

Abstract
Transmission of multiple founder variants has been associated with faster HIV disease progression. Many studies have attempted to determine the number of founder variants, mainly by analysis of sequence diversity and/or tree topology from acutely HIV-infected individuals. We hypothesized that adding sequence data collected from source partners might improve resolution and characterization of transmission events. Blood plasma samples were collected from both the source and recipient in thirty epidemiologically- and phylogenetically linked transmission pairs. All were men who have sex with men, sampled on average 70 days (range 11–170) after the recipient’s estimated date of infection. Next generation sequencing (454 FLX, Roche) of HIV-1 env (C2-V3) was performed for all samples. Inspection of sequence alignments, highlighter plots, phylogenetic tree topologies and sequence diversity were used to determine the multiplicity of founder viruses with and without the inclusion of source data. Using only recipient sequence data, we were able to resolve multiplicity in twenty-six of the thirty transmission pairs (87 percent). Among them, five presented with a high viral diversity at baseline (>0.10 subst/site), consistent with multiple founders. By incorporating sequence data collected from the source partner, we were able to characterize all thirty transmission pairs. Overall, sixteen transmission events (53.3 percent) involved multiple founders. Results obtained by combining sequence data from recipient and source were congruent for nineteen of the twenty-six (73 percent) cases where conclusions were made using only recipient sequence data. The multiplicity of founders was associated with significantly higher HIV RNA levels (P = 0.04). To further evaluate the transmission bottleneck, we focused on single founder transmissions (fourteen of the thirty), and identified four recipients (28.6 percent) that had founder variants that were inferred to arise from minority viral populations in the source. These source clades ranged from 1.0 to 5.4 percent of the sampled population. Incorporating sequence data from the source increased of the ability to determine the multiplicity of founder variants, reduced misclassification, and allowed us to infer the transmission of minority variants.
Keywords: HIV transmission, MSM, founder, deep sequencing
Introduction
A single HIV founder has been reported to establish 80 percent of infections after heterosexual (HTS) transmission (Keele et al. 2008; Abrahams et al. 2009; Haaland et al. 2009), while only 40 percent of transmissions in injection drug users are established by a single founder (Bar et al. 2010). During HIV transmission among men who have sex with men (MSM), reports are conflicting, with some demonstrating establishment of infection by single founders only 60 percent of the time (Keele et al. 2008; Li et al. 2010), while others report similar rates among MSM and HTS ( Gottlieb et al. 2008; Herbeck et al. 2011; Rolland et al. 2011). These differences are important because a greater number of founders during HIV transmission may lead to higher viral loads and faster disease progression (Janes et al. 2015). However, the method used to infer the number of viral founders can affect such conclusions. An inherent limitation in studies attempting to address the multiplicity of HIV-1 transmission variants is the numerous logistical challenges in obtaining clinical samples during the acute and early HIV-1 infection. Like all finite populations, HIV population loses genetic variation through time due to the combined effects of genetic drift and selection that occurs shortly after infection (Lynch and Conery 2003; Lemey et al. 2006a, b). Therefore, the number of transmitted lineage will always decrease after infection limiting our ability to infer the multiplicity of founder variants. In addition, most previous studies have estimated the number of founders based only on the analysis of HIV RNA populations sequenced from acutely HIV-infected individuals, i.e. the recipients (Keele et al. 2008; Salazar-Gonzalez et al. 2008; Abrahams et al. 2009; Li et al. 2010; Novitsky et al. 2011; Janes et al. 2015). In this analysis, we utilized deep sequencing (454 FLX, Roche) of HIV-1 env (C2-V3) to characterize HIV RNA populations from 30 epidemiologically- and phylogenetically linked source and recipient MSM partner pairs sampled shortly after infection. Our goal was to determine if the addition of sequence data from the source could help to more accurately determine the multiplicity of founder variants.
Materials and methods
Ethics statement
The UCSD Human Research Protections Program approved the study protocol, consent, and procedures for consent. All study participants provided voluntary, written informed consent before any study procedures were undertaken.
Study population
A total of 30 phylogenetically and epidemiologically linked MSM transmission pairs infected with HIV-1 subtype B, without evidence of superinfection were recruited from the San Diego Primary Infection Resource Consortium (SDPIRC) (Butler et al. 2010). In order to identify transmission pairs, all individuals diagnosed with primary HIV infection and enrolled in the SDPIRC were asked to recruit their most recent sexual partners, and these individuals were screened for evidence of a phylogenetically related infection. The identification of transmission pairs were performed as previously described (Novitsky et al. 2011) (Supplementary Fig. S1). The putative source partner in each transmission pair was inferred on the basis of the estimated dates of infection (EDI) of both partners using a series of well-defined stepwise rules to characterize stages of infection based on serologic and virologic criteria, as described by Le et al. (2013) (and summarized in Supplementary Table S1). For each recipient, blood samples were collected at the time of recruitment (baseline). Three identified source partners (from pairs P10–11, P15–17, and P27–29) transmitted HIV to more than one recipient partner. For each of the 25 unique source partners, biological samples were collected at the time of recruitment. All source partners had at least one blood sample collected, and a subset of eight had an additional paired semen sample. Since two of these eight individuals infected multiple recipients, seminal plasma from the source partner was available for nine transmission pairs. Semen samples were collected, prepared, and stored, as previously described (Gianella et al. 2012).
Next generation sequencing (NGS) and analysis
HIV RNA was extracted from blood and seminal plasma and NGS of PCR-amplified env C2-V3 (HXB2 coordinates 6928-7344) was performed using the Roche 454 FLX Titanium platform (Basel, Switzerland). For each sample having a minimum of 500 copies/mL, the cDNA template input was calculated assuming 43 percent reverse transcription efficiency and was expressed as the number of templates (log10) in the first round of nested polymerase chain reaction, as previously validated (Wagner et al. 2013). Read (FASTA) and quality score files produced by the 454 instruments were further analyzed using a bioinformatics pipeline.
In brief, high-quality reads were retained and aligned to HXB2 as a reference sequence (without generation of contigs) using an iterative codon-based alignment procedure. Identical sequence reads were clustered, allowing identification of non-redundant sequences. When a cluster contained a minimum of ten identical sequence reads, a haplotype was inferred, and the proportion of reads in each haplotype was collected. The final output consisted of a list of representative haplotypes and their relative frequencies (see Supplementary materials for a full description of the key steps used to generate sequence haplotypes and Supplementary Fig. S4 for the number of reads associated with each haplotype—see supplementary method). For each sample, we also computed the maximum pairwise distance (Tamura-Nei 93) between reads with at least 100 overlapping base pairs (Rolland et al. 2011).
Resolving multiplicity of transmission events
We applied a multi-step approach for resolving the multiplicity of transmission variants by combining previously published methods (Keele et al. 2008; Abrahams et al. 2009; Alizon and Fraser 2013). First from only the recipient sequence data, we analyzed the distribution of maximum pairwise viral diversity at the time of diagnosis, highlighter plots from sequence haplotypes, and maximum likelihood (ML) trees to categorize the multiplicity of founders. Given previous work from Alizon and Fraser (2013) demonstrating an intra-host evolutionary rate for env of 0.015 [range 0.0017–0.05] subs/site/year, we chose a maximum diversity cut-off of twice the maximum measured env evolutionary rate (>0.10 subst./site) to conservatively identify individuals with multiple founders (Fig. 1).
Figure 1.

Distribution of the maximum pairwise diversity of sampled sequences at the time of diagnosis among the thirty recipient partners. The maximum pairwise diversity ranged from 0.01 to 0.41 substitutions/site, with five recipients having distinctly higher maximum diversity (>0.10 substitutions/site [in red]).
For the remaining recipients, ML trees and highlighter plots were used to visually assess whether the multiplicity of transmission could be resolved (Fig. 2 and Supplementary Fig. S2). Second, to determine if the addition of sequence data from the source would affect this categorization, we analyzed the topology of ML trees inferred using paired source and recipient sequences (Fig. 3 and Supplementary Fig. S3). The inferred ML trees were rooted by (1) analyzing the topology of midpoint rooted ML trees created using only the source sequences, (2) identifying the branch closest to the midpoint root, and (3) using this branch to root our combined source and recipient partner tree. Next, we visually inferred the presence and number of phylogenetically distinct clusters of recipient sequences arising from distinct source variants. In the analysis using both recipient and source sequence data, an infection was classified as occurring from a single founder when all HIV sequences from the recipient at baseline formed a monophyletic clade with bootstrap support >70 percent without the interspersion of multiple source partner sequences.
Figure 2.
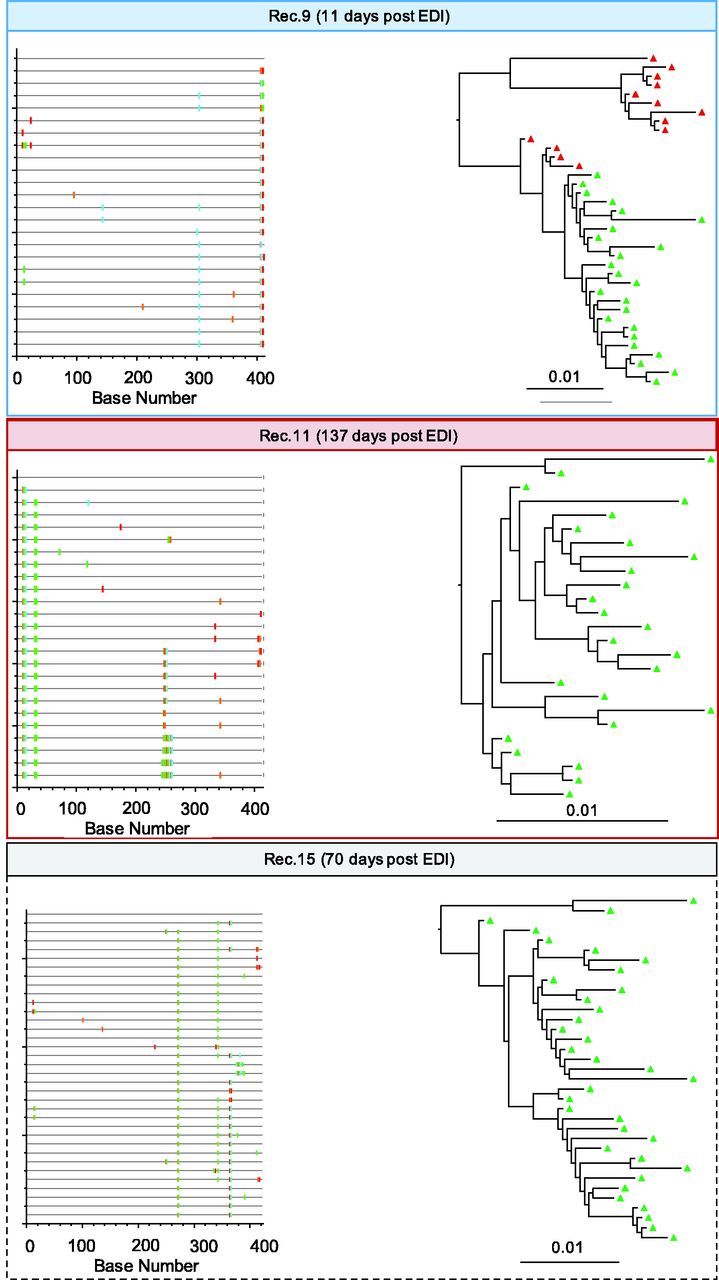
Highlighter plots and ML phylogenetic trees from baseline samples for recipients 9, 11, and 15. Baseline samples were collected after 11, 137, and 70 days respectively. Single (recipient 9) and multiple (recipient 11) founders are inferred based upon highlighter plots, tree topologies, and maximum viral diversity are indicated in red (multiple) and blue (single) squared boxes. Unresolved case (recipient 15) is squared with dashed black lines. ML trees are midpoint rooted. The scale bar represents a genetic distance of 0.01 for all pairs. EDI Estimated date of infection. The entire dataset of thirty recipients is proposed in Supplementary Figure S2.
Figure 3.

ML phylogenetic trees for transmission pairs 9, 11, and 15. Blood plasma haplotypes from the source are indicated in red. Blood plasma haplotypes collected at baseline in the recipients are colored in green. Multiple and single founders are inferred based upon highlighter plots, tree topologies and viral diversity are indicated in red (multiple) and blue (single squared boxes. The scale bar represents a genetic distance of 0.01 for all pairs. The entire dataset of 30 pairs is proposed in Supplementary Figure S3.
Characteristics of the transmitted HIV variants
In the infections in which a single founder variant was inferred, we examined the proportional representation of this variant within the sampled source population. To do this, we estimated the relative frequencies of each haplotype within the source population and ranked them based on proportional representation within the viral population (Fig. 4). Within the source viral population, haplotypes were conservatively defined as minority variants when they represented <10 percent of the observed viral population. Transmission of a minority variant was inferred when a minority source variant was most closely related to the recipient’s viral population.
Figure 4.
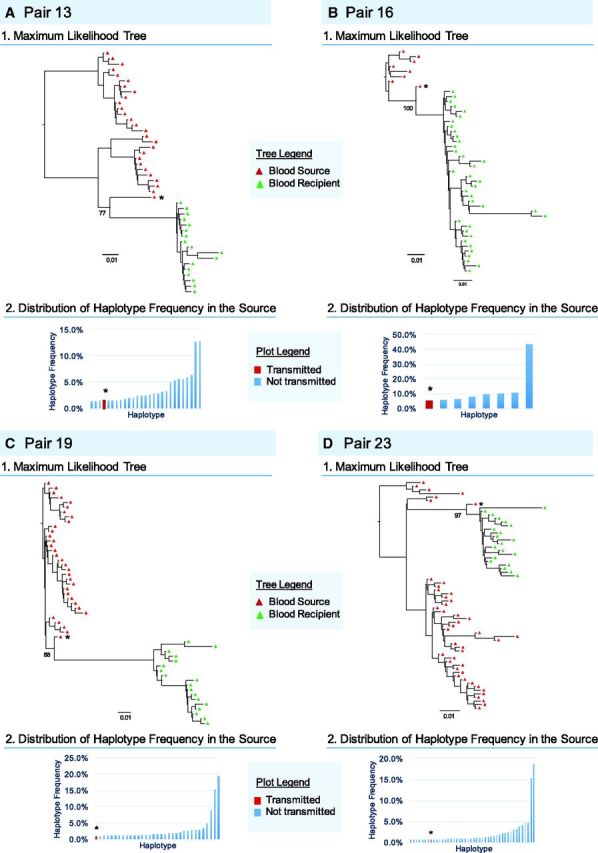
Characteristics of single transmitted HIV-1 Variants. Aligned haplotypes derived from NGS env nucleotide sequences for linked sources (blue and red triangle representing sequences derived from seminal and blood plasma RNA respectively) and recipients (green triangle) were used to generate ML trees for individual transmission pairs. Bootstrap support values are indicated. Distribution and ranking position of the transmitted (red) and not transmitted (blue) haplotypes are indicated for each source. Single transmitted founder variants were defined by a maximum pairwise diversity of <0.10 substitutions/site and a monophyletic clade visualized on the tree topology with a minimum bootstrap support of 70 percent. * The most closely related (transmitted) source variant haplotype to the recipient viral population.
Statistical analyses
Statistical analyses were performed using GraphPad Prism version 6.0c (GraphPad Software, San Diego, CA).
Results
The presence of a genetic bottleneck has been well documented during sexual transmission of HIV (Keele et al. 2008; Salazar-Gonzalez et al. 2008; Abrahams et al. 2009; Haaland et al. 2009), but the use of sequence data only from recipients may not be able to fully characterize the founding viral population in these individuals. Here, we used a uniquely well-characterized cohort of thirty phylogenetically- and epidemiologically linked MSM transmission partners to evaluate whether inclusion of source sequence data may improve our ability to deduce the multiplicity of founder HIV strains.
All thirty transmission pairs were made up of MSM, infected with HIV-1 subtype B, and were antiretroviral naïve. Among recipients, baseline blood samples were available within a mean of 70 days (range 11–170) after EDI. Mean age at baseline was 35 years (range 20–59). Mean HIV RNA level and CD4 T-cell count were 5.02 log10/mL (2.53–7.19) and 553 cells/mm3 (248–1,382), respectively, at the time of sample collection. Mean elapsed time between collection of paired source and recipient blood was 13.8 days (range 0–59 days) (Supplementary Table S2). Among source partners, the mean age was 34 years (22–51), mean CD4 T-cell count was 403 cells/mm3 (7–821), the mean HIV RNA level in blood was 4.89 log10/mL (2.60–6.17), and the mean HIV RNA in the semen was 4.07 log10/mL (1.64–6.14) at the time of sample collection (Supplementary Table S3). As expected, the phylogeny of the entire data set (Supplementary Fig. S1) showed well-supported (bootstrap >0.70) monophyletic subtrees for each transmission pair corroborating linkage defined epidemiologically by self-report.
We first used sequence data only from recently infected individuals to resolve the multiplicity of founders. Among these recently infected recipients (mean: 64 days after EDI [range 11–170]), maximum pairwise diversity ranged from 0.01 to 0.41 substitutions/site. We found no association between the maximal diversity at baseline and the time from EDI. For five recipients (Pairs 6, 11, 14, 26, and 27), the maximum diversity at time of diagnosis was distinctly higher (>0.10 subst/site), consistent with multiple founders. In the remaining twenty-five recipients, tree topologies and highlighter plots were used to assess multiplicity, allowing classification of twenty-one additional transmissions (Fig. 2 and Supplementary Fig. S2). Overall, by using sequence data from only the recipient partners, we were unable to deduce the multiplicity of founders for four cases (13.3 percent) (Recipients 15, 17, 25, and 28).
Discrepant results due to differences in methodological approaches have hindered a clear understanding of multivariant transmission. Benefitting from our uniquely characterized cohort of thirty phylogenetically and epidemiologically linked HIV-transmission pairs, we analyzed combined sequence data from the source and recipient in each transmission pair to resolve the multiplicity of founder variants. Incorporating sequence data generated from HIV RNA in both the semen (n = 9) and blood (n = 30) of the source partner, the multiplicity of founders was resolved in all thirty transmission pairs (Fig. 3 and Supplementary Fig. S3). Overall, sixteen transmissions (53.3 percent [36.1–69.8 percent CI]—Wilson 1927) involved multiple founders, an estimate similar to previous reports in MSM which evaluated data only from the recipients (36 percent, ten of twenty-eight in Li et al. 2010). Similarly, while Keele et al. (2008) reported an overall 24 percent of multiple founders (twenty of one hundred and two) in an diverse population of HTS (n = 82) and MSM (n = 20), they noted that among MSM, half had been acutely infected by more than one virus strain from their HIV-infected partner. In contrast, other studies have only observed multiple founder infections in ∼20 percent of MSM transmissions (one of nine—Herbeck et al. 2011; five of thirty-seven—Gottlieb et al. 2008, and sixteen of sixty-five—Rolland et al. 2011) of HTS transmission (22 percent, fifteen of sixty-nine in Abrahams et al. 2009).
Altogether, we found only moderate agreement between the two approaches (i.e. with and without source data) [73 percent congruent (k = 0.46, 95 percent CI = 0.12–0.8)]. More precisely, incorporating sequence data from the source partners led us to reclassify seven infections (23.3 percent), four reclassified from multiple to single founders (recipients 11 16, 18, and 24) and three from single to multiple founders (recipients 8, 10, and 31) (see Fig. 5). Interestingly, we also found that the inferred presence of multiple founders was associated with significantly higher viral load at the time of diagnosis after correction for time from the estimated date of infection (P = 0.04), consistent with a previous report (Janes et al. 2015).
Figure 5.
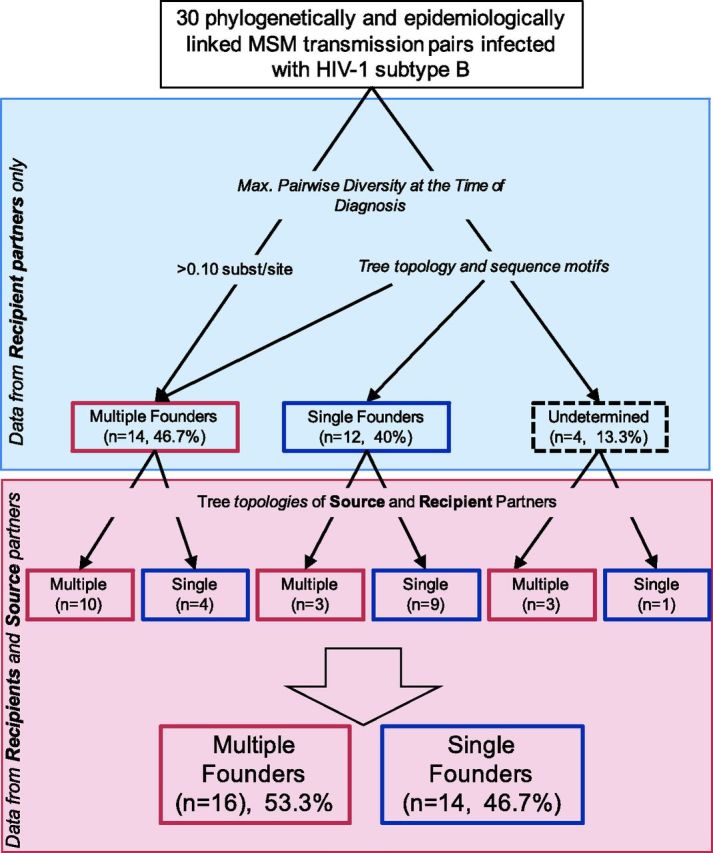
Decision tree to demonstrate how the multiplicity of HIV founder variants in 30 MSM transmission pairs was assigned. Classification (single vs multiple founder) obtained with the sequence data from the recipient only (blue) and with inclusion of the data from the source partner (red) are presented in top and bottom parts of the figure, respectively.
Having data from the source partner in each transmission pair allowed us to infer the viral population in the source from which founder variants may have arisen. Focusing on the fourteen infections with a single founder, we identified four recipients (28.6 percent) whose founders arose from minority viral populations in the source (blood or semen). These four founders were inferred to arise from populations estimated to comprise between 1.0 and 5.4 percent of the sampled viral population (Fig. 4) (Keele et al. 2008).
Discussion
An important limitation of this analysis is the limited sequence length of the analyzed fragments, which reduces the phylogenetic resolution of our data. Thus, while we were able to infer the presence of a single versus multiple founders, in order to avoid over interpreting the data, we did not attempt to quantify the number of founders in each pair. While multiple factors have been hypothesized to be involved in the selection of founder variants (Salazar-Gonzalez et al. 2008; Abrahams et al. 2009; Haaland et al. 2009), the limited length of the available env sequences also did not allow us to fully characterize any sequence signatures associated with this transmission bottleneck. Moreover, given that a period of time elapsed between transmission and sampling of recipient partner’s blood (range 11–170 days, median = 70), as well as between sampling of recipient and the source partner’s blood (range 0–59 days, median = 10), it is possible that unobserved and ongoing selective pressures and other sampling-related biases may have driven the observed level of genetic divergence and impacted our observations (Carlson et al. 2014). Other limitations of our approach include possible sampling bias since viral populations are dynamic while sampling is static, and biased measurement of viral variants during haplotype reconstruction, although the relative abundance of haplotypes is usually preserved (Jayasundara et al. 2014). Finally, we did not have enough sample to compare our approach to alternative sequencing methods.
Methods used to determine the multiplicity of founders have varied between studies (Gottlieb et al. 2008; Abrahams et al. 2009; Li et al. 2010; Herbeck et al. 2011; Novitsky et al. 2011; Rolland et al. 2011; Janes et al. 2015) but nearly all human studies have only used data generated from recipients. Although source partners are difficult to identify, and thus difficult to obtain sequence data from, we hypothesized that the addition of source data could provide insight into the dynamics of HIV-1 transmission among MSM. The results from this study suggest that including data from both source and recipient partners increases the accuracy in the determination of multiplicity of founders, and suggests that we may be underestimating the frequency of infections with multiple founders in this population. Altogether, incorporating sequence data from the source partner increased sensitivity of identifying multiplicity of founders, reduced misclassification, and allowed the identification of transmission of minority variants. While source data may not always be available, this study suggests analysis of recipient partner sequence data alone may not provide a complete picture of HIV transmission.
Supplementary data
Supplementary data are available here.
Supplementary Material
Acknowledgements
We are grateful to Martin Hoenigl for his assistance with the data collection, Caroline Ignacio and Parris Jordan for their technical support, and Demetrius Dela Cruz for his administrative assistance, as well as all the participants in the San Diego Primary Infection Research Consortium.
Funding
This work was supported by the Department of Veterans Affairs and grants from the National Institutes of Health AI093163, AI036214, AI027763, MH101012, TR000098-04AI100665, MH097520, DA034978, AI027763, AI068636, MH081482, AmFAR Grant 108537, the California HIV/AIDS Research Program (CHRP) Grant F13SD321, the James B. Pendleton Charitable Trust and the Bettencourt-Schueller Foundation.
Conflict of interest: None declared.
Data availability: The sequencing data are available on the NCBI Sequence Read Archive, accession numbers SAMN04914210 - SAMN04914278.
References
- Abrahams M. R, Anderson J. A., Giorgi E. E. et al. (2009) ‘Quantitating the Multiplicity of Infection With Human Immunodeficiency Virus Type 1 Subtype C Reveals a Non-Poisson Distribution of Transmitted Variants’, Journal of Virology, 83/8: 3556–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizon S.,, Fraser C. (2013) ‘Within-Host and Between-Host Evolutionary Rates Across the HIV-1 Genome’, Retrovirology, 10: 49.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar K. J., Li H., Chamberland A. et al. (2010) ‘Wide Variation in the Multiplicity of HIV-1 Infection Among Injection Drug Users’, Journal of Virology, 84/12: 6241–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler D. M., Delport W., Kosakovsky Pond S. L. et al. (2010) ‘The Origins of Sexually Transmitted HIV Among Men Who Have Sex With Men’, Science Translational Medicine, 2/18: 18re1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson J. M., Schaefer M., Monaco D. C., et al. (2014) ‘HIV Transmission. Selection Bias at the Heterosexual HIV-1 Transmission Bottleneck’, Science, 345/6193: 1245031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianella S., Strain M. C., Rought S. E., et al. (2012) ‘Associations Between Virologic and Immunologic Dynamics in Blood and in the Male Genital Tract’, Journal of Virology, 86/3: 1307–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb G. S., Heath L., Nickle D. C., et al. (2008) ‘HIV-1 Variation Before Seroconversion in Men Who Have Sex With Men: Analysis of Acute/Early HIV Infection in the Multicenter AIDS Cohort Study’, The Journal of Infectious Diseases, 197/7: 1011–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haaland R. E., Hawkins P. A., Salazar-Gonzalez J., et al. (2009) ‘Inflammatory Genital Infections Mitigate a Severe Genetic Bottleneck in Heterosexual Transmission of Subtype A and C HIV-1’, PLoS Pathogens, 5/1: e1000274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbeck J. T., Rolland M., Liu Y., et al. (2011) ‘Demographic Processes Affect HIV-1 Evolution in Primary Infection Before the Onset of Selective Processes’, Journal of Virology, 85/15: 7523–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes H., Herbeck J. T., Tovanabutra S., et al. (2015) ‘HIV-1 Infections With Multiple Founders Are Associated With Higher Viral Loads Than Infections With Single Founders’, Nature Medicine, 21, 1139–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayasundara D, Saeed I, Maheswararajah S. et al. (2015) ‘ViQuaS: an Improved Reconstruction Pipeline for Viral Quasispecies Spectra Generated by Next-Generation Sequencing’, Bioinformatics, 31/6: 886–96. [DOI] [PubMed] [Google Scholar]
- Keele B. F., Giorgi E. E., Salazar-Gonzalez J. F. et al. (2008) ‘Identification and Characterization of Transmitted and Early Founder Virus Envelopes in Primary HIV-1 Infection’, Proceedings of the National Academy of Sciences of the United States of America, 105/21: 7552–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le T., Wright E. J., Smith D. M. et al. (2013) ‘Enhanced CD4+ T-Cell Recovery With Earlier HIV-1 Antiretroviral Therapy’, The New Englnad Journal of Medicine, 368/3: 218–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P., Rambaut A., Pybus O. G. (2006) ‘HIV Evolutionary Dynamics Within and Among Hosts’, AIDS Review, 8/3: 125–40 [PubMed] [Google Scholar]
- Li H., Bar K. J., Wang S. et al. (2010) ‘High Multiplicity Infection by HIV-1 in Men Who Have Sex With Men’, PLoS Pathogens, 6/5: e1000890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M., Conery J. S. (2003) ‘The Origins of Genome Complexity’, Science, 302/5649: 1401–4. [DOI] [PubMed] [Google Scholar]
- Novitsky V., Wang R., Margolin L., et al. (2011) ‘Transmission of Single and Multiple Viral Variants in Primary HIV-1 Subtype C Infection’, PLoS One, 6/2: e16714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland M., Tovanabutra S., deCamp A. C. et al. (2011) ‘Genetic Impact of Vaccination on Breakthrough HIV-1 Sequences From the STEP Trial’, Nature Medicine, 17/3: 366–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar-Gonzalez J. F., Bailes E, Pham K. T. et al. (2008) ‘Deciphering Human Immunodeficiency Virus Type 1 Transmission and Early Envelope Diversification by Single-Genome Amplification and Sequencing’, Journal of Virology, 82/8: 3952–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner G. A., Pacold M. E., Vigil E. et al. (2013) ‘Using Ultradeep Pyrosequencing to Study HIV-1 Coreceptor Usage in Primary and Dual Infection’, The Journal of Infectious Diseases, 208/2: 271–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson E. B. (1927) ‘Probable Inference, the Law of Succession, and Statistical Inference’, Journal of the American Statistical Association, 22/158: 209–12. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.