

A robust and fast principle of optical hyperpolarization is proposed and demonstrated by using NV centers experimentally.
Abstract
Dynamic nuclear polarization (DNP) is an important technique that uses polarization transfer from electron to nuclear spins to achieve nuclear hyperpolarization. Combining efficient DNP with optically polarized nitrogen-vacancy (NV) centers offers promising opportunities for novel technological applications, including nanoscale nuclear magnetic resonance spectroscopy of liquids, hyperpolarized nanodiamonds as magnetic resonance imaging contrast agents, and the initialization of nuclear spin–based diamond quantum simulators. However, none of the current realizations of polarization transfer are simultaneously robust and sufficiently efficient, making the realization of the applications extremely challenging. We introduce the concept of systematically designing polarization sequences by Hamiltonian engineering, resulting in polarization sequences that are robust and fast. We theoretically derive sequences and experimentally demonstrate that they are capable of efficient polarization transfer from optically polarized NV centers in diamond to the surrounding 13C nuclear spin bath even in the presence of control errors, making the abovementioned novel applications possible.
INTRODUCTION
A key challenge in the quantum manipulation and detection of small nuclear spin ensembles is their minute level of polarization at thermal equilibrium, which is on the order of 10–5 for a magnetic field of 2 T at room temperature. Overcoming this challenge holds the key for the realization of quantum applications ranging from quantum simulators to nanoscale nuclear magnetic resonance (NMR) devices. These will be turned from laboratory demonstrations into realistic applications by polarization schemes that enable their efficient initialization and readout. An important breakthrough in this respect has been the realization that the electron spin of the nitrogen-vacancy (NV) centers in diamond (as well as color centers in silicon carbide and photoexcited triplet-state molecules) can be optically initialized nearly perfectly even at room temperature and under ambient conditions, allowing for rapid electron spin polarization to be generated optically or chemically far above thermal equilibrium and subsequently transferred to surrounding nuclei (1–6). These systems present a unique opportunity for polarizing and initializing spin baths and unlocking the potential of nanoscale applications including the initialization of quantum simulators based on nuclear spin arrays in diamonds (7, 8), magnetic resonance imaging (MRI) tracers via diamond nanoparticles and powdered diamond (9–13), and the enhancement of NMR using NV spin ensembles (14).
However, when trying to achieve nuclear hyperpolarization via NV centers or similar systems (for example, silicon carbide and photoexcited triplet-state molecules), we are faced with two key challenges. First, optically polarizable electron spins are typically higher spin systems, with S ≥ 1. The lattice-oriented zero-field splitting in the presence of external magnetic fields and disorder results in a large spectral range of the electron spin resonance, hindering effective polarization transfer to the surrounding nuclear spins. Second, many of these systems exhibit fast electron spin relaxation (for example, photoexcited triplet states) (15), requiring fast polarization transfer, or weak hyperfine coupling to the nuclear spins (for example, NV centers in diamonds, especially to external nuclear spins), limiting polarization transfer rates. Overcoming this combination of challenges presents a daunting task, as it requires a polarization transfer scheme that (i) works for a large spectral range of the electronic system and (ii) produces a fast and efficient polarization transfer from electron to nuclear spins.
Many dynamic nuclear polarization (DNP) protocols have been developed and applied over the past several decades, starting from continuous microwave (MW) irradiation (16) to more efficient pulsed schemes (17, 18). A common theme in all protocols that are effective for low electron spin concentration is the use of a long MW pulse to match the Larmor frequency of the nuclear spins to the electronic Rabi rotation in the frame of reference of the MW drive, which is well known as a Hartmann-Hahn (H-H) resonance (19). Unfortunately, owing to the weak electron-nuclear interaction, even a small detuning from this resonance inhibits the polarization transfer, which renders these schemes strongly dependent on the intensity and frequency of the MW drive as well as the orientation and transition frequency of the NV center. While modified protocols such as the integrated solid effect (ISE) (10, 18, 20) improve the robustness across larger spectral ranges, this comes at the expense of a significantly slower polarization transfer. Thus, devising a DNP protocol that is both robust and fast has remained an unmet challenge.
Here, we present a new approach for performing DNP, termed PulsePol, which combines fast polarization transfer with remarkable robustness against a broad range of experimental imperfections including power and detuning fluctuations. In sharp contrast to the schemes described above, which allow for polarization transfer only during the pulses, this is achieved by the design of sequences of short pulses (21, 22) (much shorter than the nuclear Larmor period), with extended waiting periods between the pulses, that refocus the electron-nuclear interaction such that polarization transfer is achieved through the accumulated dynamics between pulses. As we will demonstrate both theoretically and experimentally, this approach provides inherent robustness to MW errors and electron spectral width and a high degree of flexibility, which allows to take full advantage of pulse optimization methods developed in NMR (23, 24) to tailor the polarization to the specific challenges of the experimental setup.
We start by describing the theoretical framework of Hamiltonian engineering and derive the PulsePol sequence whose robustness against detuning and pulse length/strength errors we establish theoretically. We then proceed to experimentally confirm the efficiency of the protocol for a single NV center in a diamond surrounded by 13C nuclei and compare the performance of the PulsePol protocol to state-of-the-art DNP schemes. Finally, we discuss several applications of our protocol.
Theoretical framework
For simplicity, we consider a system of a single electron ( for simplicity, later realized by the |m = 0〉 and |m = − 1〉 levels of the NV center) coupled to a single nuclear , subject to intermediately applied MW pulses, which are used for control of the electron spin
| (1) |
where ωS(ωI) denotes the electron (nuclear) Larmor frequency, and is the hyperfine tensor. Hd = 2Ω(t)Sx cos(ωMWt + φ), in which ωMW is the MW frequency, φ is its phase, and the Rabi frequency Ω(t) takes the value Ω0 when the MW is on, and 0 otherwise.
For the realization of a robust and efficient interaction between the NV center and the surrounding nuclear spins, one needs to simultaneously achieve the decoupling of the NV electron spin from environmental noise while refocusing the desired interaction with the nuclear spin bath. This can be achieved by dynamical decoupling (DD) protocols (25, 26) such as Carr-Purcell-Meiboom-Gill (27) or the XY pulse family (28), where equally spaced pulses separated by a time τ such that only interactions with nuclear spins precessing at a frequency ωI = nπ/τ are preserved.
When these DD sequences achieve resonance with a nuclear spin, the resulting effective Hamiltonian that describes the time evolution is, in a suitable electronic basis, of the form Heff = αAxSxIx, where Ax denotes the x component of the hyperfine vector, and α < 1 is a constant determined by the filter function generated by the pulse sequence (29). Heff does not transfer polarization, and the concept of pulsed polarization is to engineer this Hamiltonian to produce the desired flip-flop dynamics. To this end, for a short time interval , we apply a time evolution according to Heff = αAxSxIx, and in a subsequent interval of length Δt, we let the system follow a time evolution according to Heff = αAxSyIy. This is achieved by mapping both the electronic basis by an MW pulse and the nuclear spin basis by a suitable time delay from x to y (see Fig. 1). Repeating this sequence yields a time evolution that is governed, for times exceeding and whenever the coupling strength satisfies Ax ≪ ωI, by the effective flip-flop Hamiltonian
| (2) |
which is the sum of Heff = αAxSxIx and Heff = αAxSyIy.
Fig. 1. Theoretical framework overview.
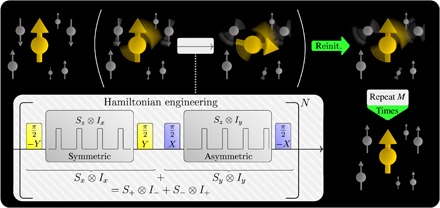
Polarization of a bath of nuclear spins (gray) by an electron spin (yellow): The initially unpolarized bath and the polarized electron spin (upper left) evolve according to an engineered Hamiltonian that describes a flip-flop interaction, and the electron spin is periodically reinitialized. After several repetitions, the nuclear spin bath is polarized (lower right). Illustration of effective Hamiltonian engineering (gray box): A combination of standard symmetric and asymmetric DD sequences with the effective Hamiltonians H ∝ Sz ⊗ Ix/y can be modified to a flip-flop Hamiltonian H ∝ Sx ⊗ Ix + Sy ⊗ Iy by introducing basis changes with π/2 pulses.
We would like to emphasize that the rapid switching in intervals Δt represents a crucial deviation from schemes that aim to achieve engineered electron-nuclear SWAP gates after a time by first applying an evolution according to exp(−iAxSxIxt/2) for half the total interaction time, followed by exp(−iAxSyIyt/2) for the second half (30), as this achieves a SWAP gate only at , while in between the evolution may include both flip-flip and flip-flop terms. Achieving flip-flop dynamics on the time scale of the Larmor frequency ωI is crucial for DNP applications, especially those in which the nuclei are in rapid motion as standard SWAP pulse sequences are slow and are not suitable for ensembles of electron spins coupled to nuclear spin baths due to coupling strength variations. Furthermore, any loss of coherence faster than the interaction time, due to noisy environment or dynamics on fast time scales (for example, molecular motion), would completely destroy any polarization transfer. These effects are mitigated when the average dynamics are achieved on the nuclear Larmor time scale, which are very fast. However, one challenge (see the Supplementary Materials) remains, as the basic scheme using the rapid alternation between AxSxIx and AxSyIy on the time scale of the Larmor frequency, and other simple variants, come at the expense of an enhanced sensitivity to pulse errors and detunings because the change between Sx and Sy dynamics typically introduce unbalanced π/2 pulses, not part of standard DD sequences.
Thus, no robust pulse sequence or tailoring of DD sequence is known to produce the fast flip-flop dynamics on very short time scales, regardless of the coupling strength while satisfying (i) that detuning errors accumulated during the free evolution are canceled (for example, by π refocusing; see the Supplementary Materials) and preferably also decoupled from unwanted noise and fluctuations and (ii) that variable pulse lengths and detuning and Rabi frequency errors are canceled at least to the first order.
We have constructed such a sequence, termed PulsePol and depicted in Fig. 2A, analytically from components chosen to satisfy the above requirements (see the Supplementary Materials for a detailed discussion). PulsePol achieves polarization transfer for a choice of pulse spacing
| (3) |
for odd n with the strongest coupling occurring for n = 3, where , and yields an effective flip-flop Hamiltonian for times longer than . Full polarization transfer between NV and nuclei is found after a time t = 2Nτ = 4π/(αAx), where N is the number of basic pulse cycles (Fig. 2A). As a consequence, PulsePol achieves polarization transfer in a time scale that is only 28% slower than that achieved with H-H coherent transfer under optimal conditions (Fig. 2B) and significantly faster than other polarization transfer schemes (for example, ISE and solid effect). Furthermore, PulsePol is robust to errors as it has several inherent advantages that enhance its robustness:
Fig. 2. PulsePol sequence and simulations of robustness.
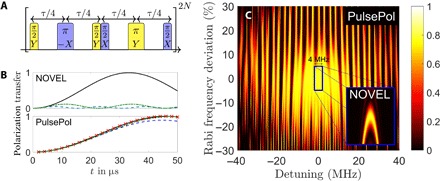
PulsePol sequence (A) and robustness compared to nuclear orientation via electron spin locking (NOVEL). (B) The polarization transfer to a single nuclear spin for NOVEL (upper graph) under perfect conditions (black line). A Δ = (2π)0.5-MHz detuning error (dashed blue line) and 2% Rabi frequency error (dashed-dotted green line) drastically reduce polarization transfer. For PulsePol (lower graph) under perfect conditions (black line), the transfer is 28% slower, as predicted by the effective Hamiltonian of Eq. 2 (red crosses). Detuning errors Δ = 0.1Ω0 = (2π)5 MHz (dashed blue line) and Rabi frequency errors δΩ = 0.1Ω0 = (2π)5 MHz (dashed-dotted green line) have a very small effect. (C) Considering a system of one electron spin and five nuclear spins, for the same parameters ωI = (2π)2 MHz and Ω0 = (2π)50 MHz, polarization transfer versus Δ and δΩ/Ω0 for the PulsePol sequence, with a comparison to a NOVEL sequence for its relevant detuning values |Δ| < (2π)2 MHz in the inset. The graphs result from averaging more than 100 realizations of the locations of the five closest nuclear spins to the NV center electron spin on a carbon lattice, and for PulsePol, a resonance shift of 2.5% and corresponding phase errors were used (see the Supplementary Materials).
(i) Strong pulses with amplitude Ω0, without the requirement of matching the H-H condition, are robust to detuning that satisfies Δ ≪ Ω0.
(ii) The PulsePol sequence cancels second-order errors in the pulse strength and first-order errors in the detuning (see the Supplementary Materials), making it robust to noise and small pulse imperfections, similar to DD sequences.
(iii) The individual pulses can be optimized by numerous methods developed in NMR, including composite pulses (23) and shaped pulses (31, 32).
(iv) Phase errors in the applied pulses can be corrected to first order by a corresponding shift in the resonance condition with no loss of robustness to detuning and Rabi frequency errors (see the Supplementary Materials for further details).
For demonstrating the robustness of the PulsePol sequence, we introduce errors into the pulses and free evolutions, with the PulsePol evolution during the pulses described as
| (4) |
including the detuning (frequency mismatch) Δ that is also present during every free evolution and accounting for deviations in pulse strength/length of the pulses by a Rabi frequency error . As the pulse duration is finite, it is subtracted from the waiting time between the pulses. Since the PulsePol sequence corrects the errors after two cycles lasting for a total of 2τ, fluctuations in Ω0 or ωS (and hence Δ), which occur on a slower time scale (for example, for X-band frequencies and 13C or 1H, this implies that 2τ < 1 μs) will have a similar effect to a constant error; thus, the following described robustness holds also for magnetic field or MW inhomogeneities.
Figure 2B demonstrates the effect of pulse errors by showing the evolution of the polarization of a single nuclear spins as quantified by 2〈Iz〉 near an initially fully polarized electron spin, when applying the PulsePol sequence and nuclear orientation via electron spin locking (NOVEL) sequence (17). The chosen parameters are typical for an NV center spin in a diamond surrounded by 13C nuclear spins. Compared to NOVEL, where small errors can almost completely eliminate the polarization transfer, PulsePol is only slightly affected by these errors.
For a detailed characterization of the robustness, we consider a system of nuclear spins coupled to an electron spin in a diamond. Because of the 1.1% natural abundance of 13C isotopes, the interaction is dominated by the closest nuclear spins, and we restrict attention of the dynamics of the five closest nuclear spins to the NV center electron spin on the lattice. The resulting polarization transfer efficiency from the fully polarized NV center is quantified by for different values of Δ, δΩ, and averaged over 100 nuclear spin configurations, as shown in Fig. 2C. As can be seen, efficient polarization transfer can be achieved in a (2π)60-MHz spectral range for the MW driving strength of Ω0 = (2π)50 MHz. Note that the periodic vertical polarization resonance lines in Fig. 2C are due to the detuning that matches a resonance condition Δτ/4 = kπ for an integer k during the free evolution.
We find that the PulsePol polarization protocol is robust for a wide range of detuning and Rabi frequency errors, sufficient for overcoming inhomogeneities and enhancing the polarization transfer efficiency in many applications (for example, most radicals or electron defects in a solid/glass). However, for overcoming the wide spectral range inherent in some systems, including NV centers in nanodiamonds and photoexcited triplet states, a wider range of detuning robustness is beneficial. PulsePol can be optimized further by making use of broadband universal rotations that have been developed in NMR spectroscopy.
Composite pulses or numerically optimized pulses, especially those derived with optimal control algorithms, have been used in NMR (24) and, more recently in electron paramagnetic resonance (EPR) (31), can be used to engineer the pulses for a required amount of robustness to detuning and pulse strength. Symmetric phase pulses (see the Supplementary Materials) (23) or the BURBOP (broadband universal rotations by optimal control) pulses defined in (24) can extend the spectral range robustness of PulsePol by a factor of 2 to around (2π)120 MHz for the same pulse duration as those used in Fig. 2. Experimental effects, such as the resonator bandwidth, can be included in the numerical optimization and accommodated for in the engineered pulses (31).
It is worth noting that, unlike traditional methods in EPR/NMR using short pulses for transferring polarization between spins [for example, pulsed ENDOR (electron nuclear double resonance) (33) and INEPT (insensitive nuclei enhanced by polarization transfer) (34)], no radio frequency pulse or nuclear spin manipulation is required in PulsePol, making it much easier to implement experimentally and mitigate issues due to nuclear rotations taking three orders of magnitude longer time.
RESULTS
The NV center with its electron spin has recently gained considerable interest as a resource spin for optical DNP for polarizing nuclear spins inside the diamond (1–5, 10) and in external molecules (6, 35–37). The NV center realizes in its ground state an electronic spin triplet (S = 1), which exhibits a zero-field splitting of D = (2π)2.87 GHz that separates the ms = 0 state energetically from the ms = ±1 states, which are split by an applied magnetic field. One of them forms with ms = 0 the desired effective two-level system shown in Fig. 3A. The NV spin can be optically polarized within 200 ns by a laser pulse that induces spin-selective relaxation into the ms = 0 sublevel of the ground state, resulting in spin polarization exceeding 92% (38).
Fig. 3. Experimental implementation via optically polarized NV centers in diamond.
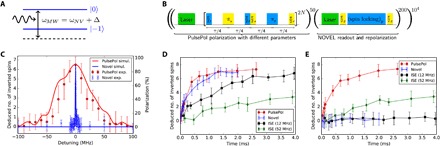
(A) Probing robustness to detuning on a single NV by detuning the MW frequency from the NV |ms = 0〉 ↔ |ms = − 1〉 transition. (B) PROPI sequence used for detecting polarization efficiency of PulsePol. (C) PROPI readout for different detunings Δ for PulsePol (red) and NOVEL (blue). The lines are the smoothed simulation result of a comparable nuclear spin bath with no free parameters. For more details, see text. Polarization buildup by consecutive polarization transfers for NOVEL, PulsePol, and ISE (D) for Δ = 0 MHz, where all sequences, (E) Δ = (2π)20 MHz, where only PulsePol and very slow ISE are able to transfer polarization.
Using this setup, the NV center can be used to polarize the surrounding nuclear spins and to use the same center to read out the polarization using polarization readout by polarization inversion (PROPI) (20) with the sequence in Fig. 3B. This allows to probe the effectiveness of polarization schemes on the level of a single NV center, providing an experimental testbed for polarization schemes. Figure 3C demonstrates the polarization efficiency of PulsePol and its robustness to detuning compared to NOVEL. The resonance shift in the free evolution time due to hardware phase errors was checked and found to be near the optimal value (see the Supplemental Materials). The robust polarization of PulsePol for about (2π)60-MHz spectral width can be seen and is in very good agreement with the theoretical simulations with five nuclear spins. One should mention that this spectral width is only limited by the power of the MW (the larger the power, the larger the width).
Previously developed methods to compensate for the lack of robustness of NOVEL and the solid effect were based on sweep-based schemes, such as ISE. However, performing this sweep induces a trade-off between its robustness to detuning and the polarization efficiency (the polarization efficiency is inversely proportional to the sweep speed; thus, larger sweep range either decreases efficiency or increases sequence time), which is not present in PulsePol. Figure 3D shows the comparison for the polarization buildup rate of PulsePol, NOVEL, and ISE [experimental parameters: Rabi frequency Ω = (2π)1.79 MHz and inverse sweep rate of 3 μs/(2π)MHz, near optimal for this NV’s coupling to the nuclear spin bath], under exact resonance conditions. For ISE, across a (2π)12-MHz bandwidth (the minimal range that is required to achieve polarization transfer for typical parameters) (20) and across a (2π)52-MHz bandwidth [which achieves polarization transfer across a (2π)40-MHz spectral range] were chosen. For the case of perfect resonance, the buildup rates for PulsePol and NOVEL are comparable because PulsePol has a factor α in its effective Hamiltonian Eq. 2 and encompasses the three resonances because of the hyperfine interaction with the nitrogen nuclear spin. However, for ISE[(2π)12 MHz] and ISE[(2π)52 MHz], the rate is significantly slower because of the quadratic dependence on the interaction with the nuclear spins in the efficiency of the MW sweep. Figure 3E shows a similar comparison with Δ = (2π)20-MHz detuning. While the performance of PulsePol and ISE[(2π)52 MHz] remains essentially unchanged, for both NOVEL and ISE[(2π)12 MHz], the NV spin no longer transfers polarization to the surrounding nuclear spin bath.
DISCUSSION
Important hyperpolarization applications with NV centers in diamonds where PulsePol is directly applicable are as follows: (i) the polarization of nuclear spins in molecules external to the diamond with an ensemble of shallow NV centers (see Fig. 4A) (6, 35, 36), where polarization transfer efficiency is the bottleneck of the achieved polarization, and (ii) the hyperpolarization of nanodiamonds as MRI biomarkers. Nanodiamonds have been previously polarized at cryogenic temperatures (1 to 3 K) and high magnetic fields (9, 11, 12). Optical polarization via NV centers at room temperature offers several key advantages such as less complex and costly experimental setups and faster polarization buildup time (10). For NV center nanodiamond polarization, an outstanding challenge is that the NV resonance can be significantly shifted because of the random lattice orientation of the nanodiamond relatively to magnetic field orientation (Fig. 4B) and (iii) the initialization of quantum simulators based on two- and three-dimensional arrays of nuclear spins (7, 8).
Fig. 4. Hyperpolarization applications with NV centers in diamonds.
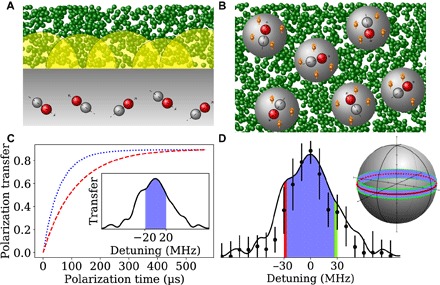
(A) Illustration of shallow NV polarization setup, where NV centers implanted ~3-nm-deep polarize-diffusing molecules in a solution outside of the diamond and (B) nanodiamonds with randomly oriented NV centers, where the polarization of 13C spins (orange) allows for the usage as MRI contrast agents. (C) Polarization transfer from near-surface NV centers to a bath of 1200 nuclear spin bath, using the same Rabi and Larmor frequency and sequence as in Fig. 2C averaged over NV centers with transition frequencies drawn from a Gaussian distribution with a (2π)20-MHz width. The simulation uses matrix product states (39) and a diffusion coefficient of D = 1.4/2.8 × 10−12m2/s (blue dotted/red dashed curve), resulting in different correlation times and efficiencies. The inset shows that the NV linewidth is well within the working range of the PulsePol protocol [Ω = (2π)50MHz]. More details of the simulation are included in the Supplementary Materials. (D) On the basis of the resilience to detunings |Δ| < (2π)30MHz for a Rabi frequency of Ω = (2π)50MHz, more than 11% of the NV orientations in nanodiamonds (azimuthal angle between 90° ± 6.5°, as shown on the sphere) contribute to polarization transfer.
Common to all these applications are the requirement of near shallow NV centers, which can exhibit between (2π)2- and (2π)20-MHz variance in the NV resonance due to surface electric charges as well as interactions with P1 centers and other surface defects (potentially more in dense ensembles). As the coupling between the shallow NV spin and nuclear spin bath is very weak, even coherent polarization transfer approaches the NV relaxation time, and ISE will not markedly increase the polarization efficiency compared with NOVEL. It is important to note that, while the speed of polarization transfer of current DNP schemes such as NOVEL in a bulk diamond is not a limiting bottleneck, the speed of polarization buildup is vital in applications such as polarization of molecules in liquid, where building up a high polarization during the relaxation time of the nuclear spins in the molecules is expected to be the main challenge. We simulate the polarization transfer from the NV center to diffusing molecules via tensor-network methods for two diffusion coefficients in Fig. 4C, exploiting the time-evolving block decimation (TEBD). Critically, PulsePol shows more than 80% polarization efficiency, whereas NOVEL (as well as ISE) is expected to perform an order of magnitude worse for this parameter regime. The inset elucidates the reason for this high efficiency, as the frequency detunings of most shallow NV centers are corrected by PulsePol.
For being applicable to nanodiamond polarization, we have strong magnetic field along the z direction of laboratory frame of reference (γeB ≫ D with γe the gyromagnetic ratio) and uniformed laboratory frame of reference for different orientations of the NV center. However, due to the large zero-field splitting, the NV detuning can reach the order of a gigahertz. In addition, the optical polarization and readout of the NV decrease for high misalignment between the magnetic field and NV axis (θ > 20°), although when approaching θ = 90°, the NV center optical polarization and readout again become efficient (10). By choosing the MW frequency near the NV resonance corresponding to 90° angle between the diamond axis and external field (Fig. 4D), more than 11% of the NV orientations can be addressed within just (2π)60 MHz (10), which, in turn, can be achieved by applying PulsePol with (2π)49-MHz Rabi frequency (for comparison, note that even in bulk diamond, only 25% NV centers participate in polarization dynamics due to four possible orientations in the lattice). The involved orientations are shown in Fig. 4D. Because of the Brownian rotation of the nanodiamonds in a solution, this wide NV addressability leads to all nanodiamonds becoming hyperpolarized under reasonable conditions (10).
CONCLUSION
In conclusion, we have introduced a framework that allows for the design of highly robust and efficient pulsed DNP schemes that transfer polarization from electron to nuclear spins via pulse sequences that create an effective evolution that is described by a flip-flop Hamiltonian. We have presented a specific example sequence, PulsePol, which not only achieves polarization transfer rates that are similar to those for existing schemes when operated under ideal conditions but also affords a flexibility that confers a remarkable robustness to detuning, spectral width, and pulse errors because PulsePol does not need to satisfy an H-H resonance for the MW amplitude. We underline the practical potential of PulsePol, with the experimental demonstration of its efficiency in transferring polarization from an optically polarized NV center in diamond to the surrounding 13C nuclear spin bath over a range of more than (2π)60-MHz detuning, where this range is only limited by the MW drive. This efficient and robust polarization transfer by PulsePol significantly enhances the potential of hyperpolarization of external molecules for nanoscale NMR or quantum simulators using near-surface NV centers and of nanodiamonds as hyperpolarized MRI markers and polarization agents.
The PulsePol sequence and the framework within which it was derived have considerable flexibility, which allows it to be used in a wide variety of DNP experiments, potentially including those conducted via radicals or at low temperatures. In addition to the advantages of robustness to MW and magnetic field inhomogeneity, the reduced sensitivity to the electron resonance frequency will enhance polarization transfer in cases where hyperfine splitting or the anisotropic g-tensor broadens the electron spectral width.
MATERIALS AND METHODS
Experiments in diamond: The PROPI sequence used consisted of two parts (Fig. 3B), which were repeated 5 × 104 times. The experiment started with a thermally polarized nuclear spin bath. With the first part of PROPI, nuclear spins were polarized into the |↑ 〉 state with 50 sequence cycles of PulsePol. Then, with the second part of PROPI, the nuclear spin bath was fully polarized into the |↓↓↓… 〉 direction with 200 cycles of a well-known polarization sequence; in this case, NOVEL with a 10-μs spin-locking time and matched H-H conditions was used. With this number of cycles, a saturation of the polarization transfer could be achieved. Hence, from the second repetition on, the first part of PROPI started with a fully polarized nuclear spin bath (|↓↓↓… 〉), which reversed nuclear spins into the opposite (|↑〉) direction up to a certain degree. The amount of transferred polarization depends on the efficiency of the sequence to be tested, for example, PulsePol, ISE, or NOVEL. The nuclear polarization signal was observed during the repolarization part (second part) by monitoring the NV’s fluorescence. A bright signal, originated from flip-flop processes between the NV electron spin and nuclear spins, saturated to a darker signal when the spin bath reached a completely polarized state. Hereby, the area below the saturation curve (first 100 of 200 cycles) gave a measure of nuclear polarization.
To determine the behavior of PulsePol with regard to detuning errors, we added a detuning Δ = ω1 ↔ 0 − ωMW between the |ms = 0〉 ↔ |ms = −1〉 transition frequency and the external MW field frequency (Fig. 3A), using a drive of approximately Ω0 = (2π)49-MHz Rabi frequency for the pulses. Additional experimental parameters: Rabi frequency Ω = (2π)1.86 MHz matching H-H conditions and a spin-locking pulse of 10 μs.
All experiments were performed at a magnetic field of approximately 1740 G and an alignment of the external field in respect to the NV axis to better than 1°. The MW was applied to the NV through an electroplated stripline on diamond. The attenuation of this stripline was frequency-dependent, such that the amplitude of the MW during PulsePol was corrected up to a deviation of the Rabi frequency of < ±5%. Potential depolarization effects in the case of severely detuned pulses were checked and found to be negligible (see the Supplementary Materials).
Polarization of molecules via shallow NV centers: To simulate the surface NV ensemble interacting with diffusing spins, we performed the following. The quantum system to be simulated comprised the electron spin of an NV center and multiple nuclear spins. The parallel and perpendicular coupling constants describing the NV spin interaction were obtained numerically by simulating the diffusion of the 1200 most strongly coupled spins, which accounts for two-thirds of the total coupling strength. We simulated 100 different NV spin systems with (2π)20-MHz SD in the NV resonance frequency to account for the broad linewidth. During evolution under PulsePol, the entanglement between the different constituents was limited and can therefore be simulated efficiently via tensor-network methods. We exploited the TEBD algorithm (39) to perform the final simulations (Fig. 4C).
Supplementary Material
Acknowledgments
We thank J. F. Haase for useful discussions. Funding: This work was supported by the European Union project, the European Research Council Synergy grant, a PhD fellowship of the Integrated Center for Quantum Science and Technology, the Humboldt Research Fellowship for Postdoctoral Researchers, the Deutsche Forschungsgemeinschaft Collaborative Research Centre 1279, and the state of Baden-Württemberg through bwHPC (Baden-Wuerttemberg high performance computing). Author contributions: I.S., M.B.P., Q.C., and Z.-Y.W. conceived the idea. B.T. carried out the simulations and analytical work with input from I.S., Q.C., and M.B.P. J.S. and S.M. performed the experiments. I.D. performed the theoretical simulations for shallow NV centers. B.N., C.M., and F.J. advised on the several aspects of experiments. I.S., B.T., and Q.C. wrote the manuscript with input from all authors. All authors reviewed the manuscript and suggested improvements. M.B.P. supervised the overall research effort. Competing interests: I.S., M.B.P., Q.C., and Z.-Y.W. are inventors on patent application PCT/EP2017/084229 related to this work. M.B.P. holds a contractual agreement as adviser to NVision Imaging Technologies and holds an observer seat on the board of NVision Imaging Technologies. F.J. holds a contractual agreement as adviser to NVision Imaging Technologies. The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/8/eaat8978/DC1
Supplementary Text
Section S1. Hamiltonian of the system
Section S2. Effective Hamiltonian of PulsePol
Section S3. Error robustness
Section S4. Finite pulses
Section S5. Composite pulses
Section S6. Effect of phase errors
Section S7. Hamiltonian with NV centers in nanodiamonds
Section S8. Depolarization behavior
Section S9. Simulation parameters for shallow NV centers
Fig. S1. MW pulse sequence for pulsed polarization transfer.
Fig. S2. Error resistance of PulsePol by using composite pulses.
Fig. S3. Effect of phase errors.
Fig. S4. Error resistance of PulsePol versus the resonance shift.
Fig. S5. Comparison between simulation results and experimental data of polarization buildup and depolarization.
REFERENCES AND NOTES
- 1.London P., Scheuer J., Cai J.-M., Schwarz I., Retzker A., Plenio M. B., Katagiri M., Teraji T., Koizumi S., Isoya J., Fischer R., McGuinness L. P., Naydenov B., Jelezko F., Detecting and polarizing nuclear spins with nuclear double resonance on a single electron spin. Phys. Rev. Lett. 111, 067601 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Scheuer J., Schwartz I., Chen Q., Schulze-Sünninghausen D., Carl P., Höfer P., Retzker A., Sumiya H., Isoya J., Luy B., Plenio M. B., Naydenov B., Jelezko F., Optically induced dynamic nuclear spin polarisation in diamond. New J. Phys. 18, 013040 (2016). [Google Scholar]
- 3.Álvarez G. A., Bretschneider C. O., Fischer R., London P., Kanda H., Onoda S., Isoya J., Gershoni D., Frydman L., Local and bulk 13C hyperpolarization in nitrogen-vacancy-centred diamonds at variable fields and orientations. Nat. Commun. 6, 8456 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.King J. P., Jeong K., Vassiliou C. C., Shin C. S., Page R. H., Avalos C. E., Wang H.-J., Pines A., Room-temperature in situ nuclear spin hyperpolarization from optically pumped nitrogen vacancy centres in diamond. Nat. Commun. 6, 8965 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drake M., Scott E., Reimer J. A., Influence of magnetic field alignment and defect concentration on nitrogen-vacancy polarization in diamond. New J. Phys. 18, 013011 (2015). [Google Scholar]
- 6.Fernández-Acebal P., Rosolio O., Scheuer J., Müller C., Müller S., Schmitt S., McGuinness L. P., Schwartz I., Chen Q., Retzker A., Naydenov B., Jelezko F., Plenio M. B., Toward hyperpolarization of oil molecules via single nitrogen vacancy centers in diamond. Nano Lett. 18, 1882–1887 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Cai J., Retzker A., Jelezko F., Plenio M. B., A large-scale quantum simulator on a diamond surface at room temperature. Nat. Phys. 9, 168–173 (2013). [Google Scholar]
- 8.T. Unden, N. Tomek, T. Weggler, F. Frank, P. London, J. Zopes, C. Degen, N. Raatz, J. Meijer, H. Watanabe, K. M. Itoh, M. B. Plenio, B. Naydenov, F. Jelezko, Coherent control of solid state nuclear spin nano-ensembles; arXiv:1802.02921 (2018).
- 9.Rej E., Gaebel T., Boele T., Waddington D. E. J., Reilly D. J., Hyperpolarized nanodiamond with long spin-relaxation times. Nat. Commun. 6, 8459 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Q., Schwarz I., Jelezko F., Retzker A., Plenio M. B., Optical hyperpolarization of 13C nuclear spins in nanodiamond ensembles. Phys. Rev. B 92, 184420 (2015). [Google Scholar]
- 11.Kwiatkowski G., Jähnig F., Steinhauser J., Wespi P., Ernst M., Kozerke S., Direct hyperpolarization of micro- and nanodiamonds for bioimaging applications—Considerations on particle size, functionalization and polarization loss. J. Magn. Reson. 286, 42–51 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Bretschneider C. O., Akbey Ü., Aussenac F., Olsen G. L., Feintuch A., Oschkinat H., Frydman L., On the potential of dynamic nuclear polarization enhanced diamonds in solid-state and dissolution 13C NMR spectroscopy. ChemPhysChem 17, 2691–2701 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Ajoy A., Liu K., Nazaryan R., Lv X., Zangara P. R., Safvati B., Wang G., Arnold D., Li G., Lin A., Raghavan P., Druga E., Dhomkar S., Pagliero D., Reimer J. A., Suter D., Meriles C. A., Pines A., Orientation-independent room temperature optical 13C hyperpolarization in powdered diamond. Sci. Adv. 4, eaar5492 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.I. Schwartz, J. Rosskopf, S. Schmitt, B. Tratzmiller, Q. Chen, L. P. McGuinness, F. Jelezko, M. B. Plenio, Blueprint for nanoscale NMR; arXiv:1706.07134 (2017). [DOI] [PMC free article] [PubMed]
- 15.Tateishi K., Negoro M., Nishida S., Kagawa A., Morita Y., Kitagawa M., Room temperature hyperpolarization of nuclear spins in bulk. Proc. Natl. Acad. Sci. U.S.A. 111, 7527–7530 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abragam A., Proctor W., A new method of dynamic polarization of atomic nuclei in solids. Compt. Rend. 246, 2253 (1958). [Google Scholar]
- 17.Henstra A., Dirksen P., Schmidt J., Wenckebach W. T., Nuclear spin orientation via electron spin locking (NOVEL). J. Magn. Reson. 77, 389–393 (1988). [Google Scholar]
- 18.Henstra A., Dirksen P., Wenckebach W. T., Enhanced dynamic nuclear polarization by the integrated solid effect. Phys. Lett. A 134, 134–136 (1988). [Google Scholar]
- 19.Hartmann S. R., Hahn E. L., Nuclear double resonance in the rotating frame. Phys. Rev. 128, 2042 (1962). [Google Scholar]
- 20.Scheuer J., Schwartz I., Müller S., Chen Q., Dhand I., Plenio M. B., Naydenov B., Jelezko F., Robust techniques for polarization and detection of nuclear spin ensembles. Phys. Rev. B 96, 174436 (2017). [Google Scholar]
- 21.Machnes S., Plenio M. B., Reznik B., Steane A. M., Retzker A., Superfast laser cooling. Phys. Rev. Lett. 104, 183001 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Machnes S., Cerrillo J., Aspelmeyer M., Wieczorek W., Plenio M. B., Retzker A., Pulsed laser cooling for cavity optomechanical resonators. Phys. Rev. Lett. 108, 153601 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Shaka A. J., Pines A., Symmetric phase-alternating composite pulses. J. Magn. Reson. 71, 495–503 (1987). [Google Scholar]
- 24.Kobzar K., Ehni S., Skinner T. E., Glaser S. J., Luy B., Exploring the limits of broadband 90° and 180° universal rotation pulses. J. Magn. Reson. 225, 142–160 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Yang W., Wang Z.-Y., Liu R.-B., Preserving qubit coherence by dynamical decoupling. Front. Phys. 6, 2–14 (2011). [Google Scholar]
- 26.Souza A. M., Álvarez G. A., Suter D., Robust dynamical decoupling. Philos. Trans. Ser. A Math. Phys. Eng. Sci. 370, 4748–4769 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Carr H. Y., Purcell E. M., Effects of diffusion on free precession in nuclear magnetic resonance experiments. Phys. Rev. 94, 630 (1954). [Google Scholar]
- 28.Maudsley A. A., Modified Carr-Purcell-Meiboom-Gill sequence for NMR fourier imaging applications. J. Magn. Reson. 69, 488–491 (1986). [Google Scholar]
- 29.Taminiau T. H., Cramer J., van der Sar T., Dobrovitski V. V., Hanson R., Universal control and error correction in multi-qubit spin registers in diamond. Nat. Nanotechnol. 9, 171–176 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Wang Z.-Y., Casanova J., Plenio M. B., Delayed entanglement echo for individual control of a large number of nuclear spins. Nat. Commun. 8, 14660 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spindler P. E., Zhang Y., Endeward B., Gershernzon N., Skinner T. E., Glaser S. J., Prisner T. F., Shaped optimal control pulses for increased excitation bandwidth in EPR. J. Magn. Reson. 218, 49–58 (2012). [DOI] [PubMed] [Google Scholar]
- 32.J. Casanova, Z.-Y. Wang, I. Schwartz, M. B. Plenio, Shaped pulses for energy efficient high-field NMR at the nanoscale; arXiv:1805.01741 (2018).
- 33.Davies E. R., A new pulse endor technique. Phys. Lett. A 47, 1–2 (1974). [Google Scholar]
- 34.Morris G. A., Freeman R., Enhancement of nuclear magnetic resonance signals by polarization transfer. J. Am. Chem. Soc. 101, 760–762 (1979). [Google Scholar]
- 35.Chen Q., Schwarz I., Jelezko F., Retzker A., Plenio M. B., Resonance-inclined optical nuclear spin polarization of liquids in diamond structures. Phys. Rev. B 93, 060408(R) (2016). [Google Scholar]
- 36.Broadway D. A., Tetienne J.-P., Stacey A., Wood J. D. A., Simpson D. A., Hall L. T., Hollenberg L. C. L., Quantum probe hyperpolarisation of molecular nuclear spins. Nat. Commun. 9, 1246 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abrams D., Trusheim M. E., Englund D. R., Shattuck M. D., Meriles C. A., Dynamic nuclear spin polarization of liquids and gases in contact with nanostructured diamond. Nano Lett. 14, 2471–2478 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Waldherr G., Beck J., Steiner M., Neumann P., Gali A., Frauenheim T., Jelezko F., Wrachtrup J., Dark states of single nitrogen-vacancy centers in diamond unraveled by single shot NMR. Phys. Rev. Lett. 106, 157601 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Schollwoeck U., The density-matrix renormalization group in the age of matrix product states. Ann. Phys. Rehabil. Med. 326, 96–192 (2011). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/8/eaat8978/DC1
Supplementary Text
Section S1. Hamiltonian of the system
Section S2. Effective Hamiltonian of PulsePol
Section S3. Error robustness
Section S4. Finite pulses
Section S5. Composite pulses
Section S6. Effect of phase errors
Section S7. Hamiltonian with NV centers in nanodiamonds
Section S8. Depolarization behavior
Section S9. Simulation parameters for shallow NV centers
Fig. S1. MW pulse sequence for pulsed polarization transfer.
Fig. S2. Error resistance of PulsePol by using composite pulses.
Fig. S3. Effect of phase errors.
Fig. S4. Error resistance of PulsePol versus the resonance shift.
Fig. S5. Comparison between simulation results and experimental data of polarization buildup and depolarization.