae were identified to generate a multiple alignment through multiple iterations of PSI-BLAST searches and the alignment was annotated with predicted secondary structure and confidence values from PSIPRED (Jones, 1999). Based on the predicted secondary structure, a profile hidden Markov model (HMM) was constructed, which was then compared with HMMs from the Protein Data Bank (PDB) using the HHsearch software (Soding, 2005). Using position-specific gap penalties, secondary structure similarity was scored and used to construct local alignments with the Aae sequence and the matching PDB database sequences. The best-matched proteins from the PDB database were selected as templates for comparative modeling with the MODELLER software (Sali & Blundell, 1993). The resulting structure models were viewed with PyMOL version 1.0 (DeLano, 2002). Using this analysis, we predicted that amino acids 623-912 would be of sufficient length to display the passenger domain completely outside the surface.
Bacterial strains, media and growth conditions
E. coli strains DH5α and Rosetta (DE3) pLysS were used for cloning and protein expression, respectively. The E. coli strains were grown in LB broth supplemented with kanamycin (DH5α; 30 μg/mL), or kanamycin/chloramphenicol (pLysS; 30 μg/mL each), and induced with 0.1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) when appropriate. S. epidermidis (NJ 9709; (Kaplan et al., 2004)) strains were grown in Trypticase soy broth (TSB; Becton Dickinson) supplemented with 6 g of yeast extract and 8 g of glucose per liter (Kaplan et al., 2003a). A. pleuropneumoniae (IA5) was grown in TSB with 6 g of yeast extract supplemented with 10% sodium bicarbonate, 20% dextrose and NAD.
Recombinant DNA techniques
First, the N-terminal portion of Aae that encodes the signal sequence (corresponding to bp 1 to 159 in GenBank accession no. AY487820) was amplified by PCR by using genomic DNA isolated from A. actinomycetemcomitans strain IDH781 using the primers P1 and P2 (Table 1). The PCR product was digested with NdeI and KpnI and ligated into the NdeI/KpnI sites of the T7 expression vector pET29b (Novagen), resulting in plasmid pKD10. Secondly, to generate a plasmid containing the T7-tag (MASMTGGQQMG) and the Aae β-domain (bp 1869 and 2736), the IDH781 genomic DNA was amplified using the primers P5 (forward with KpnI site) and P4 (reverse with XhoI site). The forward primer, P5, has the DNA sequence corresponding to the T7 tag built into it. The resulting product was double digested with KpnI and XhoI, and ligated into the KpnI/XhoI sites of pKD10. The resulting plasmid, pKD11, has the following sequence: Aae signal sequence, T7 tag, and the β-domain of Aae. This construct was used to verify whether or not the β-domain (as determined in the Bioinformatics section above) is of sufficient length to display a foreign peptide (T7 tag) outside the cell. Nucleotide sequencing was used to confirm the insert sequence.
Table 1.
Aae hybrid-specific PCR Primers used in the study.
| Name | Sequence (5′-3′) | Orientation | Restriction site* |
|---|---|---|---|
| P1 | GGAATTCCATATGTGTTCTTTTTCAAAA AAGTACTGC |
Forward | Nde1 |
| P2 | CGGGGTACCCGCAAATGCCGGTTGATTAAA | Reverse | Kpn1 |
| P3 | CGCGGATCCAAATTAGGCAGAACAAGTTTC | Forward | BamH1 |
| P4 | CCGCTCGAGTTACCAGTAGTAATTCAGTTTTAC | Reverse | Xho1 |
| P5 | GCGCGCGGTACCATGGCGAGCATGACCGGCGGC CAGCAGATGGGCATATATAACCCAAATGAAATT |
Forward | Kpn1 |
| P6 | GCGCGCGGTACCAATTGTTGCGTAAAAGGCAATTCC | Forward | Kpn1 |
| P7 | CGCGGATCCCTCATCCCCATTCGTCTTATG | Reverse | BamH1 |
Corresponds to the underlined sequence in the primers
To generate a plasmid containing the sequence: Aae signal sequence, DspB, and the β-domain of Aae, the following protocol was used. First, the gene encoding DspB was amplified using primer pairs, P6 and P7, with KpnI and BamHI sites using the construct pRC3 (Kaplan et al., 2003b). The PCR product was digested with KpnI and BamHI, and ligated into the KpnI/BamHI sites of pKD10 resulting in the plasmid pKD2. Secondly, the Aae β-domain (corresponding to bp 1869 and 2736) was amplified by PCR using primer pairs, P3 (forward) and P4 (reverse), introducing BamHI and XhoI sites, respectively by using the genomic DNA isolated from A. actinomycetemcomitans strain IDH781. The PCR product was double digested with BamHI and XhoI, and ligated into the plasmid BamHI/XhoI sites of pKD2. This resulting plasmid (pKD3) was sequenced to confirm the insert sequences corresponding to the signal peptide, DspB as well as the β-domain of Aae.
Immunofluorescence microscopy
Recombinant DspB (Kaplan et al., 2003b) was used in the immunization of specific-pathogen-free female New Zealand White rabbits (Pocono Rabbit Farm and Laboratory, Canadensis, PA.) to obtain a rabbit anti-DspB polyclonal antibody. Rosetta (DE3) pLysS containing pKD3 and pKD11 were grown overnight in LB broth containing kanamycin and chloramphenicol at 37°C. The overnight cultures were transferred to fresh medium (1:100) containing kanamycin and the flasks were incubated at 37°C with agitation (200 rpm) until the optical density of the culture at 600 nm reached 0.6 (ca. 3 h). At this time, IPTG was added to a final concentration of 0.1 mM, and the flask was incubated for an additional 2 h with agitation. Cells were spun down for 5 min at 4000 × g and washed with PBS buffer (3x). Cells (1×107) were fixed in 0.3% glutaraldehyde and incubated overnight at 4°C (Freeman et al., 1996), centrifuged at 4°C for 5 min at 4000 × g and washed with PBS (3x). The cells were spread on poly L-lysine coated cover slips and dried at room temperature. Cover slips were incubated with 5% normal goat serum (NGS) followed by primary antibody (rabbit anti-T7 polyclonal antibody raised against the T7 peptide (Convance, CA)) for pKD11 incorporated Rosetta (DE3) pLysS (1:200 dilution) or rabbit anti DspB polyclonal antibody for pKD3 and control (1:500) in PBS with 5% NGS and incubated for 1h. After washing (3x for 5 min in PBS), secondary antibody (1:100 dilution, Alexa Fluor 488-goat anti rabbit IgG (Invitrogen)) in PBS with 5% NGS was added to the cover slips and incubated for 1h in the dark. Cover slips were washed 3x for 5 min in PBS, dried at room temperature, mounted on microscope slides with VECTASHIELD H-1000 (Vector Labs, Burlingame, CA) and examined by fluorescent microscopy using an Olympus BX50WI microscope using anti DspB polyclonal antibody.
Flow-cytometric analysis and FACS
Cultures of E. coli strains (Rosetta (DE3) pLysS containing pKD3 and Rosetta (DE3) pLysS containing pET29b) were grown overnight at 37°C and subcultured 1:50 at 37°C (100 mL) until the optical density at 600 nm (OD600) reached 0.6. The cells were further grown for an additional 1 hr after induction with 0.1 mM IPTG. Cells were pelleted by centrifugation and resuspended in 1 μL of PBS. To 10 μL of cells, 1 μl of the anti-rabbit DspB antibody was added and the cells were incubated for 5 min at room temperature and pelleted. After washing the pellet with 500 μl of PBS, cells were centrifuged and resuspended in 10 μL of PBS containing secondary antibody (1:100 dilution, Alexa Fluor 488-goat anti rabbit IgG (Invitrogen)). After 5 min of incubation at room temperature and addition of 500 μL of PBS, cells were pelleted and resuspended in 100 μL of PBS for flow cytometry analysis (NJMS Flow Cytometry And Immunology Core Laboratory, RBHS). A total of 300,000 events were collected on a BD FACSCalibur. Parameters were set as follows: forward scatter at E02 with a gain of 5.00, side scatter 906 with a gain of 1.00 (both in linear mode) and FL1 (fluorescein isothiocyanate) at a voltage of 777 (LOG mode).
Surface localization of DspB
The Rosetta (DE3) pLysS E. coli cells expressing DspB (pKD3) or pET29b (control cells) were fractionated into cytoplasmic and membrane fractions by sonication (Georgiou et al., 1996) and analyzed using SDS-PAGE (10%) by Coomassie staining and immunoblot. Western blotting (Burnette, 1981) was performed by electrotransfer of proteins from SDS-PAGE gels to Immobilon-P membranes (Millipore). After blocking with 5% (w/v) non-fat milk in Tris buffered saline with Tween 20, and washing, the blots were incubated with polyclonal rabbit anti-DspB, washed with PBS, and then incubated with goat anti-rabbit IgG conjugated with horseradish peroxidase (Promega). Finally, the blots were developed using the Pierce Supersignal (ThermoScientific).
Enzyme assays
The substrate 4-methylumbelliferyl-β-D-glucosamine (Sigma) was used to determine the enzyme activity (Manuel et al., 2007) of the displayed DspB. The DspB expressing E. coli cells (pKD3) were washed (3x), pelleted, weighed, and resuspended in 50 mM sodium phosphate buffer (pH 5.8). In a typical experiment, 10 μL of the cells were used (corresponding to 107 cfu). As a negative control, T7-expressing cells (pKD11) were used, while purified recombinant DspB was used as a positive control. Typically, enzyme reactions were carried out in 100 μL suspended mixtures using a substrate concentration (1 to 5 mM) in 50 mM sodium phosphate buffer (pH 5.8). All assays were carried out in 96 well microtiter plates placed in a 37°C incubator. The reactions were terminated at various times by adding 10 μL of 10N NaOH. Additionally, wells having suspended E. coli cells without addition of NaOH or without addition of the substrate were also monitored. The increase in fluorescence resulting from the release of 4-methylumbelliferone (excitation at 360 nm and emission at 460 nm) in each well was measured using a fluorescence/luminescence/visible light microplate reader (BIO-TEK Instruments). All assays were performed in triplicate on at least three separate occasions, which exhibited similar results with minimal variations among them.
Biofilm detachment assay
The biofilm of S. epidermidis strain NJ 9709 or A. pleuropneumoniae strain IA5 was prepared as previously described in a 96 well plate (Kaplan et al., 2003b; Kaplan et al., 2004; Manuel et al., 2007). The A. pleuropneumoniae biofilm was grown in a 96-well plate for 20 hours at 37 °C 10% CO2 incubator. Rosetta (DE3) pLysS cells containing pKD3 were grown and treated with IPTG as described above for immunofluorescence microscopy. Cells were spun down for 5 minutes at 4000 × g, washed 3x with PBS and resuspended in 500 μL PBS. Wells with S. epidermidis biofilm or A. pleuropneumoniae biofilm were treated with varying amounts of Rosetta (DE3) pLysS cells containing pKD3 (105 to 108 in 200 μL). Mock treated (PBS) wells (negative control), DspB treated wells (positive control) and bacteria treated wells were incubated for 1h at 37°C, washed and stained with crystal violet for 1 minute. After washing and drying, 33% acetic acid was added to each well and the plates were scanned using a microplate reader (Bio-Rad) at 590 nm. Optical intensities of the residual biofilm representing the biofilm detachment were recorded (Kaplan et al., 2003b; Kaplan et al., 2004; Manuel et al., 2007). This experiment was performed in triplicate on at least three separate occasions with consistent results.
RESULTS
Identification and modeling of Aae β-domain
Since the C-terminal domain of autotransporters are vital to the export of the passenger domain, we sought to determine the correct size of this domain so that only a few N-terminal amino acids will exude out of the pore. If the estimated length of the C-terminus is too short, the foreign protein/peptide will be prevented from being brought outside the barrel completely. Our intent was to keep the hybrid size as small as possible without affecting the transport function. To do this, we used the server HHpred (Soding et al., 2005), which detected and aligned homologous sequences in the Protein Data Bank (PDB). The C-terminal domain of Aae had best matches with a probability of > 90% (hits) to the E. coli EspP (Barnard et al., 2007) and to the Neisseria meningitidis NalP (Oomen et al., 2004) autotransporters. These proteins form an outer membrane pore composed of antiparallel β-strands with the N-terminal part of the chain threaded through the barrel. The HHpred server modeled Aae based on these hits and produced a barrel structure for Aae (data not shown). From this analysis, we selected residues 623 through 912 (290 residues) to be the appropriate length/size for our purpose to generate a hybrid protein with DspB as the passenger domain (Fig. 1).
Figure 1.

Domain organization of the autotransporter Aae from A. actinomycetemcomitans. Unlike other autotransporters, Aae is lacking a protease like domain at the N-terminus of the extruded passenger domain. The numbers refer to the aa position of the start and end of the signal sequence and the β-domain, respectively. In the middle, the T7-tag containing hybrid with the Aae signal sequence (SP) and associated restriction enzymes sites are shown. When fused together, the hybrid protein will lack the passenger domain of Aae completely and in its place will have the T7 tag. At the bottom, the DspB/Aae hybrid with associated restriction sites are shown. This hybrid will lack the Aae passenger domain but will have DspB as the new passenger domain. In the truncated mutants, the signal sequence and the passenger domains are linked and the dashed line is used to keep the β-domains aligned in the figure.
Construction of recombinant fusion protein encoding genes
In order to determine whether or not the 290 residues selected for Aae β domain was large enough to be able to extrude the full passenger protein, a T7 tag as the passenger domain was first used (Fig. 1). Since the Aae has three domains, we retained the Aae signal sequence and the selected C-terminal domain in the construct, but used the T7 tag instead of the passenger domain. This was cloned into a pET29b plasmid (pKD11). A second construct (pKD3) was created after confirming the exposure of the T7 tag outside membrane (see below) wherein DspB was used as the passenger domain. The use of Aae signal sequence is apparently important since constructs using the signal sequence of DspB instead of Aae were not successful in displaying either the T7 or DspB (data not shown). The signal sequence of DspB is smaller (20 aa residues) compared to that of Aae (53 aa residues).
Visualization with immunofluorescence microscopy
The display of the T7 tag and DspB with the 290 C-terminal residues of the Aae β-domain was confirmed by immunofluorescence microscopy (Fig. 2). Control cells of Rosetta (DE3) pLysS containing pET29b displayed no fluorescence (Fig. 2a) with or without IPTG (data not shown). Interestingly, the Rosetta (DE3) pLysS mobilized with either pKD11 (T7 tag; Fig. 2b) or pKD3 (DspB; Fig. 2c) were positively stained irrespective of whether or not IPTG was used for induction. The results for the T7-tag hybrid pKD11 cells suggest that the length of the Aae β-domain identified by the bioinformatics analysis is sufficient to bring out even the 11-mer T7 peptide outside of the cell surface.
Figure 2.

Immunofluorescence of Rosetta (DE3) pLysS cells. a) Control Rosetta (DE3) pLysS cells transformed with pET29b. b) Rosetta (DE3) pLysS cells mobilized with pKD11 encoding the T7 tag. c) Rosetta cells with pKD3 encoding dispersin B. Shown on the left are the phase contrast images and on the right, immunofluorescence images. The bar indicates 5 μm. Green fluorescence was due to Alexa Fluor 488 labeled secondary antibody.
The cellular localization of the fusion protein was also analyzed by fluorescence-activated cell sorting (FACS), which clearly demonstrated that the DspB could be detected on the surface of pKD3 cells (Fig. 3). The peak fluorescence channel of the pKD3 strain that corresponds to a measure of the number of surface-exposed DspB molecules per cell turned out to be approximately 2208.
Figure 3.

FACS analysis of cell surface display of DspB as passenger domain. A) FACS histogram of Rosetta cells with pET29b and B) with pKD3. Induced cells were successively incubated with ant-rabbit DspB antibody and Alexa Fluor 488-goat anti rabbit IgG.
Surface localization of DspB
In order to assess whether or not the expressed hybrid protein containing DspB is localized in the membrane fractions, the E. coli cells (Rosetta (DE3) pLysS containing pKD3 and Rosetta (DE3) pLysS containing pET29b with or without IPTG induction) were fractionated into cytoplasmic and membrane fractions. These extracts were probed with rabbit anti DspB polyclonal antibody (Fig. 4). The cells with pKD3 showed the presence of DspB in both the cytoplasmic as well as membrane fractions; however, the membrane fractions showed a much higher presence of DspB. The E. coli cells with pET29b did not show any bands. These results clearly suggest that pKD3 cells are capable of exporting DspB to the membrane while fused to the Aae β-domain (~70 kD).
Figure 4.
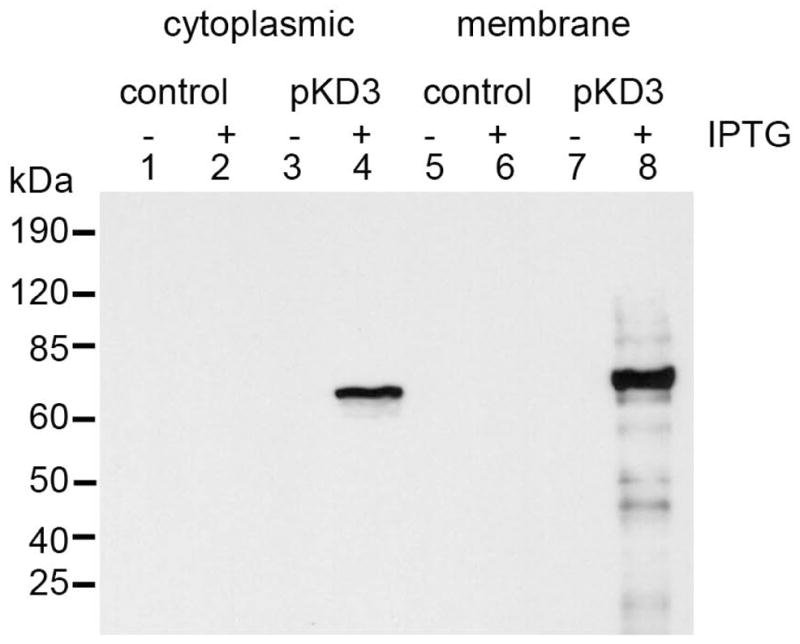
Western blot analysis of DspB surface localization. The cytoplasmic as well as membrane fractions were boiled in sample buffer. The samples were run on 10% SDS-PAGE, transferred to a membrane and probed using polyclonal anti-DspB antibody. Lanes 1, 2, 5 and 6 were from control Rosetta (DE3) pLysS cells. Lanes 3, 4, 7 and 8, are from the pKD3 cells. Odd numbered lanes were not induced with IPTG (0.1 mM) whereas even numbered lanes were induced with IPTG.
Enzyme assays
To assess whether or not the displayed DspB was enzymatically active, we tested the catalytic activity of DspB using a fluorescent substrate (Fig. 5). Only pKD3 cells, which were designed to express DspB on the surface, showed enzyme activity whereas the pKD11 control cells did not. These results suggest that the DspB was enzymatically active on the surface. The number of cells in each of the wells was approximately 1.0 × 107 cells. Interestingly, pKD3 cells that were uninduced with IPTG did not show any activity (data not shown).
Figure 5.

Hydrolytic activity of surface displayed DspB. The hydrolytic activity of DspB was measured using 4-methylumbeliferyl-β-D-glucopyranoside as substrate in 50 mM phosphate buffer using 10 μL of cells (107 cfu) from a wet weight of 0.2 g/mL at pH 5.8. The control cells (pKD11) did not have any activity. The activity of purified recombinant DspB (8 μg per well) is shown for comparison. The DspB reactions were carried out for 15 min.
Biofilm detachment assay
The activity of the surface-displayed DspB/Aae hybrid was then tested for its ability to detach a S. epidermidis biofilm using a 96-well microtiter plate assay (Kaplan et al., 2003b; Kaplan et al., 2004; Manuel et al., 2007). As shown in Fig. 6a, only E. coli cells transformed with pKD3 construct exhibited detachment whereas the control cells did not. The absorbance of the residual biofilm was measured after the addition of acetic acid (Fig. 6b). The amount of biofilm detachment by the DspB displaying E. coli cells when compared with purified DspB (1 mg/mL to 1 pg/mL) suggests that a minimum of 1.0 × 107 cells/200 μL with plasmid pKD3 are needed for any observable biofilm detachment. The number of pKD3 E. coli cells to reduce the biofilm by 50% was estimated to be about 5×107 cells/200 μL. The DspB displaying pKD3 cells were also able to detach biofilms of A. pleuropneumoniae (Fig. 7). We have shown earlier that DspB detaches A. pleuropneumoniae biofilms (Kaplan et al., 2004). A minimum of 1.0 × 107 pKD3 cells was also required for observable biofilm detachment. As with S. epidermidis biofilms, the number of pKD3 E. coli cells to detach 50% of A. pleuropneumoniae biofilms was estimated to be 108 cells (Fig. 7b). Control E. coli cells did not detach biofilms.
Figure 6.

a) S. epidermidis biofilm detachment by Rosetta (DE3) pLysS cells displaying DspB on their surface. The wells containing E. coli cells had a range of CFUs from 1.0×107 to 1.0×109. The bottom wells contained purified DspB. The wells were then rinsed with water and stained with crystal violet. b) OD of the rows of wells in a) corresponding to pKD3+IPTG (filled squares), pKD3-IPTG (open squares), Rosetta+IPTG (filled circles), and Rosetta-IPTG (open circles). The number of cells required for 50% detachment was estimated at 5×107 cells.
Figure 7.

a) A. pleuropneumoniae biofilm detachment by Rosetta (DE3) pLysS cells (pKD3) displaying DspB on their surface shown in triplicate wells. Note that the biofilm is completely detached with 5×108 cells after 1 hr at 37°C. Control E. coli cells (pKD11) did not detach biofilm while 5 μg of DspB completely detached the biofilm. b) Dose response curve using the absorbance of residual biofilm after treatment with acetic acid. The number of cells required for 50% biofilm detachment is estimated to be 108 cells.
DISCUSSION
The results from our study support our hypothesis that the β-domain of the autotransporter Aae is able to display foreign proteins that remain functional on the surface of E. coli. This is the first time that the autotransporter from the periodontal pathogen A. actinomycetemcomitans has been utilized as a translocator unit for surface display. The size of the β-domain selected through in silico analysis is sufficient for this purpose. Several notable points emerge from our study. The Aae β-domain we have identified consists of only 291 amino acids and unlike other trimeric autotransporters, is a single chain and thus well suited for such purpose. The display of DspB on the surface was confirmed using immunofluorescence in which we noted that DspB was detected regardless of whether or not IPTG was present (Fig. 2; IPTG absent data not shown). To ensure that the display of DspB is not an artifact, and that the immunofluorescence did not arise due to fixing of the cells with glutaraldehyde, we provide data on the results of two subsequent experiments. Firstly, western blot analysis showed that neither the cytoplasmic nor the membrane fractions of uninduced cells had detectable DspB. As shown in Fig. 4, only IPTG induced cells were positive for the presence of DspB. Thus, the expressed protein is properly shuttled to the membrane where it could be processed for display. Secondly, and more importantly, the FACS analysis (Fig. 3) strongly suggests that the E. coli cells decorating the enzyme on their surface are intact.
In the biofilm detachment assay, only those bacteria that were induced with IPTG were capable of detaching the S. epidermidis biofilm. The surface mobilized enzyme was able to hydrolyze a substrate (Fig. 5) and detach S. epidermidis (Fig. 6) or A. pleuropneumoniae biofilms (Fig. 7) suggesting that DspB is catalytically active and is in the correctly folded conformation on the surface. The detachment ability of cells that were not induced was minimal and comparable to the control Rosetta (DE3) pLysS cells. Thus, a possible explanation for the display of immunofluorescence in the uninduced cells could be that DspB is mobilized to the cell surface through leaky expression from T7 promoter.
It is known that the success of mobilizing a functional protein to the outer membrane surface depends on the protein’s ability to be folded correctly in the periplasm. Dispersin B apparently does so partly due to the absence of disulfide bonds, which increases the efficiency of transport since their presence could pose complications during the folding stages in the periplasm. For those proteins that do possess disulfide bonds, techniques such as utilizing site directed mutagenesis to remove the cysteines, using an oxidoreducatase negative mutant (dsbA), or adding 2-mercaptoethanol have been proposed as a remedy (Rutherford et al., 2006; Rutherford & Mourez, 2006). While we have demonstrated that the Aae β-domain is capable of exporting a protein with no disulfide bonds, studies to demonstrate the capability of the Aae β-domain to successfully bring out proteins with disulfide bonds to the surface need to be explored. Nevertheless, the use of the Aae C-terminal β-domain for surface display offers many new possibilities in clinical and environmental applications in biofilm degradation, vaccinology, or biocatalysis. The use of pKD3 containing cells to degrade different bacterial biofilms shows great promise in the use of bacterial cells displaying antibiofilm enzyme(s) on their surface for biofilm degradation of pathogenic bacteria. In summary, we have shown that foreign peptides and proteins could be successfully displayed as an Aae hybrid and that use of EPS degrading enzymes could detach biofilms.
Acknowledgments
This work was supported by the USPHS Grant DE16291 (NR) from the National Institute of Dental and Craniofacial Research.
Abbreviations
- DspB
dispersin B
- NGS
normal goat serum
- IPTG
Isopropyl β-D-1-thiogalactopyranoside
- EPS
extracellular polysaccharide
References
- Barnard TJ, Dautin N, Lukacik P, Bernstein HD, Buchanan SK. Autotransporter structure reveals intra-barrel cleavage followed by conformational changes. Nat Struct Mol Biol. 2007;14:1214–1220. doi: 10.1038/nsmb1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker S, Theile S, Heppeler N, et al. A generic system for the Escherichia coli cell-surface display of lipolytic enzymes. FEBS Lett. 2005;579:1177–1182. doi: 10.1016/j.febslet.2004.12.087. [DOI] [PubMed] [Google Scholar]
- Burnette WN. “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate--polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal Biochem. 1981;112:195–203. doi: 10.1016/0003-2697(81)90281-5. [DOI] [PubMed] [Google Scholar]
- Costerton JW, Lewandowski Z, Caldwell DE, Korber DR, Lappin-Scott HM. Microbial biofilms. Annu Rev Microbiol. 1995;49:711–745. doi: 10.1146/annurev.mi.49.100195.003431. [DOI] [PubMed] [Google Scholar]
- Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- Cotter SE, Surana NK, St Geme JW., 3rd Trimeric autotransporters: a distinct subfamily of autotransporter proteins. Trends Microbiol. 2005;13:199–205. doi: 10.1016/j.tim.2005.03.004. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOl User’s Manual. San Carlos, CA: Delano Scientific; 2002. [Google Scholar]
- Fine DH, Velliyagounder K, Furgang D, Kaplan JB. The Actinobacillus actinomycetemcomitans autotransporter adhesin Aae exhibits specificity for buccal epithelial cells from humans and old world primates. Infect Immun. 2005;73:1947–1953. doi: 10.1128/IAI.73.4.1947-1953.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman A, Abramov S, Georgiou G. Fixation and stabilization of Escherichia coli cells displaying genetically engineered cell surface proteins. Biotechnol Bioeng. 1996;52:625–630. doi: 10.1002/(SICI)1097-0290(19961205)52:5<625::AID-BIT10>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Georgiou G, Stephens DL, Stathopoulos C, Poetschke HL, Mendenhall J, Earhart CF. Display of beta-lactamase on the Escherichia coli surface: outer membrane phenotypes conferred by Lpp’-OmpA’-beta-lactamase fusions. Protein Eng. 1996;9:239–247. doi: 10.1093/protein/9.2.239. [DOI] [PubMed] [Google Scholar]
- Henderson IR, Cappello R, Nataro JP. Autotransporter proteins, evolution and redefining protein secretion: response. Trends Microbiol. 2000;8:534–535. doi: 10.1016/s0966-842x(00)01884-9. [DOI] [PubMed] [Google Scholar]
- Hendrixson DR, de la Morena ML, Stathopoulos C, St Geme JW., 3rd Structural determinants of processing and secretion of the Haemophilus influenzae hap protein. Mol Microbiol. 1997;26:505–518. doi: 10.1046/j.1365-2958.1997.5921965.x. [DOI] [PubMed] [Google Scholar]
- Hendrixson DR, St Geme JW., 3rd The Haemophilus influenzae Hap serine protease promotes adherence and microcolony formation, potentiated by a soluble host protein. Mol Cell. 1998;2:841–850. doi: 10.1016/s1097-2765(00)80298-1. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Wang X, Hinnebusch BJ, Preston JF, 3rd, Romeo T. Depolymerization of beta-1,6-N-acetyl-D-glucosamine disrupts the integrity of diverse bacterial biofilms. J Bacteriol. 2005;187:382–387. doi: 10.1128/JB.187.1.382-387.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izano EA, Amarante MA, Kher WB, Kaplan JB. Differential roles of poly-N-acetylglucosamine surface polysaccharide and extracellular DNA in Staphylococcus aureus and Staphylococcus epidermidis biofilms. Appl Environ Microbiol. 2008;74:470–476. doi: 10.1128/AEM.02073-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izano EA, Wang H, Ragunath C, Ramasubbu N, Kaplan JB. Detachment and killing of Aggregatibacter actinomycetemcomitans biofilms by dispersin B and SDS. J Dent Res. 2007;86:618–622. doi: 10.1177/154405910708600707. [DOI] [PubMed] [Google Scholar]
- Johansen C, Falholt P, Gram L. Enzymatic removal and disinfection of bacterial biofilms. Appl Environ Microbiol. 1997;63:3724–8. doi: 10.1128/aem.63.9.3724-3728.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- Jose J. Autodisplay: efficient bacterial surface display of recombinant proteins. Appl Microbiol Biotechnol. 2006;69:607–614. doi: 10.1007/s00253-005-0227-z. [DOI] [PubMed] [Google Scholar]
- Jose J, Jahnig F, Meyer TF. Common structural features of IgA1 protease-like outer membrane protein autotransporters. Mol Microbiol. 1995;18:378–380. doi: 10.1111/j.1365-2958.1995.mmi_18020378.x. [DOI] [PubMed] [Google Scholar]
- Jose J, Meyer TF. The autodisplay story, from discovery to biotechnical and biomedical applications. Microbiol Mol Biol Rev. 2007;71:600–619. doi: 10.1128/MMBR.00011-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose J, von Schwichow S. Autodisplay of active sorbitol dehydrogenase (SDH) yields a whole cell biocatalyst for the synthesis of rare sugars. ChemBioChem. 2004;5:491–499. doi: 10.1002/cbic.200300774. [DOI] [PubMed] [Google Scholar]
- Kaplan JB, Meyenhofer MF, Fine DH. Biofilm growth and detachment of Actinobacillus actinomycetemcomitans. J Bacteriol. 2003a;185:1399–1404. doi: 10.1128/JB.185.4.1399-1404.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan JB, Ragunath C, Ramasubbu N, Fine DH. Detachment of Actinobacillus actinomycetemcomitans biofilm cells by an endogenous beta-hexosaminidase activity. J Bacteriol. 2003b;185:4693–4698. doi: 10.1128/JB.185.16.4693-4698.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan JB, Ragunath C, Velliyagounder K, Fine DH, Ramasubbu N. Enzymatic detachment of Staphylococcus epidermidis biofilms. Antimicrob Agents Chemother. 2004;48:2633–2636. doi: 10.1128/AAC.48.7.2633-2636.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauser T, Pohlner J, Meyer TF. Extracellular transport of cholera toxin B subunit using Neisseria IgA protease beta-domain: conformation-dependent outer membrane translocation. EMBO J. 1990;9:1991–1999. doi: 10.1002/j.1460-2075.1990.tb08327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattemann CT, Maurer J, Gerland E, Meyer TF. Autodisplay: functional display of active beta-lactamase on the surface of Escherichia coli by the AIDA-I autotransporter. J Bacteriol. 2000;182:3726–3733. doi: 10.1128/jb.182.13.3726-3733.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuel SG, Ragunath C, Sait HB, Izano EA, Kaplan JB, Ramasubbu N. Role of active-site residues of dispersin B, a biofilm-releasing beta-hexosaminidase from a periodontal pathogen, in substrate hydrolysis. FEBS J. 2007;274:5987–5999. doi: 10.1111/j.1742-4658.2007.06121.x. [DOI] [PubMed] [Google Scholar]
- Oomen CJ, van Ulsen P, van Gelder P, Feijen M, Tommassen J, Gros P. Structure of the translocator domain of a bacterial autotransporter. EMBO J. 2004;23:1257–1266. doi: 10.1038/sj.emboj.7600148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parise G, Mishra M, Itoh Y, Romeo T, Deora R. Role of a putative polysaccharide locus in Bordetella biofilm development. J Bacteriol. 2007;189:750–760. doi: 10.1128/JB.00953-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragunath C, Shanmugam M, Bendaoud M, Kaplan JB, Ramasubbu N. Effect of a biofilm-degrading enzyme from an oral pathogen in transgenic tobacco on the pathogenicity of Pectobacterium carotovorum subsp. carotovorum. Plant Pathol. 2011;61:346–354. [Google Scholar]
- Ramasubbu N, Thomas LM, Ragunath C, Kaplan JB. Structural analysis of dispersin B, a biofilm-releasing glycoside hydrolase from the periodontopathogen Actinobacillus actinomycetemcomitans. J Mol Biol. 2005;349:475–486. doi: 10.1016/j.jmb.2005.03.082. [DOI] [PubMed] [Google Scholar]
- Rose JE, Meyer DH, Fives-Taylor PM. Aae, an autotransporter involved in adhesion of Actinobacillus actinomycetemcomitans to epithelial cells. Infect Immun. 2003;71:2384–2393. doi: 10.1128/IAI.71.5.2384-2393.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford N, Charbonneau ME, Berthiaume F, Betton JM, Mourez M. The periplasmic folding of a cysteineless autotransporter passenger domain interferes with its outer membrane translocation. J Bacteriol. 2006;188:4111–4116. doi: 10.1128/JB.01949-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford N, Mourez M. Surface display of proteins by gram-negative bacterial autotransporters. Microb Cell Fact. 2006;5:22. doi: 10.1186/1475-2859-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Skillman KM, Barnard TJ, Peterson JH, Ghirlando R, Bernstein HD. Efficient secretion of a folded protein domain by a monomeric bacterial autotransporter. Mol Microbiol. 2005;58:945–958. doi: 10.1111/j.1365-2958.2005.04885.x. [DOI] [PubMed] [Google Scholar]
- Slots J, Reynolds H, Genco RJ. Actinobacillus actinomycetemcomitans in human periodontal disease: a cross-sectional microbiological investigation. Infect Immun. 1980;29:1013–1020. doi: 10.1128/iai.29.3.1013-1020.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gerven N, Sleutel M, Deboeck F, De Greve H, Hernalsteens JP. Surface display of the receptor-binding domain of the F17a-G fimbrial adhesin through the autotransporter AIDA-I leads to permeability of bacterial cells. Microbiology. 2009;155:468–476. doi: 10.1099/mic.0.022327-0. [DOI] [PubMed] [Google Scholar]