

Abstract
Background & objectives:
Insulin resistance associated with hyperinsulinaemia and overexpression of insulin receptors (IRs) have been intricately linked to the pathogenesis and treatment outcomes of the breast carcinoma. Studies have revealed that upregulated expression of IRs in breast cancer pathogenesis regulates several aspects of the malignant phenotype, including cell proliferation and metastasis. This study was aimed to investigate the pivotal role of an IR antagonist S961 on IR signalling and other biological parameters in MCF-7, MDA-MB-231 and T47D cell lines.
Methods:
The effect of human insulin and S961 on growth, proliferation rate and clonogenic potential of breast cancer cells was evaluated by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide] assay and clonogenic assay. The mRNA expression of IR isoforms (IR-A and IR-B) was measured in the breast carcinoma cells using quantitative PCR.
Results:
The study revealed that breast cancer cells predominantly expressed IR-A isoform and showed extensive growth and proliferation owing to IR overexpression. It was found that S961 downregulated the IRs (IR-A and IR-B) with nanomolar dose and efficiently blocked expression of IRs even in the presence of insulin. IR mRNA expression levels were significantly downregulated in the continued presence of S961. S961 also inhibited cellular proliferation and colony formation in breast tumour cells.
Interpretation & conclusions:
IR antagonist, S961 showed distinct antagonism in vitro and appeared to be a powerful therapeutic modality that might provide insight into the pathogenesis of impaired IR signalling.
Keywords: Breast cancer, hyperinsulinaemia, insulin receptor, S961
Breast cancer is the most common malignant disease and second leading cause of cancer mortality in females worldwide after lung cancer. In India, breast cancer has ranked number one cancer among females with an age-adjusted rate as high as 25.8 per 100,000 women and mortality 12.7 per 100,000 women1. Several established risk factors are associated with the pathophysiology of breast cancer; one of them is the metabolic syndrome, which is often associated with hyperinsulinaemia2. Metabolic syndrome induces the change in several hormonal signalling systems, including insulin, insulin-like growth factors (IGFs) and oestrogen that severely affect breast cancer risk3. High level of circulating insulin is regarded as the significant marker of greater breast cancer risk in type 2 diabetes mellitus (T2DM) patients4. The role of hyperinsulinaemia in the development and progression of breast cancer has been shown by several follow up and epidemiological studies5,6. Clinical studies also suggested that hyperinsulinaemia was intricately linked with 10-20 per cent elevated risk of breast cancer7.
Insulin acting as a mitogen promotes neoplasm in the epithelial tissues through insulin receptor (IR) signalling. Insulin signalling initiates at the level of the IR. This receptor occurs in two isoforms, IR-A and IR-B8. The physiological roles of both the IR isoforms are likely based on their different binding affinities for IGF-II, rather than on their slightly different binding affinities for insulin. Isoform IR-A is predominantly expressed during embryogenesis and foetal life and isoform B is expressed in adult differentiated insulin target tissues. While isoform IR-B has a high affinity for insulin and is responsible for most of the metabolic effects of insulin, isoform IR-A has a high affinity additionally for IGF-II and contributes to cell proliferation. Isoform IR-A is aberrantly expressed in many cancer cells8. IRs are overexpressed in several breast cancers resulting into transformation in the phenotype of breast epithelial cells and IR overexpression in breast cancer is positively correlated with malignant phenotype9. Aberrant IR expression favours cancer resistance to both conventional and targeted therapies. Therefore, a clear understanding of IR signalling mechanism has important implications for cancer prevention measures, which should include efficient control of insulin resistance and associated hyperinsulinaemia10. Moreover, IRs could be potential molecular targets for anticancer therapies. Currently, our understanding of these molecular targets is insufficient, but the requirement of modulating the activity of IRs according to specific requirements of the disease must now be considered11. Developing novel and improved therapeutic tools is need of the hour for patients with impaired insulin secretory function that can specifically and selectively target the IRs as an ultra-long-acting modulator, mimic the regulatory actions of insulin and implicate least side effects. A biosynthetic IR antagonist S961 (single-chain peptide of 43 amino acids) has been shown to exhibit high affinity and selectivity for the IR, along with in vitro and in vivo activity12. Another study has demonstrated that S961 has a potential to improve glucose disposal through receptor-independent mechanisms providing direct evidence to the crucial role in IR signalling13.
In the present study, an attempt was made to investigate the effect of IR antagonist S961 on the expression of IRs in three diverse breast carcinoma cell lines MCF-7, MDA-MB-231 and T47D. Furthermore, the effect of S961 on the insulin-induced cell proliferation and colony formation of breast tumour cells was also explored.
Material & Methods
The cell culture chemicals were purchased from Gibco, Invitrogen (USA). MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide] was procured from Molecular Probes, Life Technologies (USA). Primers were purchased from Imperial Life Sciences (P) Limited, New Delhi). Kits for RNA isolation and cDNA synthesis were purchased from Ambion (Thermo Fisher Scientific, USA). SYBR green was obtained from Applied Biosystems (USA). IR antagonist S961 was kindly provided by Dr. Lauge Schäffer (Novo Nordisk, Copenhagen, Denmark).
Cell culture: This study was conducted at Cell culture laboratory, Centre for Biosciences, Central University of Punjab, Bhatinda, Punjab, India, during May 2012-August 2014. Diverse human breast carcinoma cell lines with variable genetic and hormonal background (MCF-7, MDA-MB-231 and T47D) were obtained from the cell repository of the National Centre for Cell Sciences (NCCS), Pune, Maharashtra, India. MCF-7, MDA-MB-231 and T47D cells were cultured and maintained in Dulbecco's modified eagle's medium (DMEM), L-15 medium and RPMI 1640 medium (Gibco, Invitrogen, USA), respectively, supplemented with 10 per cent foetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin. MCF-7 and T47D cells were incubated in a humidified atmosphere of 5 per cent CO2 at 37°C, while MDA-MB-231 cells were maintained at 37°C in CO2 free atmosphere.
Cell proliferation assay: The MTT assay was used to evaluate the influence of human insulin on the proliferation of MCF-7, MDA-MB-231 and T47D cell lines. The insulin-mediated proliferation rate of cells treated with 1 or 10 nM of the S961 antagonist for 24 h was also established. Briefly, cells were plated into 96-well flat-bottomed plates (Corning, USA) at a density of 1×104 cells per well and incubated for 24 h at 37°C to allow surface attachment. Cells were synchronized by serum starvation and stimulated for 24 h in their respective media containing 0.1 per cent FBS supplemented with increasing concentrations of insulin (10 and 100 nM). In another set of experiment, two hours before insulin stimulation, the media was replaced with starvation medium containing S961 at the concentrations at 1 or 10 nM. The cells were incubated at 37°C in the humidified atmosphere for two hours, and then insulin was administered, and the assay was carried out for 24 h. After 24 h, the medium was removed and fresh MTT solution (0.5 mg/ml in medium) was added per well and cells were incubated at 37°C in the dark for four hours. The supernatant was discarded and the reduced MTT, formazan crystals formed by metabolically active cells were solubilized in dimethyl sulphoxide (DMSO). The absorbance was measured at 570 nm using microplate reader (Synergy H1, BioTek, USA). Results were expressed as the average of three independent experiments.
Colony formation assay: Cells were seeded into 6-well flat-bottom plates at a density of 800 cells/well in two ml of culture medium containing 10 per cent FBS. After 24 h, cultures were replaced with fresh medium containing 0.5 per cent FBS (control), insulin (100 nM), and insulin (100 nM) in the continued presence of S961 (10 nM) and grown for two weeks in 37°C humidified atmosphere. The cell clones were fixed with the methanol-acetic acid solution (3:1v/v) for five minutes. Fixed cells were stained with 0.5 per cent crystal violet and 25 per cent methanol solution for 15 min, followed by three to five rinses with tap water to remove excess dye. The colony numbers were counted by ImageJ software (National Institutes of Health, USA).
Cellular morphological analysis: For cytomorphological analysis, 2×105 cells were seeded in 6-well culture plate and treated for 24 h with 0.5 per cent FBS supplemented media (control), insulin (100 nM) and insulin (100 nM) in the presence of antagonist S961 (10 nM). Cells were photographed with phase-contrast microscope (Magnus Analytics, New Delhi) at ×100 magnification.
RNA isolation and cDNA synthesis: Total RNA was extracted from untreated control cells and cells treated with insulin for 24 h in the continued presence of S961, using the RNA isolation kit according to the manufacturer's instructions. The concentration and purity of RNA samples were measured using the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA). For complementary DNA synthesis, 1 μg of total RNA was reverse transcribed by cDNA synthesis kit using M-MULV reverse transcriptase, random hexamers and oligo (dT). The whole process yielded 20 μl of cDNA, which was further processed for quantitative PCR (q-PCR) analysis.
Quantitative polymerase chain reaction (q-PCR): The effect of IR analogue on IR-A and IR-B expression was assessed by q-PCR14, performed using SYBR Green Master Mix on Applied Biosystems StepOnePlus Real-Time System. The 20 μl PCR mixture contained 10 μl of ×2 SYBR Green Master Mix, 2 μl of 10× cDNA sample and primers sequences of human beta-actin (5’- CATTAAGGAGAAGCTGTGCT-3’ and 3’-TTGAAGGTAGTTTCGTGGAT-5’), IR A (5’-GGAAGAGACTGGCACTGAGG-3’ and 3’-CTGACGGGGACAACTCATCT-5’) and IR B (5’-CTACCTGCGCAAGCAGAAG-3’ and 3’-TTGATGTTCAGGCAGCAGTC-5’). The amplification conditions consisted of an initial incubation at 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec, 58°C for 30 sec and 72°C for 30 sec. The fluorescence threshold cycle (CT) values for all specified genes were normalized against CT values for endogenous control beta-actin and a relative fold change in mRNA expression with respect to a control sample was calculated by the 2−ΔΔCT method15.
Statistical analysis: Statistical analysis was performed using Sigma plot statistical software (Systat Software Inc., USA). Student's t test was used to examine the difference between the two groups. For multiple groups, one-way analysis of variance (ANOVA) followed by post hoc Tukey's test was performed.
Results
Effect of insulin and S961 on proliferation of breast cancer cells: Insulin treatment led to significant increase in the growth and proliferation of 24 h serum-starved breast cancer cells in a concentration-dependent manner. Insulin treatment (10 to 100 nM) increased the MCF-7 cells proliferation by 2.75 to 4.73 folds over control (Fig. 1A). A similar type of trend was observed in MDA-MB-231 (1.07 fold at 100 nM insulin) and T47D cells (1.47 fold at 100 nM insulin) as the proliferation rate was increased over control in response to insulin (Fig. 1B and C). However, all the three cell lines demonstrated differences in their individual cell proliferative effect towards insulin treatment. MCF-7 was found to be the most sensitive and MDA-MB-231 be the least sensitive cell line towards insulin treatment among all tested cancer cell lines. To access the role of IR in breast carcinoma cells proliferation, cells were treated with IR antagonist S961. The tumour cells growth rate was significantly reduced in the S961-treated cells compared to control cells. Insulin antagonist S961 effectively inhibited insulin-influenced cell proliferation in vitro. At a concentration of 10 nM of S961, the growth rate of MCF-7, MDA-MB-231 and T47D cells were inhibited up to 82, 47 and 42 per cent, respectively, as compared to insulin-treated cells (100 nM), in 24 h cell proliferation assay (Fig. 1A-C).
Fig. 1.
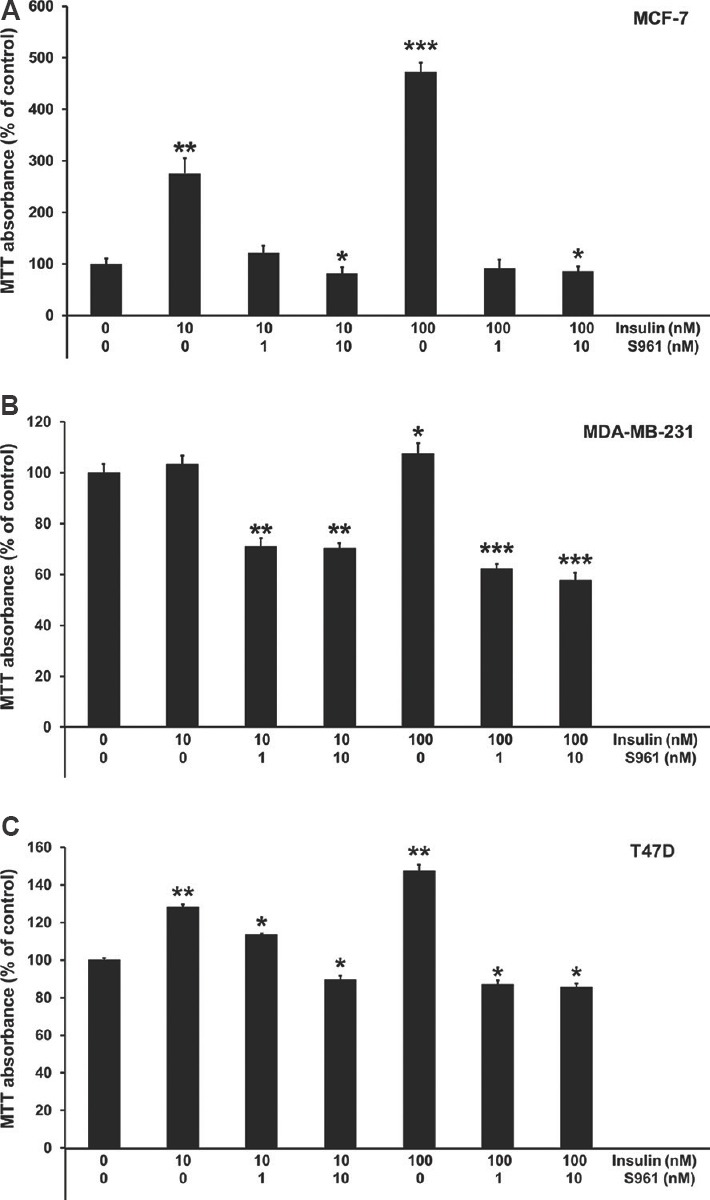
Effect of human insulin (10 and 100 nM) and insulin receptor antagonist S961 (1 and 10 nM) on proliferation of MCF-7 (A), MDA-MB-231 (B) and T47D (C) cells measured by MTT assay. All data are represented as means±standard deviation, n=3. P *≤0.05, **≤0.01 and ***≤0.001 vs control.
Effect of S961 on clonogenic potential of breast cancer cells: All cell lines showed an increase in cell colonies when treated with insulin (100 nM) as compared to control cells. However, S961 reduced insulin-induced colony formation. The numbers of colonies formed were reduced in the presence of S961 (10 nM) in all three cell lines, as shown in Table I.
Table I.
Total number of colonies formed by breast tumour cells in response to insulin and S961

On treatment with S961 (10 nM), colony formation was reduced up to 34, 16 and 26 per cent in MCF-7, MDA-MB-231 and T47D cells, respectively, as compared to the untreated controls. Of the three cell lines examined, S961 was somewhat less active against MDA-MB-231.
Cytomorphological analysis: The cytomorphological examination of breast cancer cells showed that cells were in their typical epithelial morphology in untreated controls of MCF-7, MDA-MB-231 and T47D (Fig. 2). Excessive increase in cell number was observed as cells were treated with 100 nM insulin. However, treatment with IR antagonist S961 (10 nM) efficiently reduced the cell density even in the presence of mitogenic insulin (100 nM).
Fig. 2.

Cytomorphological analysis. MCF-7, MDA-MB-231 and T47D cells were treated with insulin (100 nM) alone, or insulin (100 nM) + S961 (10 nM) for 24 h. The cells were photographed by phase-contrast microscope (×100).
Effect of S961 on the insulin receptor isoforms expression: Fig. 3A-C shows bar plot visualization of log10 fold change in expression levels of IR isoforms (IR-A and IR-B) in response to S961 in MCF-7, MDA-MB-231 and T47D cells, respectively. qRT-PCR analysis revealed that IR isoform expression levels were markedly inhibited by S961 (1 nM and 10 nM), compared to their respective insulin-treated group. Under standard culture condition, all three breast cancer cell lines predominantly expressed IR-A isoform as compared to the IR-B. S961 at 1 and 10 nM concentrations downregulated IR-A expression up to 1.027 and 3.925 log10 folds, respectively, in MCF-7 cells as compared to their insulin-treated counterparts. The same scenario was observed in MDA-MB-231 and T47D cells, where S961 significantly decreased expression of IR-A by 1.121 and 3.14 log10 folds at 10 nM doses, respectively.
Fig. 3.
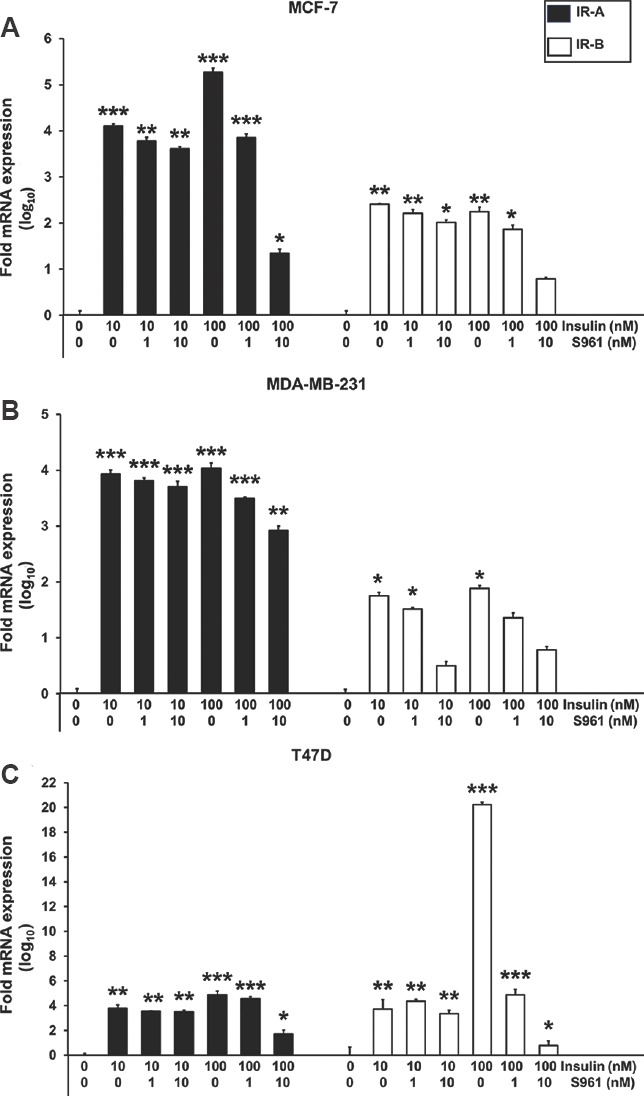
Effects of insulin and S961 on insulin receptor (IR-A and IR-B) mRNA expression in breast cancer cells. The mRNA levels of insulin receptor isoforms in MCF-7 cells (A) MDA-MB-231 cells (B) and T47D cells (C) were measured by quantitative PCR analysis. All data are represented as means±standard deviation, n=3, P*< 0.05, **<0.01 and ***<0.001 vs control.
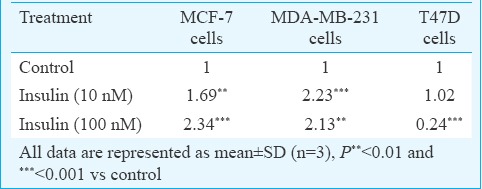
Expression profiling of IR-B isoform demonstrated that the IR-B was exclusively suppressed by 1.46, 1.1 and 19.46 log10 folds in response to S961 (10 nM) in MCF-7, MDA-MB-231 and T47D cells, respectively, as compared to their insulin-treated control counterparts. To evaluate relative expression levels, ratios between both IR isoforms (IR-A/IR-B) were calculated (Table II). On analyzing the data, the ratio of IR isoform A to B was found significantly different in favour of IR isoform A in most of the instances.
Table II.
Insulin receptor (IR)-A/IR-B ratio of the analyzed mRNA expressions
Discussion
Insulin resistance, hyperinsulinaemia and changes in the signalling of growth hormones elevate the risk of breast cancer. Both IR and IGF1R belong to tyrosine kinase receptor subfamily, sharing great structural homology, especially in the tyrosine kinase domain16. IR activates the PI3K/Akt and mitogen-activated protein kinase (MAPK) signalling pathways, regulating several fundamental processes such as cell cycle, cell migration, proliferation, differentiation, metabolism and body growth17,18. Insulin induces mitogenic impact on cellular proliferation in the breast tissue. In the present study, it was found that breast carcinoma cells were responsive to insulin, elevating their growth and proliferation to several folds. This elevation may be attributed to upregulation of insulin signalling, owing to overexpression of IR. The data indicated that all the breast cancer cell lines demonstrated a significant mitogenic response to 100 nM insulin. However, each cell line showed a differential pattern in their proliferation response to insulin. The mitogenic response was different in different cells. The possible reason behind this differential response pattern of breast tumour cells lies in their heterogeneous molecular and functional characteristics and metabolic phenotypes. MCF-7 (oestrogen/progesterone receptor positive) and T47D (oestrogen/progesterone/androgen receptor positive) cell lines are non-invasive, oestrogen-dependent, hormone-responsive breast cancer cell lines, whereas MDA-MB-231 [oestrogen/progesterone/human epidermal growth factor receptor (HER-2) negative] is a triple-negative, highly invasive, oestrogen-independent, hormone-insensitive, late-stage breast cancer cell line19.
IR exists in two isoforms - IR-A and IR-B, generated by alternative splicing of the 36-base pair exon 11. IR-B is associated with stronger insulin binding, while isoform IR-A binds both insulin and IGF20. Studies found IR-A often aberrantly expressed in tumour cells, thus increasing its responsiveness to insulin and IGF and explaining the cancer-promoting effect of hyperinsulinaemia observed in type 2 diabetes patients. In comparison, IR-B is less mitogenic than IR-A, enhancing the metabolic effects of insulin8,21. In the current study, IR-A was overexpressed comparatively higher to IR-B in breast cancer cells. Furthermore, the ratio between IR-A and IR-B was found in favour of isoform IR-A suggesting that in the breast cancer cells a higher IR-A to IR-B ratio showed a positive correlation with their responsiveness to insulin-induced cell growth owing to the high mitogenic activity of IR-A as compared to IR-B.
Over the past several years, most efforts were invested in developing strategies and tools for specific targeting the IGF1R in cancer therapy, sparing the IR, due to concern about metabolic abnormalities secondary to IR inhibition4. Several phase 2 and phase 3 randomized clinical trials using anti-IGF1R-specific antibodies failed to deliver any benefit to cancer patients and ended with unacceptable side effects and limited efficacy22. One mechanistic explanation for this failure is that the IR may deliver mitogenic signals independently by acting as a compensatory pathway to IGF1R loss and IGF1R inhibition23,24.
Studies showed crosstalk between tyrosine kinase receptors induced resistance towards tumour therapy when specific targeting of only a single receptor was performed25. Zhang et al26 exhibited an enhancement in IR signalling following IGF1R downregulation in several breast cancer cell lines, indicating the necessity to target both receptors in order to disrupt the malignant phenotype. Another study has demonstrated a reduction in mammary tumour growth rate by dual inhibition of both IR and IGF1R using short hairpin RNA technology4. Another similar study has concluded that targeting of both IGF1R and IR function inhibits proliferation in endocrine-resistant breast cancer cells27. An improved understanding of the IR expression and function involving signal diversification could lead to the development of novel therapeutic options for management of hyperinsulinemia-induced tumour progression.
The significant outcome of the present study was that S961 markedly reduced the hyperelevated insulin levels and improved insulin sensitivity. A significant decrease was observed in the insulin concentration-proliferation response after S961 treatment. Furthermore, S961 demonstrated both rapid and sustained antagonism of the IR, downregulating IRs expression with a single treatment in vitro. Downregulation of IR by S961 may result in inhibition of cell proliferation and suppression of clonogenic potential of tumour cells. The results, therefore, indicate that IR antagonist S961 may have the potential to be a novel agent for the regulation of hyperinsulinemia and IR. The major limitation of the study was the lack of advanced experimental set-up that is, Western blotting to make the mechanistic explanation of signalling mechanism involved in the biological responses of the S961 more reliable and authentic. Specific changes regarding the effect of S961 on IR turnover would require further investigation and would be of considerable interest for future studies.
Acknowledgment
The authors thank Dr Lauge Schäffer (Novo Nordisk, Copenhagen, Denmark) for providing IR antagonist S961 as a gift sample.
Footnotes
Financial support & sponsorship: This work was financially supported by the Central University of Punjab, Bathinda, India as part of Ph.D. thesis. The first author (PS) acknowledges Central University of Punjab for providing financial support as Ph.D. fellowship.
Conflicts of Interest: None.
References
- 1.Malvia S, Bagadi SA, Dubey US, Saxena S. Epidemiology of breast cancer in Indian women. Asia-Pac J Clin Oncol. 2017;13:289–95. doi: 10.1111/ajco.12661. [DOI] [PubMed] [Google Scholar]
- 2.Pothiwala P, Jain SK, Yaturu S. Metabolic syndrome and cancer. Metab Syndr Relat Disord. 2009;7:279–88. doi: 10.1089/met.2008.0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xue F, Michels KB. Diabetes, metabolic syndrome, and breast cancer: A review of the current evidence. Am J Clin Nutr. 2007;86:s823–35. doi: 10.1093/ajcn/86.3.823S. [DOI] [PubMed] [Google Scholar]
- 4.Rostoker R, Abelson S, Bitton-Worms K, Genkin I, Ben-Shmuel S, Dakwar M, et al. Highly specific role of the insulin receptor in breast cancer progression. Endocr Relat Cancer. 2015;22:145–57. doi: 10.1530/ERC-14-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, et al. Diabetes and cancer: A consensus report. Diabetes Care. 2010;33:1674–85. doi: 10.2337/dc10-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peairs KS, Barone BB, Snyder CF, Yeh HC, Stein KB, Derr RL, et al. Diabetes mellitus and breast cancer outcomes: A systematic review and meta-analysis. J Clin Oncol. 2011;29:40–6. doi: 10.1200/JCO.2009.27.3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alimova IN, Liu B, Fan Z, Edgerton SM, Dillon T, Lind SE, et al. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle. 2009;8:909–15. doi: 10.4161/cc.8.6.7933. [DOI] [PubMed] [Google Scholar]
- 8.Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev. 2009;30:586–623. doi: 10.1210/er.2008-0047. [DOI] [PubMed] [Google Scholar]
- 9.Sciacca L, Costantino A, Pandini G, Mineo R, Frasca F, Scalia P, et al. Insulin receptor activation by IGF-II in breast cancers: Evidence for a new autocrine/paracrine mechanism. Oncogene. 1999;18:2471–9. doi: 10.1038/sj.onc.1202600. [DOI] [PubMed] [Google Scholar]
- 10.Yee D. Insulin-like growth factor receptor inhibitors: Baby or the bathwater? J Natl Cancer Inst. 2012;104:975–81. doi: 10.1093/jnci/djs258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vigneri R, Squatrito S, Frittitta L. Selective insulin receptor modulators (SIRM): A new class of antidiabetes drugs? Diabetes. 2012;61:984–5. doi: 10.2337/db12-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schäffer L, Brand CL, Hansen BF, Ribel U, Shaw AC, Slaaby R, et al. A novel high-affinity peptide antagonist to the insulin receptor. Biochem Biophys Res Commun. 2008;376:380–3. doi: 10.1016/j.bbrc.2008.08.151. [DOI] [PubMed] [Google Scholar]
- 13.Vikram A, Jena G. S961, an insulin receptor antagonist causes hyperinsulinemia, insulin-resistance and depletion of energy stores in rats. Biochem Biophys Res Commun. 2010;398:260–5. doi: 10.1016/j.bbrc.2010.06.070. [DOI] [PubMed] [Google Scholar]
- 14.Sharma P, Kumar S. Metformin inhibits human breast cancer cell growth by promoting apoptosis via a ROS-independent pathway involving mitochondrial dysfunction: Pivotal role of superoxide dismutase (SOD) Cell Oncol (Dordr) 2018 doi: 10.1007/s13402-018-0398-0. doi: 10.1007/s13402-018-0398-0. [DOI] [PubMed] [Google Scholar]
- 15.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protoc. 2008;3:1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 16.Wang CF, Zhang G, Zhao LJ, Qi WJ, Li XP, Wang JL, et al. Overexpression of the insulin receptor isoform A promotes endometrial carcinoma cell growth. PLoS One. 2013;8:e69001. doi: 10.1371/journal.pone.0069001. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Arcaro A, Guerreiro AS. The phosphoinositide 3-kinase pathway in human cancer: Genetic alterations and therapeutic implications. Curr Genomics. 2007;8:271–306. doi: 10.2174/138920207782446160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Munshi A, Ramesh R. Mitogen-activated protein kinases and their role in radiation response. Genes Cancer. 2013;4:401–8. doi: 10.1177/1947601913485414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh B, Rani M, Singh J, Moudgil L, Sharma P, Kumar S, et al. Identifying the preferred interaction mode of naringin with gold nanoparticles through experimental, DFT and TDDFT techniques: Insights into their sensing and biological applications. RSC Advances. 2016;6:79470–84. [Google Scholar]
- 20.Malakar P, Chartarifsky L, Hija A, Leibowitz G, Glaser B, Dor Y, et al. Insulin receptor alternative splicing is regulated by insulin signaling and modulates beta cell survival. Sci Rep. 2016;6:31222. doi: 10.1038/srep31222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belfiore A, Frasca F. IGF and insulin receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia. 2008;13:381–406. doi: 10.1007/s10911-008-9099-z. [DOI] [PubMed] [Google Scholar]
- 22.Ekyalongo RC, Yee D. Revisiting the IGF-1R as a breast cancer target. NPJ Precision Oncology. 2017;1:14. doi: 10.1038/s41698-017-0017-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Y, Yee D. Targeting insulin and insulin-like growth factor signaling in breast cancer. J Mammary Gland Biol Neoplasia. 2012;17:251–61. doi: 10.1007/s10911-012-9268-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guha M. Anticancer IGF1R classes take more knocks. Nat Rev Drug Discov. 2013;12:250. doi: 10.1038/nrd3992. [DOI] [PubMed] [Google Scholar]
- 25.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 26.Zhang H, Pelzer AM, Kiang DT, Yee D. Down-regulation of type I insulin-like growth factor receptor increases sensitivity of breast cancer cells to insulin. Cancer Res. 2007;67:391–7. doi: 10.1158/0008-5472.CAN-06-1712. [DOI] [PubMed] [Google Scholar]
- 27.Chan JY, LaPara K, Yee D. Disruption of insulin receptor function inhibits proliferation in endocrine-resistant breast cancer cells. Oncogene. 2016;35:4235–43. doi: 10.1038/onc.2015.488. [DOI] [PMC free article] [PubMed] [Google Scholar]