

Abstract
RBCs are the most abundant circulating cells in humans and typically comprise 35% to 45% of the blood volume (hematocrit). Anemia is associated with an increase in bleeding, and epidemiological studies have shown an association between an elevated hematocrit and thrombosis. RBCs may contribute to hemostasis and thrombosis via mechanisms that include platelet margination leading to an increase in the near-wall platelet concentration, blood viscosity, thrombin generation, and platelet activation. In this issue of the JCI, Klatt et al. report that binding of the Fas ligand FasL on the surface of platelets to its cognate receptor FasR on the surface of RBCs increases thrombin generation in vitro and thrombosis in mouse models. This represents a new mechanism by which RBCs contribute to thrombosis.
RBCs in thrombin generation
The role of RBCs in hemostasis and thrombosis is complex. While RBCs are directly involved in increasing thrombin generation and platelet activation, they are also indirectly involved in affecting platelet margination and altering blood viscosity. Therefore, it is often difficult to determine the relative contributions of these different mechanisms to hemostasis and thrombosis.
RBCs express the Fas ligand FasL, and the death receptor FasR (1). Activation of FasR on RBCs leads to a loss of membrane asymmetry and exposure of negatively charged phospholipids, such as phosphatidylserine (PS), on the cell surface. PS provides an “eat me” signal that directs the removal of aged RBCs from the circulation. Indeed, aged RBCs have higher levels of surface PS compared with young RBCs. Platelets express FasL and induce apoptosis of neural cells and apoptosis in a stroke model (2).
In the current issue of the JCI, Klatt et al. (3) present the interesting observation that platelets induce PS exposure on RBCs via an interaction between FasL on platelets and FasR on RBCs. Although ADP stimulation of platelets has been shown to increase surface expression of FasL (2), Klatt et al. (3) found no effect on RBC PS exposure after inhibition of platelet activation with clopidogrel, an inhibitor of ADP signaling via the ADP receptor P2Y12.
The presence of PS and other negatively charged phospholipids on cell surfaces facilitates the assembly of coagulation factor complexes by binding to the γ-carboxyglutamic acid residue–rich domain present in various coagulation factors (4). Activated platelets expose PS and play a key role in thrombin generation. Several studies have suggested a role for a subpopulation of PS-positive (PS+) RBCs in thrombin generation (5). It is estimated that approximately 0.5% of normal RBCs display PS on their surface. In sickle cell disease, there is an increase in PS+ RBCs that correlates with biomarkers of coagulation activation (6). Addition of RBCs to platelet-rich plasma has been shown to increase thrombin generation (7). Klatt et al. (3) also found that addition of RBCs to platelet-rich plasma increased thrombin generation. Similarly, Walton et al. (8) observed an increase in thrombin generation with a higher hematocrit in combination with a low (200 × 106/ml) but not high (450 × 106/ml) platelet count.
Klatt et al. (3) found that RBCs enhanced deposition of platelet aggregates on a collagen surface under high (1,700 s–1) and low (150 s–1) shear rates. The greatest enhancement was observed under low shear that corresponds to the flow rate in veins. Interestingly, a pan-caspase inhibitor did not reduce the deposition of platelet aggregates or PS exposure on RBCs. This is surprising because caspase 3 has been shown to be required for PS exposure on RBCs after activation of FasR (1).
RBCs and hemostasis
Recent studies have shown that thrombosis can be distinguished from hemostasis. For instance, factor XII contributes to thrombosis but has no role in hemostasis (9). Hemostatic plugs contain a core of densely packed, highly activated platelets covered by a shell of loosely packed, less activated platelets (10). Few RBCs are present in hemostatic plugs, suggesting that they contribute to hemostasis indirectly. This may occur by increasing platelet margination towards the wall of the injured vessel or by biochemical activation of resident platelets (Figure 1A).
Figure 1. Composition of hemostatic plugs and arterial and venous thrombi.
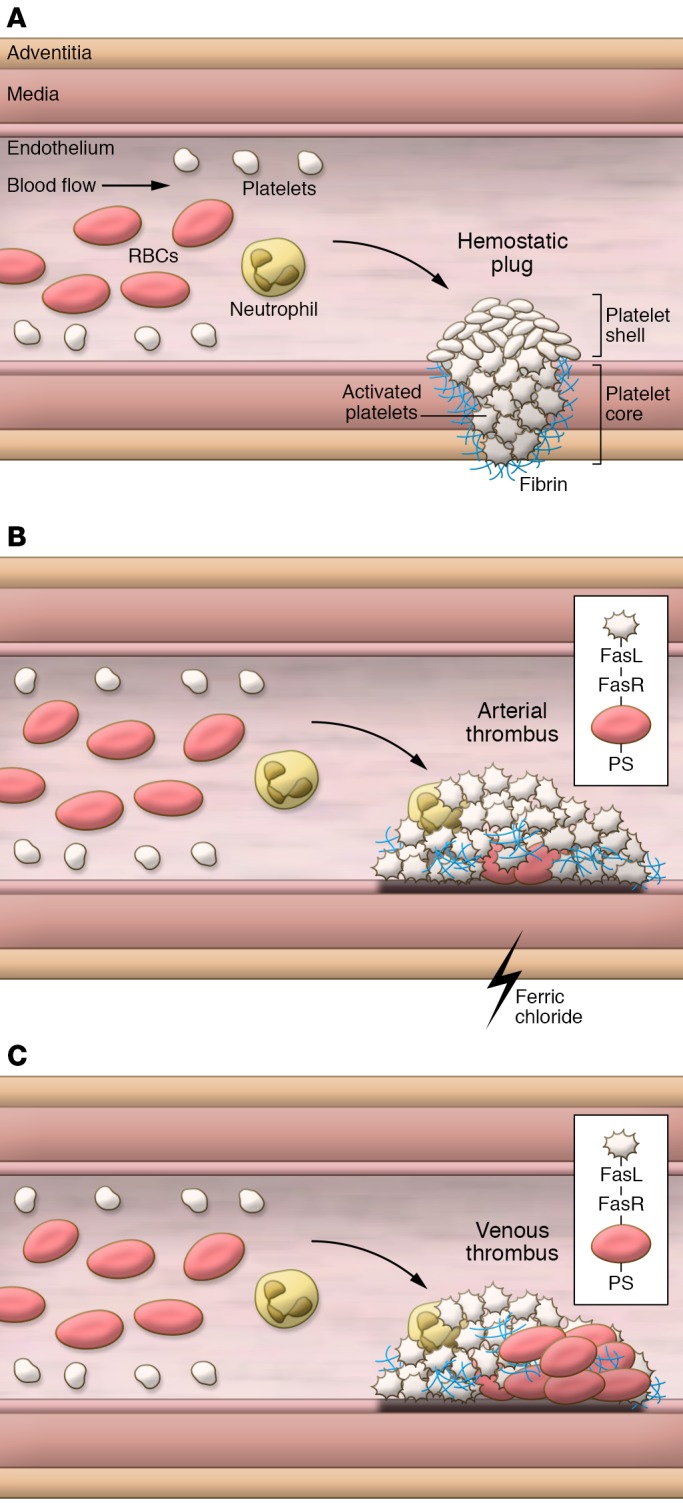
(A) Hemostatic plugs consist of a core of highly activated platelets surrounded by fibrin and a shell of less activated platelets. RBCs force the smaller platelets to the vessel wall where they can monitor vascular integrity. (B) Arterial thrombi are platelet rich with few RBCs. In the ferric chloride model of carotid artery thrombosis the first event is binding of RBCs to the endothelium and subsequent recruitment of platelets followed by formation of large platelet aggregates. Platelet-RBC interactions mediated by FasL-FasR may contribute to thrombosis in the mouse ferric chloride model. (C) Venous thrombi contain red regions composed of RBCs and fibrin and white regions composed of platelets. Platelet-RBC interactions via FasL-FasR may enhance thrombosis in the mouse inferior vena cava model.
Anemia is associated with increased bleeding. An early study found that transfusion of RBCs into patients with severe anemia and normal platelet counts corrects the bleeding time (11). RBC transfusion also shortens the bleeding time in anemic and thrombocytopenic rabbits (12). Klatt et al. (3) found that anemic mice (21%–28% hematocrit) had prolonged bleeding in a tail transection model compared with normal mice (hematocrit 35%–45%), whereas an increase in hematocrit (>50%) did not affect bleeding. Similarly, Walton et al. (8) found that mice with an increased hematocrit (~47%) had a normal tail bleeding time.
RBCs and thrombosis
Arterial thrombi are platelet-rich (so-called “white clots”) and typically form after rupture of atherosclerotic plaques (ref. 13 and Figure 1B). Platelets provide the cellular component to these thrombi that form under high shear. Whelihan and Mann (5) concluded that, due to the low number of RBCs in arterial thrombi, RBCs would not play a direct role in thrombin generation. Indeed, Walton et al. (8) did not detect increased circulation of thrombin-antithrombin complexes in a mouse model of elevated hematocrit and arterial injury. In contrast, venous thrombi are fibrin- and RBC-rich (so-called “red clots”) and form in-valve pockets on an intact endothelium under low shear (ref. 14 and Figure 1C). Human venous thrombi consist of a “red” part enriched in RBCs and fibrin and a “white” part enriched in platelets (14). Since RBCs comprise approximately 50% of the mass of venous thrombi, they may contribute to thrombin generation in this scenario. Interestingly, crosslinking of fibrin is required to retain RBCs in venous clots during platelet-mediated clot retraction (15).
Several epidemiological studies have examined the associations between elevated hematocrit and both arterial and venous thrombosis (16). One study reported that patients with a high hematocrit had a 1.5-fold increased risk of venous thromboembolism compared with patients with a low hematocrit (17). Another study found that perioperative RBC transfusion was associated with venous thromboembolism (18).
The ferric chloride carotid artery thrombosis model is the most commonly used mouse model of thrombosis (19). Importantly, one study showed that in this model, RBCs are the first cells to adhere to the damaged endothelium, recruit platelets, and form nuclei for the formation of large platelet complexes that ultimately occlude blood flow (20). Of note, a platelet-rich thrombus is formed in this model with very few RBCs incorporated as islands (ref. 13 and Figure 1B).
Klatt et al. (3) found that anemic mice exhibited a prolonged occlusion time in the ferric chloride carotid artery thrombosis model (15% ferric chloride for 3 minutes) but the occlusion time was not affected by an increased hematocrit. In contrast, Walton et al. (8) used a similar thrombosis model (10% ferric chloride for 2 minutes) and observed a significant shortening of the occlusion time in mice with a high hematocrit. Use of a less severe model may explain why Walton et al. (8) observed an effect of high hematocrit on thrombosis. The two studies conclude that hematocrit influences platelet adhesion and aggregate formation at the injured vessel (3), and that RBCs promote arterial thrombosis via a platelet-dependent increase in thrombus growth (8). However, it is not clear how the low number of RBCs in the thrombi in this model affects its growth.
Role of FasL and FasR in the formation of thrombi
Lastly, Klatt et al. (3) analyzed the role of FasL and FasR in a ferric chloride mesenteric arteriole model and an inferior vena cava stenosis model. Thrombi formed in the inferior vena cava model have red and white regions similar to human venous thrombi (21). In the mesenteric arteriole model, wild-type mice all occluded within 40 minutes, whereas no occlusion was observed with FasL- or FasR-knockout mice. Interestingly, the phenotype of FasL-knockout mice was more pronounced than FasR-knockout mice. Mice with a platelet-specific deletion of FasL had results similar to FasL-knockout mice in the mesenteric arteriole model, indicating that FasL on platelets was contributing to thrombosis in this model. Since there are few RBCs in the arterial thrombus, it is possible that loss of either FasL or FasR disrupts the early recruitment of platelets to the RBCs bound to the endothelium. In the inferior vena cava model, FasL- and FasR-knockout mice had a significant decrease in incidence, whereas only FasL-knockout mice had a decrease in thrombus weight. These results suggest that FasL may bind to other receptors that affect thrombosis.
In summary, Klatt et al. (3) describe a new interaction between platelets and RBCs that is mediated by FasL and FasR. Although this interaction increases PS exposure on RBCs and increases thrombin generation in vitro, it is unclear if this interaction increases thrombin generation in vivo. It is easier to understand how an interaction between platelets and RBCs can contribute to venous thrombosis because they contain large numbers of RBCs. FasL and FasR were shown to contribute to ferric chloride–induced thrombosis in mesenteric arterioles. However, RBCs appear to play an unusual role in this model by binding to the injured endothelium and providing an initial site for platelet recruitment. It will be important to show that platelet binding to RBCs via FasL and FasR also contributes to other arterial thrombosis models. Klatt et al. (3) conclude that RBCs significantly contribute to thrombus formation and that interfering with the interaction between FasL and FasR may represent a novel antithrombotic strategy. Further studies are needed to better understand the role of RBCs in arterial and venous thrombosis before proposing them as targets for antithrombotic therapy.
Acknowledgments
I would like to thank Alisa Wolberg, Yohei Hisada, and Jens Posma for helpful comments and the John C. Parker Professorship for funding.
Version 1. 08/13/2018
Electronic publication
Version 2. 08/31/2018
Print issue publication
Footnotes
Conflict of interest: The author has declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(9):3747–3749. https://doi.org/10.1172/JCI122881.
See the related article at Platelet-RBC interaction mediated by FasL/FasR induces procoagulant activity important for thrombosis.
References
- 1.Mandal D, Mazumder A, Das P, Kundu M, Basu J. Fas-, caspase 8-, and caspase 3-dependent signaling regulates the activity of the aminophospholipid translocase and phosphatidylserine externalization in human erythrocytes. J Biol Chem. 2005;280(47):39460–39467. doi: 10.1074/jbc.M506928200. [DOI] [PubMed] [Google Scholar]
- 2.Schleicher RI, et al. Platelets induce apoptosis via membrane-bound FasL. Blood. 2015;126(12):1483–1493. doi: 10.1182/blood-2013-12-544445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klatt C, et al. Platelet-RBC interaction mediated by FasL/FasR induces procoagulant activity important for thrombosis. J Clin Invest. 2018;128(9):3906–3925. doi: 10.1172/JCI92077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Owens AP, 3rd, Mackman N. Microparticles in hemostasis and thrombosis. Circ Res. 2011;108(10):1284–1297. doi: 10.1161/CIRCRESAHA.110.233056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whelihan MF, Mann KG. The role of the red cell membrane in thrombin generation. Thromb Res. 2013;131(5):377–382. doi: 10.1016/j.thromres.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 6.Setty BN, Rao AK, Stuart MJ. Thrombophilia in sickle cell disease: the red cell connection. Blood. 2001;98(12):3228–3233. doi: 10.1182/blood.V98.12.3228. [DOI] [PubMed] [Google Scholar]
- 7.Whelihan MF, Zachary V, Orfeo T, Mann KG. Prothrombin activation in blood coagulation: the erythrocyte contribution to thrombin generation. Blood. 2012;120(18):3837–3845. doi: 10.1182/blood-2012-05-427856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walton BL, et al. Elevated hematocrit enhances platelet accumulation following vascular injury. Blood. 2017;129(18):2537–2546. doi: 10.1182/blood-2016-10-746479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Renne T, Gailani D. Role of factor XII in hemostasis and thrombosis: clinical implications. Expert Rev Cardiovasc Ther. 2007;5(4):733–741. doi: 10.1586/14779072.5.4.733. [DOI] [PubMed] [Google Scholar]
- 10.Welsh JD, Poventud-Fuentes I, Sampietro S, Diamond SL, Stalker TJ, Brass LF. Hierarchical organization of the hemostatic response to penetrating injuries in the mouse macrovasculature. J Thromb Haemost. 2017;15(3):526–537. doi: 10.1111/jth.13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hellem AJ, Borchgrevink CF, Ames SB. The role of red cells in haemostasis: the relation between haematocrit, bleeding time and platelet adhesiveness. Br J Haematol. 1961;7:42–50. doi: 10.1111/j.1365-2141.1961.tb00318.x. [DOI] [PubMed] [Google Scholar]
- 12.Blajchman MA, Bordin JO, Bardossy L, Heddle NM. The contribution of the haematocrit to thrombocytopenic bleeding in experimental animals. Br J Haematol. 1994;86(2):347–350. doi: 10.1111/j.1365-2141.1994.tb04737.x. [DOI] [PubMed] [Google Scholar]
- 13.Mackman N. Triggers, targets and treatments for thrombosis. Nature. 2008;451(7181):914–918. doi: 10.1038/nature06797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sevitt S. The structure and growth of valve-pocket thrombi in femoral veins. J Clin Pathol. 1974;27(7):517–528. doi: 10.1136/jcp.27.7.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aleman MM, et al. Factor XIII activity mediates red blood cell retention in venous thrombi. J Clin Invest. 2014;124(8):3590–3600. doi: 10.1172/JCI75386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Byrnes JR, Wolberg AS. Red blood cells in thrombosis. Blood. 2017;130(16):1795–1799. doi: 10.1182/blood-2017-03-745349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Braekkan SK, Mathiesen EB, Njølstad I, Wilsgaard T, Hansen JB. Hematocrit and risk of venous thromboembolism in a general population. The Tromso study. Haematologica. 2010;95(2):270–275. doi: 10.3324/haematol.2009.008417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goel R, et al. Association of perioperative red blood cell transfusions with venous thromboembolism in a North American Registry [published online ahead of print June 13, 2018]. JAMA Surg . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Owens AP, Lu Y, Whinna HC, Gachet C, Fay WP, Mackman N. Towards a standardization of the murine ferric chloride-induced carotid arterial thrombosis model. J Thromb Haemost. 2011;9(9):1862–1863. doi: 10.1111/j.1538-7836.2011.04287.x. [DOI] [PubMed] [Google Scholar]
- 20.Barr JD, Chauhan AK, Schaeffer GV, Hansen JK, Motto DG. Red blood cells mediate the onset of thrombosis in the ferric chloride murine model. Blood. 2013;121(18):3733–3741. doi: 10.1182/blood-2012-11-468983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brill A, et al. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood. 2011;117(4):1400–1407. doi: 10.1182/blood-2010-05-287623. [DOI] [PMC free article] [PubMed] [Google Scholar]