

Abstract
Purpose
Enzalutamide resistance could result from raised androgens and be overcome by combination with abiraterone acetate. PLATO (ClinicalTrials.gov identifier: NCT01995513) interrogated this hypothesis using a randomized, double-blind, placebo-controlled design.
Patients and Methods
In period one, men with chemotherapy-naïve metastatic castration-resistant prostate cancer received open-label enzalutamide 160 mg daily. Men with no prostate-specific antigen (PSA) increase at weeks 13 and 21 were treated until PSA progression (≥ 25% increase and ≥ 2 ng/mL above nadir), then randomly assigned at a one-to-one ratio in period two to abiraterone acetate 1,000 mg daily and prednisone 5 mg twice daily with either enzalutamide or placebo (combination or control group, respectively) until disease progression as defined by the primary end point: progression-free survival (radiographic or unequivocal clinical progression or death during study). Secondary end points included time to PSA progression and PSA response in period two.
Results
Of 509 patients enrolled in period one, 251 were randomly assigned in period two. Median progression-free survival was 5.7 months in the combination group and 5.6 months in the control group (hazard ratio, 0.83; 95% CI, 0.61 to 1.12; P = .22). There was no difference in the secondary end points. Grade 3 hypertension (10% v 2%) and increased ALT (6% v 2%) or AST (2% v 0%) were more frequent in the combination than the control group.
Conclusion
Combining enzalutamide with abiraterone acetate and prednisone is not indicated after PSA progression during treatment with enzalutamide alone; hypertension and elevated liver enzymes are more frequent with combination therapy.
INTRODUCTION
Enzalutamide, a potent androgen receptor (AR) antagonist, and abiraterone acetate (administered in combination with prednisone and hereafter referred to as abiraterone), a potent inhibitor of cytochrome P450 (CYP17A1) that suppresses androgen synthesis, induce declines in prostate-specific antigen (PSA) in up to 90% of patients when administered alone to men with metastatic castration-resistant prostate cancer (mCRPC).1,2 Both improve survival and are approved for the first-line treatment of chemotherapy-naïve mCRPC. However, resistance develops after a median of approximately 18 months, and strategies that prolong treatment benefit are urgently required. A rise in PSA, suggesting reactivation of AR target genes, is often associated with clinical progression.3 Several mechanisms have been implicated in driving resistance and can be broadly divided into categories that are either dependent on or independent of ligand-binding domain-driven reactivation of AR signaling.3 The former can include an adaptive feedback loop of increased serum and tissue androgens and AR levels after AR antagonism with enzalutamide4 that in vitro modeling suggests could result in outcompeting of enzalutamide at the AR.5 A single-center, single-arm study of the combination of abiraterone and enzalutamide reported manageable toxicity and no meaningful pharmacokinetic drug-drug interactions.6
PLATO was designed to investigate enzalutamide resistance, which is thought to be attributable to ligand-binding domain reactivation of AR signaling. We hypothesized that cross-resistance may exist between single-agent enzalutamide and abiraterone, making their use in sequence of limited benefit, and that a rise in androgens in patients receiving enzalutamide may result in reactivation of AR, so that disrupting this adaptive feedback loop by combining with abiraterone would reinduce enzalutamide sensitivity, resulting in prolonged patient benefit and tumor responses versus abiraterone alone. Abiraterone effectively inhibits androgen synthesis,7 but we hypothesized that raised levels of progesterone, synthetic glucocorticoids, and other potential ligands with abiraterone in the presence of high AR levels after enzalutamide treatment would result in continued AR activity and primary resistance (Fig 1A).8-10 To test these hypotheses, we used a novel trial design whereby patients received open-label enzalutamide and at predefined PSA progression were randomly assigned to receive abiraterone plus placebo or abiraterone plus enzalutamide (Fig 1B). Combining abiraterone with enzalutamide would not inhibit reactivation of AR independent of the ligand-binding domain, including resistance mediated by AR splice variants or glucocorticoid receptors.11,12 To attempt enrichment for cancers more likely to benefit from the combination of enzalutamide and abiraterone, we excluded patients who had a rise in PSA at week 13 or 21. Here we describe the final analysis of the trial.
Fig 1.

(A) Scientific hypotheses underlying PLATO trial and (B) PLATO trial design. Actual patient numbers at each trial milestone are included on the bottom row of panel B, and target numbers are included in brackets. More details on period one patient disposition are provided in the Data Supplement. AR, androgen receptor; PFS, progression-free survival; PSA, prostate-specific antigen. (*) Random assignment was stratified by confirmed PSA response at week 13 in period one (≥ 0% to < 30% v ≥ 30%).
PATIENTS AND METHODS
Study Design
This trial comprised consecutive periods of open-label treatment with enzalutamide 160 mg orally once daily (period one) followed by randomized, double-blind treatment in period two (Fig 1B). Men with no PSA increase from baseline at weeks 13 and 21 in period one continued treatment until PSA progression (defined as a ≥ 25% increase and ≥ 2 ng/mL above nadir that required confirmation by a second consecutive PSA assessment ≥ 3 weeks later). They were then randomly assigned in period two at a one-to-one ratio to receive enzalutamide 160 mg daily or blinded enzalutamide placebo (capsules identical in appearance to enzalutamide) orally once daily in combination with open-label abiraterone 1,000 mg orally once daily and open-label prednisone 5 mg orally twice daily. Randomization was stratified by PSA ≥ 30% decline at period one week 13 (≥ 0 to < 30% v ≥ 30%).
Patients not eligible for period two were allowed to continue open-label enzalutamide in period one until loss of clinical benefit as assessed by the investigator and thereafter underwent safety follow-up. In period two, discontinuation because of PSA progression was discouraged, and administration of study drugs continued until disease progression as defined by the primary end point, intolerable toxicity, or patient withdrawal, whichever occurred first. Patients who chose to discontinue before meeting the primary end point were censored. PLATO was approved by the independent board at each participating site and conducted according to the provisions of the Declaration of Helsinki and Good Clinical Practice Guidelines of the International Conference on Harmonisation. All patients provided written informed consent before participating.
Patients
Eligible patients starting period one had histologically confirmed, asymptomatic or minimally symptomatic prostate cancer (defined as a score of < 4 on Brief Pain Inventory–Short Form [BPI-SF] question three), Eastern Cooperative Oncology Group performance status of ≤ 1, and chemotherapy-naïve disease that progressed during androgen-deprivation therapy as defined by the Prostate Cancer Clinical Trials Working Group 2.13 For periods one and two, patients were required to have: confirmation of metastases using whole-body radionuclide bone scan and computed tomography or magnetic resonance imaging; castrate levels of testosterone (≤ 1.73 nmol/L) and prior bilateral orchiectomy or inhibition of gonadotropin-releasing hormone throughout the study; no history of predisposition to seizures or known brain metastases; no clinically significant cardiovascular disease; and no prior treatment of prostate cancer with cytotoxics, aminoglutethimide, ketoconazole, abiraterone, or enzalutamide or an investigational agent that inhibited the AR or androgen synthesis. Additionally, for period two, patients were required to have confirmed PSA progression and were excluded if they had cancer-related pain requiring chronic opiate use (excluding acetaminophen acetate fixed-dose combinations), which was defined as daily use for > 7 consecutive days or > 10 days within a 14-day period.
Assessments
During period one, PSA values were monitored at screening, baseline, weeks 13 and 21, and every 4 weeks thereafter. During period two, PSA values were monitored at screening, weeks 9 and 13, and every 8 weeks thereafter. Radiographic images were assessed at screening in periods one and two, every 8 weeks in period two after random assignment for 32 weeks, and every 12 weeks thereafter or earlier than scheduled if progression was clinically suspected. Radiographic disease progression was defined in soft tissue using Response Evaluation Criteria in Solid Tumors (version 1.1) and in bone using Prostate Cancer Clinical Trials Working Group 2 criteria as done previously.1 Radiologic assessments were conducted by designated readers at individual sites. Pain was assessed using the BPI-SF questionnaire, and quality of life was assessed using the Functional Assessment of Cancer Therapy–Prostate (FACT-P) questionnaire. Use of cytotoxic chemotherapy, cancer vaccines, or experimental therapies for the treatment of cancer was prohibited during the study. Use of bisphosphonates or denosumab was allowed if initiated ≥ 4 weeks before random assignment and the dose was stable. After discontinuation of period two study drugs, patients had a safety follow-up assessment at 4 weeks and were monitored at 4-week intervals for 16 weeks.
Outcomes
Primary and secondary end points were assessed using data obtained in period two. The primary end point was progression-free survival (PFS), defined as the time from random assignment to the first of the following events assessed by the investigator: radiographic progression, unequivocal clinical progression, or death during study (ie, death from random assignment to within 112 days [ie, four cycles] of treatment discontinuation without objective evidence of radiographic progression). Unequivocal clinical progression was defined as any of the following: new onset of prostate cancer pain requiring chronic opiate use as defined previously, deterioration of Eastern Cooperative Oncology Group performance status to ≥ 3 as a result of prostate cancer, initiation of cytotoxic chemotherapy for prostate cancer, or radiation therapy or surgical intervention because of complications of tumor progression. Secondary end points were time to PSA progression, PSA response of ≥ 50%, PSA response of ≥ 30%, rate of pain progression, objective response rate, time to first use of subsequent antineoplastic therapy, time to degradation of FACT-P score, and safety using the National Cancer Institute Common Terminology Criteria for Adverse Events (version 4). Assessments of safety included adverse events, clinical laboratory tests, physical examinations, vital signs, and 12-lead ECGs.
Statistical Analyses
Estimated median PFS was 5 and 8 months for the control and combination groups, respectively. An estimated 175 events were required to achieve a target hazard ratio (HR) of approximately 0.63 with a two-sided 5% type I error rate and approximately 85% power. Approximately 500 patients with mCRPC were to be enrolled, with 83% expected to have no rise in PSA at weeks 13 and 21. Of these, approximately two thirds were expected to have PSA progression by planned closure of random assignment, and approximately 10% were expected to drop out of the study. Therefore, approximately 250 patients with PSA progression were expected to be randomly assigned to double-blind treatment in period two, with approximately 70% of these expected to demonstrate radiographic or unequivocal clinical progression or death during study to achieve the target number of 175 events. Estimates of median PFS were determined using the Kaplan-Meier method. Prespecified sensitivity analyses using the same methods were designed to assess the interaction of five predefined factors on the primary end point of PFS. If the primary end point was met, the key secondary end points (time to PSA progression and PSA response of ≥ 50%) would be tested using a gatekeeping procedure to control for experiment-wise type I error.14 P values for other secondary efficacy analyses were exploratory, and no adjustment for multiplicity was to be performed. All efficacy analyses were performed in the intention-to-treat population, and differences in time to event were calculated using the log-rank test stratified by period one week 13 PSA decline (≥ 0% to < 30% v ≥ 30%). HRs were calculated based on a Cox regression model with treatment as the only covariate and stratified by period one week 13 PSA decline (≥ 0% to < 30% v ≥ 30%), with < 1.00 favoring the combination group. All safety analyses were performed in the population receiving any study treatment.
RESULTS
Patient Characteristics and Disposition
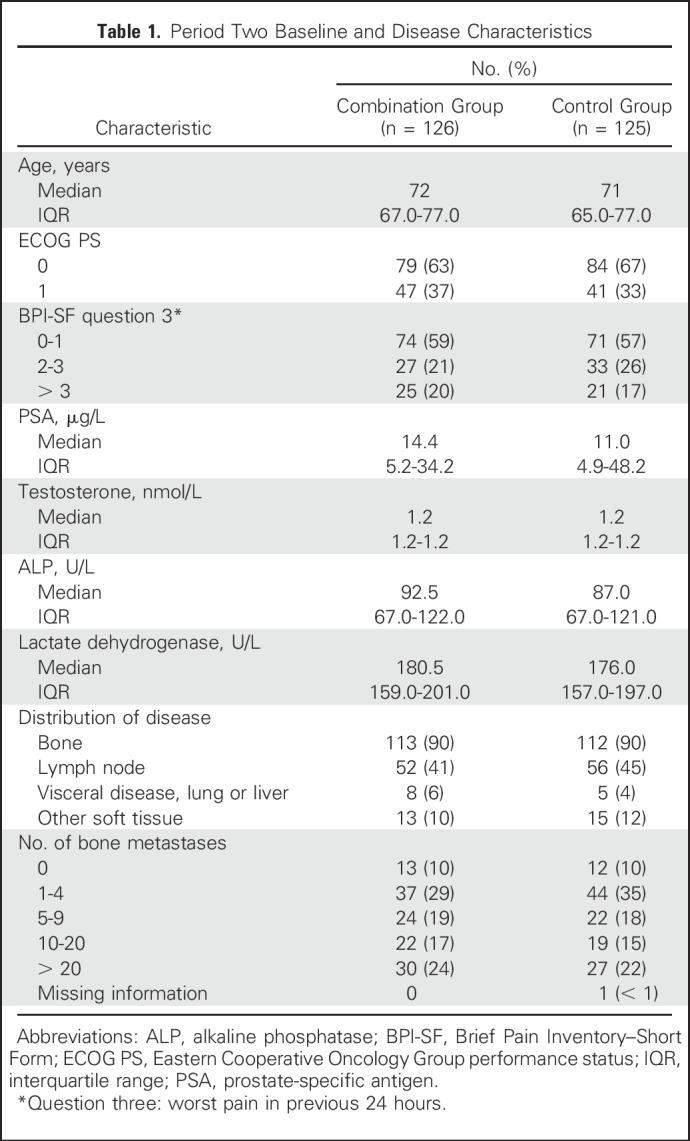
A total of 509 patients enrolled in period one at 51 sites in North America, Europe, and Australia and received open-label single-agent enzalutamide. Baseline patient characteristics for period one are listed in the Data Supplement; 370 (73%) of 509 patients had received a first-generation antiandrogen, 151 (30%) were receiving a stable dose of bisphosphonates or denosumab, and 421 (83%) had bone metastases on bone scan. Median duration of treatment with single-agent enzalutamide in period one was 9.1 months; 377 (74%) and 340 patients (67%) had a PSA decline of ≥ 30% and ≥ 50%, respectively, at week 21 (Data Supplement). Overall, 408 patients had stable or decreased PSA levels at weeks 13 and 21 and were therefore eligible to enter period two when they developed PSA progression (Data Supplement). Of these, 251 patients had confirmed PSA progression, met period two eligibility criteria, and were randomly assigned to one of the two treatment groups before the cutoff date of October 7, 2016. A total of 126 were assigned to enzalutamide plus abiraterone and prednisone (combination group), and 125 were assigned to placebo plus abiraterone and prednisone (control group; Fig 1B). All except one patient in each treatment group received study treatment (Fig 2). Patient characteristics at screening for period two by treatment group are listed in Table 1. At the data cutoff for the primary analysis, of the 258 patients who were not randomly assigned, 84 (33%) continued enzalutamide and 174 (67%) discontinued period one, primarily because of disease progression (Data Supplement). Of the 251 patients randomly assigned, 27 (21%) of 126 patients in the combination group and 18 (14%) of 125 patients in the control group continued treatment (Fig 2).
Fig 2.

Patient disposition in period two. ECOG PS, Eastern Cooperative Oncology Group performance status. (*) Reasons included clinical disease progression and rising prostate-specific antigen. (†) Reasons included rising prostate-specific antigen and disease metastasis.
Table 1.
Period Two Baseline and Disease Characteristics
Primary End Point
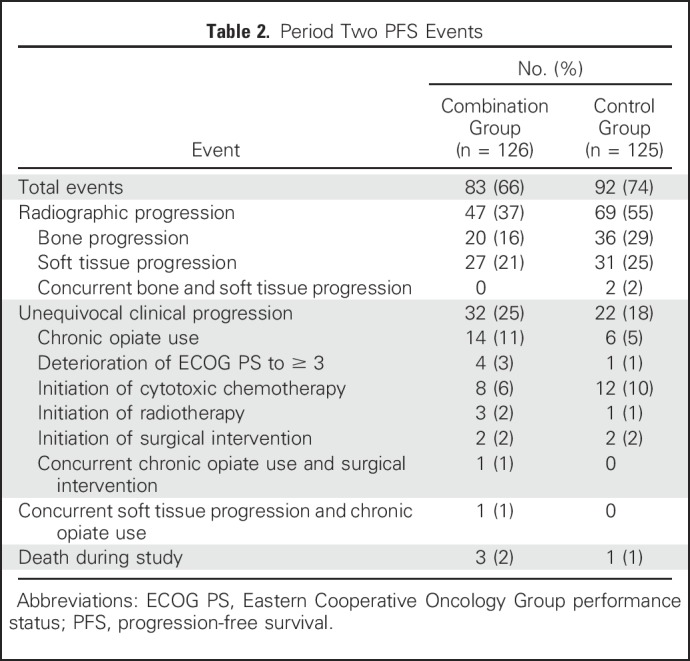
Median treatment duration in period two was 5.6 months for both groups. Median PFS was 5.7 and 5.6 months for the combination and control groups, respectively (HR, 0.83; 95% CI, 0.61 to 1.12; P = .22; Fig 3). A predefined analysis of the PFS event by unequivocal clinical or radiographic progression or death suggested a difference in the proportion of events by treatment group, with a higher rate of clinical progression events in the combination group and a higher rate of radiologic events in the control group (Table 2).
Fig 3.

Kaplan-Meier estimates for progression-free survival (PFS). Dotted line represents median. HR, hazard ratio.
Table 2.
Period Two PFS Events
Sensitivity Analyses
In a predefined sensitivity analysis to determine the impact of unequivocal clinical progression on the duration of PFS, which included only events of radiographic progression or death during study and censored patients with unequivocal clinical progression, median PFS was 10.0 and 7.0 months for the combination and control groups, respectively (HR, 0.67; 95% CI, 0.47 to 0.94; P = .02; Data Supplement). The results of the other prespecified sensitivity analyses, including those to determine the impact of antineoplastic therapies, treatment discontinuation, unconfirmed bone disease progression, and disease progression at unscheduled visit, were not statistically significant (Data Supplement). Predefined subgroup analyses showed that the treatment effects were broadly consistent across subgroups; differences were observed for those receiving opiates or those with visceral disease, but these groups were small (Data Supplement).
Secondary End Points
Because the primary end point was not met, all analyses of secondary end points were exploratory. In period two, 124 (98%) of 126 patients in the combination group and 122 (98%) of 125 patients in the control group had a baseline and at least one postbaseline PSA assessment. One (1%) of 124 patients in the combination group and three (2%) of 122 patients in the control group had a confirmed reduction in baseline PSA of ≥ 50%; Fig 4A). Median time to PSA progression was 2.8 months for both groups (HR, 0.87; 95% CI, 0.62 to 1.24; P = .45; Fig 4B). There was no difference between the two groups with regard to rate of pain progression, objective response rate, or time to first use of subsequent antineoplastic therapy (Data Supplement). FACT-P score degradation was observed in 67 (53%) of 126 patients in the combination group and 54 (43%) of 125 patients in the control group. There was no difference in median time to degradation of FACT-P score (4.6 months; 95% CI, 3.7 to 6.5 v 6.4 months; 95% CI, 5.5 to 13.9, respectively; HR, 1.40; 95% CI, 0.97 to 2.03; P = .07).
Fig 4.

(A) Best single prostate-specific antigen (PSA) change in period two and (B) Kaplan-Meier estimate for time to PSA progression. Dotted lines represent (A) PSA declines of 30%, 50%, and 90% and (B) median. HR, hazard ratio.
Safety
In period one, 35 (7%) of 509 patients had an adverse event that led to discontinuation of enzalutamide. The most common enzalutamide-related adverse events of any grade were fatigue in 173 patients (34%) and hot flushes in 74 (15%; Data Supplement). In period two, 102 (41%) of 249 patients had at least one grade ≥ 3 adverse event (56 [45%] of 125 in the combination group; 46 [37%] of 124 in the control group). The two most common grade ≥ 3 events were hypertension and a rise in ALT in 12 (10%) and seven patients (6%) in the combination group, respectively, and in two (2%) and three patients (2%) in the control group (Table 3). Eleven patients (9%) in the combination group and five patients (4%) in the control group had an adverse event that was the primary reason for discontinuation of enzalutamide or placebo. Twelve patients (10%) receiving the combination and four patients (3%) in the control group had an adverse event that was the primary reason for discontinuation of abiraterone. Nineteen patients (8%) had an adverse event leading to abiraterone dose reduction (11 [9%] receiving the combination; eight [6%] in the control group), and 60 patients (24%) had an adverse event leading to temporary interruption of abiraterone (37 [30%] receiving the combination; 23 [19%] in the control group).
Table 3.
TEAEs by Decreasing Frequency

DISCUSSION
PLATO evaluated reinduction of sensitivity to AR antagonism by combining enzalutamide with abiraterone to treat patients with a rising PSA while receiving enzalutamide. The comparator arm was common practice in several parts of the world, where abiraterone is often used after enzalutamide. Because the PLATO design randomly assigned patients who had received single-agent enzalutamide until confirmed PSA progression, a primary end point was used that included unequivocal clinical progression. This differed from the regulatory COU-AA-302 and PREVAIL trials, which mandated radiographic progression,1,2 and was considered necessary because of the ethical and pragmatic challenges of continuing AR-targeting therapy in men with a rising PSA and worsening symptoms, given the risk of patients becoming too unwell for the next proven effective line of treatment with docetaxel. In fact, approximately one fifth of patients at screening before random assignment in period two had a pain score of > 3 on question three (worst pain in the previous 24 hours) of the BPI-SF.
PLATO did not meet its primary end point. Consequently, the gatekeeping procedure was not used, and secondary analyses were not adjusted for multiplicity and were considered exploratory. These included a sensitivity analysis for radiographic PFS that showed a nominally significant difference benefiting the combination group. This may have been subject to multiple biases, including more clinical progression events (25% v 18%) before confirmation of radiographic progression for the combination group versus the control group.
A small increase in the risk of hepatic impairment and uncontrolled hypertension was reported in the combination group. Although in general the toxicity was manageable, more patients in the combination group than in the control group discontinued treatment because of adverse events, which also may have contributed to symptomatic worsening and earlier treatment discontinuation. Pharmacokinetic data were not evaluated for drug-drug interactions; however, the rates of hypokalemia and liver function abnormalities were similar to those in previous studies of single-agent abiraterone,15,16 suggesting that exposure to active abiraterone metabolites was not compromised.
PLATO also reported limited benefit with abiraterone after enzalutamide. This was not a primary aim of the trial but is concordant with several retrospective studies17-19 published after PLATO was designed that support our hypothesis of low response rates for abiraterone after enzalutamide. Other trials have reported limited benefit with enzalutamide after abiraterone.20 Several clinical trials have been initiated to evaluate the combination of CYP17A1 inhibition and AR antagonism, both in patients with mCRPC (ClinicalTrials.gov identifiers: NCT01650194 and NCT01949337) and in hormone-naïve men starting long-term androgen-deprivation therapy (ClinicalTrials.gov identifier: NCT02268175),21 with the first of these expected to be reported in 2018.
Overall, results from PLATO suggest that enzalutamide should not be continued in combination with abiraterone after PSA progression during enzalutamide monotherapy. Because of the exclusion of patients who had a PSA rise at 13 or 21 weeks while receiving single-agent enzalutamide (8% of patients; Data Supplement), the lack of representation of long-term single-agent enzalutamide responders (17% continued single-agent enzalutamide at study closure; Data Supplement), and the absence of a molecular biomarker for enriching for sensitive patients, PLATO may have missed a signal of benefit in these subgroups. Ongoing exploratory biomarker analyses from PLATO using plasma DNA data as described previously10,22 aim to identify molecularly selected patients who may benefit from the combination of enzalutamide with abiraterone.
ACKNOWLEDGMENT
We thank all the patients who participated in the trial, and their families. We also acknowledge Stephanie Vadasz and Shannon Davis of Ashfield Healthcare Communications for medical writing and editorial support, which was funded by Medivation, a Pfizer company, and Astellas Pharma.
Appendix
List of PLATO Collaborators
Australia - E. Abdi, S. Begbie, I. Davis, A. Guminski, H. Gurney, A. Hayden, P. Mainwaring, G. Marx, F. Parnis, D. Pook, J. Shapiro, M. Stockler, C. Underhill, H. Woo; Belgium - B. Tombal, H. Van Poppel, P. Werbrouck; Denmark - M. Borre, P. Iversen, P. Ostri; Finland - K. Taari, T. Tammela, M. Vaarala; France - Y. Loriot; Italy - U. De Giorgi, R. Passalacqua, G. Scagliotti, C. Sternberg; Slovakia - V. Balaz, M. Brezovky, F. Goncalves, J. Kliment; Spain - A. Alcaraz, M. Domenech Santasusana, A. Font, E. Gallardo, M.A. Gonzalez del Alba Baamonde, M. Guix Arnau, D. Olmos Hidalgo; Sweden - O. Andrén, A. Bjartell, J.E. Damber; United Kingdom - G. Attard, P. Hoskin, H. Payne, A. Protheroe, J. Tanguay; United States - R. Given, M.E. Taplin
Footnotes
Supported by Medivation, a Pfizer company, and Astellas Pharma, which also funded medical writing and editorial support.
Presented at the European Society for Medical Oncology Congress, Vienna, Austria, September 25-29, 2015, and American Society of Clinical Oncology Annual Meeting, Chicago, IL, June 2-5, 2017.
Clinical trial information: NCT01995513.
AUTHOR CONTRIBUTIONS
Conception and design: Gerhardt Attard, Corina Andresen-Daniil, Mary-Ellen Taplin
Provision of study materials or patients: Gerhardt Attard, Michael Borre, Howard Gurney, Yohann Loriot, Mary-Ellen Taplin
Collection and assembly of data: Gerhardt Attard, Corina Andresen-Danil, Ranjith Kalleda, Trinh Pham, Mary-Ellen Taplin
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Abiraterone Alone or in Combination With Enzalutamide in Metastatic Castration-Resistant Prostate Cancer With Rising Prostate-Specific Antigen During Enzalutamide Treatment
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Gerhardt Attard
Honoraria: Janssen, Astellas Pharma
Consulting or Advisory Role: Janssen-Cilag, Veridex, Ventana Medical Systems, Astellas Pharma, Medivation, Novartis, Millennium Pharmaceuticals, Abbott Laboratories, ESSA Pharma, Bayer HealthCare Pharmaceuticals
Speakers’ Bureau: Janssen, Astellas Pharma, Takeda Pharmaceuticals, Sanofi, Ventana Medical Systems
Research Funding: Janssen (Inst), Arno Therapeutics (Inst), Innocrin Pharma (Inst)
Patents, Royalties, Other Intellectual Property: Institute of Cancer Research rewards to inventors list of abiraterone acetate
Travel, Accommodations, Expenses: Janssen, Astellas Pharma, Medivation, Ventana Medical Systems, Abbott Laboratories, Bayer HealthCare Pharmaceuticals, ESSA Pharma, Janssen (I), Astellas Pharma (I)
Michael Borre
Honoraria: Astellas Pharma, Janssen, BiPar/Sanofi
Travel, Accommodations, Expenses: Astellas, Medivation
Howard Gurney
Honoraria: Pfizer
Consulting or Advisory Role: Bristol-Myers Squibb (Inst), Ipsen (Inst), Merck Sharp & Dohme (Inst), AstraZeneca (Inst), Janssen-Cilag (Inst)
Travel, Accommodations, Expenses: Astellas, Medivation, Bristol-Myers Squibb/Sanofi
Yohann Loriot
Consulting or Advisory Role: Sanofi, Janssen, Astellas Pharma, Pfizer, Roche, AstraZeneca, Seattle Genetics, Merck Sharp & Dohme
Research Funding: Sanofi (Inst), Merck Sharp & Dohme (Inst), Janssen (Inst)
Travel, Accommodations, Expenses: Roche, Merck Sharp & Dohme, AstraZeneca
Corina Andresen-Daniil
Employment: Pfizer
Stock or Other Ownership: Pfizer
Ranjith Kalleda
Employment: Pfizer
Stock or Other Ownership: Pfizer
Travel, Accommodations, Expenses: Pfizer
Trinh Pham
Employment: Aimmune Therapeutics, Pfizer
Stock or Other Ownership: Aimmune Therapeutics, Pfizer
Mary-Ellen Taplin
Honoraria: Medivation, Janssen-Ortho, Clovis Oncology, Bayer HealthCare Pharmaceuticals
Consulting or Advisory Role: Medivation, Janssen-Ortho, Sanofi, Bayer HealthCare Pharmaceuticals, Guidepoint Global, Best Doctors, UpToDate, Clovis Oncology, Research to Practice, DAVA Oncology, Myovant
Research Funding: Janssen-Ortho (Inst), Medivation (Inst), Bayer HealthCare Pharmaceuticals (Inst)
Travel, Accommodations, Expenses: Medivation, Janssen Oncology
REFERENCES
- 1.Beer TM, Armstrong AJ, Rathkopf DE, et al. : Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med 371:424-433, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ryan CJ, Smith MR, Fizazi K, et al. : Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): Final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol 16:152-160, 2015 [DOI] [PubMed] [Google Scholar]
- 3.Attard G, Cooper CS, de Bono JS: Steroid hormone receptors in prostate cancer: A hard habit to break? Cancer Cell 16:458-462, 2009 [DOI] [PubMed] [Google Scholar]
- 4.Efstathiou E, Titus M, Wen S, et al. : Molecular characterization of enzalutamide-treated bone metastatic castration-resistant prostate cancer. Eur Urol 67:53-60, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richards J, Lim AC, Hay CW, et al. : Interactions of abiraterone, eplerenone, and prednisolone with wild-type and mutant androgen receptor: A rationale for increasing abiraterone exposure or combining with MDV3100. Cancer Res 72:2176-2182, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. doi: 10.1016/j.euo.2019.01.008. Efstathiou E, Titus MA, Wen S, et al: Enzalutamide (ENZA) in combination with abiraterone acetate (AA) in bone metastatic castration resistant prostate cancer (mCRPC). J Clin Oncol 32, 2014 (suppl; abstr 5000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Attard G, Reid AHM, Yap TA, et al. : Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol 26:4563-4571, 2008 [DOI] [PubMed] [Google Scholar]
- 8.Carreira S, Romanel A, Goodall J, et al. : Tumor clone dynamics in lethal prostate cancer. Sci Transl Med 6:254ra125, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen EJ, Sowalsky AG, Gao S, et al. : Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin Cancer Res 21:1273-1280, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romanel A, Gasi Tandefelt D, Conteduca V, et al. : Plasma AR and abiraterone-resistant prostate cancer. Sci Transl Med 7:312re10, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arora VK, Schenkein E, Murali R, et al. : Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 155:1309-1322, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, Chan SC, Brand LJ, et al. : Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res 73:483-489, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scher HI, Halabi S, Tannock I, et al. : Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: Recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol 26:1148-1159, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marcus R, Peritz E, Gabriel KR: On closed testing procedures with special reference to ordered analysis of variance. Biometrika 63:655-660, 1976 [Google Scholar]
- 15.de Bono JS, Logothetis CJ, Molina A, et al. : Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 364:1995-2005, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ryan CJ, Smith MR, de Bono JS, et al. : Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med 368:138-148, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bianchini D, Lorente D, Rodriguez-Vida A, et al. : Antitumour activity of enzalutamide (MDV3100) in patients with metastatic castration-resistant prostate cancer (CRPC) pre-treated with docetaxel and abiraterone. Eur J Cancer 50:78-84, 2014 [DOI] [PubMed] [Google Scholar]
- 18.Loriot Y, Bianchini D, Ileana E, et al. : Antitumour activity of abiraterone acetate against metastatic castration-resistant prostate cancer progressing after docetaxel and enzalutamide (MDV3100). Ann Oncol 24:1807-1812, 2013 [DOI] [PubMed] [Google Scholar]
- 19.Noonan KL, North S, Bitting RL, et al. : Clinical activity of abiraterone acetate in patients with metastatic castration-resistant prostate cancer progressing after enzalutamide. Ann Oncol 24:1802-1807, 2013 [DOI] [PubMed] [Google Scholar]
- 20. de Bono JS, Chowdhury S, Feyerabend S, et al: Antitumour activity and safety of enzalutamide in patients with metastatic castration-resistant prostate cancer previously treated with abiraterone acetate plus prednisone for ≥24 weeks in Europe. Eur Urol [epub ahead of print on August 22, 2017] [DOI] [PubMed]
- 21.Attard G, Sydes MR, Mason MD, et al. : Combining enzalutamide with abiraterone, prednisone, and androgen deprivation therapy in the STAMPEDE trial. Eur Urol 66:799-802, 2014 [DOI] [PubMed] [Google Scholar]
- 22.Conteduca V, Wetterskog D, Sharabiani MTA, et al. : Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: A multi-institution correlative biomarker study. Ann Oncol 28:1508-1516, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]