

Abstract
Background
MicroRNAs (miR) are small non-coding RNAs that regulate diverse biological functions. The bicistronic gene miR-143/145 determines cell fate and phenotype of vascular smooth muscle cells (VSMC), in part, by destabilizing Elk-1 mRNA. The transcription factor c-Myb also regulates differentiation and proliferation of VSMC, and here we test whether these effects may be mediated by miR-143/145.
Methods & results
Flow cytometry of cardiovascular-directed d3.75 embryoid bodies (EBs) isolated smooth muscle progenitors with specific cell surface markers. In c-myb knockout (c-myb -/-) EB, these progenitors manifest low levels of miR-143 (19%; p<0.05) and miR-145 (6%; p<0.01) expression as compared to wild-type (wt) EB. Primary VSMC isolated from transgenic mice with diminished expression (c-myblx/lx) or reduced activity (c-mybh/h) of c-Myb also manifest low levels of miR-143 (c-myblx/lx: 50%; c-mybh/h: 41%), and miR-145 (c-myblx/lx: 49%; c-mybh/h: 56%), as compared to wt (P<0.05). Sequence alignment identified four putative c-Myb binding sites (MBS1-4) in the proximal promoter (PP) of the miR-143/145 gene. PP-reporter constructs revealed that point mutations in MBS1 and MBS4 abrogated c-Myb-dependent transcription from the miR-143/145 PP (P<0.01). Chromatin immunoprecipitation (ChIP) revealed preferential c-Myb binding at MBS4 (p<0.001). By conjugating Elk-1 3’-untranslated region (UTR) to a reporter and co-transducing wt VSMC with this plus a miR-143-antagomir, and co-transducing c-myblx/lx VSMC with this plus a miR-143-mimic, we demonstrate that c-Myb’s ability to repress Elk-1 is mediated by miR-143.
Conclusion
c-Myb regulates VSMC gene expression by transcriptional activation of miR-143/145.
Introduction
MicroRNAs (miR) are conserved, small, non-coding RNAs that are 20–25 nt in length [1]. RNA polymerase II transcribes pri-miR that are processed by the nuclear RNase complex Drosha–DGCR8 into pre-miR, that are then translocated from the nucleus by Exportin5. In cytoplasm, Dicer processes pre-miR into mature miR that are subsequently loaded into RNA-induced silencing complex (RISC) [1]. miR modulate gene expression by destabilizing mRNAs or disrupting their translation, and thus serve as important regulators of events such as proliferation and differentiation [2,3]. In the vasculature, miR have been implicated in the differentiation of smooth muscle cell progenitors [4], and in the proliferation of adult vascular smooth muscle cells (VSMC) [5]. However, little is known about the transcriptional machinery that regulates miR in the production of VSMC from stem/progenitor cells or to what extent they contribute to the pathophysiology of cardiovascular disease. Understanding these regulatory processes may present them as therapeutic targets.
Embryonic stem cells (ESCs) can be differentiated into SMC lineages [6,7]. Although the precise roles of miR networks in their specification to SMC remain unclear, expression of miR-1, -10, -26a and -143/145 have been implicated [8–11]. In particular, the bicistronic gene miR-143/145 appears to regulate VSMC differentiation [12], with miR-143 and miR-145 destabilizing specific mRNAs important to the VSMC phenotype, such as platelet-derived growth factor receptor alpha (PDGFRα) and Kruppel-like factor 4 (KLF4) [13]. While miR-143 and miR-145 KO mice are viable, they have decreased levels of the VSMC markers SM α-actin, SM-MHC and smoothelin, and their phenotype includes reduced media thickness, arterial hypotension, and blunted responses to vasopressors [14]. Hence, miR-143 and miR-145 are important for VSMC differentiation, maintenance, and cell function with effects on integrated vascular pathophysiology.
The transcription factor c-Myb, first described as a key regulator of proliferation and differentiation of hematopoietic cell types [15,16], also plays an important role in the generation of VSMC from ESCs. ESCs give rise to vascular endothelial growth factor receptor type-2 (VEGFR2)-expressing progenitors, which can produce VSMC in a process involving c-Myb. Indeed, vegfr2 KO embryoid bodies (EB) have decreased expression of Pim-1 kinase, a known modulator of DNA binding sites for c-Myb, resulting in limited formation of cells expressing SM-actin [17,18]. Furthermore, we found that c-myb-/- ESC could be differentiated into cardiomyocytes but not contractile SMC in EB [19], and demonstrated that c-Myb not only activates VEGFR2 expression but also enables the subsequent capacity of VEGFR2+ progenitors to differentiate into VSMC [20]. Thus, both c-Myb and miR-143/145 serve important roles in ESC to VSMC differentiation that appear to have similar, or complementary, effects.
The roles of c-Myb and miR-143/145 in VSMC expansion holds true in the pathological state. Expansion of the VSMC compartment clearly underlies the pathogenesis of both vessel responses to injury and atherosclerosis [21,22]. The mechanisms by which this occurs include: proliferation of normally quiescent synthetic VSMC, the phenotypic modulation of differentiated, contractile VSMC to a synthetic cell type or a proliferative cell type [23], and the proliferation and subsequent differentiation of vessel-resident (or perhaps even circulating) progenitors of VSMC [14,24]. Supporting the latter, we recently showed that c-Myb regulates the proliferation and differentiation of adult Sca1+ adventitial VSMC progenitors, with the differentiation of this progenitor critically mediated by c-Myb-dependent transcriptional activation of myocardin [25]. Interestingly, miR-143 and miR-145 KO mice have impaired VSMC proliferation and medial thickening after carotid ligation [5], sharing a similar phenotype to that of mice with VSMC-specific expression of dominant-negative form of c-Myb (Myb-Engrailed) [26], and that of mice homozygous for a hypomorphic point mutation in the c-Myb transactivation domain (c-mybh/h) [25]. Given these observations, we hypothesized that the effect of c-Myb may be mediated, at least in part, by its ability to regulate expression of miR-143 and miR-145 in both embryonic and adult models of VSMC differentiation.
Materials and methods
ESC culture
Cytogenetic studies confirmed early (<10) passage c-myb-/- ESCs of the CCE genetic background to harbor normal chromosomes (data not shown), and all studies used ESC from this stage. wt and c-myb-/- CCE ESCs were maintained as previously described [20].
Mouse cell lines and cell culture
Primary VSMC were isolated as described [27]. Primary VSMC were isolated from homozygous LoxP (Myb knockdown strain) mice (c-myblx/lx) and their wt littermate controls. LoxP allele carrying mice [16] have a neomycin resistance (neoR) cassette inserted into the c-myb locus at intron-6 and also have LoxP sites inserted into introns-2 and -6 of the c-myb locus (c-myblx). Insertion of the neoR cassette into intron-6 of the c-myb gene results in an alternatively spliced event where mis-splicing of exon5 to the neoR cassette occurs with 90% probability and normal exon5-exon6 event splicing occurs with a 10% probability. Thus, the c-myblx allele produces only 5–10% of the wild-type level of full-length c-Myb protein, as validated by immunoblots of fetal liver protein extracts from E11 embryos probed with a c-Myb specific monoclonal antibody [16]. c-myblx/lx mice have a significantly smaller body size and a reduced lifespan; with most animals unable to survive past 4–6 months of age. A mouse carotid VSMC line derived from c-myblx/lx mice was immortalized as previously described [27]. Primary mouse carotid VSMC were cultured in DMEM with 10% fetal bovine serum, 50ng/mL rat recombinant PDGF-ßß (Sigma-Aldrich, Mississauga, ON), and 1% penicillin-streptomycin in humidified atmosphere with 5% CO2
Site directed mutagenesis
To disrupt c-Myb binding site in the miR-143/145 promoter, MBS1 (ΔMBS1: aatTAACtgcatgct to aatTGGGtgcatgc), MBS2 (ΔMBS2: ggtCAACaggcattg to ggtCGGGaggcattg), MBS3 (ΔMBS3: atTAACtgcatgc to aatTGGGtgcatgc), and MBS4 (ΔMBS4: tgtCAACagcttgaa to tgtCGGGagcttgaa) were mutated using the Quickchange II Site Directed Mutagenesis Kit (#200523) and primers described in S1 Table. Each clone was confirmed with sequencing.
Quantitative real-time RT-PCR
For RNA extraction and qRT-PCR of mRNA transcripts, total RNA from EBs and monolayers of mouse carotid VSMCs were isolated using the miRvana system (Lifesystems). miRNA was tested for purity (UV absorption ratio, A260/A280 > 1.8). DNase I (Fermentas) treatment was performed at room temperature for 30 min. RNA was reverse transcribed using qScript cDNA SuperMix (Quanta Biosciences). For microRNA qPCR, PerfeCTa SYBR Green SuperMix qPCR was employed with primers corresponding to the exact sequences of each individual microRNA (Integrated DNA Technologies, S2 Table) and the universal reverse primer supplied by the manufacturer (Quanta Biosciences). Samples were analyzed in triplicate. Final primer concentrations used were 0.2 μM with 60°C annealing temperature. Standard curves were generated for all primers and samples were run in triplicate. qPCR for mRNA and miRNA were performed on Roche LightCycler 480 System as per the manufacturer’s instructions, and normalized to expression of the housekeeping genes GAPDH or U6.
Fluorescence-activated cell sorting (FACS)
FACS was performed as previously described [20]. In brief, EBs at indicated time were trypsinized and DNase treated. The cells were incubated with FITC-conjugated anti-mouse VEGFR2 (Flk1) (eBioscience) and PE-conjugated anti-mouse PDα (eBioscience) at 4°C for 30 min. The resultant cells were washed and sorted for each fraction (V-/P-, V+/P-, V+/P+, V-/P+) by flow cytometry performed with ARIA-RITT and LSR-SC (Becton-Dickinson). The FACS profiles were analyzed by FlowJo software (Ashland).
Chromatin-immunopreciptiation (ChIP)
ChIP-IT Express Enzymatic Kit (Active Motif) was used as per the manufacture’s protocol. Mouse carotid VSMC and EB were fixed with 1% formaldehyde at room temperature for 10 min. Fixation was stopped with 125 mM glycine. Chromatin was isolated using a dounce homogenizer and centrifugation. After resuspending in digestion buffer, chromatin was enzymatically digested into 400-1500bp fragments, which were IPed overnight at 4°C with rabbit anti-c-Myb antibody (2μg, SC-517X, Santa Cruz Biotechnology), anti-H3 antibody or control rabbit serum (2μg, Santa Cruz Biotechnology). Chromatin-antibody complexes were pulled down with protein G-conjugated magnetic beads (Dynal, Invitrogen). IPed chromatin was eluted from the beads, reverse-crosslinked, treated with RNase-A and proteinase-K. ChIP was analyzed qualitatively by PCR and gel electrophoresis, and quantitatively with real time PCR with primers flanking putative c-Myb binding sites on a GeneAmp PCR System 9700 (Applied Biosystems) under the following conditions: 94°C 3 min, 94°C 30 sec, 57°C 30 sec, 72°C 30 sec for 40 cycles, and 72°C 30 sec. qPCR for ChIP were performed on Roche LightCycler 480 System. After determining the concentration of input chromatin by UV spectrometry, a standard curve was constructed for each set of primers. In this manner, qRT-PCR of each putative c-Myb binding site enabled us to quantify the absolute amount of c-Myb and/or H3 binding from the IPed fragment, which was then normalized to qRT-PCR results for ChIP performed with rabbit serum. The primer sequences used are provided in the S3 Table. qPCR data were analyzed by one-way ANOVA.
Cell transfection and reporter gene assays
Immortalized VSMC, a mouse hemangioendothelioma endothelial cell line (EOMA), or human nasopharyngeal carcinoma (CNE2) cells were seeded at a density of 2.5 × 104/well in a 6-well plate. At 18 h post seeding, the cells were washed once with phosphate-buffered saline, pH 7.4, and replaced with 2 ml of complete medium. Plasmids were transfected into EOMA using Jet Prime (Polyplus) and grown in high glucose Dulbecco's modified Eagle's medium (DMEM, Roche Applied Science) containing 5 u/ml penicillin, 50 μg/ml streptomycin, and 10% fetal bovine serum (FBS). The cells were incubated for 4 h with a total of 2 μg of plasmid DNA (1.5 μg of promoter-, 0.5 μg of various expression plasmids or empty vectors, and 0.05 μg of pRLTK-Renilla luciferase as an internal control). Media was subsequently removed and replaced with DMEM with 10% FBS and 5 u/ml penicillin. After 24 h, extracts (100 μl/well) were prepared for measurement of luciferase activity using Dual Luciferase Report (Promega, E1910). Cell lysates were plated onto a 96-well plate (Corning, 3917) and assayed on a PHERSTAR (BMG Labtech). Relative luminescence units over two ranges were recorded: Range-1: 2.5–11.5 secs or cycles 6–24 (Firefly luminescence) and Range-2: 14.5–23.5 secs or cycles 30–48 (Renilla luminescence). The level of firefly luciferase activity was normalized to control Renilla luciferase activity. Measurements were made from a minimum of three independent transfections. Results were reported as mean ± SEM, and all variables were analyzed by Students’ t-test, with significance defined as P ≤ 0.05.
Immunoblots
At specified time intervals and after serum starvation and/or transient transfection, immortalized mouse carotid VSMC were harvested with PBS and pelleted by centrifugation for 10 min in a JS-4.2 rotor at 3000 rpm. The pellet was resuspended in hypotonic buffer (20 mM HEPES, pH 7.9, 25% glycerol, 1.5 mM MgCl2, 0.8 M KCl, 0.2 mM EDTA, 0.2 mM PMSF, 0.5 mM DTT) and incubated on ice for 30 min. Cell lysis was confirmed with microscopy and the cytoplasmic extract was removed by centrifugation at 18,000 RCF for 30 min at 4’C. The cell extracts were normalized using Lowry method and loaded on a 10% Tris Glycine gel and subjected to electrophoresis for 90 min at 125 V. Proteins were transferred to PVDF membranes and blocked in 5% milk with 1% BSA. The membrane was incubated with primary antibody (Elk-1 (1:1,000, Santacruz, sc-355), c-Myb (1:500, Santacruz, sc-517), sm22alph (1:1000, Santacruz, sc-53015) or GAPDH (1:10,000, Santacruz, sc-47724)) overnight at 4’C. The membranes were washed and incubated with donkey anti rabbit IgG (BioRad, #1706515) for 1 h at RT. The membranes were again washed and then incubated with Western ECL Lighting (Perkin-Elmer) for 1 min and detected on MiniBis Pro (Frogga Bio).
Results
c-Myb increases miR-143 and miR-145 expression in VSMC progenitor cells
To assess the expression of miR-143 and -145 in ESC-derived VSMC progenitor cells, embryoid bodies (EB) from wt and c-myb-/- mouse ESC were differentiated into VSMC progenitor cells under serum-free cardiovascular-directed conditions, and subsequently analyzed by flow cytometry for cell surface markers VEGFR2 (V) and PDGFRα (P) [20] to identify VSMC and non-VSMC progenitor populations. Given that c-Myb expression had been shown to peak at d3.75 of this protocol [20], miR-143 and miR-145 expression levels were also measured at this time point. c-myb-/- EB-derived V+/P- progenitors, which generate functional smooth muscle cells [20], manifest markedly reduced expression levels of miR-143 (19%; p = 0.03) and miR-145 (6%; p = 0.003) when normalized to wt EB (i.e. 81% and 94% reduction in miR-143 and miR-145, respectively). By contrast, V-/P+ progenitors, which do not yield functional smooth muscle cells [20], had no change in miR-143/145 expression in c-myb-/- EB vs. wt (n = 3) (Fig 1A).
Fig 1. c-Myb-regulates expression of miR-143 and miR-145 in mouse embryonic stem cell-derived VEGFR2+ progenitors and adult mouse carotid vascular smooth muscle cells.

(A) Flow cytometry of cardiovascular-directed d3.75 embryoid bodies (EBs), derived from wild-type (wt) or c-myb-/- (knockout) mouse embryonic stem cells, was used to isolate progenitors with cell surface markers VEGFR2 (V) and PDGFRα (P). The known vascular smooth muscle cell (VSMC) progenitor V+/P- derived from c-myb-/- EB had lower expression levels of miR-143 (p<0.05*) and miR-145 (p<0.01**) than V+/P- derived from wt EB. The non-SMC progenitor cells V-/P+ showed no such difference in miR-143 and miR-145 expression, compared to V+/P-. (B) Primary mouse carotid VSMCs isolated from c-myblx/lx mice have reduced expression levels of c-myb (p<0.05*), as well as miR-143 (p<0.01**) and miR-145 (p<0.001***) as compared to VSMCs isolated from wt mice. (C) Carotid VSMCs isolated from mice homozygous for a hypomorphic c-myb allele (c-mybh/h) have no differences in c-myb expression, but their known reductions in c-Myb activity were associated with reduced expression levels of miR-143 (p<0.01**) and miR-145 (p<0.001***) versus wt. qPCR analysis was performed with n = 3 biological samples and three technical repeats. (D) VSMC from c-myblx/lx mice show reduced expression levels of VSMC marker, Sm22α versus wt. (E) Densitometry reveals c-myblx/lx mice have significantly reduced expression of Sm22a relative to GAPDH compared to wt (p<0.05*, n = 4).
We next tested the relationship between c-Myb expression or activity and miR-143 and miR-145 expression levels in primary VSMC isolated from the adult mouse carotid artery. Primary carotid VSMCs isolated from homozygous c-myblx/lx mice expressed lower levels of c-myb than VSMC isolated from wt littermates (Fig 1B). These carotid VSMC from c-myblx/lx mice also showed 50% lower miR-143 (p<0.01) and 49% less miR-145 (P<0.001) expression than primary carotid VSMC isolated from wt littermates. We next examined the relationship between c-myb and miR-143/145 in mice homozygous for a c-Myb hypomorphic allele (c-mybh/h), which carries a single point mutation in the activation domain of c-Myb but does not affect c-Myb expression levels. Compared to wt, aortas of c-mybh/h mice show 41% less miR-143 (p<0.01) expression and 56% less miR-145 (p<0.01) expression than wt controls (Fig 1C).
To further examine the relationship between c-Myb, miR-143 and miR-145, and VSMC, we next assessed expression levels of the VSMC marker Sm22α in c-myblx/lx mice. Sm22α protein expression was 71% lower in primary carotid VSMC from c-myblx/lx vs. wt mice (Fig 1D and 1E and S1 Fig). Taken together, these data consistently suggest that c-Myb expression and activity correlate with expression levels of miR-143 and miR-145 in ESC-derived VSMC progenitors and adult VSMC, as well as with expression levels of the VSMC marker Sm22α.
Specific DNA-binding sites on the miR-143/145 promoter mediate c-Myb-dependent transcriptional activation
An in silico analysis with MatInspector identified four conserved putative c-Myb-binding sites (MBS) in the proximal promoter (PP) of the miR-143/145 gene (Fig 2A). These MBS were located within a 4kb segment (S2 and S3 Figs). This PP includes the CArG box that putatively binds myocardin and an adjacent SMAD-binding element previously shown to regulate miR-143 and miR-145 expression [28]. The functional importance of these putative MBS was studied using site-directed mutagenesis of a promoter-luciferase reporter construct transfected into (a) carotid VSMC from wt mice, (b) EOMA, an endothelial cell line with no detectable c-myb expression in which c-Myb was also over-expressed, and (c) CNE2, which is known to express high levels of c-Myb [29].
Fig 2. miR-143/145 promoter activity requires c-Myb binding sites (MBS).

Promoter activity was determined by co-transfection with promoter-luciferase constructs and renilla control plasmid. Site-directed mutagenesis revealed MBS to be important for reporter gene activation when compared to intact proximal promoter. (A) Proximal promoter (PP) with c-Myb binding site (MBS) positions shown relative to predicted transcriptional start. (B) Promoter luciferase constructs were transfected in c-myblx/lx VSMC or wt controls. Compared to proximal promoter, MBS1 (p<0.001+++) and MBS4 (p<0.001+++) were required for c-Myb-dependent transcription in c-myblx/lx VSMC (n = 3), both MSB1 (p<0.01**) and MBS4 (p<0.05*) were also required for transcriptional activity in wt VSMC (n = 3). Importantly, activation in wt was always greater than in c-myblx/lx VSMC (p<0.05). (C) In EOMA, an endothelial cell line that does not express c-Myb, MBS1 (p<0.001***/+++), MBS2 (p<0.05*/+), and MBS4 (p<0.001***/+++) were relevant to transcriptional activity when compared to promoter, regardless of c-Myb expression (n = 12). Co-transfection with exogenous c-Myb is however, associated with increased transcriptional activity (p<0.05, n = 3). (D) The human nasopharyngeal carcinoma cell line (CNE2) is known to express c-Myb. Intact MBS1 (p<0.05*) and MBS4 (p<0.05*, n = 3) were also necessary for miR-143/145 promoter activity in this cell line.
In VSMC, the wt PP-reporter construct showed endogenous transcriptional activity (Fig 2A). Mutation of MBS1 and MBS4 in the PP caused a significant decrease in transcriptional activity in immortalized wt and c-myblx/lx VSMC (p<0.001 and p<0.001, respectively) (Fig 2B). Importantly, wt VSMC manifest higher levels of PP-reporter activity as compared to c-myblx/lx VSMC (p<0.05). Next, EOMA cells cotransfected with c-Myb show significantly greater miR-143/145 promoter activity as compared to EOMA cells without c-Myb expression (p<0.05), and point mutations in MBS1 (p<0.001), MBS2 (p<0.05) and MBS4 (p<0.001) were found to be functionally important in this cell line (Fig 2C).
Using CNE2 cells, Wang et al. had demonstrated that c-Myb bound the miR-143 and miR-145 promoter, but purportedly functioned as a repressor [29]. In our analysis, c-Myb binding sites MBS1, MBS2 and MBS3 were identical to those identified by Wang et al. (S2 Fig), while MBS4 was not identified in their analysis. However, in our hands, c-Myb activates the miR-143/145 promoter in CNE2 cells, with point mutations in MBS1 (p<0.05) and MBS4 (p<0.05) significantly reducing this activity (Fig 2D). Finally, further supporting c-Myb-dependent transcriptional activation of the miR-143/145 gene, qRT-PCR analysis of VSMC isolated from c-myblx/lx mice revealed reduced pre-miR-143 and pre-miR-145 levels as compared to wt controls (S4 Fig). Thus, we demonstrate, with multiple cell lines from different species, that c-Myb is functioning as a transcriptional activator of the miR-143/145 gene.
c-Myb binds to the promoter of the miR-143/145 gene
Chromatin immunoprecipitation (ChIP) analysis was used to detect histone H3 and c-Myb binding at the miR-143/145 promoter in carotid VSMC (Fig 3A). c-Myb was easily IPed on the miR-143 and miR-145 promoter. By PCR and agarose gel electrophoresis, c-Myb appears to bind MBS4 and MBS2 (Fig 3B) (p<0.01). By contrast, histone H3 occupancy was not statistically distinct at any MBS (data not shown). When c-Myb binding is normalized to histone H3 binding, c-Myb can be shown to preferentially bind only MBS4 in wt VSMC (one-way ANOVA, p<0.001) (Fig 3C). As a negative control (NC), a promoter without any putative c-Myb binding sites was subjected to ChIP with the same c-Myb antibody (Fig 3A). In this experiment, no c-Myb binding can be detected on this NC promoter (Fig 3B). These data reveal that c-Myb targets miR143/145 by preferential binding of MBS4.
Fig 3. Chromatin immunoprecipitation (ChIP) of c-Myb on the miR-143/145 promoter of immortalized mouse carotid artery vascular smooth muscle cells (VSMC).

Cells were fixed, lysed and chromatin was fragmented into oligonucleosomes (200-1500bp) then immunoprecipitated overnight at 4°C with rabbit anti-c-Myb antibody (2mg, SC-517X, Santa Cruz), anti-histone H3 (2mg, Santa Cruz) or control rabbit serum (2mg, Santa Cruz). (A) The miR-143 and miR-145 promoter contains four c-Myb binding sites (MBS). (B) ChIP and PCR revealed specificity of c-Myb binding to MBS in carotid VSMC. As a negative control, ChIP was performed on a promoter situated ~15kb upstream of miR-143/145 with no known or predicted MBS. (C) qPCR reveals differential binding of c-Myb on predicted MBS of the miR-143/145 promoter. c-Myb preferentially binds MBS2 and MBS4 in carotid VSMC (p<0.01, one-way ANOVA). When normalized to histone H3 density, c-Myb preferentially binds MBS4 (p<0.001***, one-way ANOVA). ChIP was performed in n = 3 biological samples. qPCR analysis had three technical repeats for each biological sample.
c-Myb regulates Elk-1 expression via miR-143
The activity and expression of Elk-1, a known target of miR-143 [12] that is involved in the regulation of VSMC proliferation [30], were assessed to investigate the c-Myb-mediated regulation by miR-143 on downstream VSMC regulators. By conjugating the 3’ untranslated region of Elk-1 to a luciferase reporter (3’UTR-Elk-1-luc), the stability of this mRNA, as assessed by luciferase activity, can be used to gauge the consequences of transfection with a miR-143 mimic and conversely with an antagomir to miR-143. In this manner, the effect of c-Myb activity on miR-143-driven 3’UTR-Elk-1-luciferase activity was assayed in c-myblx/lx and wt VSMC. Reflecting reduced miR-143 destabilization of 3’UTR-Elk-1-luc, luciferase activity was greater in c-myblx/lx than wt VSMC (p<0.01) (Fig 4A).
Fig 4. c-Myb represses Elk-1 expression via miR-143/145.
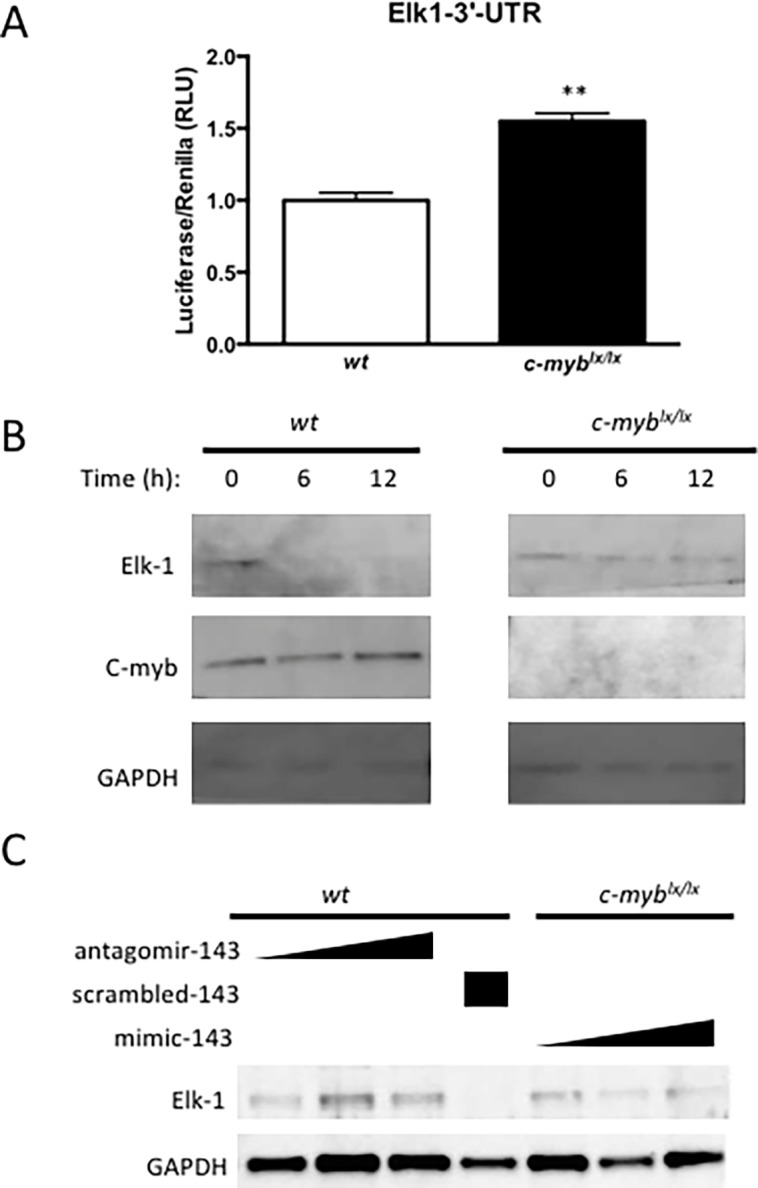
(A) Elk1 has previously been validated as a miR-143/145-repressed gene. Elk1-3’UTR-Luciferase construct was transfected into wt and c-myblx/lx carotid artery VSMC. Luciferase activity was assayed on PHERASTAR and found to be augmented in c-myblx/lx VSMC, which have known reductions in c-Myb expression (p<0.01**). (B) Time course analysis of c-Myb and Elk-1 expression. After serum starvation and cell cycle arrest, c-Myb levels are diminished. Following addition of 10% fetal bovine serum, c-Myb expression peaks at 12 h. As c-Myb expression level rise, Elk-1 expression levels decrease and are undetectable at 12 h. Note that c-Myb expression is not detectable in c-myblx/lx VSMC released from serum starvation, and Elk-1 expression remains stable. (C) miR-143 mediates c-Myb repression of Elk1. The miR-143 antagomir augments Elk-1 protein expression in wt cells that express c-Myb, while the mimic abrogates protein expression in c-myblx/lx VSMC.
Upon serum starvation-induced cell cycle arrest, immunoblots reveal reduced c-Myb expression in VSMC. These levels of c-Myb expression rapidly increase upon the addition of fetal bovine serum (FBS) and peak at 12 h (Fig 4B). Coincident with this, c-Myb-driven miR-143-mediated Elk-1 protein expression is dramatically reduced in serum-stimulated wt VSMC. By contrast, c-myblx/lx VSMC with undetectable levels of c-Myb do not suppress Elk-1 expression following serum stimulation (Fig 4B). These presumed miR-143-mediated effects on Elk-1 expression were then tested with miR-143 mimic and antagomir experiments in c-myblx/lx and wt VSMC. miR-143 antagomir abrogates the effect of c-Myb of Elk-1- expression in wt VSMC. By contrast, a scrambled oligonucleotide (negative control for the antagomir) had no such effect. Conversely, c-myblx/lx VSMC transfected with a miR-143 mimic show reduced Elk-1 expression, even in the absence of normal c-Myb levels (Fig 4C). These data demonstrate that c-Myb represses Elk-1 via miR-143.
Discussion
We have identified c-Myb as a transcription factor that regulates miR-143 and miR-145 expression levels in both ESC and adult VSMC. c-Myb is required for miR-143 and miR-145 expression in ESC-derived VEGFR2+ progenitor cell populations. In adult mouse carotid VSMC, c-myblx/lx VSMC with diminished levels of c-Myb expression have reduced levels of miR-143 and miR-145. Of the multiple MBS identified by in silico analysis of the PP of the miR-143/145 gene, we show that while MBS1 and MBS4 are important for transcriptional activation, only MBS4 is found to bind c-Myb in wt VSMC. Our study also demonstrates that c-Myb-dependent regulation of miR-143 has functional consequences in VSMC, with c-Myb-dependent regulation of Elk-1 gene expression and subsequent translation being directly mediated by a miR-143-responsive destabilizing motif in its 3’-UTR.
c-Myb activates transcription on the miR-143 and miR-145 promoter
Although a recent study has suggested that c-Myb can bind and regulate the miR-143/145 promoter in a nasopharyngeal carcinoma cell line (CNE2), they found c-Myb to be acting as a transcriptional repressor [29]. While c-Myb is capable of acting as both a transcriptional activator [31] and repressor [27], in our hands the miR-143/145 gene is activated by c-Myb in (a) in EB-derived VSMC progenitors, (b) adult VSMC, (c) the EOMA cell line transduced to express c-Myb, and (d) the CNE2 cell line. In EOMA, a cell line with no c-Myb expression, MBS mutations have decreased transactivation, which may be caused by other transcription factors that overlap with the c-Myb binding site. With respect to the CNE2 cell line, our data contradict those of Wang et. al. [29].
Wang et. al. describe c-Myb activating miR-143/145 in their abstract, but the results and subsequent discussion suggests c-Myb is a repressor. The data supporting the role of c-Myb as a repressor is based on correlations among nasopharyngeal cancer lines with high c-Myb and low miR-143/145 levels as determined by qRT-PCR. Anti-c-Myb siRNA caused a decrease in c-Myb mRNA and an increase in miR-143 expression, although anti-c-Myb siRNA may de-repress c-Myb autoregulatory repression mediated by dimerization [32]. Although Wang et al. also showed that c-Myb binds to the miR-143/145 promoter, their study did not capture MBS4. By contrast, our data clearly show MBS4 to be the most important binding site for c-Myb, with point mutations in MBS4 abrogating c-Myb-dependent transcriptional activation of the luciferase reporter. Our data also demonstrated that MBS1 and MBS2 were important for transcriptional activity, but MBS1 did not bind c-Myb. We speculate that MBS1 is in close proximity to the transcriptional start site and may not be associated with the active promoter. Curiously, Wang et al. described luciferase reporter experiments but did not provide data supporting this claim in either their manuscript or supplemental data online.
c-Myb-directed VSMC progenitor differentiation via miR-143/145
Previous studies from our lab show high c-Myb expression in VEGFR2+ progenitor cells and that c-Myb is crucial for their differentiation from ESCs, with c-myb-/- EB yielding greatly reduced numbers of VEGFR2+ progenitors [20]. In the current study, we go on to show that c-Myb mediates the effect of these miR, and that VEGFR2+ progenitor cells derived from c-myb-/- EB have reduced expression of miR-143/145. Complementing our present work, other studies have implicated miR-143/145 in the formation of VEGFR2+ progenitors, suggesting that miR-143/145 may be sufficient to drive differentiation of ESC into VSMC progenitors [5,12,33].
c-Myb regulates VSMC proliferation through miR-143 and miR-145
Several studies have demonstrated a role for increased c-Myb expression in remodeling responses following vascular injury [26,34]; however, there is discordance over the role of miR-143 and miR-145 in similar models. In a carotid ligation mouse model, Xin et al. demonstrated that miR-143 and miR-145 knockout mice have reduced neointima formation after vascular injury [5]. Conversely, Cheng et al. showed in a balloon injury rat model that a miR-145 mimic has the same effect [35]. In Apolipoprotein E-deficient mice, atherosclerotic lesions are associated with low miR-145 expression and plaque stability is improved with mimic therapy [36]. As such, we posit that the effects of miR-143 and miR-145 may be model- and context-specific and may not precisely reflect similar processes mediated by c-Myb.
The E26 transformation specific (ETS) family of transcription factors function in cell differentiation, proliferation, and angiogenesis [37]. ETS transcription factor Elk-1 antagonizes the effect of miR-145 and myocardin. Elk-1 blocks myocardin and promotes VSMC proliferation, while miR-145 and myocardin promote VSMC differentiation [37]. In the current study, we demonstrate that c-Myb-driven repression of Elk-1 is mediated by miR-143. Elk-1 is a known target of miR-143, functioning to prevent VSMC proliferation and to maintain quiescence [12].
In pathological conditions such as atherosclerosis and vessel injury, which are associated with a shift from quiescent to proliferative VSMC phenotypes, c-Myb expression levels increase [26,38], while miR-143 and miR-145 expression levels are reduced. These observations suggest directionally distinct regulation of c-Myb and miR-143/145 in diseased models, as opposed to our current demonstration of directionally parallel changes in c-Myb expression and activity and miR-143/145 expression. In this context, Elk-1 would be de-repressed, which would inhibit myocardin and promote the synthetic VSMC phenotype [12,35] that characterizes these conditions. Other results by Xin et al. and Cheng et al. add to the notion that c-Myb-dependent regulation of miR-143/145 is context-dependent [5,35]. One explanation stems from the potential role played by c-Myb in post-transcriptional regulation of miR by Dicer, which may itself be perturbed in atherosclerosis [39]
Supporting information
(TIFF)
The putatively common miR-143 and -145 promoter was analyzed using JASPAR in VSMC derived from mouse, rat, and human. This analysis revealed four putative c-Myb binding sites.
(TIFF)
(TIFF)
(TIFF)
(TIFF)
(TIFF)
(TIFF)
Acknowledgments
M.C was supported by the Clinician Scientist Training Program and University of Toronto Clinician Investigator Program. Some of the equipment used in this study was supported by The 3D (Diet, Digestive Tract and Disease) Centre funded by the Canadian Foundation for Innovation and Ontario Research Fund, project number 19442 and 30961.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This study was funded by a CIHR Grant # MOP-14648 to Mansoor Husain. M.C was supported by the Clinician Scientist Training Program and University of Toronto Clinician Investigator Program. Some of the equipment used in this study was supported by The 3D (Diet, Digestive Tract and Disease) Centre funded by the Canadian Foundation for Innovation and Ontario Research Fund, project number 19442 and 30961. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Battistella M & Marsden PA (2015) Advances, nuances, and potential pitfalls when exploiting the therapeutic potential of RNA interference. Clin Pharmacol Ther 97, 79–87. 10.1002/cpt.8 [DOI] [PubMed] [Google Scholar]
- 2.Valencia-Sanchez MA, Liu J, Hannon GJ & Parker R (2006) Control of translation and mRNA degradation by miRNAs and siRNAs. Genes & development 20, 515–24. [DOI] [PubMed] [Google Scholar]
- 3.Vasudevan S, Tong Y & Steitz JA (2007) Switching from repression to activation: microRNAs can up-regulate translation. Science 318, 1931–4. 10.1126/science.1149460 [DOI] [PubMed] [Google Scholar]
- 4.Ivey KN, Muth A, Arnold J, King FW, Yeh RF, Fish JE, et al. (2008) MicroRNA regulation of cell lineages in mouse and human embryonic stem cells. Cell stem cell 2, 219–29. 10.1016/j.stem.2008.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF, et al. (2009) MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes & development 23, 2166–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kattman SJ, Witty AD, Gagliardi M, Dubois NC, Niapour M, Hotta A, et al. (2011) Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell stem cell 8, 228–40. 10.1016/j.stem.2010.12.008 [DOI] [PubMed] [Google Scholar]
- 7.Kattman SJ, Huber TL & Keller GM (2006) Multipotent flk-1+ cardiovascular progenitor cells give rise to the cardiomyocyte, endothelial, and vascular smooth muscle lineages. Developmental cell 11, 723–32. 10.1016/j.devcel.2006.10.002 [DOI] [PubMed] [Google Scholar]
- 8.Liu GC, Fang F, Zhou J, Koulajian K, Yang S, Lam L, et al. (2012) Deletion of p47phox attenuates the progression of diabetic nephropathy and reduces the severity of diabetes in the Akita mouse. Diabetologia 55, 2522–32. 10.1007/s00125-012-2586-1 [DOI] [PubMed] [Google Scholar]
- 9.Huang H, Xie C, Sun X, Ritchie RP, Zhang J & Chen YE (2010) miR-10a contributes to retinoid acid-induced smooth muscle cell differentiation. The Journal of biological chemistry 285, 9383–9. 10.1074/jbc.M109.095612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leeper NJ, Raiesdana A, Kojima Y, Chun HJ, Azuma J, Maegdefessel L, et al. (2011) MicroRNA-26a is a novel regulator of vascular smooth muscle cell function. Journal of cellular physiology 226, 1035–43. 10.1002/jcp.22422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie C, Huang H, Sun X, Guo Y, Hamblin M, Ritchie RP, et al. (2011) MicroRNA-1 regulates smooth muscle cell differentiation by repressing Kruppel-like factor 4. Stem cells and development 20, 205–10. 10.1089/scd.2010.0283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, et al. (2009) miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 460, 705–10. 10.1038/nature08195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis-Dusenbery BN, Chan MC, Reno KE, Weisman AS, Layne MD, Lagna G et al. (2011) down-regulation of Kruppel-like factor-4 (KLF4) by microRNA-143/145 is critical for modulation of vascular smooth muscle cell phenotype by transforming growth factor-beta and bone morphogenetic protein 4. The Journal of biological chemistry 286, 28097–110. 10.1074/jbc.M111.236950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang Z, Wang A, Yuan F, Yan Z, Liu B, Chu JS, et al. (2012) Differentiation of multipotent vascular stem cells contributes to vascular diseases. Nature communications 3, 875 10.1038/ncomms1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verbeek W, Gombart AF, Chumakov AM, Muller C, Friedman AD & Koeffler HP (1999) C/EBPepsilon directly interacts with the DNA binding domain of c-myb and cooperatively activates transcription of myeloid promoters. Blood 93, 3327–37. [PubMed] [Google Scholar]
- 16.Emambokus N, Vegiopoulos A, Harman B, Jenkinson E, Anderson G & Frampton J (2003) Progression through key stages of haemopoiesis is dependent on distinct threshold levels of c-Myb. Embo J 22, 4478–88. 10.1093/emboj/cdg434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Winn LM, Lei W & Ness SA (2003) Pim-1 phosphorylates the DNA binding domain of c-Myb. Cell Cycle 2, 258–62. [PubMed] [Google Scholar]
- 18.Zippo A, De Robertis A, Bardelli M, Galvagni F & Oliviero S (2004) Identification of Flk-1 target genes in vasculogenesis: Pim-1 is required for endothelial and mural cell differentiation in vitro. Blood 103, 4536–44. 10.1182/blood-2003-11-3827 [DOI] [PubMed] [Google Scholar]
- 19.Kolodziejska KM, Noyan-Ashraf MH, Nagy A, Bacon A, Frampton J, Xin HB, et al. (2008) c-Myb-dependent smooth muscle cell differentiation. Circ Res 102, 554–61. 10.1161/CIRCRESAHA.105.162628 [DOI] [PubMed] [Google Scholar]
- 20.Ishida M, El-Mounayri O, Kattman S, Zandstra P, Sakamoto H, Ogawa M, et al. (2012) Regulated expression and role of c-Myb in the cardiovascular-directed differentiation of mouse embryonic stem cells. Circ Res 110, 253–64. 10.1161/CIRCRESAHA.111.259499 [DOI] [PubMed] [Google Scholar]
- 21.Handa S, Sadi AM, Cybulsky MI, Stewart DJ & Husain M (2008) Region-specific patterns of vascular remodelling occur early in atherosclerosis and without loss of smooth muscle cell markers. Atherosclerosis 196, 617–23. 10.1016/j.atherosclerosis.2007.06.032 [DOI] [PubMed] [Google Scholar]
- 22.Regan CP, Adam PJ, Madsen CS & Owens GK (2000) Molecular mechanisms of decreased smooth muscle differentiation marker expression after vascular injury. J Clin Invest 106, 1139–1147. 10.1172/JCI10522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hultgardh-Nilsson A & Durbeej M (2007) Role of the extracellular matrix and its receptors in smooth muscle cell function: implications in vascular development and disease. Curr Opin Lipidol 18, 540–5. 10.1097/MOL.0b013e3282ef77e9 [DOI] [PubMed] [Google Scholar]
- 24.Naito H, Kidoya H, Sakimoto S, Wakabayashi T & Takakura N (2012) Identification and characterization of a resident vascular stem/progenitor cell population in preexisting blood vessels. EMBO J 31, 842–855. 10.1038/emboj.2011.465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shikatani EA, Chandy M, Besla R, Li CC, Momen A, El-Mounayri O, Robbins CS, et al. (2016) c-Myb Regulates Proliferation and Differentiation of Adventitial Sca1+ Vascular Smooth Muscle Cell Progenitors by Transactivation of Myocardin. Arterioscler Thromb Vasc Biol 36, 1367–1376. 10.1161/ATVBAHA.115.307116 [DOI] [PubMed] [Google Scholar]
- 26.You XM, Mungrue IN, Kalair W, Afroze T, Ravi B, Sadi AM, et al. (2003) Conditional expression of a dominant-negative c-Myb in vascular smooth muscle cells inhibits arterial remodeling after injury. Circulation research 92, 314–21. [DOI] [PubMed] [Google Scholar]
- 27.Afroze T, Yang LL, Wang C, Gros R, Kalair W, Hoque AN, et al. (2003) Calcineurin-independent regulation of plasma membrane Ca2+ ATPase-4 in the vascular smooth muscle cell cycle. Am J Physiol Cell Physiol 285, C88–95. 10.1152/ajpcell.00518.2002 [DOI] [PubMed] [Google Scholar]
- 28.Long X & Miano JM (2011) Transforming growth factor-beta1 (TGF-beta1) utilizes distinct pathways for the transcriptional activation of microRNA 143/145 in human coronary artery smooth muscle cells. The Journal of biological chemistry 286, 30119–29. 10.1074/jbc.M111.258814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang W, Wu S, Shi Y, Miao Y, Luo X, Ji M, et al. (2015) c-MYB regulates cell growth and DNA damage repair through modulating MiR-143. FEBS Lett 589, 555–564. 10.1016/j.febslet.2015.01.012 [DOI] [PubMed] [Google Scholar]
- 30.Kawai-Kowase K & Owens GK (2007) Multiple repressor pathways contribute to phenotypic switching of vascular smooth muscle cells. Am J Physiol Cell Physiol 292, C59–69. 10.1152/ajpcell.00394.2006 [DOI] [PubMed] [Google Scholar]
- 31.Sakura H, Kanei-Ishii C, Nagase T, Nakagoshi H, Gonda TJ & Ishii S (1989) Delineation of three functional domains of the transcriptional activator encoded by the c-myb protooncogene. Proc Natl Acad Sci U S A 86, 5758–5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nomura T, Sakai N, Sarai A, Sudo T, Kanei-Ishii C, Ramsay RG, et al. (1993) Negative autoregulation of c-Myb activity by homodimer formation through the leucine zipper. J Biol Chem 268, 21914–21923. [PubMed] [Google Scholar]
- 33.Yamaguchi S, Yamahara K, Homma K, Suzuki S, Fujii S, Morizane R, et al. (2011) The role of microRNA-145 in human embryonic stem cell differentiation into vascular cells. Atherosclerosis 219, 468–474. 10.1016/j.atherosclerosis.2011.09.004 [DOI] [PubMed] [Google Scholar]
- 34.Simons M, Edelman ER, DeKeyser JL, Langer R & Rosenberg RD (1992) Antisense c-myb oligonucleotides inhibit intimal arterial smooth muscle cell accumulation in vivo. Nature 359, 67–70. 10.1038/359067a0 [DOI] [PubMed] [Google Scholar]
- 35.Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q, et al. (2009) MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circulation research 105, 158–66. 10.1161/CIRCRESAHA.109.197517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lovren F, Pan Y, Quan A, Singh KK, Shukla PC, Gupta N, et al. (2012) MicroRNA-145 targeted therapy reduces atherosclerosis. Circulation 126, S81–90. 10.1161/CIRCULATIONAHA.111.084186 [DOI] [PubMed] [Google Scholar]
- 37.Rao VN, Huebner K, Isobe M, ar-Rushdi A, Croce CM & Reddy ES (1989) elk, tissue-specific ets-related genes on chromosomes X and 14 near translocation breakpoints. Science 244, 66–70. [DOI] [PubMed] [Google Scholar]
- 38.Simons M & Rosenberg RD (1992) Antisense nonmuscle myosin heavy chain and c-myb oligonucleotides suppress smooth muscle cell proliferation in vitro. Circ Res 70, 835–843. [DOI] [PubMed] [Google Scholar]
- 39.Hartmann P, Zhou Z, Natarelli L, Wei Y, Nazari-Jahantigh M, Zhu M, et al. (2016) Endothelial Dicer promotes atherosclerosis and vascular inflammation by miRNA-103-mediated suppression of KLF4. Nat Commun 7, 10521 10.1038/ncomms10521 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIFF)
The putatively common miR-143 and -145 promoter was analyzed using JASPAR in VSMC derived from mouse, rat, and human. This analysis revealed four putative c-Myb binding sites.
(TIFF)
(TIFF)
(TIFF)
(TIFF)
(TIFF)
(TIFF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.