

SUMMARY
Protein kinase C (PKC) isozymes are commonly recognized as oncoproteins based on their activation by tumor-promoting phorbol esters. However, accumulating evidence indicates that PKCs can be inhibitory in some cancers, with recent findings propelling a shift in focus to understanding tumor suppressive functions of these enzymes. Here, we report that PKCα acts as a tumor suppressor in PI3K/AKT-driven endometrial cancer. Transcriptional suppression of PKCα is observed in human endometrial tumors in association with aggressive disease and poor prognosis. In murine models, loss of PKCα is rate limiting for endometrial tumor initiation. PKCα tumor suppression involves PP2A-family-dependent inactivation of AKT, which can occur even in the context of genetic hyperactivation of PI3K/AKT signaling by coincident mutations in PTEN, PIK3CA, and/or PIK3R1. Together, our data point to PKCα as a crucial tumor suppressor in the endometrium, with deregulation of a PKCα→PP2A/PP2A-like phosphatase signaling axis contributing to robust AKT activation and enhanced endometrial tumorigenesis.
Graphical Abstract
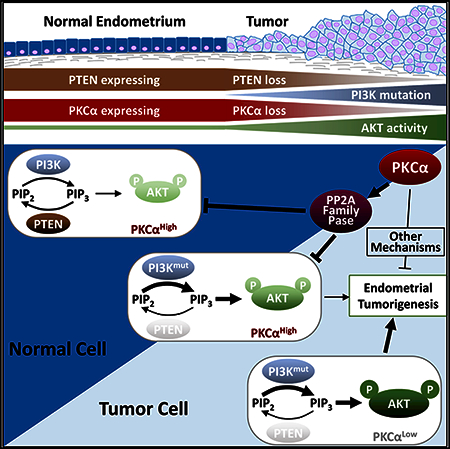
In Brief
Hsu et al. find that PKCα is frequently lost in human and murine endometrial tumors and that PKCα deficiency enhances PI3K/ AKT-driven endometrial neoplasia. PKCα suppresses aberrant AKT activity via a PP2A family phosphatase-dependent mechanism. Thus, PKCα loss appears to cooperate with PI3K/AKT perturbations to hyperactivate AKT and promote endometrial tumorigenesis.
INTRODUCTION
Protein kinase C (PKC) serine/threonine kinases are a family of signaling intermediates that act downstream of cell-surface receptors and the second messenger diacylglycerol to control multiple cellular responses, including proliferation, differentiation, migration, survival, and tumorigenesis (Black and Black, 2013; Garg et al., 2014). The identification of PKC as the major cellular receptor for tumor promoting phorbol esters provided the first link between these kinases and cancer (Castagna et al., 1982). In the years following this discovery, research focused on the oncogenic properties of PKCs, and considerable effort was dedicated to their development as therapeutic targets (Mochly-Rosen et al., 2012). However, it has become increasingly apparent that there is significant complexity in the roles of PKC isozymes in tumorigenesis (Garg et al., 2014). The PKC family consists of ten isozymes divided into three classes based on differences in structure and regulation: conventional or classical PKCs (cPKCs; α, βI, βIl, and γ), novel PKCs (nPKCs; δ, ε, η, and θ) and atypical PKCs (aPKCs; ζ and λ/ɩ). While there is strong evidence for pro-oncogenic functions of some PKCs (e.g., PKCε and PKCɩ), studies from our laboratory and others have demonstrated antiproliferative and tumor-suppressive activity of these kinases in several systems (Garg et al., 2014; Pysz et al., 2009; Hill et al., 2014; Osterand Leitges, 2006; Black, 2001). A tumor-suppressive role of PKCs was further highlighted by a recent comprehensive analysis of cancer-associated PKC mutations, which showed that 28 of 46 mutations tested led to loss of kinase function (Antal et al., 2015). Much of the complexity regarding the involvement of PKCs in cancer arises from their pleiotropic and context-dependent effects on cellular function. Contradictory data on PKC isozyme levels in tumors, combined with the use of a limited number of cell lines to assess PKC isozyme function and a lack of correlation between in vitro and in vivo data, have added to the confusion (Garg et al., 2014). Thus, understanding of the roles of PKCs in tumorigenesis will require comprehensive and coordinated in vivo and in vitro studies of individual isozymes in a specific cancer type.
Endometrial cancer (EC) is the most common gynecological malignancy in the United States, with an estimated 61,380 new cases and 10,920 deaths in 2017 (Siegel et al., 2017). With an overall 5-year survival rate of >80%, EC has attracted less public attention than other cancers. However, advanced and recurrent disease is refractory to treatment, and the prognosis for these patients is dismal, with survival estimates of less than 1 year (Engelsen et al., 2009). Because of its high incidence, EC is the sixth leading cause of cancer death in women, accounting for more deaths than melanoma, cervical cancer, glioblastoma, all lymphomas, or all leukemias (Siegel et al., 2017). Alarmingly, the incidence and mortality for EC are on the rise, with a >50% increase since 2005. Since obesity is a major risk factor for the disease (Fader et al., 2009), EC will become an even greater health concern as the effects of increased societal obesity become evident in coming years.
EC has historically been classified into two histopathological subtypes. Type I tumors (85%−90% of cases) are of endometrioid histology, while type II non-endometrioid tumors are predominantly of serous histology (Suarez et al., 2017). Recent genomic analysis has recognized four molecular subgroups (POLE ultramutated, microsatellite instability hypermutated, copy number low, and copy number high) that are distinct from their histological classification (Kandoth et al., 2013). A common feature of these subgroups is the prevalence of mutations in the phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway (Dedes et al., 2011; Hong et al., 2015). Activation of PI3K by growth factor receptors generates phosphatidylinositol-3,4,5-trisphosphate (PIP3), which recruits AKT to the plasma membrane, where it is phosphorylated and activated by PDK1 (T308) and mTORC2 (S473) (Manning and Toker, 2017). AKT directly or indirectly inactivates inhibitors of cell-cycle progression, survival, glycolysis, angiogenesis, and translation (e.g., p27, FOXO1, BAD, and 4E-BP1), thus unlocking key processes involved in oncogenesis (Manning and Toker, 2017). PI3K/AKT signaling is negatively regulated by the tumor suppressor PTEN, a lipid phosphatase that opposes the activity of PI3K by dephosphorylating PIP3 (Manning and Toker, 2017; Georgescu, 2010). The most common alterations in EC are loss-of-function mutations in PTEN and mutation or amplification of the catalytic subunit of PI3K, PIK3CA (Dedes et al., 2011; Hong et al., 2015). Mutations in PIK3R1, the regulatory subunit of PI3K, AKT, or factors that crosstalk with PI3K/AKT signaling, such as KRAS and EGFR, are also observed. The importance of PI3K/AKT pathway alterations in uterine tumorigenesis is highlighted by the increased incidence of EC in Cowden syndrome patients, who carry germline mutations in PTEN (Hollander et al., 2011), and the predisposition of mice with deletion or loss-of-function mutations in Pten to uterine neoplasia (Podsypanina et al., 1999; Stambolic et al., 2000). Notably, alterations in PI3K/AKT pathway components are not mutually exclusive in EC, with multiple mutations frequently coexisting at higher than predicted rates (Oda et al., 2005). The association between accumulation of multiple pathway mutations and tumor progression, combined with the sensitivity of EC cells to PI3K/AKT pathway inhibition (Hayes et al., 2006; Weigelt et al., 2013), indicates that strong hyperactivation of PI3K/AKT signaling is critical for driving EC tumorigenesis. A better understanding of the dysregulation of PI3K/AKT signaling in EC may, therefore, point to new therapeutic targets for this disease.
PKCα is a ubiquitously expressed PKC isozyme that has been implicated in control of cell proliferation, differentiation, survival, and motility (Garg et al., 2014). The effects of this kinase appear to be context dependent, with evidence for tumor-suppressive (e.g., in colorectal, lung, and basal cell cancers) and tumor-promoting (glioma and breast cancers) activity in different tumor types (Pysz et al., 2009; Oster and Leitges, 2006; Neill et al., 2003; Hill et al., 2014; Tam et al., 2013; Cameron et al., 2008). Using patient samples, animal models, and a panel of human EC cell lines, our in-depth studies show that PKCα plays a key tumor-suppressive role in the endometrial epithelium that is at least partially mediated by its ability to suppress the PI3K/AKT signaling pathway.
RESULTS
Loss of PKCα Expression Is Associated with High-Grade EC
As a first step toward understanding the role of PKCα signaling in EC, we used immunohistochemistry (IHC) to profile its expression in a panel of 436 human endometrial tumors (330 endometrioid and 106 non-endometrioid) in tissue microarray (TMA) format. While PKCα was detected in normal secretory and proliferative endometrium, the enzyme was absent or markedly reduced in >60% of endometrioid ECs and >50% of all non-endometrioid tumors, with the proportion of PKCα-deficient tumors ranging from 31% of malignant mixed Mullerian tumors (MMMT) to 70% of clear cell ECs (Figures 1A and 1B). The availability of frozen tissue for a subset of these ECs allowed parallel quantification of PKCα mRNA by qRT-PCR. A significant correlation was noted between PKCα mRNA and protein levels (Figure 1C), indicating that downregulation of PKCα protein in human EC is controlled primarily at the level of mRNA expression. Analysis of The Cancer Genome Atlas (TCGA) data confirmed that PKCα mRNA is markedly downregulated in human ECs (p = 2.77e−9) (and pointed to altered expression of several additional PKC family members in these tumors) (Figure S1A).
Figure 1. PKCα Expression Is Lost in EC.
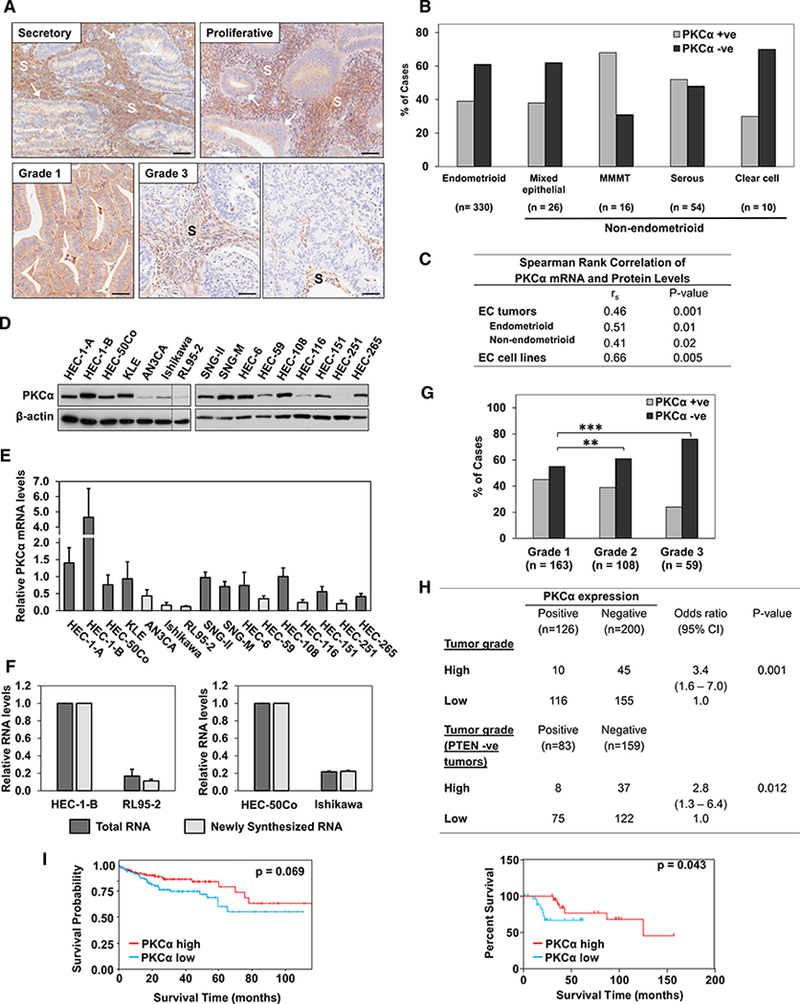
(A) IHC analysis of PKCα in normal human secretory and proliferative endometrium (arrows), a PKCα-retaining grade 1 endometrioid tumor, and grade 3 endometrioid tumors lacking PKCα. PKCα signal in the stroma (S) serves as a positive control for IHC staining. Scale bars, 100 μm.
(B) PKCα expression status (IHC staining) in EC histological subtypes.
(C) Correlation between PKCα mRNA (qRT-PCR) and protein (IHC) levels in 26 endometrioid and 26 non-endometrioid tumors and 16 EC cell lines.
(D and E) PKCα protein (immunoblot [IB]) (D) and mRNA(qRT-PCR normalized against18S rRNA) (E) levels in EC cell lines. Vertical line indicates rearrangement of lanes from a single membrane for clarity. Dark bars, PKCαhigh; light bars, PKCαlow.
(F) PKCα status is an indicator of risk for high-grade disease (table) and survival of patients with endometrioid tumors (graph). Grade 1 and 2 tumors are considered low grade, and grade 3 tumors are high grade.
(G) Survival probability of EC patients in TCPA endometrial dataset with high and low PKCα protein expression.
(H) PKCα mRNA expression and synthesis (EU-labeling) in PKCαhigh and PKCαlow EC cells (normalized against GAPDH mRNA).
(I)PKCα expression status in endometrioid ECs of different grades.
**p < 0.01, ***p < 0.005 (2-sided Fisher’s exact test). Error bars represent SEM (n = 3 biological replicates, except in F [n = 2 biological replicates]).
To further examine the regulation of PKCα in EC, we determined its expression in a panel of 16 well-characterized human EC cell lines (Table S1)(Weigelt et al., 2013). Six of these cell lines expressed markedly reduced levels of PKCα (Figures 1D and 1E), and a significant correlation was observed between PKCα protein and mRNA levels across the panel of EC cell lines (Figure 1C). To test for changes in mRNA transcription, RNA was pulse-labeled with 5-ethynyluridine (EU) in two PKCαhigh (HEC-1-B; HEC-50Co) and two PKCαlow (RL95–2; Ishikawa) EC cell lines. PKCαlow cells incorporated markedly lower amounts of EU into PKCα mRNA than PKCαhigh cells, with the extent of labeling closely paralleling the differences in overall levels of PKCα mRNA in the cells (Figure 1F). Together, these data confirm that PKCα is downregulated in a significant fraction of human ECs and EC cell lines and that PKCα deficiency primarily reflects a reduction in the transcription rate of the PKCα gene.
Further analysis of the TMA data revealed a positive correlation between decreased levels of PKCα and disease aggressiveness in the endometrioid EC subtype, with 56% of grade 1,61% of grade 2, and 76% of grade 3 lesions showing loss of the enzyme (Figure 1G). A significant association was also observed between loss of PKCα and risk of high-grade endometrioid disease (odds ratio [OR] = 3.4) (Figure 1H, table). PKCα deficiency also correlated with disease severity when only PTEN negative endometrioid tumors were considered (OR = 2.8), indicating that PKCα loss is a risk factor for aggressiveness in human EC even in the presence of activating mutations in the PI3K/AKT pathway. Consistent with this conclusion, low PKCα expression was indicative of reduced survival in patients with endometrioid EC (Figure 1H, graph). Similarly, reverse phase protein array analysis of 244 EC cases (~80% endometrioid; Liang et al., 2012) from The Cancer Proteome Atlas (TCPA) showed that PKCα expression trends with enhanced survival of EC patients (Figure 1I). In contrast, the expression of other PKC isozymes either showed no association with survival or correlated with a less favorable prognosis (e.g., PKCε and PKCɩ; Figure S1). Collectively, the data show that loss of PKCα can be an early event in EC and that PKCα deficiency is associated with more aggressive disease, pointing to a potential tumor-suppressive function of PKCα signaling in the endometrium.
PKCα Expression Is Lost in Mouse Models of Endometrial Neoplasia
Next, we characterized PKCα expression in tissues from mouse models of endometrial neoplasia. In the normal murine endometrium, PKCα was detectable in all four phases of the estrous cycle (Figure S2; data not shown). PKCα expression was relatively low and predominantly cytoplasmic in metestrus, diestrus, and early proestrus. However, endometrial glands showed robust expression and plasma membrane association of PKCα in late proestrus and estrus (arrows). The proestrus-to-estrus transition was also accompanied by increased expression and strong plasma membrane translocation of PKCα in a majority of cells of the luminal epithelial compartment (Figure S2, arrows). Since membrane association is indicative of PKC activation (Black and Black, 2013), these data suggest that PKCα is predominantly active during late proestrus and estrus phases. Membrane-associated PKCα generally coincided with reduced staining for the proliferation marker Ki67 (Figure S3, arrows), with cytoplasmic PKCα staining coinciding with stronger Ki67 signal in both proestrus and estrus (Figure S3, arrowheads). These results, which were confirmed by immunofluorescence analysis of frozen uterine tissues (Figure S2), suggest that PKCα may be involved in antiproliferative signaling in the endometrium and are thus consistent with a tumor-suppressive function of the enzyme in this tissue.
The expression of PKCα in endometrial lesions was explored in mice with single-allele knockin of Pten mutations seen in Cowden syndrome (frameshift/truncation PtenαΔ4−5/+ mutation, and missense PtenG129E/+ or PtenC124R/+ mutations). These mice develop endometrial hyperplasia with high penetrance upon loss of expression of the wild-type (WT) Pten allele (Figure 2; Wang et al., 2010). Loss of the WT allele is reflected in absence of PTEN staining in PtenΔ4−5/+ lesions and a reduction in PTEN staining in PtenG129E/+ or PtenC124R/+ lesions (Figures 2A–2C). As expected, loss of PTEN function was accompanied by a marked increase in AKT activity, as indicated by enhanced pAKTSer473 signal. Remarkably, these lesions uniformly lacked PKCα protein expression, with a perfect correspondence between PTEN deficiency, AKT activation, and PKCα loss (arrows and boxed areas). The reduced levels of PKCα protein were associated with decreased mRNA expression, as revealed by qRT-PCR analysis of normal and tumor tissues obtained by laser capture microdissection (LCM) (Figures 2E and S4). Interestingly, the lesions also expressed increased levels of inhibitor of DNA binding 1 (Id1), a gene known to be suppressed by PKCα signaling (Figure S5) (Hao et al., 2011). PKCα loss was also observed in uterine tumors of mice with cre-mediated conditional endometrial deletion of Pten in the epithelium and stroma (Ptenpr−/−) or the epithelium alone (Ptenltf−/−) regulated by the progesterone receptor promoter or the lactoferrin promoter, respectively (Figure 2F) (Daikoku et al., 2008, 2014). The loss of PKCα in multiple in vivo models of PI3K/ AKT-driven endometrial neoplasia further supports a role for PKCα deficiency in uterine tumorigenesis.
Figure 2. PKCα Expression Is Lost in Mouse Models of Endometrial Neoplasia.
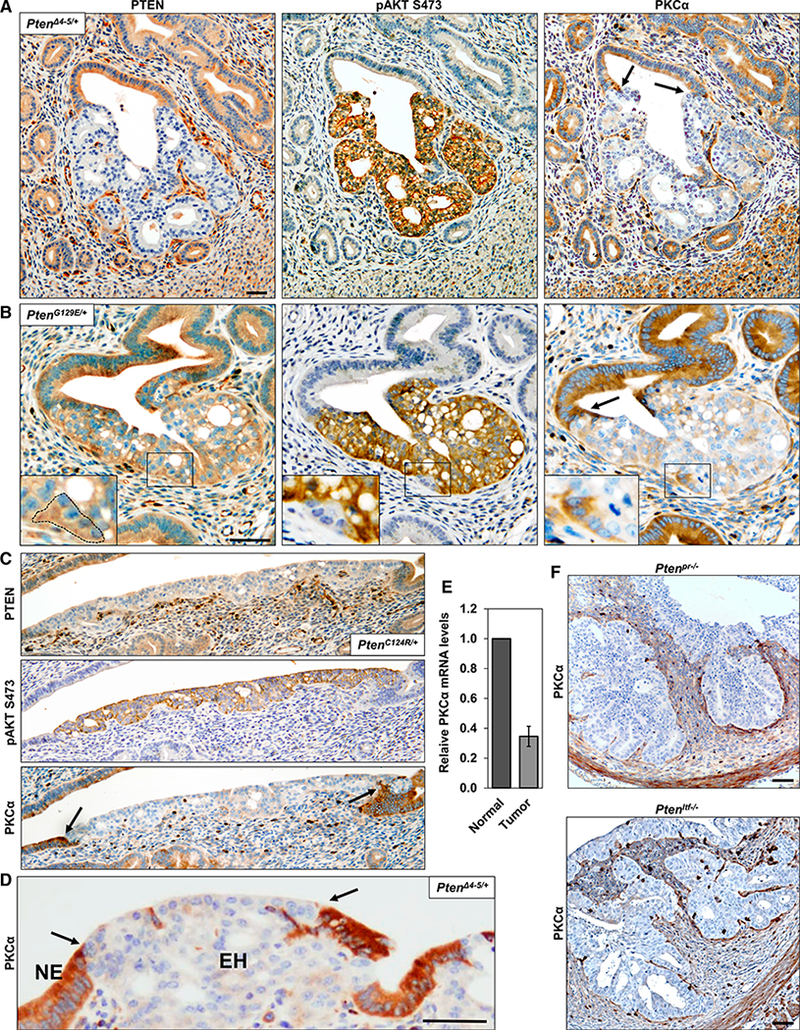
(A-D and F) IHC analysis of PTEN, pAKT, and PKCα expression in consecutive sections of uterine lesions arising in mutant Pten allelic-knockin mice (PtenΔ4−5/+ [A and D], PtenG129E/+ [B], and PtenC124R/+ [C]) and conditional Pten-knockout mice (F). Arrows indicate boundary between normal endometrial tissue (NE) and endometrial hyperplasia (EH). Inserts in (B) show a higher magnification of boxed areas; note the correspondence between a WT PTEN retaining region delineated by a dotted line, the absence of pAKT, and retention of PKCα. Scale bars, 50 μm.
(E) PKCα mRNA expression (normalized against GAPDH) in normal and neoplastic uterine tissue collected from PtenΔ4−5/+ mice by LCM.
PKCα Deficiency in EC Is Not a Direct Result of PTEN Loss/AKT Activation and Does Not Appear to Drive Loss of PTEN Expression
To determine if PKCα deficiency in EC is a direct consequence of PTEN loss of function/AKT activation, PTEN was stably knocked down in three PTEN WT, PKCαhigh human EC cell lines (Figure 3A). While the functional consequences of PTEN knockdown were confirmed by increased AKT phosphorylation, loss of PTEN did not affect the levels of PKCα (or of any other PKC isozyme) (Figure 3B). Similarly, inhibition of aberrant AKT activity in PTEN mutant/PKCαlow EC cell lines by restoration of PTEN expression (Figure 3C) or treatment with AKT or PI3K inhibitors (MK-2206 or LY294002, respectively) (Figure 3D) failed to affect PKCα levels. Collectively, these data indicate that PKCα deficiency is not simply a passenger event downstream of PI3K/AKT hyperactivation and suggest that loss of the enzyme may contribute directly to endometrial tumor development. To test the possibility that PKCα deficiency drives loss of PTEN in endometrial cells, we explored the effects of PKCα knockdown in PTEN WT, PKCαhigh EC cell lines. As shown in Figure 3E, PKCα knock-down failed to affect PTEN levels in these cells.
Figure 3. PKCα Deficiency in EC Is Not a Direct Result of PTEN Loss/AKT Activation and Does Not Promote PTEN Loss.
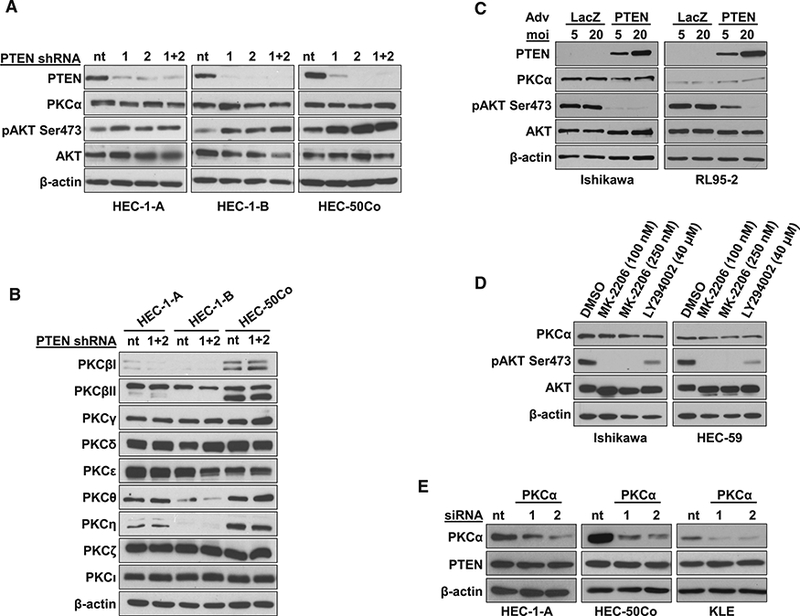
(A and B) IB analysis of indicated proteins in EC cells expressing two different PTEN shRNAs (1 and 2) or non-targeting shRNA (nt).
(C) IB analysis of PTEN-null EC cells with restored PTEN expression.
(D) IB analysis of EC cells treated with AKT inhibitor MK-2206 or PI3K inhibitor LY294002.
(E) IB analysis of PTEN in PKCα-knockdown EC cells.
Data are representative of 3 biological replicates.
PKCα Inhibits Endometrial Tumorigenesis In Vivo and In Vitro
To directly examine the effects of PKCα deficiency on endometrial tumor development, we generated Prkca+/+;PtenΔ4−5/+ and Prkca−/−;PfenΔ4−5/+ mice. Uterine tissue was collected at 1,3,5, 7, and 9 months, and hyperplastic lesions were identified by pAKTSer473 staining and histopathological analysis (Wang et al., 2002) (Figure 4A, top and middle panels). Quantification of the proportion of pAKT positive endometrium pointed to a statistically significant 3-fold increase in tumor burden in PKCα−/− animals at both 1 and 3 months compared with PKCα+/+ littermate controls (Figure 4A). However, this difference diminished at later times. Blinded histological analysis of H&E-stained sections by a pathologist (Y.M.S.) confirmed this conclusion (not shown). Since hyperplastic lesions arising in Prkca+/+,PtenΔ4−5/+ mice uniformly lacked PKCα (Figure 2), the increased tumor burden in Prcka−/−;PtenΔ4−5/+ mice indicates that PKCα loss is a rate-limiting step in endometrial tumorigen-esis in this model.
Figure 4. PKCα Inhibits Endometrial Tumorigenesis In Vivo and In Vitro.
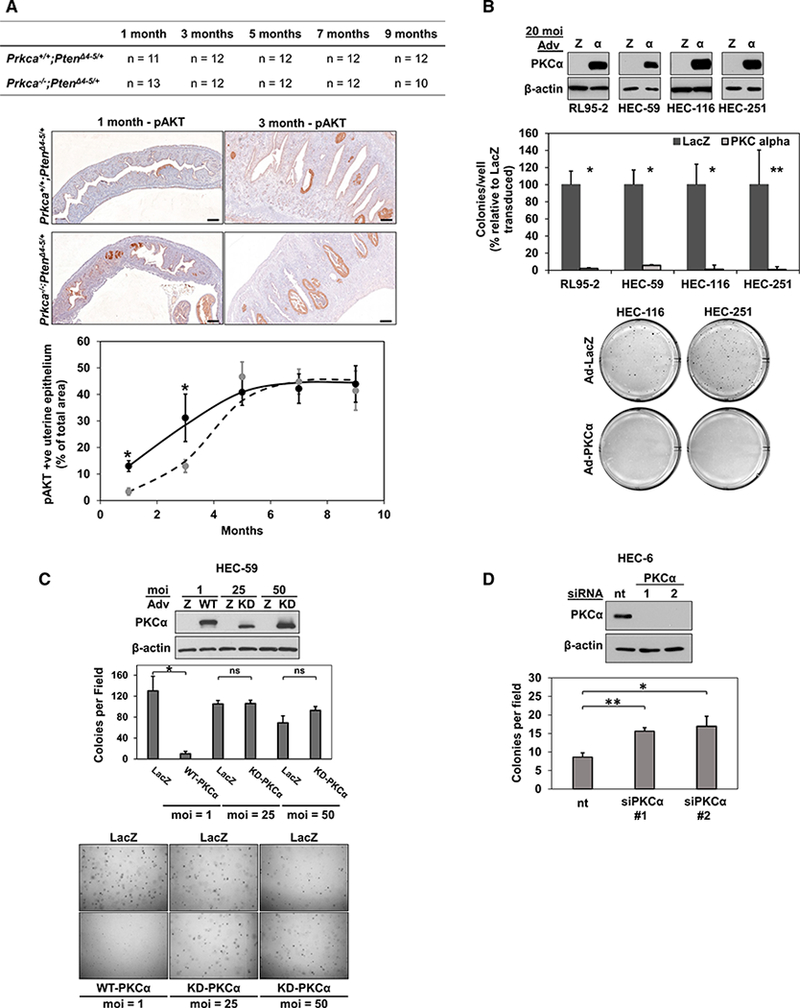
(A) Quantification of relative levels of hyperplastic uterine epithelium identified by pAKTS473 staining, as shown in representative images at 1 and 3 months. Dotted line, Prkca+/+;PtenΔ4−5/+; solid line, Prkca−/−;PtenΔ4−5/+. Scale bars, 200 μm.
(B) IB analysis and anchorage-independent growth (colony formation in soft agarose) of PKCαlow EC cells transduced with 20 MOI adenovirus expressing LacZ or PKCα.
(C) Anchorage-independent growth of HEC-59 cells transduced with WT-PKCα-or KD-PKCα. The higher mobility of KD-PKCα in the IB reflects absence of priming phosphorylation (Pysz et al., 2009).
(D) Anchorage independent growth of HEC-6 cells transfected with two different PKCα siRNAs (1 and 2) or nt siRNA.
*p < 0.05; **p < 0.01; ns, not significant. Error bars represent SEM for each animal cohort in (A) or 3 biological replicates in (B)-(D).
To test the effects of PKCα on the transformed phenotype of human EC cells, PKCα expression was restored in PKCαlow cell lines by adenoviral transduction or silenced in PKCαhigh cells using siRNA, and anchorage-independent growth was assessed by colony formation in soft agarose. Exogenous PKCα markedly inhibited colony formation of PKCαlow EC cells, and colonies that did form were substantially smaller than those of LacZ-transduced cells (Figure 4B). One infection unit/cell (MOI) of PKCα adenovirus was sufficient to suppress anchorage-independent growth of these cells (Figure 4C), pointing to strong tumor-suppressive activity of PKCα in human EC cells. The requirement for PKCα kinase activity in these effects was tested using kinase-dead enzyme (KD-PKCα). Since KD-PKCα is unstable in cells (Pysz et al., 2009), 25 or 50 MOI KD-PKCα adenovirus was used to ensure levels of mutant protein expression comparable with WT PKCα at 1 MOI. KD-PKCα failed to affect colony formation, even at the highest level of expression (Figure 4C). The effects of PKCα knockdown were examined in PKCαhigh HEC-6 cells, which grow less efficiently in soft agarose than other EC cells in our panel. Notably, PKCα depletion led to an ~2-fold increase in colony formation of these cells (Figure 4D). Thus, PKCα kinase activity is a potent inhibitor of the transformed phenotype of human EC cells.
PKCα Activity Inhibits PI3K/AKT Signaling in EC Cells via a PHLPP-1/2-Independent, PP2A-Family-Dependent Mechanism
Since a majority of ECs are highly dependent on PI3K/AKT signaling (Weigelt et al., 2013), we next examined the effects of PKCα activity on this pathway using a panel of human EC cell lines harboring different PI3K/AKT pathway mutations (Figure 5A). Treatment with the PKC agonists phorbol 12-myristate 13-acte-tate (PMA) orthe short-chain diacylglycerol 1,2-dioctanoylglycerol (DiC8) markedly decreased AKT activity in PKCαhigh cells (Figures 5B and 5C), as indicated by reduced levels of pAKTSer473 and decreased phosphorylation of the AKT substrate PRAS40. Full activation of AKT requires phosphorylation at S473 and T308 residues, and both of these residues are targeted by PKC activation in EC cells (Figure 5D). While PKC agonists failed to affect AKT phosphorylation in PKCαlow cells (Figure 5E), adenoviral restoration of PKCα rescued the effect (Figure 5F). In contrast, knockdown of PKCα in PKCαhigh cells with two different siRNAs prevented PKC agonist-induced AKT hypophosphorylation (Figure 5G). Since PKCα siRNA had no notable effect on the expression of other PKC isozymes (Figure 5H), these data confirm a requisite role for PKCα in PKC agonist-induced suppression of AKT activity in EC cells. The ability of the cPKC inhibitor, Go6976, to block PKCα-induced AKT hypophosphorylation in PKCαhigh cells established the requirement for PKCα catalytic activity (Figure 5I), which was sustained for at least 6 hr in the cells (Figure S6).
Figure 5. PKCα Activity Inhibits PI3K/AKT Signaling in EC Cells.
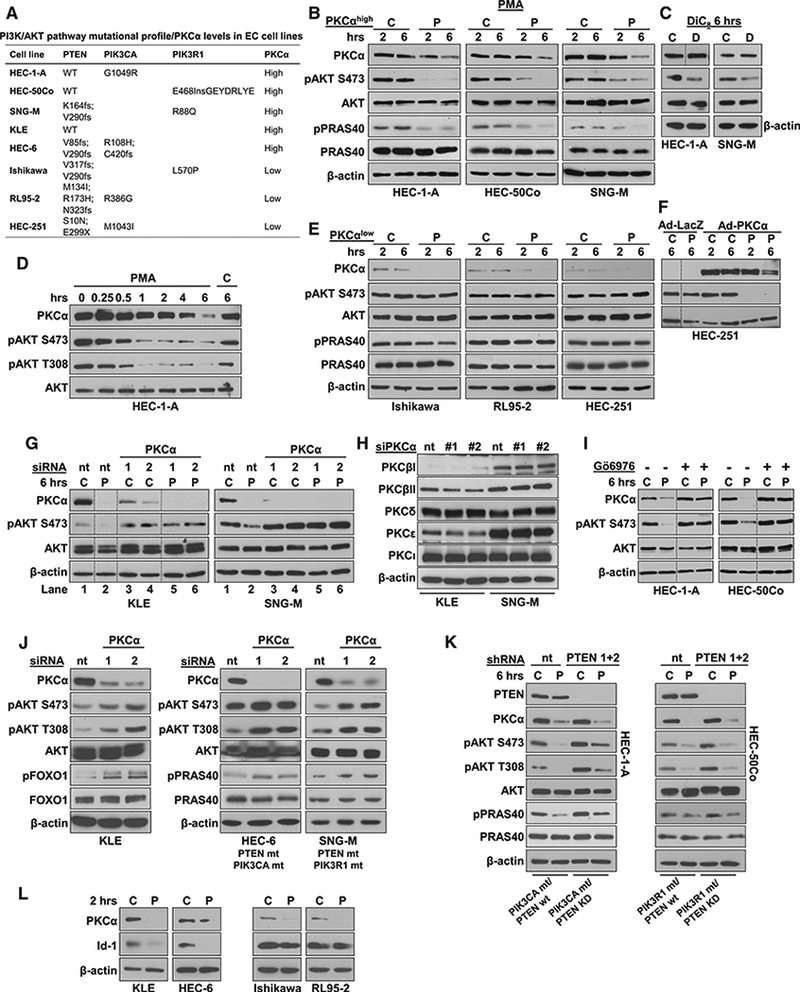
(A) PI3K/AKT pathway mutations and PKCα expression in EC cell lines. (B-D) IB analysis of PKCαhigh EC cells treated with PMA for 2 or 6 hr (B), DiC8 for 6 hr (C), or PMA for various times as indicated (D). C, vehicle control; D, DiC8; P, PMA.
(E and F) IB analysis of PKCαlow EC cells (E), or HEC-251 cells transduced with 1 MOI of LacZ or PKCα adenovirus (F), treated with PMA.
(G) PKCαhigh EC cells transfected with nt or PKCα siRNAs (1,2) and subjected to PMA treatment.
(H) PKC isozyme expression in PKCα-knockdown EC cells.
(I) EC cells pretreated with GÖ6976 (2.5 μM, 30 min) to inhibit classical PKCs prior to addition of PMA.
(J) Induction of pAKT activity in PKCα-knockdown EC cells.
(K)PMA treatment of EC cells with stable expression of nt or PTEN shRNA.
(L) Down regulation of Id1 in PMA-treated PKCαhlgh (left) but not PKCαlow (right) EC cells.
(L) C, vehicle-treated. P, PMA (100 nM). D, DiC8 (200μg/ml, replenished every 30 min). (M) In (B), (E), (J), and (K), effects on AKT activity are confirmed by analysis of the phosphorylation status of the AKT substrates, PRAS40 or FOXO1. Vertical lines in (C), (N) (F),(G), and (I) indicate rearrangement of lanes from a single membrane for clarity. Results are representative of at least 3 biological replicates.
Next, we determined if PKCα knockdown can affect basal levels of AKT activity in EC cells in the context of different levels of PI3K/AKT pathway hyperactivation. Loss of PKCα resulted in increased steady-state levels of AKT activity (as confirmed by increased pFOXOI) in KLE cells, which have no known alterations in the AKT/PI3K pathway, pointing to a tonic repressive effect of the enzyme on PI3K/AKT signaling in these cells (Figure 5J, left panel). Remarkably, PKCα knockdown also upregulated basal AKT activity in HEC-6 and SNG-M cells, two PTEN mutant cell lines that naturally harbor coincident mutations in the catalytic (PIK3CA) or regulatory (PIK3R1) subunits of PI3K, respectively (Weigelt et al., 2013) (Figure 5J, middle and right panels; also see Figure 5G, compare lane 1 with lanes 3–6 in both panels). The ability of PKCα to override hyperactive PI3K/AKT signaling was further investigated by silencing PTEN in two PTEN WT, PKCαhigh EC cell lines (HEC-1 -A and HEC-50Co) that harbor activating mutations in PIK3CA or PIK3R1, respectively (Figure 5A). As expected, PTEN knockdown further increased PI3K/AKT signaling, as indicated by enhanced basal levels of pAKTSer473, pAKTThr308, and pPRAS40 (although consistently observed, this effect was modest, as might be expected in cell lines that already harbor PI3K subunit mutations) (Figure 5K). Importantly, PKCα signaling was able to suppress AKT activity even in the face of further activation of the pathway. These data, combined with evidence for downregulation of the PKCα target gene Id1 (Figure 5L), a master regulator of tumor aggressiveness (Hao et al., 2011), further support a role for PKCα as a tumor suppressor in EC.
Since PP2A and PH domain leucine-rich repeat protein phosphatases (PHLPPs) have been identified as the main phosphatases that reverse AKT phosphorylation (Van Kanegan et al., 2005; Gao et al., 2005), we investigated their involvement in PKCα-mediated AKT hypophosphorylation in EC cells. These phosphatases are expressed (Figures 6A and 6B) and functional in EC cells. A role for PHLPP1/2 in regulation of AKT in EC cells was confirmed by the ability of the PHLPP1/2 inhibitors NCS45586 and NCS117079 (Sierecki et al., 2010) to increase basal AKT phosphorylation (Figure 6C). Enhanced AKT phosphorylation was similarly observed following inhibition of PP1 and PP2A/PP2A-like phosphatases with calyculin A or of PP2A family phosphatases alone with okadaic acid (Figure 6E). PMA was still able to suppress pAKT in the presence of the PHLPP1/2 inhibitors, indicating that the effects of PKCα are independent of PHLPP1/2 (Figure 6C). Since PP2A immunopre-cipitation phosphatase assays combined with PKCα knockdown determined that PKCα activates PP2A in EC cells (Figure 6D), we explored the effects of PP2A inhibitors. Both calyculin A and okadaic acid blocked PMA-induced AKT hypophosphorylation, pointing to a mechanism dependent on PP2A or a PP2A-like phosphatase (Figure 6E). Together, these results indicate that PKCα inhibits PI3K/AKT signaling through a PHLPP-independent, PP2A-family-dependent mechanism.
Figure 6. PKCα Inhibits AKT Phosphorylation via a PHLPP-1/2-In-dependent, PP2A-like Phosphatase-Dependent Mechanism.
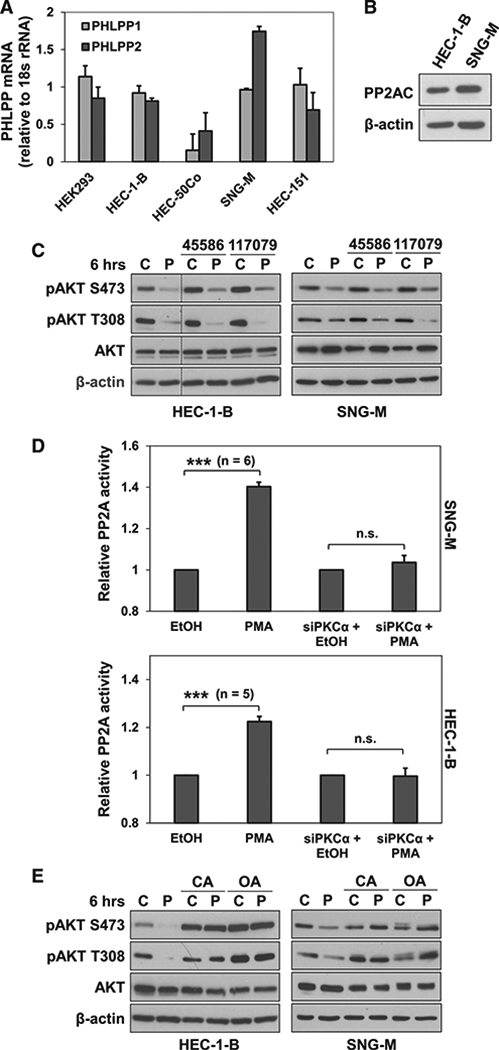
(A) PHLPP 1/2 mRNA expression in EC cells (HEK293 cells; positive control).
(B) Expression of PP2AC in EC cells.
(C) IB analysis of EC cells treated with PMA(P) or vehicle (C) in the presence of the PHLPP 1/2 inhibitors NSC45586 or NSC117079 (50 μM). Vertical lines indicate rearrangement of lanes from a single membrane for clarity.
(D) PP2A activity in EC cells treated with PMA in the presence or absence of PKCα knockdown.
(E) EC cells treated with PMA in the presence of calyculin A (CA; 10 nM) or okadaic acid (OA; 1 μM).</p/.Results are representative of at least 3 biological replicates. Error bars represent SEM. ***p = 0.005; n.s., not significant.
PKCα Loss Is Associated with Increased AKT Activity in Endometrial Lesions In Vivo
While universal loss of PKCα was seen in uterine lesions from PtenΔ4−5/+, PfenG129E/+, and PfenC124R/+ mouse models, PKCα-retaining areas were detected in lesions arising in the Ptenpr−/− mouse. This allowed comparison of AKT activity in PKCα-retaining and PKCα-deficient cells in situ. pAKT staining was stratified as none/low, medium, or high, and the association between pAKT staining intensity and the presence or absence of PKCα signal was quantified in serial sections. PKCα expression was associated with significant differences in AKT staining intensity (p < 0.005), with PKCα-negative areas predominantly exhibiting high intensity pAKT staining and PKCα-positive areas largely coinciding with low-intensity pAKT staining (Figure 7A, compare areas indicated with block and open arrows). This correspondence supports a role for PKCα in suppression of PI3K/ AKT signaling in endometrial tumor cells in vivo as well as in vitro.
Figure 7. Inhibition of AKT Activity Is an Important Component of the PKCα Tumor-Suppressive Axis in EC Cells.
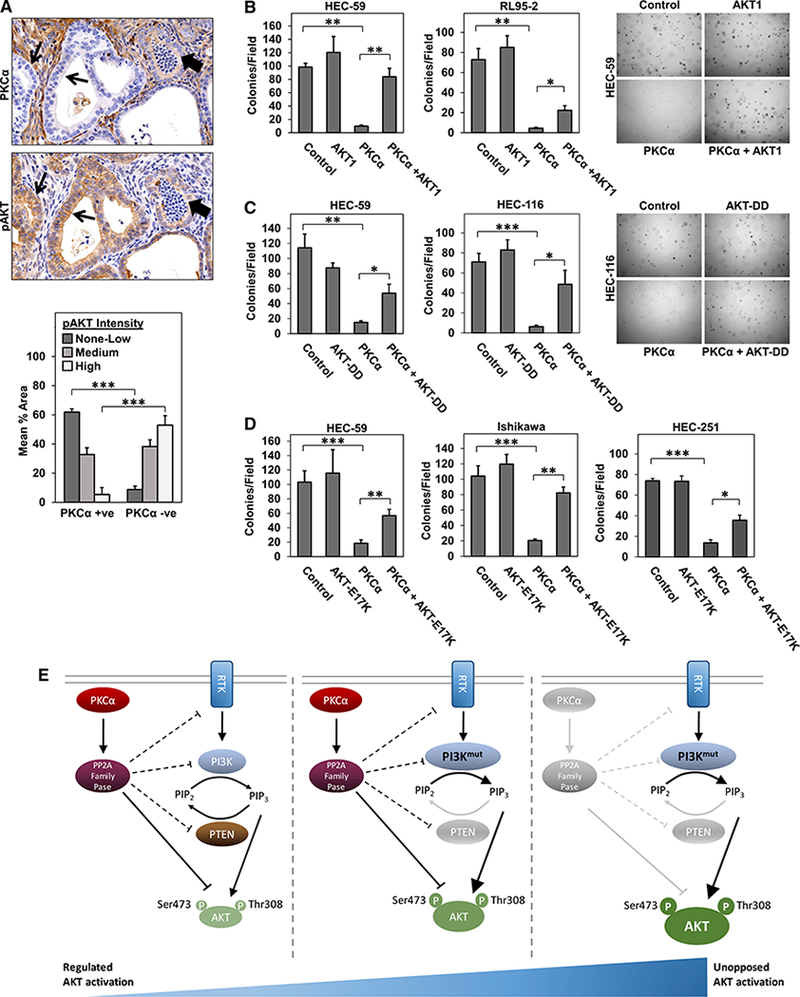
(A) Serial sections of uterine tissue from Ptenpr−/− mice immunostained for PKCα or pAKTSer473. Quantification of pAKT staining intensity (none/low, medium, or high) in PKCα-positive and negative areas. Data are representative of 3 independent experiments.
(B-D) Colony formation in soft agarose of EC cells expressing LacZ and/or empty vector (controls), myr-AKT1 (B), AKT-DD (C), AKT-E17K (D), and/or PKCα as indicated.
(A) Schematic representation of PI3K/PTEN and PKCα as independent regulators of AKT activation in endometrial tumorigenesis. The PKCα/PP2A inhibitory module may intersect the PI3K/AKT pathway at the level of AKT and/or upstream of the kinase.
*p < 0.05; **p < 0.01; ***p < 0.005. Error bars represent SEM from 3 independent experiments.
Inhibition of AKT Activity Is a Component of the PKCα Tumor-Suppressive Axis in EC Cells
The role of inhibition of PI3K/AKT signaling in the tumor-suppressive effects of PKCα in EC cells was tested using constructs expressing constitutively active AKT1: myristoylated AKT1 (myr-AKT), AKT-DD (T308D, S473D), and AKT-E17K(an active mutant identified in EC). All three constructs were tested in HEC-59 cells and at least one additional PKCαlow EC cell line (RL95–2, HEC-116, Ishikawa, and/or HEC-251 EC cells). Constructs were introduced into cells by adenoviral or retroviral transduction, and the ability of exogenous PKCα to inhibit anchorage-independent growth was assessed. Expression of constitutively active AKT had minor, if any, effects on anchorage-independent growth of EC cells on its own (Figures 7B–7D). In contrast, all three constructs had a significant effect in cells transduced with PKCα, restoring both colony number and overall colony size, albeit to different extents in different cell lines (Figures 7B–7D). While the inability of constitutively active AKT to fully reverse the effects of PKCα points to the involvement of additional mechanisms (e.g., Id1 suppression), these data indicate that inhibition of AKT signaling is an important contributor to the tumor-suppressive actions of PKCα in the endometrium.
DISCUSSION
The current study provides evidence for a tumor-suppressive role of PKCα signaling in PI3K/AKT-driven EC, via a mechanism that at least partially involves PP2A-family-mediated inactivation of AKT. The role of PKCα in tumorigenesis appears to be multifaceted. While the enzyme has been linked to tumor promotion in some systems, our studies add to a growing list of tumor types (e.g., colon and lung) in which PKCα signaling has tumor suppressive effects.
PKCα Is Downregulated Early in Endometrial Tumorigenesis
Our IHC and qRT-PCR analysis of human ECs revealed loss of PKCα protein and mRNA in a majority of cases. Notably, more than half of grade 1 ECs showed deficiency of the enzyme, indicating that PKCα suppression can occur early during human endometrial tumorigenesis. This notion is supported by our analysis of mouse models of PI3K/AKT-driven endometrial neoplasia, which demonstrated loss of PKCα expression in pre-malignant hyperplasia regardless of the nature of PTEN dysregulation. Additional supportive evidence is provided by previous transcriptome analysis of early Type I ECs, which demonstrated a 3-fold down-regulation of PKCα mRNA in tumors relative to normal endometrium (Saghir et al., 2010; Risinger et al., 2013), by stage-specific TCGA data, and by IHC analysis of a limited set of grade 1 ECs, which revealed regions of low or undetectable PKCα protein in a subset of lesions (Haughian et al., 2009).
Although loss of PKCα mRNA and protein has been noted in other tumor types, such as colon cancer (Verstovsek et al., 1998), non-small cell lung cancer (Hill et al., 2014), basal cell carcinoma (Neill et al., 2003), and T cell acute lymphoblastic leukemia (T-ALL) (Milani et al., 2014), the mechanisms underlying PKCα deficiency have yet to be defined. Our qRT-PCR and IHC analysis argues that decreased mRNA expression is the major mechanism involved in disabling PKCα functions in the endometrium. We further demonstrate that PKCα loss in EC can be mediated by transcriptional repression of the PKCα gene.
Loss of PKCα Is Rate Limiting for Endometrial Tumor Development and Is Associated with More Aggressive EC
The precise correspondence between PKCα loss, PTEN deficiency, and AKT activation in murine endometrial lesions suggested that transcriptional suppression of PKCα could be a direct result of hyperactivation of the PI3K/AKT pathway, at least in some ECs. However, modulation of PTEN expression or AKT activity in human EC cells excluded this possibility, indicating that PKCα downregulation is not merely a passenger effect of aberrant PI3K/AKT signaling; rather, it is an alteration that is selected for because of specific contributions to endometrial tumor development. Anchorage-independent colony-formation assays provided direct evidence for a tumor-suppressive function of PKCα in human endometrial cells; restoration of PKCα expression and activity profoundly inhibited colony formation of human EC cells in soft agarose, while PKCα knockdown enhanced the transformed properties of EC cells that retained expression of the enzyme. The importance of early loss of PKCα was highlighted by the marked acceleration in uterine hyperplastic transformation observed in PKCα`-deficient PtenΔ4−5/+ mice, a finding that pointed to abrogation of PKCα signaling as a rate-limiting step for tumor initiation in the endometrium. In advanced cases of human EC, the proportion of PKCα-negative tumors increased to >75%, suggesting that PKCα deficiency defines a subset of ECs with high risk of progression, a notion that is supported by our TMA analysis and TCPA data pointing to an association between reduced PKCα expression and decreased survival of EC patients. The finding that PKCα deficiency is associated with a 3-fold increase in risk of high-grade disease is consistent with previous reports in other tumor types. Genetic deletion of PKCα led to increased tumor burden, adenoma-to-carcinoma progression, and reduced survival in the APCMin/+ mouse model of intestinal neoplasia (Oster and Leitges, 2006) and KRas-mediated models of lung tumorigenesis (Hill et al., 2014). Low PKCα expression identified a new subgroup of childhood T-ALL with an extremely poor outcome (Milani et al., 2014) and appears to enhance the tumorigenic potential of Gli1 in basal cell carcinoma (Neill et al., 2003). It will be interesting to determine if common mechanisms mediate PKCα loss in these tumor types.
PKCα-Mediated Tumor Suppression in the Endometrium Involves Inhibition of PI3K/AKT Signaling
A majority of ECs harbor coexisting alterations in two or more PI3K/AKT pathway components, with additive effects on AKT activation that are crucial for EC development (Weigelt et al., 2013; Oda et al., 2005; Chen et al., 2006). Remarkably, knockdown of PKCα in cells with genetic hyperactivation of the PI3K/AKT pathway further enhanced the activity of AKT, highlighting the ability of PKCα deficiency to contribute to the strength of PI3K/AKT signaling in EC cells. The demonstration that PKCα-negative uterine regions in Ptenpr−/− mice are associated with significantly higher levels of pAKT than PKCα-retaining regions argues that PKCα loss also contributes to PI3K/AKT hyperactivation during endometrial tumorigenesis in vivo.PKCα activity may also restrain AKT in the normal endometrium, since increased expression and membrane association of the enzyme coincide with the reported downregulation of AKT activity during the proestrus-to-estrus transition in rats (Dery et al., 2003). These findings, together with the demonstration that agonist-induced PKCα activation suppresses AKT in EC cells, identify PKCα as an inhibitor of PI3K/AKT signaling in the endometrium that is capable of disabling the AKT oncoprotein, even in the context of multiple activating mutations in upstream signaling intermediates (e.g., PTEN and PIK3CA in HEC-251 cells and PTEN, PIK3CA, and KRAS in SNG-M cells). Inactivation of AKT appears to be a component of the tumor-suppressive actions of PKCα in endometrial cells, since constitutively active AKT diminished the inhibitory effects of PKCα on anchorage-independent growth of EC cells. Together, our findings support the idea that disruption of PKCα signaling may be critical for achieving the robust PI3K/AKT pathway activation that is required to drive EC development and progression (Figure 7E).
Crosstalk between PKCα and the PI3K/AKT pathway has been noted in other systems (Guan et al., 2007; Tanaka et al., 2003). PKCα can inactivate upstream modulators of PI3K such as growth factor receptors and scaffolding proteins (Koese et al., 2013; Oriente et al., 2005) and can dampen PI3K activity directly (Sipeki et al., 2006; Hoshino et al., 2012). Here, we demonstrate that PKCα activates the serine/threonine phosphatase PP2A in EC cells and inactivates AKT via a PP2A- or PP2A-like phosphatase-dependent mechanism. PKCα-induced AKT inactivation may involve direct effects of PP2A family phosphatases on activating phosphorylation at T308 and S473 (Manning and Toker, 2017), although potential effects on upstream regulators of AKT activity remain to be formally excluded (Figure 7E). (It should be noted that although PP2A may preferentially dephosphorylate AKT on the T308 site, it can also dephosphorylate AKT on the S473 site, as described in a variety of biological systems [Liao and Hung, 2010].) A role of PP2A in the antitumor effects of PKCα in the endometrium is consistent with known functions of the phosphatase as a bona fide tumor suppressor, with key activities in maintenance of cellular homeostasis (Perrotti and Neviani, 2013). Future studies will explore the specific PP2A family members involved as well as the specific holoenzyme complex(es) responsible for mediating PKCα-induced inactivation of AKT.
Together, our findings indicate that disruption of a PKCα⊣AKT signaling module plays a role in the development of EC, contributing to robust, unopposed AKT activation and promoting transformation of endometrial cells (Figure 7E). These studies provide a foundation for exploring the prognostic value of PKCα expression in EC and the potential of harnessing PKCa→PP2A family phosphatase signaling for therapeutic benefit in a subset of ECs.
EXPERIMENTAL PROCEDURES
A detailed description of methodology and analysis is provided in Supplemental Experimental Procedures.
Human Tissues
Endometrial tumors and normal tissue were collected by C.D.M. with approval from the Ohio State University and Roswell Park Comprehensive Cancer Center institutional review boards. Analysis of patient survival in relation to PKCα protein status used the MDACC-endometrial-L3-S40 dataset (TCPA).
Cell Lines and Reagents
The sources of human EC cell lines and reagents used in the study are detailed in Supplemental Experimental Procedures.
Mice
All animal husbandry was in compliance with regulations and protocols approved by Institutional Animal Care and Use Committees at the University of Nebraska Medical Center, Ohio State University, and Cincinnati Children’s Hospital Medical Center. Phases of the estrous cycle in female mice were determined by vaginal smears as described previously (Byers et al., 2012). Allele-specific mutant Pten-knockin mice were generated and characterized as described previously (Wang et al., 2010), and generation and genotyping of Prkca+/+;PtenΔ4−5/+ and Prkca−/−PtenΔ4−5/+ mice were performed as detailed in Supplemental Experimental Procedures. Pten conditional-knockout animals were generated by mating Ptenflf and PR-Cre or Ltf-Cre animals (Daikoku et al., 2008, 2014). Uterine tissues were collected from 1- to 9-month-old female mice for analysis.
LCM
Normal and neoplastic uterine tissue was collected from frozen sections by LCM and analyzed for PKCα mRNA levels by qRT-PCR.
IHC
Tissue sections were immunostained for PKCα, pAKTSer473, PTEN, Ki67, and Id1 using published protocols. Immunostaining was quantified using DEFINIENS software. For quantitative comparison of pAKT and PKCα levels, staining for these markers in consecutive sections was overlaid using Adobe Photoshop CC, and areas of negative/low, medium, and high pAKT staining within PKCα-positive and PKCα-negative regions were determined.
Western Blotting
Western blot analysis was performed as described previously (Frey et al., 2000). Information on antibodies is provided in Supplemental Experimental Procedures. All immunoblots are representative of at least three biological replicates.
RNAi
Lentiviral vectors expressing PTEN small hairpin RNA (shRNA) were used to generate stable PTEN-knockdown EC cells. Transient knockdown of PKCα was achieved by transfection of siRNA using RNAiMAX transfection reagent. Non-targeting shRNA and small interfering RNA(siRNA)were used as controls.
PP2A Activity Assay
PP2A activity was measured using a PP2A Immunoprecipitation Phosphatase Assay Kit (Millipore) as described elsewhere (Guan et al., 2007).
Anchorage-Independent Growth Assays
Transduction of cells with adenoviral and retroviral vectors expressing LacZ, PKCα, kinase-dead PKCα, and/or constitutively active AKT1 (myr-AKT, AKT-DD [T308D, S473D], and AKT-E17K) and analysis of growth in soft agarose are described in Supplemental Experimental Procedures.
RNA Isolation and qRT-PCR Analysis
RNA isolation was performed using the illustra RNAspin Mini RNA isolation kit (GE Healthcare) or Trizol reagent (Invitrogen) and qRT-PCR reactions used Brilliant II SYBR Green QRT-PCR 1-Step Master Mix, with primers listed in Supplemental Experimental Procedures.
Nascent RNA Capture
Isolation of nascent RNA was performed using the Click-iT Nascent RNA Cap-ture Kit (Life Technologies). Quantification of PKCα and GAPDH mRNA by qRT-PCR used iScript cDNA Synthesis and iTaq Universal SYBR Green Supermix Kits (Bio-Rad).
Statistical Analysis
Data are presented as the mean ± SEM of three or more independent experiments unless otherwise stated. Statistical analysis was performed using Excel, GraphPad Prism, or SAS software version 9.3. p values of less than 0.05were considered statistically significant. Significance was assessed using Student’s t test for comparison of means, chi-square or Fisher’s exact test for categorical analysis of two groups, Wilcoxon rank-sum test for nonparametic comparison of two samples, two-way ANOVA test for multiple comparisons, Spearman’s rank correlation for comparison of ordinal categorical expression data, and log-rank test for survival data. A linear mixed-effects model was used to determine the association between PKCα and pAKT staining in Ptenpr−/− endometrium. Fixed effects were included for analysis of PKCα and pAKT levels and their interaction. A random effect was included for the slide. Pairwise comparisons were adjusted for multiple comparisons with Tukey’s method. Model assumptions were examined with residual plots.
Supplementary Material
Highlights.
PKCα deficiency is a common characteristic of human and murine uterine neoplasms
Loss of PKCα is rate limiting for endometrial tumorigenesis in Pten mutant mice
PKCα suppresses AKT activity via a mechanism dependent on a PP2A family phosphatase
Deregulation of a PKCα⊣AKT signaling axis contributes to endometrial neoplasia
ACKNOWLEDGMENTS
We thank Kathryn Curry, Angela Omilian, and Elizabeth Brese for expert technical assistance and the UNMC Tissue Facility (Lijun Sun and Jiang Jiang) for services. This research was supported by NIH grants CA036727 and CA016056 (Cancer Center grants), DK060632 and CA191894 (J.D.B.), and HD068524 and CA077839 (S.K.D.). A.H.H. was supported by a UNMC Dean for Graduate Studies Fellowship.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, six figures, and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.06.067.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Antal CE, Hudson AM, Kang E, Zanca C, Wirth C, Stephenson NL, Trotter EW, Gallegos LL, Miller CJ, Furnari FB, et al. (2015). Cancer-associated protein kinase C mutations reveal kinase’s role as tumor suppressor. Cell 160, 489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JD (2001). Protein kinase C isozymes in colon carcinogenesis: guilt by omission. Gastroenterology 120, 1868–1872. [DOI] [PubMed] [Google Scholar]
- Black AR, and Black JD (2013). Protein kinase C signaling and cell cycle regulation. Front. Immunol 3, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers SL, Wiles MV, Dunn SL, and Taft RA (2012). Mouse estrous cycle identification tool and images. PLoS ONE 7, e35538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron AJ, Procyk KJ, Leitges M, and Parker PJ (2008). PKC alpha protein but not kinase activity is critical for glioma cell proliferation and survival. Int. J. Cancer 123, 769–779. [DOI] [PubMed] [Google Scholar]
- Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, and Nishizuka Y (1982). Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J. Biol. Chem 257, 78477851. [PubMed] [Google Scholar]
- Chen ML, Xu PZ, Peng XD, Chen WS, Guzman G, Yang X, Di Cristo-fano A, Pandolfi PP, and Hay N (2006). The deficiency ofAkt1 is sufficient to suppress tumor development in Pten+/− mice. Genes Dev. 20, 1569–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daikoku T, Hirota Y, Tranguch S, Joshi AR, DeMayo FJ, Lydon JP, Ellenson LH, and Dey SK (2008). Conditional loss of uterine Pten unfailingly and rapidly induces endometrial cancer in mice. Cancer Res. 68, 5619–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daikoku T, Ogawa Y, Terakawa J, Ogawa A, DeFalco T, and Dey SK (2014). Lactoferrin-iCre: a new mouse line to study uterine epithelial gene function. Endocrinology 155, 2718–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedes KJ, Wetterskog D, Ashworth A, Kaye SB, and Reis-Filho JS (2011). Emerging therapeutic targets in endometrial cancer. Nat. Rev. Clin. Oncol 8, 261–271. [DOI] [PubMed] [Google Scholar]
- Dery M-C, Leblanc V, Shooner C, and Asselin E (2003). Regulation ofAkt expression and phosphorylation by 17β-estradiol in the rat uterus during estrous cycle. Reprod. Biol. Endocrinol 1, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelsen IB, Akslen LA, and Salvesen HB (2009). Biologic markers in endometrial cancer treatment. APMIS 117, 693–707. [DOI] [PubMed] [Google Scholar]
- Fader AN, Arriba LN, Frasure HE, and von Gruenigen VE (2009). Endometrial cancer and obesity: epidemiology, biomarkers, prevention and survivorship. Gynecol. Oncol 114, 121–127. [DOI] [PubMed] [Google Scholar]
- Frey MR, Clark JA, Leontieva O, Uronis JM, Black AR, and Black JD (2000). Protein kinase C signaling mediates a program of cell cycle withdrawal in the intestinal epithelium. J. Cell Biol 151, 763–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Furnari F, and Newton AC (2005). PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol.Cell 18, 13–24. [DOI] [PubMed] [Google Scholar]
- Garg R, Benedetti LG, Abera MB, Wang H, Abba M, and Kazanietz MG (2014). Protein kinase C and cancer: what we know and what we do not. Oncogene 88, 5225–5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu MM (2010). PTEN tumor suppressor network in PI3K-Akt pathway control. Genes Cancer 1, 1170–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan L, Song K, Pysz MA, Curry KJ, Hizli AA, Danielpour D, Black AR, and Black JD(2007). Protein kinase C-mediated down-regulation of cy-clin D1 involves activation of the translational repressor 4E-BP1 via a phosphoinositide 3-kinase/Akt-independent, protein phosphatase 2A-dependent mechanism in intestinal epithelial cells. J. Biol. Chem 282, 14213–14225. [DOI] [PubMed] [Google Scholar]
- Hao F, Pysz MA, Curry KJ, Haas KN, Seedhouse SJ, Black AR, and Black JD (2011). Protein kinase Cα signaling regulates inhibitor of DNA binding 1 in the intestinal epithelium. J. Biol. Chem. 286, 18104–18117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haughian JM, Reno EM, Thorne AM, and Bradford AP (2009). Protein kinase C alpha-dependent signaling mediates endometrial cancer cell growth and tumorigenesis. Int. J. Cancer 125, 2556–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes MP, Wang H, Espinal-Witter R, Douglas W, Solomon GJ, Baker SJ, and Ellenson LH (2006). PIK3CA and PTEN mutations in uterine endometrioid carcinoma and complex atypical hyperplasia. Clin. Cancer Res. 12, 5932–5935. [DOI] [PubMed] [Google Scholar]
- Hill KS, Erdogan E, Khoor A, Walsh MP, Leitges M, Murray NR, and Fields AP (2014). Protein kinase Ca suppresses Kras-mediated lung tumor formation through activation of a p38 MAPK-TGFß signaling axis. Oncogene 33, 2134–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander MC, Blumenthal GM, and Dennis PA (2011). PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer 11, 289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong B, Le Gallo M, and Bell DW (2015). The mutational landscape of endometrial cancer. Curr. Opin. Genet. Dev. 30, 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino D, Jourquin J, Emmons SW, Miller T, Goldgof M, Costello K, Tyson DR, Brown B, Lu Y, Prasad NK, et al. (2012). Network analysis of the focal adhesion to invadopodia transition identifies a PI3K-PKCα invasive signaling axis. Sci. Signal 5, ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, Benz CC, et al. ; Cancer Genome Atlas Research Network (2013). Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koese M, Rentero C, Kota BP, Hoque M, Cairns R, Wood P, Vilà de Muga S, Reverter M, Alvarez-Guaita A, Monastyrskaya K, et al. (2013). Annexin A6 is a scaffold for PKCα to promote EGFR inactivation. Oncogene 32, 2858–2872. [DOI] [PubMed] [Google Scholar]
- Liang H, Cheung LWT, Li J, Ju Z, Yu S, Stemke-Hale K, Dogruluk T, Lu Y, Liu X, Gu C, et al. (2012). Whole-exome sequencing combined with functional genomics reveals novel candidate driver cancer genes in endometrial cancer. Genome Res. 22, 2120–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, and Hung M-C (2010). Physiological regulation of Akt activity and stability. Am. J. Transl. Res. 2, 19–42. [PMC free article] [PubMed] [Google Scholar]
- Manning BD, and Toker A (2017). AKT/PKB signaling: navigating the network. Cell 169, 381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani G, Rebora P, Accordi B, Galla L, Bresolin S, Cazzaniga G, Bul-dini B, Mura R, Ladogana S, Giraldi E, et al. (2014). Low PKCα expression within the MRD-HR stratum defines a new subgroup of childhood T-ALL with very poor outcome. Oncotarget 5, 5234–5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochly-Rosen D, Das K, and Grimes KV (2012). Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov 11, 937–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neill GW, Ghali LR, Green JL, Ikram MS, Philpott MP, and Quinn AG (2003). Loss of protein kinase Calpha expression may enhance the tumorigenic potential of Gli1 in basal cell carcinoma. Cancer Res. 63, 4692–4697. [PubMed] [Google Scholar]
- Oda K, Stokoe D, Taketani Y, and McCormick F (2005). High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res. 65, 10669–10673. [DOI] [PubMed] [Google Scholar]
- Oriente F, Andreozzi F, Romano C, Perruolo G, Perfetti A, Fiory F, Miele C, Beguinot F, and Formisano P (2005). Protein kinase C-alpha regulates insulin action and degradation by interacting with insulin receptor substrate-1 and 14–3-3 epsilon. J. Biol. Chem. 280, 40642–0649. [DOI] [PubMed] [Google Scholar]
- Oster H, and Leitges M (2006). Protein kinase C alpha but not PKCzeta suppresses intestinal tumor formation in ApcMin/+ mice. Cancer Res. 66, 6955–6963. [DOI] [PubMed] [Google Scholar]
- Perrotti D, and Neviani P (2013). Protein phosphatase 2A: a target for anticancer therapy. Lancet Oncol. 14, e229–e238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, Cordon-Cardo C, Catoretti G, Fisher PE, and Parsons R (1999). Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc. Natl. Acad. Sci. USA 96, 1563–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pysz MA, Leontieva OV, Bateman NW, Uronis JM, Curry KJ, Threadgill DW, Janssen KP, Robine S, Velcich A, Augenlicht LH, et al. (2009). PKCalpha tumor suppression in the intestine is associated with transcriptional and translational inhibition of cyclin D1. Exp. Cell Res 315, 1415–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risinger JI, Allard J, Chandran U, Day R, Chandramouli GV, Miller C, Zahn C, Oliver J, Litzi T, Marcus C, et al. (2013). Gene expression analysis of early stage endometrial cancers reveals unique transcripts associated with grade and histology but not depth of invasion. Front. Oncol 3, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saghir FS, Rose IM, Dali AZ, Shamsuddin Z, Jamal AR, and Mokhtar NM (2010). Gene expression profiling and cancer-related pathways in type I endometrial carcinoma. Int. J. Gynecol. Cancer 20, 724–731. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2017). Cancer Statistics, 2017. CA Cancer J. Clin 67, 7–30. [DOI] [PubMed] [Google Scholar]
- Sierecki E, Sinko W, McCammon JA, and Newton AC (2010). Discovery of small molecule inhibitors of the PH domain leucine-rich repeat protein phosphatase (PHLPP) by chemical and virtual screening. J. Med. Chem 53, 68996911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipeki S, Bander E, Parker PJ, and Faragó A (2006). PKCalpha reduces the lipid kinase activity of the p110alpha/p85alpha PI3K through the phosphorylation of the catalytic subunit. Biochem. Biophys. Res. Commun 339, 122–125. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Tsao MS, Macpherson D, Suzuki A, Chapman WB, and Mak TW (2000). High incidence of breast and endometrial neoplasia resembling human Cowden syndrome in pten+/− mice. Cancer Res. 60, 3605–3611. [PubMed] [Google Scholar]
- Suarez AA, Felix AS, and Cohn DE (2017). Bokhman redux: endometrial cancer ‘‘types’’ in the 21st century. Gynecol. Oncol. 144, 243–249. [DOI] [PubMed] [Google Scholar]
- Tam WL, Lu H, Buikhuisen J, Soh BS, Lim E, Reinhardt F, Wu ZJ, Krall JA, Bierie B, Guo W, et al. (2013). Protein kinase C a is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell 24, 347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Gavrielides MV, Mitsuuchi Y, Fujii T, and Kazanietz MG (2003). Protein kinase C promotes apoptosis in LNCaP prostate cancer cells through activation of p38 MAPK and inhibition of the Akt survival pathway. J. Biol. Chem 278, 33753–33762. [DOI] [PubMed] [Google Scholar]
- Van Kanegan MJ, Adams DG, Wadzinski BE, and Strack S (2005). Distinct protein phosphatase 2A heterotrimers modulate growth factor signaling to extracellular signal-regulated kinases and Akt. J. Biol. Chem 280, 36029–36036. [DOI] [PubMed] [Google Scholar]
- Verstovsek G, Byrd A, Frey MR, Petrelli NJ, and Black JD (1998). Co-lonocyte differentiation is associated with increased expression and altered distribution of protein kinase C isozymes. Gastroenterology 115, 75–85. [DOI] [PubMed] [Google Scholar]
- Wang H, Douglas W, Lia M, Edelmann W, Kucherlapati R, Podsypanina K, Parsons R, and Ellenson LH (2002). DNA mismatch repair deficiency accelerates endometrial tumorigenesis in Pten heterozygous mice. Am. J. Pathol 160, 1481–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Karikomi M, Naidu S, Rajmohan R, Caserta E, Chen HZ, Rawahneh M, Moffitt J, Stephens JA, Fernandez SA, et al. (2010). Allele-specific tumor spectrum in pten knockin mice. Proc. Natl. Acad. Sci. USA 107,5142–5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelt B, Warne PH, Lambros MB, Reis-Filho JS, and Downward J (2013). PI3K pathway dependencies in endometrioid endometrial cancer cell lines. Clin. Cancer Res 19, 3533–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.